

I. Introduction
The main task of the medicinal chemist is to design molecules that interact specifically with derailed or degenerating processes in a diseased organism, translating the available knowledge of pathobiochemical and physiological data into chemically useful information and structures (1). Current knowledge of the biological and chemical processes underlying diseases is vast and rapidly expanding. In particular the unraveling of the genome in combination with, for instance, the rapid development of structural biology has led to an explosion in available information and identification of new targets for chemotherapy. The task of translating this wealth of data into active and selective new drugs is an enormous, but realistic, challenge. It requires knowledge from many different fields, including molecular biology, chemistry, pharmacology, physiology, and medicine and as such requires a truly interdisciplinary approach.
Ultimately, the goal is to design molecules that satisfy all the requirements for a candidate drug to function therapeutically. Therapeutic activity can then be achieved by an understanding of and control over structure and reactivity of the candidate drug through molecular manipulation.
Metals in Medicine
Our involvement in drug design comes from our interest in the development of metal-based chemotherapeutics. The use of metals in medicine is as old as human civilization and has, in more recent years, led to the establishment of the field of medicinal inorganic chemistry. Initially fuelled by the discovery of the cytotoxic activity of cisplatin, the world’s most widely used chemotherapeutic anticancer drug today, the field has developed into a thriving area of research, now with several other notable successes such as the gadolinium-based MRI agents. We and others have recently discussed the use of metals in medicine in more comprehensive overviews (2-6). The use of metals is attractive as they offer a unique spectrum of reactivity and indeed a structural diversity which is not readily available to the more common organic-based drugs. The wide range of coordination numbers and geometries, accessible redox states, thermodynamic and kinetic characteristics, and the intrinsic properties of both the cationic metal ion and ligand itself offer the medicinal chemist a wide spectrum of reactivities that can be exploited. The (transition) metals provide a rich palette from which to choose from and offer the real possibility of the discovery of truly novel drugs with new mechanisms of action (2).
In our group, a major part of our research is devoted to the design of new anticancer drugs. Our recent efforts towards the discovery of new platinum-, ruthenium- and osmium-based anticancer agents provide the topic for this account and a section is devoted to each metal. We focus on recent results from our lab in the context of other developments and related research in this field (hence our coverage of the field is focused on these areas and is not comprehensive).
Platinum and Ruthenium Anticancer Drugs
In the field of anticancer drug design and in particular with metallochemotherapeutics, DNA has most commonly served as the major target, either by direct interaction with the drug or indirectly by inhibition of DNA synthesis and replication. The anticancer agents thus work by inhibiting cell growth and/or by hastening cell death. The cytotoxic effects of complexes are experienced by the rapidly dividing tumor cells, but this mode of action invariably means that normal cells (especially rapidly dividing healthy cells of bone marrow, gut, and the skin epithelium) are affected as well. This lack of selectivity of cancer drugs is one of the main problems of cancer chemotherapy. Addressing this problem of selectivity, together with that of (multi)drug resistance, is one of the major goals of research in this field. The genomic revolution has identified many new targets, for instance elevated levels of particular enzymes in tumor cells or their involvement in pathways significant for proliferation, that hold some promise for the development of more selective drugs. Other strategies to alleviate systemic toxicity and resistance make use of prodrugs that can either be activated locally by an internal stimulus, e.g. a physiological difference in the environment (pH, salt concentration, redox potential) or an enzyme-catalyzed chemical transformation, or, alternatively, by an external stimulus, such as light. Examples of both approaches will be discussed.
Considerations for Metal-Based Drug Design
Regardless of the target or approach chosen, many considerations for drug design apply to all metal-based therapeutics. As with organic drugs, the ADME properties (Absorption, Distribution, Metabolism, and Excretion) are of prime importance as the pharmacological and pharmacokinetic parameters of the drug must be such for the drug to be able to reach the site of action in a timely manner, in a sufficient concentration, and be effectively cleared from the system (1). Additionally, for a potent and selective drug, the possibility for strong association with the intended target preferably via multiple specific interactions should be incorporated into the design of the drug. Metal-based drugs have additional possibilities for such molecular recognition, as reactive sites might be available that can result in direct coordination to the target. In this respect, stereochemical arguments play an important role as well.
Some aspects of drug design need to be addressed specifically for metal-based therapeutics. The reactivity that offers so many options for new mechanisms of action, also poses some inherent difficulties in delivering the drug to its target. It is therefore important to constrain the reactivity of the complex sufficiently for it to reach its target, without being deactivated by reactions with the large number of biomolecules encountered along the way. If they are designed to bind directly to the target then the compounds should also not be too stable, as too high kinetic inertness in general will render them non-toxic. This is a feature that, however, can be harnessed for the design of activatable prodrugs. The prodrug approaches briefly mentioned above attempt to address these challenges. Other characteristics specific to metal-containing compounds that can have a major influence on their cytotoxic properties include geometrical isomerism and redox properties.
To understand fully processes such as molecular recognition, reactivity and bioactivity, it is therefore imperative to obtain a detailed insight into the thermodynamic and kinetic properties of the metal-based anticancer agent at hand. An important part of our work is concerned with the study of the coordination chemistry of the anticancer drugs under physiological conditions and in the presence of biomolecules that can either be the intended target or a possible influence on deactivation or activation of the drug. Identification of the key processes for activity, such as hydrolysis or photoreduction, and subsequent manipulation by systematic ligand variation proved to be valuable for obtaining potent anticancer agents. Indeed, the following sections will show that is possible to control metal-ion reactivity for drug design.
II. Platinum Anticancer Prodrugs: a Photoactivation Strategy
Introduction to Platinum Anticancer Drugs
Platinum anticancer drugs such as the archetypical cisplatin and second generation drugs carboplatin and oxaliplatin (Figure 1) are widely employed in cancer chemotherapy and are amongst the most effective chemotherapeutic agents in clinical use. Cisplatin is particularly active against testicular cancer and, if tumors are discovered early, an impressive cure rate approaching 100% is achieved. The clinical use of cisplatin against this and other malignancies is, however, limited. The low efficacy stems from a combination of uni- and multi-cellular resistance, poor whole-body or cellular pharmacokinetic profiles, and systemic toxicity (7). Dose-limiting side-effects include neuro-, hepato- and nephrotoxicity. Hence, much effort has been devoted to the development of new platinum drugs and to the elucidation of cellular responses to them to alleviate these limitations (8, 9). An excellent review on the current status of platinum-based cancer chemotherapy is available (10). Of the various strategies that have been developed (11), those using platinum(IV) complexes as prodrugs appear to have particular potential (7). Some general properties of metal-based prodrugs are briefly discussed below.
Figure 1.

Molecular structures of a) some platinum(II)-based anticancer drugs currently used in the clinic; b) satraplatin, a platinum(IV) prodrug currently in clinical trials.
II. A. Prodrug Strategies
Prodrugs are derivatives of drug molecules that undergo an enzymatic or other chemical transformation in vivo to release the active parent drug (12), or indeed to generate an active species which could not be delivered by itself (e.g. because it is too reactive). Prodrug strategies are aimed at improving physicochemical, biopharmaceutical and pharmacokinetic properties of pharmacologically-active agents (12). Major applications include the ability to improve oral bioavailability, for instance by increasing aqueous solubility or lipophilicity, and to achieve site-selective drug delivery. The latter is a central aim in cancer therapy: targeting an inactive prodrug selectively to tumor cells, followed by its release, so avoiding toxicity to normal, healthy tissue (12).
Prodrug strategies are common for metal-based anticancer drugs as well; in fact cisplatin itself can be regarded as a prodrug since hydrolysis of the Pt–Cl bonds activates the drug for DNA binding (Figure 2). Prodrug activation mechanisms of metal-based prodrugs are usually different from organic prodrug activation mechanisms, which typically involve bioconversion by enzymes (e.g. esterases, phophodiesterases, reductive enzymes). Metal-based prodrugs are commonly activated by ligand substitution, a change in oxidation state, a photochemical process, or combinations of these (Figures 2 and 3). Ligand substitution can result in the creation of a (more) reactive site on the metal, as occurs during hydrolysis of cisplatin. Alternatively, substitution can result in the controlled release of a biologically active ligand, a typical example being nitric oxide delivery by photolabile metal nitrosyls (13), or CO delivery by metal carbonyls (14). Drug activation by substitution requires detailed knowledge of exchange kinetics. The ligand exchange rates depend not only on the metal employed and its oxidation state, but also on the other ligands in the complex (often stereospecifically) and can vary over many orders of magnitude. For example, the exchange rates for aqua ligands on metal ions vary over some nineteen orders of magnitude, even though the M–OH2 bonds can have similar thermodynamic strengths (15, 16). Consideration of exchange kinetics is therefore of prime importance in drug design. The timescale of substitution needs to be long enough to allow the drug to reach its target site before it is activated, but activation needs to take place before excretion, leaving a relatively narrow window. Furthermore the ligand exchange rates can depend on pH, the stereochemistry of the complex (cf. hydrolysis rates of cisplatin vs its geometric isomer transplatin), and the other ligands coordinated to the metal, so providing many options to exert control over this process. Such considerations have played a major role in the development of ruthenium- and osmium-arene anticancer complexes in our group, a topic that will be discussed further in sections III and IV.
Figure 2.

Ligand substitution as a prodrug strategy for metallochemotherapeutics; a) general scheme of prodrug activation by ligand substitution; hydrolysis of a metal-halide bond is a typical activation pathway of metal-based anticancer drugs, as exemplified by the activation of cisplatin (b) and a ruthenium-arene complex (c).
Figure 3.

Photoactivation of Pt(IV) complexes as a prodrug strategy for metallochemotherapeutics; a) general scheme of prodrug activation by photoreduction; b) photosubstitution and photoisomerization are competing photoreaction pathways, which can result in different reactive species upon reduction; c) an example of a photoactive platinum(IV) diazido complex developed in our lab.
A redox activation mechanism usually involves a change in oxidation state of the metal (but could also be ligand-centered). Typically, the oxidation state changes from a state in which ligand exchange is very slow, i.e. the complex is kinetically inert, to a state in which ligand exchange can occur. Again, reduction can either result in a reactive metal complex, such as reduction of platinum(IV) complexes to yield reactive platinum(II) species (7), or in controlled release of bio-active ligands. An example of the latter is Co(III) acting as a chaperone for an analogue of marimastat, a cytotoxic inhibitor of matrix metalloproteinases, which is then released upon reduction of the metal to Co(II) (17). The redox change can be triggered by the properties of the local environment. The cobalt chaperone complexes, for instance, exploit high level hypoxia, a property both common in and unique to solid tumors, for activation, as low levels of oxygen prevent reoxidation to the inert Co(III) complex. Other physiological characteristics that can be used to advantage are the reducing environment in the cell, tissue permeability, and pH (18). These properties differ between healthy and tumor cells, which provides a strategy for tumor-selective activation of the prodrug. Cancer cell selectivity can furthermore be achieved by targeting of biomarkers and receptors that are specifically expressed on tumor cell surfaces. Additionally, levels of certain enzymes are often elevated in these cells and this can also be exploited in targeted prodrug-tumor delivery. Conjugation of a bioactive tumor-targeting group to the metal-based prodrug thus provides another interesting strategy for tumor cell-selective activation to combat systemic toxicity.
Photoactivation as a Prodrug Strategy
Alternatively, prodrug activation by ligand substitution or redox change can also be brought about by photochemical means (19). In general, the combination of light and metal complexes has been widely employed in medicine (20). Phototherapy of cancer, i.e. the treatment of a tumor with a drug and light, is particularly attractive as it allows for local treatment of the tumor (i.e. site-specific activation of the photodrug), thereby minimizing the side-effects of chemotherapy. Recent advances in laser and fiber optic technologies have made it possible now to reach almost any tissue in the body with light of highly defined intensity and wavelength, expanding the applicability of this approach beyond only those cancers that are easily accessible. A key parameter in these studies is the wavelength of the light. In general, for efficient phototherapy, light with wavelengths between 300 and 900 nm should be employed, as shorter wavelengths are typically damaging to tissue, DNA and proteins, and longer wavelengths are of insufficient energy for chemical transformation (19). Within this window, longer wavelengths are preferred if deep penetration into tissue is required. Photodynamic therapy (PDT) is a common technique used in the clinic that selectively damages tissue of easily accessible cancers (e.g. skin, neck and head, mouth, oesophagus, and bladder) by using a photosensitizing drug and light. In PDT, a non-toxic photosensitizer catalytically generates cytotoxic singlet oxygen after electronic excitation by light. This oxygen-dependence for cytotoxicity is a major drawback of PDT as many malignant and most aggressive cancer cells are hypoxic (21). There is therefore an incentive to develop photochemotherapy that is not dependent on oxygen. We have recently reviewed the different approaches to photochemotherapy with metal-based anticancer complexes (19).
Pt(IV) Prodrugs
Platinum(IV) complexes have been widely studied as potential prodrugs that avoid the limitations of the cisplatin class of anticancer drugs. Indeed, the Pt(IV) compound satraplatin [Pt(cha)Cl2(OAc)2(NH3)] (cha, cyclohexylamine) is currently in clinical trials for treatment of hormone-refractory prostate cancer (Figure 1) (22). Satraplatin is the first orally-bioavailable platinum derivative under active clinical investigation and is particularly attractive because of the convenience of administration, milder toxicity profile, and lack of cross-resistance with cisplatin. These results are promising and support the idea that platinum(IV) complexes offer the opportunity to overcome some of the problems associated with cisplatin and its analogues.
In general, the high kinetic inertness of Pt(IV) complexes lowers reactivity and the prospect of side reactions (7). In fact, substitution reactions of the ligands take place very slowly or not at all under physiological conditions. Hence, intracellular reduction to Pt(II), for instance by cellular reducing agents, is thought to be essential for cytotoxic activity. In addition, the octahedral geometry of Pt(IV) introduces two extra ligand sites, which offer many possibilities for drug design. They can be used to modify the pharmacokinetic parameters of the prodrugs, including the rate of reduction and lipophilicity, and to allow inclusion of biologically-active ligands that either target the complexes to tumor cells or, alternatively, are themselves cytotoxic upon release (7). Recent advances in the design and development of Pt(IV) anticancer complexes have been reviewed (7).
An interesting alternative mechanism of activation is the photochemical reduction of Pt(IV) to Pt(II) (Figure 3). In addition to photoreduction, photosubstitution and photoisomerization can also occur, making the photochemistry of Pt complexes difficult to predict and a careful analysis of the photoproducts imperative (21). We have been involved particularly in the development of photochemotherapeutic agents based on Pt(IV) and the study of their photodecomposition and (subsequent) interactions with biomolecules (21). Our aim is the development of Pt(IV) prodrugs which are stable towards reduction, non-toxic in the dark and activated only by light and not by cellular processes or responses to other stimuli, such as pH or biological reducing agents. In the following sections a detailed overview is given on how rational ligand variation led to control over metal ion reactivity and to the development of highly cytotoxic, selectively activatable platinum anticancer drugs.
Ultimately, for Pt(IV) anticancer drugs, a combination of incorporation of bioactive ligands that specifically target cancer cells, control over ligand exchange kinetics, and selective activation by light would allow for temporal and spatial control of drug delivery and activation.
II. B. Photoactivated Platinum Anticancer Drugs
Pt(IV)-Iodido Complexes
The first photoactive Pt(IV) prodrugs were reported by Bednarski et al. in the 1990’s (23). The complex trans,cis-[PtCl2(en)I2] (1; en = ethylenediamine) (Figure 4) can be photolyzed with visible light to give species that bind to DNA and inhibit the growth of human cancer cells in vitro. The chelating ligand en was chosen as a non-labile ligand (as opposed to NH3) to avoid photoisomerization to the thermodynamically more stable but originally considered inactive trans-isomer (vide infra). This compound, however, suffered from a very limited stability in serum even in the absence of light. Consequently, no difference in dark and light toxicity was seen towards human cancer cells. This was most likely due to the relatively high reduction potential of this complex, which leads to facile intracellular reduction (21). Electron transfer to the metal can readily occur through donation to the coordinated iodido ligands (24). The nature of the axial ligand exerts a major influence on the reduction potential and indeed, altering these ligands led to complexes with better dark stability (23). In the dark, the complex cis,trans-[Pt(en)I2(OAc)2] (2) (Figure 4) showed no binding of Pt to CT DNA after 6 h, but, on irradiation (λirr > 375 nm) resulted in 63% of the platinum being bound to CT DNA after 6 h, suggesting photoreduction to Pt(II) and loss of the iodido ligands. The electronic spectra of diiodido-Pt(IV) complexes are dominated by relatively low-energy LMCT transitions, which allow irradiation of these complexes with visible light. Cancer cell growth inhibition studies showed a small, but significant difference in light and dark cytotoxicity, with a 35% greater growth inhibition activity with irradiation. The complex cis,trans-[Pt(en)I2(OH)2] (3) (Figure 4), on the other hand, gave rise to very limited DNA platination on irradiation and a smaller enhancement of antiproliferative activity (22%). The non-photolyzed complex could, however, be chemically reduced quite readily by addition of the major cellular reducing agent glutathione (γ-l-Glu-l-Cys-Gly; GSH). This led to the conclusion that photolysis resulted predominately in photosubstitution rather than photoreduction in this case, illustrating the influence of the axial ligands on photodecomposition pathways (23). As intracellular reduction mechanisms are important for Pt(II) and Pt(IV) drugs alike, the chemical reduction of cis,trans-[Pt(en)I2(OH)2] (3) by GSH was further explored under biologically relevant conditions (24), which led to the unexpected detection of a long-lived chelate-ring-opened Pt(II) complex capable of forming DNA-Pt adducts. These initial results showed for the first time that it is feasible to design photoactivated platinum anticancer drugs by proper choice of the coordinating ligands (23). Cisplatin exerts its cytotoxic effect through binding to DNA, which is thought to be a major target of the Pt(IV) cisplatin-type prodrugs after activation. It is therefore important to gain insight into the reactivity of photoactive prodrugs towards DNA and model compounds, such as nucleobases and nucleotides. In further collaboration with the Bednarski group, we therefore studied the interaction of the photoactive platinum complexes 2 and 3 with nucleotides. Stereospecific reactions between guanosine 5′-monophosphate (5′-GMP) and cis,trans-[Pt(en)I2(OH)2] (3) or cis,trans-[Pt(en)I2(OAc)2] (2) could be induced by visible light (λirr 457.9 nm) and photoactivation could be controlled by the axial ligands, allowing fine-tuning of the photoreactivity of this class of complexes (25). NMR studies on nucleotide binding of 15N-labelled complexes confirmed the previously-observed differences in DNA binding properties of the two complexes and the suggested differences in the photolysis mechanism. 1D 1H, 2D [1H, 15N] HSQC and 2D [1H, 15N] HSQC-TOCSY NMR studies of photoreactions of cis,trans-[Pt(en)I2(OH)2] (3) showed that neither the dihydroxido complex nor its photosubstitution product mer-[Pt(en)I(OH)3] react with 5′-GMP. On the contrary, similar experiments on cis,trans-[Pt(en)I2(OAc)2] (2) revealed two steps in the photoactivation. First, photosubstitution of the labile iodido ligands resulted in formation of two complexes, trans-[Pt(en)I(OAc)2(OH)] (2s1) and cis,trans-[Pt(en)(OH)2Y2] (2s2) (Y might be I− or OH−). The latter complex does not react any further, but kinetic analysis revealed that in a second photoactivation step trans-[Pt(en)I(OAc)2(OH)] (2s1) is photoreduced and the bis-nucleotide adduct [Pt(en)(5-GMP-N7)2] (2rG) is observed.
Figure 4.

Photoactive Pt(IV)-iodido complexes. a) molecular structures of complexes 1-3; b) influence of visible light on CT DNA binding of 2 and cytotoxicity of 2 against a TCCSUP human bladder cell line (data from ref. 23); c) NMR studies showed that photosubstitution precedes photoreduction in the reaction of 2 with 5′-GMP upon irradiation.
The iodido-Pt(IV) complexes thus provided a proof-of-principle being photoactive, but the complexes still suffered from slow photoreactions and, importantly, limited stability in the dark especially against biological reducing agents such as glutathione, which results in undesired toxicity of the anticancer agents in the dark.
Pt(IV)-Azido Complexes
We sought to address this issue and reasoned that replacement of the iodido ligands for other photolabile ligands could improve stability against reducing agents. Pt(IV)-azido coordination complexes are well known to be photoactive (26, 27) and previous studies on Pt(IV)-azido photochemistry had shown a two-electron reduction from Pt(IV) to Pt(II) with concomitant liberation of two unstable azide radicals, which rapidly decompose in water to form N2 (Figure 5) (27). Halide radicals, on the contrary, do not readily decompose in water, and therefore recombination to generate the starting material is a competing process. Azides therefore promised to be an attractive alternative to iodides for our drug design.
Figure 5.
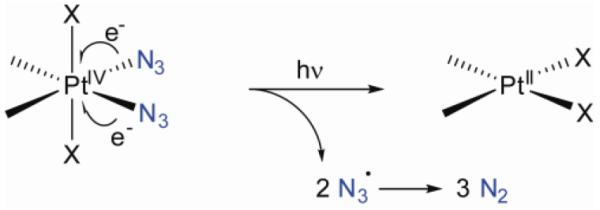
Originally perceived, possible mechanism for photoactivation of platinum(IV) diazido complexes.
We synthesized and reported the first crystal structures of Pt(IV)–diazidodiam(m)ine compounds and showed that they also could be activated with visible light to give rapid and stereospecific binding of the nucleotide 5′-GMP (28) (Figure 6). The X-ray crystal structures of cis,cis,trans-[Pt(N3)2(NH3)2(OH)2] (4) and cis,trans-[Pt(en)(N3)2(OH)2] (5) show that the metal ion has a close to octahedral coordination geometry with almost linear azides in cis-position in the equatorial plane. Importantly, the complexes are stable in human blood plasma and the presence of even 5 mM GSH has little effect on their stability over a period of several weeks. No reactions with either 5′-GMP or the dinucleotide d(GpG) occurred over a week at 298 K in the dark, further illustrating that these complexes possess the low chemical reactivity that is desired in the dark. The electronic absorption spectra of complexes 4 and 5 contain a strong LMCT transition centered on 255 nm, but tailing into the visible. Irradiation of cis,trans-[Pt(en)(N3)2(OH)2] (5) (λirr 457.9 nm, 15 mW) in the presence of 5′-GMP resulted in metal coordination of 60% of the nucleotide present after 20 h and the bis-nucleotide Pt(II) adduct [PtII(en)(5′-GMP-N7)2]2+ was identified as the major product. Photoreaction with d(GpG) resulted in the rather clean, rapid and stereospecific formation of [PtII(en){d(G1pG2-N71,N72)2}]2+ (charges on the nucleotide are not considered). Similar photoreactivity was observed for cis,cis,trans-[Pt(N3)2(NH3)2(OH)2] (4). Photolysis of the complexes in water alone revealed that the photoreaction pathway involved both photoisomerization and photoreduction to highly reactive Pt(II) species of the kind implicated as capable of killing cancer cells. High power red light (λirr 647.1 nm, 75 mW), which penetrates tissue more effectively, was also able to activate the complexes.
Figure 6.

a) Molecular structures of Pt(IV)-diazido complexes 4-7; b) X-ray crystal structures of a cis- (5) and trans-diazido complex (7).
DNA platination experiments with cis,trans-[Pt(N3)2(OH)2(en)] (5) were performed both in the dark and with irradiation in collaboration with the group of Brabec (29). DNA platination of a 212 base pair (bp) fragment of plasmid DNA and transcription mapping by RNA polymerase in vitro revealed that whereas no DNA binding occurred in the dark, DNA binding in the irradiated sample occurred to the same extent and produced similar stop sites as cisplatin, appearing at guanine residues mainly contained in GG sequences (29). These results indicate that the prodrugs indeed form cisplatin-like reactive intermediates upon irradiation, although other reactive species might be formed as well.
These promising results prompted us, in collaboration with the groups of Woods and Bednarski, to study the photocytotoxicity of platinum diazido complexes (30). The effect of photoactivation of cis,trans-[Pt(en)(N3)2(OH)2] (5) and cis,cis,trans-[Pt(N3)2(NH3)2(OH)2] (4) on the growth of 5637 human bladder cancer cells was quite dramatic. The complexes were non-toxic in the dark (IC50 > 300 μM), yet irradiation resulted in selective inhibition of cell growth, with IC50 values of 63 (5) and 49 (4) μM (Figure 7). Growth inhibition by cisplatin, on the other hand, was unaffected by light. Importantly, the platinum diazido complexes were equally cytotoxic to 5637 and cisplatin-resistant 5637 cell lines, showing no cross-resistance with cisplatin. This latter result suggests that the mechanism of action of the photoactive drug is actually different from cisplatin. This is an important observation, as overcoming tumor resistance is a major goal in anticancer drug design. Photoactivation of the prodrug resulted in dramatic changes in cell morphology, i.e. shrinking of the cancer cells, loss of adhesion, packing of nuclear material and disintegration of the nuclei. Such changes in morphology are not observed with cisplatin, providing further evidence that the light-activated complexes cause a very different kind of cytotoxicity than cisplatin (30). This suggests that additional cytotoxic species not available to cisplatin might be generated. CT DNA platination studies showed that photolysis rates matched closely the rates of irreversible DNA platination, indicating that the photolysis products react directly with DNA and notably faster than cisplatin, for which the platination rate is determined by slow hydrolysis steps.
Figure 7.

Effect of light on the IC50 values for the inhibition of cell growth of various cancer cell lines by platinum(IV) diazido complexes. a) toxicity of the cis-complexes 4 and 5 on human bladder cancer cell lines; b) comparison of cytotoxicities of the cis- and trans-isomers 4 and 6 in the dark and upon irradiation; cisplatin is included for comparison (data from ref 30).
It is important to note here that the activity of the platinum diazido complexes does not depend on the presence of oxygen, in contrast to conventional PDT, which is a potential advantage, as some tumors are oxygen-deficient.
The photoactive platinum compounds discussed so far have their labile, photo-induced electron-donating ligands (I−, N3−) and their nitrogen donor ligands (NH3, en) positioned cis with respect to each other. They can therefore, at least in principle, be regarded as prodrugs of cisplatin-like complexes. It is well established that whereas cisplatin is highly cytotoxic to cancer cells, its trans-isomer, transplatin, is relatively non-toxic and clinically ineffective (31). Our observation that the trans-isomer of a platinum(IV) diazido complex, trans,trans,trans-[Pt(N3)2(NH3)2(OH)2] (6), is as cytotoxic when photoactivated as cisplatin was therefore somewhat unexpected and quite remarkable (Figure 7) (32). The stereochemistry of the octahedral complex was again confirmed by X-ray crystallography, and bond lengths and angles comparable to the cis-isomer were found. The trans complex has a much higher aqueous solubility than its cis-isomer. This is noteworthy as not only the reactivity of the drug needs to be tuned, but also other parameters, such as (oral) bioavailability.
Complex 6 exhibits an intense LMCT absorption band at 285 nm, which is shifted to longer wavelength and is more intense compared to the cis-isomer. Irradiation led to the disappearance of this band, indicating loss of azide. Photoactivation of the trans-isomer initially resulted in the appearance of new Pt(IV) complexes (probably substitution of N3 for OH), as judged by 2D [1H, 15N] HSQC NMR, and after 60 min peaks for Pt(II) species appeared, including trans-[Pt(NH3)2(OH2)2]2+ (6r). However, after 120 min of irradiation the majority of Pt was still in the +4 oxidation state. One of the new Pt(IV) complexes was identified as the monoazido species trans,mer-[Pt(N3)(NH3)2(OH)3] (6s), which indicates that the azides do not necessarily leave the platinum metal center together (33). Remarkably, photoreduction proceeded much faster in the presence of 5′-GMP, and after 1 h over 75% had converted to the major product, the bis-GMP adduct trans-[PtII(5′-GMP-N7)2(NH3)2]2+. The absence of the range of side-products observed on photoactivation of trans,trans,trans-[Pt(N3)2(NH3)2(OH)2] (6) alone, suggests that 5′-GMP can readily trap reactive intermediates produced during the photoexcitation process. No reaction with 5′-GMP is observed without irradiation. These results illustrate the importance of studying interactions with biomolecules as part of the attempt to elucidate the mechanism of action of these new anticancer complexes. The photoreactivity products are remarkable in that they not only form very fast compared to reactions of transplatin, but also the formation of bis-guanine adducts is rarely observed for transplatin. The lack of progression from mono- to bis-guanine adducts is thought to be responsible for its lack of anticancer activity (32).
Cytotoxicity studies (32) on HaCaT cells showed that both the cis- and trans-isomers of [Pt(N3)2(NH3)2(OH)2] were equally active upon irradiation and as effective as cisplatin (Figure 7). Again, the complexes are non-toxic in the absence of light. A ‘comet’-assay showed that the complexes produced DNA cross-linking in living cells on irradiation. An appreciation of the intensity of exposure to light in these experiments, can be obtained from the comparison that the intensity of the UVA irradiation used (5 J cm−2 for 50 min) is equivalent to about 15-60 min of exposure to sunlight at midday in the UK. These results suggest that further studies also on the trans-isomers of the platinum diazido complexes are merited.
As systematic ligand variation provides an attractive method to explore and indeed tune the properties of metal-based anticancer drugs (see also the section on ruthenium-arenes, section III), we tried to improve further our platinum diazido prodrug design by varying one of the ammines in trans,trans,trans-[Pt(N3)2(OH)2(NH3)2] (6) for different N donor ligands, such as aliphatic amines (methylamine, ethylamine, cyclohexylamine) and aromatic N donors (pyridine, picolines, quinoline and thiazole) (34). Replacement of the ammine by the π-acceptor pyridine ligand had a dramatic effect. The complex trans,trans-[Pt(N3)2(NH3)(OH)2(py)] (7) was found to be highly phototoxic (35). In the dark, no cytotoxicity towards HaCaT keratinocytes, SH-SY5Y neuroblastoma, cisplatin-sensitive A2780 and cisplatin–resistant A2780cis human ovarian cancer cells was observed, but upon irradiation 7 strongly reduced the viability of the cancer cells (Figure 8). In the A2780 cell line, the complex was 80 × more toxic than cisplatin under identical conditions, and ca. 15 × more effective against the cisplatin resistant A2780cis cell line (33). The trans diazido-Pt(IV) complex therefore has remarkable cytotoxic properties.
Figure 8.

Complex 7 is non-toxic in the dark, yet shows remarkable cytotoxicity with irradiation against several cancer cell lines; cisplatin is included for comparison (data from ref 35).
Complex 7 is stable in water, does not react with 5′-GMP in the dark, and only 5% of Pt(IV) was reduced by GSH after 21 days. Irradiation of the complex alone in water yielded little reduction to Pt(II) and photosubstitution of one or both of the azides was observed. In the presence of 5′-GMP, however, rapid photoreduction resulted in the formation of the mono-adduct (SP-4-4)-[PtII(5′-GMP-N7)(N3)(NH3)(py)]+ (7rG) and bis-adduct trans-[PtII(5′-GMP-N7)2(NH3)(py)]2+ (7rG2) (Figure 9). These results again argue for stepwise (photo)dissociation of the two azides, rather than reductive elimination of both azides in a concerted step.
Figure 9.

a) [1H, 15N] HSQC NMR spectra of 15N-labeled 7 and 2 mol equiv of 5′-GMP after 1 min and 30 min of irradiation (*, 195Pt satellites); b) time dependent decrease in concentration of 7 and formation of 7rG and 7rG2 c) quantification of in vitro DNA repair synthesis using an extract prepared from the repair-proficient HeLa cell line. Cisplatin was taken as 100%. Adapted from ref 35.
Several biological assays pointed to an unusual mechanism of action for 7, which is clearly different from cisplatin, as expected from its remarkable cytotoxic activity. For example, different levels of p53 and caspase activity were observed. Unusual crosslinks were detected upon DNA platination, mainly intrastrand trans-guanine Pt(II) adducts. Importantly, DNA repair synthesis of the platinated lesion resulting from photo-platination was markedly lower than for cisplatin or transplatin (Figure 9) (33). All these results show that trans,trans-[Pt(N3)2(NH3)(OH)2(py)] (7) exerts a rather unique cytotoxic effect and render the complex a candidate for use in photoactivated cancer chemotherapy.
A quick survey of the photochemistry of the different complexes described above shows that the mechanism of photoactivation and the subsequent nature of the observed photoproducts varies from complex to complex and from one geometric isomer to another. Photochemical pathways often involve a combination of photosubstitution, photoisomerization and photoreduction steps. In general, photolysis is rather slow in water and many different products are obtained if the complex is irradiated alone. The presence of nucleophilic biomolecules, on the other hand, can have a major influence, as photoreduction is usually rapid and accompanied by simpler reaction pathways. NMR methods including 2D [1H, 15N] NMR spectroscopy in particular, have been instrumental in studying the photochemical decomposition of the platinum(IV) prodrugs. However, full understanding of the mechanistic pathways of photoactivation has not yet been achieved and remains an important goal of this research. The influence of biomolecules other than the nucleobase derivatives is still relatively unexplored and deserves more attention. Photochemically induced interactions with abundant extra- and intracellular constituents, e.g. proteins such as serum albumin and cytochrome c, are currently underway in our laboratory. Elucidation of photoactivation pathways and, as a result, predictable photochemistry would greatly aid further improvement of drug design.
All things considered, the originally proposed mechanism of a concerted, photoinduced reductive elimination of the two azide ligands to give highly reactive Pt(II) species and dinitrogen seems somewhat oversimplified. Although this mechanism appears to operate under some conditions, other pathways of activation should be considered as well. Indeed, recent photodecomposition studies paint a more complex picture (36). NMR studies of the UVA-induced photodecomposition of cis,cis,trans-[Pt(N3)2(NH3)2(OH)2] (4) under anaerobic conditions do indeed confirm N2 formation and photoreduction to Pt(II), but a tentative assignment of the major product (ca 56%) as trans-[Pt(NH3)2(OH2)2]2+, suggests that photoisomerization accompanies photoreduction (36). Surprisingly, O2 was also detected as a product in the sealed NMR tube, suggesting that alternative pathways can operate (Figure 10). Evidence for the involvement of nitrene intermediates was also obtained. Trapping experiments with dimethylsulfide gave rise to an unusual N,N’-bis(ethyl)-sulfurousdiamide ligand involving an apparently unprecedented carbon-carbon bond formation. This is an intriguing observation since dialkylsulfides (e.g. methionine derivatives) are readily available in biological components. The production of dioxygen and detection of oxidized sulfur species points to the photochemical generation of reactive oxygen species (ROS). Hydroxyl radicals might, for instance, result from homolytic cleavage of Pt(IV)-OH bonds upon reduction. The formation of nitrenes and ROS might contribute to the mechanism of action of these photoactivated platinum anticancer drugs.
Figure 10.

Possible photoactivation pathway of 4: mechanism for nitrene formation and subsequent trapping with dimethylsulfide (DMS) results in C–C bond formation, DMS oxidation and oxygen evolution.
In addition to key factors that have guided improvements in our design of drugs, such as stability in the dark and controlled photoactivation, efforts are aimed particularly at shifting the absorption band relevant for photoactivation towards higher wavelengths for better tissue penetration, while maintaining the stability of the complexes towards reducing agents and alike. The latter is key for non-toxicity in the absence of light and hence for site-selective activation. As ligand variation can give only a limited bathochromic shift, other strategies need be explored as well. An interesting alternative pathway for photoaction would be multiphoton excitation (19). This way, for molecules with a large multiphoton cross-section, a higher energy transition may be obtained by the absorption of more than one photon of lower energy (19, 37). Preliminary studies on some of the platinum(IV) diazido complexes showed no photoactivation by two- or three-photon absorption (33). Current efforts are therefore directed at increasing the multiphoton cross-section to achieve photoactivation by this method.
In summary, the overview given here of our work on photoactive platinum anticancer drugs shows how stepwise improvements transformed an interesting lead compound into a highly active new anticancer drug. Rational drug design allowed us to tackle initial problems of the photoactive prodrugs, such as the limited stability against reducing agents and non-selective activation of the drug leading to cytotoxicity also in the absence of light. Systematic ligand variation resulted in a high degree of control over the reactivity of the prodrug complex and the discovery of a stable, non-toxic and selectively photoactivated potent prodrug that is a promising candidate for photoactivated cancer chemotherapy.
III. Ruthenium-Arene Anticancer Drugs
The field of bioorganometallic chemistry (38) has now established itself as an exiting, vibrant area of research at the interface of biology and organometallic chemistry (39). The compatibility of organometallic compounds with aqueous conditions, water being the essential biological component, has long been considered improbable, many organometallic compounds being sensitive to water and oxygen. Yet, the gradual discovery of different biomolecules with metal-carbon bonds such as methylcobalamin and the enzyme acetyl-CoA synthase has shown that organometallic chemistry and biology are not mutually exclusive. Indeed, the development of aqueous organometallic chemistry shows that these limitations can be overcome and has expanded the toolbox of the medicinal chemist by introducing many structures (M–C bonds with σ–, π–, and/or δ–bonding) and reactivities unique to organometallics (40). This expansion of the field is reminiscent of the introduction of inorganic drugs to medicinal chemistry by way of cisplatin in the early ‘70s, which has had an enormous impact on medicine and resulted in an active and successful research community. Notable examples of the application of organometallic compounds to medicinal chemistry include the work of Jaouen on tamoxifen-derivatives as chemotherapeutics against breast cancer (41) and the development of organometallic 99mTc complexes as radioimaging agents by Alberto (42). Our contribution to this field has been the study of ruthenium-arene and recently osmium-arene complexes as potential anticancer drugs (43, 44).
Introduction to Ruthenium Anticancer Drugs
The discovery of the antitumoral properties of cisplatin (Figure 1), as discussed above, marked the advent of modern medicinal inorganic chemistry (45). The subsequent clinical success of cisplatin and related platinum-based anticancer drugs showed the viability of this approach, but the limitations associated with the platinum drugs (mainly required or inherent drug resistance and severe dose-limiting side effects) provided the impetus for the search for alternative chemotherapeutic strategies based on different complexes with other metals. The transition metals offer a rich palette to choose from and indeed many different transition metal compounds have been explored for potential anticancer activity. Ruthenium, however, stands out as a particularly attractive alternative to platinum. The rich and well-established synthetic and coordination chemistry of ruthenium compounds combined with the fact that the metal has several oxidation states available under physiological conditions, e.g. Ru(II), Ru(III) and Ru(IV) (46), make ruthenium compounds in general well-suited for medicinal applications. Indeed, ruthenium compounds have been investigated as immunosuppressants, nitric oxide scavengers, antimicrobial agents and antimalarials (44, 47). The potential ability of ruthenium to mimic iron in binding to various biomolecules, such as human serum albumin and the iron-transport protein transferrin, may aid a more effective delivery of ruthenium complexes to cancer cells as such rapidly dividing cells have a greater demand for iron and over-express transferrin receptors (46). This property may be responsible for the markedly lower toxicity of ruthenium-based anticancer agents compared to platinum drugs (48).
Finally, a key advantage of ruthenium-based metallodrugs is the ability to tune the metal-ligand exchange kinetics over many orders of magnitude via ligand variation. Such influence over kinetic stability is critical for drug development and our work on ruthenium-arene anticancer drugs that is described in this section shows that such control can indeed be exerted.
The first report on the anticancer properties of ruthenium was published in 1976 when the Ru(III) compound fac-[RuCl3(NH3)3] (Figure 11) was found to induce filamentous growth of E. coli at concentrations comparable to those at which cisplatin generates similar effects (49). This Ru(III) complex and related compounds such as cis-[RuCl2(NH3)4]Cl illustrated the potential anticancer activity of ruthenium complexes, but insolubility prevented further pharmacological use. Since these initial studies, other Ru(III) complexes have been studied for potential anticancer activity, and two compounds, NAMI-A (50) and KP1019 (51), are currently undergoing clinical trials. Remarkably, although the structures of NAMI-A ([Him][trans-RuCl4(dmso)(im)], im = imidazole) and KP1019 ([Hind][trans-RuCl4(ind)2], ind = indazole) are quite similar (Figure 11), they exhibit contrasting therapeutic effects. KP1019 is significantly cytotoxic to cancer cells, whereas NAMI-A is virtually devoid of cytotoxicity in vitro, but has antimetastatic activity in vivo. Understandably, this remarkable difference has spurred a widespread interest in the chemistry of these and related Ru(III) complexes, and elucidation of their mechanisms of action. Several reviews on this topic are available (46, 48, 52, 53).
Figure 11.

Molecular structures of a) fac-[RuCl3(NH3)3], the first reported ruthenium complex with anticancer activity, and b) NAMI-A and KP1019, two ruthenium compounds currently in clinical trials.
These relatively inert Ru(III) compounds are essentially prodrugs (see section on prodrugs above, section II.A): they are ‘activated by reduction’ in vivo to their more labile and reactive Ru(II) counterparts, which in turn are responsible for the observed cytotoxicity (cf. the Pt(IV)/Pt(II) redox couple) (54, 55). With this in mind, we concentrated on the design of ruthenium(II) anticancer complexes. Initial studies showed that aminophosphine ruthenium(II) complexes were cytotoxic to cancer cells (56), but these complexes suffered from low aqueous solubility and were not further pursued (57, 58). We synthesized the organometallic ruthenium(II) half-sandwich complex [Ru(η6-benzene)Cl(Me2NCH2CH2PPh2)](PF6) in which Ru(II) is stabilized not only by the phosphine but also by an η6-bonded arene ligand. This complex has the characteristic ‘piano-stool’ geometry typical of half-sandwich Ru(II)-arene complexes (57). This structural element turned out to be key to our later discovery of potent organometallic ruthenium anticancer drugs. The inclusion or an η6-arene in our ruthenium anticancer drug design held some promise, since arene ligands are known to stabilize ruthenium in its +2 oxidation state and they provide the complex with a hydrophobic face, which might enhance biomolecular recognition processes and transport of ruthenium through cell membranes. In an attempt to increase the aqueous solubility of the ruthenium-arene complexes (an advantage for clinical use) the aminophosphine ligand was replaced with N,N-bidentate ethylenediamine (en), which indeed resulted in complexes that are reasonably soluble in water (59). Most importantly, the new complexes showed significant inhibition of human ovarian cancer cell growth and thus led to our discovery of a new family of organometallic ruthenium(II)-arene anticancer drugs (59). The study of this family of ruthenium anticancer drugs is the topic of this section.
Other active areas of research into the anticancer properties of ruthenium(II) complexes, include, amongst several other examples, the related work on RAPTA ruthenium-arene complexes by Dyson and coworkers (46, 60). The recent work by Meggers et al. (61) shows an interesting departure from the central paradigm of utilizing the transition metal in metallotherapeutics for its inherent reactivity. Their approach focuses on the use of the metal as a building block, relying on the kinetic inertness of certain coordination/organometallic bonds, for well-defined three-dimensional bioactive constructs, which are not accessible through purely organic, carbon-based compounds (61). The potential of this strategy is underlined by the reported organometallic indolocarbazole ruthenium(II) complexes which were designed as staurosporine mimetics and are highly cytotoxic towards human melanoma cancer cells (62).
III. A. General Features of Ru-Arene Anticancer Drugs
General Structural Features
The general structure of half-sandwich ruthenium(II)-arene complexes is shown in Figure 12. The structural, stereochemical and electronic features of metal-arene complexes have been discussed (63). A typical ‘piano-stool’ geometry consists of an η6-arene occupying three coordination sites of the pseudo-octahedral complex, leaving the three ‘legs’ X, Y and Z available for coordination. The sites X and Y can be taken up by two monodentate ligands, but are more commonly occupied by a bidentate ligand L with e.g. nitrogen and/or oxygen donor atoms (e.g. NN, NO, or OO coordination), typical examples being ethylenediamine (en) or acetylacetonate (acac) (Figure 12). The chelating nature of the bidentate ligand seems to be advantageous for anticancer activity. Linking two chelating ligands together allows for the synthesis of dinuclear ruthenium-arene complexes.
Figure 12.

a) General structure of the half-sandwich, piano-stool ruthenium-arene complexes; b) X and Y are commonly occupied by a bidentate ligand L giving a monofunctional complex; c) tethering of a monodentate ligand to the arene results in a bifunctional complex.
The sixth and final coordination site Z is usually a halide and endows the molecule with a reactive site. The arene ligand, on the other hand, is relatively inert towards displacement under physiological conditions (vide infra for some exceptions). As a result of this single-site reactivity, the complexes are considered monofunctional, in this case meaning that the metal is capable of forming one direct coordinative bond with a biological target. Other additional non-covalent interactions, such as hydrogen bonding and π-π stacking, are available to some of the complexes and can be advantageous for high cytotoxicity. Depending on the nature of both the chelating ligand L and ligand Z, the complexes are either neutral, or mono- or dications and as such isolated as salts. The charge on the complex and the nature of the anion can have a pronounced influence on important pharmacological properties, such as aqueous solubility and partition coefficients.
The general structure allows the nature of the arene, the type of chelate and the monodentate ligand to be varied. This gives access to a vast library of compounds that can be synthesized and screened for anticancer activity (see Figure 13 for the cross section of ruthenium-arene anticancer agents discussed in this section). Indeed, variation of each of the building blocks allows us to modify key thermodynamic and kinetic parameters, and in this way tailor the pharmacological properties of the complexes. The general reactivity will be discussed in a following section.
Figure 13.

Structural diversity: some ruthenium-arene anticancer agents from the Sadler lab.
Whereas the general structure described above can yield monofunctional adducts with biomolecules (vide infra), it is interesting to explore bifunctional ruthenium-arene complexes as well, as a different profile of reactivity can be expected. The use of a monodentate ligand to achieve this is, however, unadvisable as such complexes readily undergo substitution reactions and are, as such, unstable in solution. Tethering the monodentate ligand to the arene provides an attractive strategy (Figure 12), such chelation stabliizes the molecule towards substitution. In this way, we have synthesized some nitrogen-containing, bifunctional tethered Ru(II) arene complexes (64). Alternatively, if such a N-donor tethered ruthenium-arene is complexed with a bidentate chelating ligand, e.g. en, a molecular switch can be designed to work by selective by selective ring-opening of the tether under certain conditions (65).
We have also recently explored some ruthenium-arene complexes that depart markedly from the general structure described above. For instance, full sandwich ruthenium complexes have been synthesized, in which the positions X, Y, and Z are taken by an η6-arene ring of a biologically-active ligand, such as aspartame, to assess the influence of a metal-complex as a modulating substituent on the properties of the bioactive ligand (66).
Synthesis
The various synthetic routes to these complexes have been recently reviewed (67). Usually, the synthesis involves the reaction of dimeric Ru(II)-arene complexes of the type [Ru(η6-arene)Cl2] with an appropriate (chelating) ligand. The dimers are generally prepared by the redox reaction of RuCl3 and a cyclic 1,4-diene as arene precursor, but other routes are available as well. The redox reaction between RuCl3 and a cyclic 1,4-diene usually gives the ruthenium-arene dimers in high yield and purity, but this synthetic route is limited by the commercial availability or ease of synthesis of diene precursors. Thermal displacement of a coordinated arene provides another valuable synthetic route. The latter method has been employed for the synthesis of ruthenium-arenes with sterically demanding arenes such as hexamethylbenzene (hmb) and for the synthesis of amine-tethered complexes (64, 68). We have also synthesized the 106Ru radiolabeled complex [106RuCl(en)(η6-fluorene)](PF6) (8) to facilitate pharmacological (ADME) studies (69).
Stereochemistry
It is important to note here that the ruthenium-arene complexes are inherently chiral if the three legs of the piano-stool are non-equivalent. For instance, the use of an N,O-bidentate mixed chelate such as glycine or hydroxyquinoline gives rise to a chiral ruthenium centre. Alternatively, the coordination of a secondary amine, such as in N-ethyldiaminoethane (Et-en), to a Ru(II)-arene leads to the formation of four diastereoisomers with stereogenic centers at nitrogen and ruthenium. This realization is important as the enantiomers (diastereomers), in principle, have different biological properties. Knowledge of epimerization rates and, if possible, resolution of the enantiomers may then be important for formulation and mechanism of action. In this light, the stereochemistry of [Ru(η6-bip)Cl(Et-en)](PF6) (9) (bip, biphenyl) was studied in detail (70). The synthesis of [Ru(η6-bip)Cl(Et-en)](PF6) (9) gave rise to two diastereomers (with relative configurations R*RuR*N (A) and S*RuR*N (B), see Figure 14), which were separable by crystal picking. Dissolution of the diastereomerically-pure compounds resulted in the dynamic interconversion of the A or B diastereomers, resulting in an equilibrium mixture of 72:28 % of R*RuR*N (A) and S*RuR*N (B). The preferred isomer has the ethyl-group pointing away from the arene ring, thus minimizing steric repulsion. Our studies thus suggested that isomer A (R*RuR*N) is thermodynamically preferred, and that each ruthenium complex is in dynamic equilibrium between the A (R*RuR*N) and B (S*RuR*N) configurations. Interestingly, the reaction of the diastereomeric mixture with 9-ethylguanine (9-EtG) resulted in the formation of a single diastereomeric pair B (S*RuR*N), apparently made possible by the facile epimerization at the nitrogen center (Figure 14). This dynamic chiral recognition of guanine provides the possibility of highly diastereoselective DNA recognition by ruthenium anticancer agents by an induced fit mechanism (70).
Figure 14.

Dynamic chiral recognition of 9-ethylguanine by chiral ruthenium-arene complex 9.
Hydrophobicity
An important pharmacological characteristic of the complexes is their hydrophobicity (partition coefficients), as it is strongly related to both influx and efflux of anticancer drugs. Transport and sequestration of ruthenium complexes, i.e. drug uptake, into tumor cells may involve both active and passive diffusion, and may be mediated by protein receptors. Active drug efflux on the other hand is sometimes mediated by P-glycoprotein, a membrane protein that is overexpressed in cancer cells. Hydrophobicity is thought to be a major determinant in substrate specificity and is therefore another structural parameter that can be explored and, in principle, optimized (71). Additionally, hydrophobic interactions are important in binding to biomolecules such DNA and proteins.
A convenient and easily accessible way to quantify hydrophobicity is the determination of the octanol/water partition coefficient (log P) and we have determined the hydrophobicity of 13 selected ruthenium-arene complexes (71). As expected, hydrophobicity increases with increase of the size of the coordinated arene ring, but decreases significantly when the chloride is replaced by neutral ligands such as pyridine and 4-cyanopyridine. The latter observation is somewhat counterintuitive at first inspection, but correlates with replacement of anionic chloride to yield a dicationic complex. The hydrophobicity correlates significantly with the biological activity of these complexes (71).
III. B. Cytotoxicity Studies: Towards Establishing Structure-Activity Relationships
Our in vitro and in vivo activity studies have been carried out in collaboration with Jodrell’s group at the Western General Hospital in Edinburgh and with the help of Oncosense Ltd, and more recently also with the group of Brabec.
Initial cancer cell growth inhibition tests done on the human ovarian cancer cell line A2780 revealed the cytotoxic activity of some of the tested ruthenium-arene complexes (59). IC50 values as low as 6 μM were obtained for the complexes [Ru(η6-bip)Cl(en)](PF6) (10) and [Ru(η6-bip)Cl(Et-en)](PF6) (9). This was an order of magnitude higher than cisplatin (0.5 μM in the same test), but comparable to the second generation anticancer drug carboplatin, illustrating the potential of this new family of metallo-anticancer drugs. The complexes [Ru(η6-bip)Cl(en)](PF6) (10) and [Ru(η6-bip)Cl(Et-en)](PF6) (9) did not inhibit the catalytic activity of topoisomerase I or II over the concentration range of observed cytotoxicity (1-50 μM), making it unlikely that the anticancer activity is related to inhibition of these enzymes (59). Complexes with other arenes showed slight differences in activity, which already indicated that the structure of the arene is important for cytotoxicity. Indeed, increasing the size and overall hydrophobicity of the arene increased cytotoxicity and IC50 values of 2 and 0.4 μM were found for [RuCl(η6-dha)(en)](PF6) (11) (dha, dihydroanthracene) and [RuCl(en)(η6-tha)](PF6) (12) (tha, tetrahydroanthracene), respectively, the latter thus being equipotent to cisplatin (Figure 15) (67, 72). Complexes with three monodentate ligands, i.e. devoid of the chelate, were essentially inactive. These complexes are probably too reactive towards components of the cell culture medium and/or the cells and do not reach the target site. Indeed, the bifunctional tethered complexes [Ru(η6:η1-C6H5CH2(CH2)nNH2)Cl2] (n = 1,2) (13a,b) possessing two reactive Ru–Cl bonds, are also not cytotoxic against the A2780 cell line (64). A more hydrophobic arene and a single ligand exchange site therefore seem associated with high cytotoxicity.
Figure 15.

Trends illustrating the influence of the arene, the chelate and the leaving group on the cytotoxicity of ruthenium-arene complexes developed in the Sadler lab. The complexes are not cross-resistant with cisplatin.
Cross resistance profiles of selected Ru(II) arene complexes in cisplatin-resistant A2780cis cells and multi-drug-resistant A2780AD cells showed a relatively high degree of cross-resistance in A2780AD. By contrast, the organometallic complexes tested were completely non-cross resistant in A2780cis cells (72) (Figure 15).
Cross resistance to [Ru(η6-bip)Cl(en)]PF6 in A2780AD fell from a factor of 38 to only three-fold upon co-administration of verapamil, indicating that P-glycoprotein mediated active efflux of the anticancer drug was pre-dominantly responsible for the observed cross resistance and could be abrogated by addition of the competitive inhibitor. Such behavior is common for lipophilic positively-charged drugs.
The patterns of activity established in vitro for 10 were mirrored to a large degree in vivo, with high activity in an A2780 xenograft together with non-cross-resistance in an A2780cis xenograft and a lack of activity in the A2780AD xenograft (72). [Ru(η6-bip)Cl(en)](PF6) (10) was further evaluated against a 13-cell line panel (Freiburg screen). The results demonstrated a broad spectrum of activity with a “compare-negative” score versus other common anticancer compounds suggesting a unique mode of action (73). Particular sensitivity was noted in a breast cancer cell line (401NL) and a non-small cell lung cancer (NSCLC) cell line (LXFL 529L). The activity of [Ru(η6-bip)Cl(en)](PF6) (10) in two other NSCLC cell lines (H520 (3.5 μM) and A549 (3 μM)) was similar to cisplatin (9.5 μM and 2.6 μM, respectively). [RuCl(en)(η6-tha)](PF6) (12) was even more potent with IC50 values of 0.53 and 0.5 μM, respectively. In vivo experiments showed anti-tumor activity of both complexes against an A549 xenograft. A preliminary assessment of the potential therapeutic index for these agents, showed that liver toxicity is a concern (73).
We recently tested a more extended range of these organometallic Ru(II) complexes complexes for cytotoxicity to elaborate on the structure-activity relationships (67). Some general trends could be discerned, but the structure-activity relationship turned out to be quite complex. Variation of the arene ring revealed that inclusion of polar substituents, such as amides, esters and alcohols lowered cytotoxicity. Nonpolar, sterically-demanding substituents, however, resulted in more potent complexes with IC50 values as low as 3 μM (compared to 0.5 μM for cisplatin). Fused ring systems showed good activity, with the polycyclic aromatic hydrocarbons being the most active, emphasizing the importance of lipophilicity and possible hydrophobic interactions with the nucleobases of DNA (vide infra).
Data for a series of [Ru(η6-arene)Cl(en)](PF6) complexes with the isomeric p-, o-, and m-terphenyls mirrored these observations (74). The complex with the most extended arene (p-terphenyl, 14) was the most potent, with potency similar to cisplatin, but is not cross-resistant, and a much higher activity than its isomeric complexes. Again, no cross-resistance with cisplatin was observed for these complexes (74).
Variation of the chelated ligand resulted in complexes with vastly differing cytotoxicities. Aliphatic diamines generally showed good cytotoxicity (e.g. 9-12), provided they contain a primary amine for stereospecific hydrogen bonding with guanine (vide infra). 1,2-Diaminobenzene (dab) complexes such as [Ru(η6-bip)Cl(dab)]+ (15) also showed good activity and, interestingly, dab complexes overcome the cross-resistance to A2780AD, previously observed for en-containing complexes. Bipyridine and phenanthroline derivatives are inactive, again perhaps indicating the importance of NH groups.
Surprisingly, replacement of the N,N-chelating ligand en by N,O-chelating ligands of some amino acids (amino acidates) proved detrimental for activity. Reactivity studies indicated that fast hydrolysis rates and a relatively large proportion of a reactive aquated species at physiological chloride concentration might be responsible for the lack of activity. Finally, complexes with O,O-chelating ligands such as acac and its derivatives (16a,b) showed good to moderate activity, despite the lack of hydrogen bond donors. Some of the trends described above, but not all, can be explained by the general reactivity profile of ruthenium-arene complexes, which will be discussed in more detail below.
In the series of diamine complexes, changing the monodentate ligand Z from chlorido to iodido or even the pseudohalide N3− has little effect on activity, despite significant differences in rate and extent of hydrolysis they are all potential leaving groups (59, 75). This contrasts dramatically with the chlorido and iodido ruthenium(II) arene complexes that contain σ-donor/π-acceptor 2-phenylazopyridines (azpy) as N,N-chelating ligands (17a,b). These chlorido complexes are inactive against the A2780 and A549 cell lines (76). Some of the corresponding phenylazopyridine/iodido complexes, on the other hand, proved highly cytotoxic to the same cell lines (see section on catalytic anticancer drugs and Figure 23) (77). These complexes exhibit a different mechanism of cancer cell cytotoxicity, involving catalytic redox reactions (vide infra), in which the reactivity is ligand-based rather than metal-based. The complexes provide a nice example of how metal ions can also tune the reactivity of coordinated ligands, rather than the other way round. The use of a transition metal as a modulating substituent of a ligand is an interesting strategy and should be explored further. It takes advantage of properties of the metal center other than its direct reactivity and is in this way related to the metal-as-scaffold approach in which the metal serves the structural function of spatially orienting the coordinated ligands (61).
Figure 23.

a) The [Ru(η6-arene)(azpy)I]+ complex (17a) is a catalytically active ruthenium-arene anticancer agent; (b) remarkable influence of the leaving group on cytotoxic activity; c) [Ru(η6-arene)(azpy)I]+ complexes can act as catalysts for the oxidation of the major intracellular reducing agent glutathione to glutathione disulfide with concomitant production of reactive oxygen species. Data from ref 77.
The cytotoxicity data cited above and illustrated in Figure 15 show that ligand variation can have dramatic effects on the biological activity of these ruthenium-arenes. Certain trends can be discerned and such structure-activity relationships then carry some predictive value for further drug design. An important next step is then to rationalize these relationships by correlating biological activity to chemical reactivity. Hence, much attention has been given to the study of the reactivity of this family of ruthenium-arene anticancer drugs and their interaction with biomolecules that are considered a possible target or that can be encountered by the complex before reaching its (final) target.
III. C. Reactivity of Ruthenium-Arene Anticancer Drugs
Aquation
The principal reactivity of our family of ruthenium-arene complexes is the exchange of the leaving group Z, usually a halide, with water to form the more reactive aqua complexes (Figure 16). This latter species undergoes substitution reactions, for instance DNA nucleobase binding, much faster than the corresponding chlorido precursors and is therefore considered as the active species that exerts the pharmacological effect. The ruthenium-arene anticancer complexes [Ru(η6-arene)Cl(L)] can therefore be regarded as prodrugs (Figure 2). Understanding the thermodynamic and kinetic parameters that govern prodrug activation by aquation have been an important part of our research, as detailed knowledge of the factors that control such ligand substitutions under physiological conditions is very valuable in drug design. The aqueous chemical reactivity of the complexes can be chosen so as to balance the inertness required for the drug to reach its target site and minimize attack on other sites, yet allow activation necessary for binding to the target (75).
Figure 16.

General reactivity of the ruthenium(II)-arenes. Hydrolysis of the Ru–Z bond gives the more reactive aqua species. The pKa of the coordinated water molecule is important, as the hydroxido complex is less reactive. The different structures are exemplified by the reactivity of [Ru(η6-bip)Cl(en)]+ (10) for which Z = Cl.
The rate of hydrolysis depends strongly on the nature of all three building blocks, i.e. the leaving group, the coordinated arene and the chelate, and can be varied over several orders of magnitude, opening a time-window of activation. A detailed study of the aquation and the reverse, anation reactions of three [Ru(η6-arene)Cl(en)](PF6) complexes (arene = bip (10), dha (11), and tha (12)) showed that the rates of aquation (kH2O 1.23 – 2.59 × 10−3 s−1, at 298 K and I = 0.1 M) with half-lives of < 10 min are an order of magnitude (> 20 times) faster than that of cisplatin (78). The anation reactions in the presence of 100 mM NaCl were very rapid (kCl 0.127 – 0.306 M−1 s−1, at 298 K and I = 0.1 M), resulting in small equilibrium constants for aquation (7.3 – 9.7 × 10−3 M). This is significant for the speciation of the ruthenium complexes under physiological conditions (Figure 17). The results indicate that in blood plasma, where the [Cl−] is high (about 104 mM), the complexes would exist primarily in their chlorido forms (> 89%). In contrast, the chloride concentrations are much lower in the cell cytoplasm (about 23 mM) and cell nucleus (4 mM) which would increase the amount of more reactive aqua species to ca. 30% and 70 %, respectively. As a result, the extracellular suppression of aquation followed by activation upon entering the cell provides a selective mechanism of activation.
Figure 17.

Speciation of [Ru(η6-bip)Cl(en)]+ (10) [5 μM] in blood plasma, cytoplasm and nucleus at equilibrium, based on the equilibrium constants of aquation, pKa, [Cl−] and pH of the environments. Data from ref 78.
The mean values of the activation parameters for both aquation and anation show that ΔS‡ is negative, indicative of an associative pathway (78). Density functional theory calculations also suggest that aquation occurs via a more associative pathway in an Ia ↔ Id mechanistic continuum for which bond-making is of greater importance than bond-breaking (75). The electron-accepting effect of the strong π-acid arene ligands is thought to be responsible for the shift towards a more associative Ia pathway, as it increases the charge on the metal (75). Ru(II) in {Ru(η6-arene)} complexes may therefore behave more like a Ru(III) center, which usually reacts via associative pathways (79).
Both aquation and anation reactions are about twice as fast for the dha (11) and tha (12) complexes compared to the biphenyl complex 10. Consequently, the hydrolysis rates decrease with increase in the electron-accepting ability of the arene, an observation that was later confirmed for a more extensive series of compounds (75). We also studied the influence of the type of leaving group on the hydrolysis rate and found that it has a large effect. For the halides, the aquation rate was found to decrease in the order Cl ≈ Br > I. Replacement of the chloride ligand with the pseudohalide N3 even slowed down the hydrolysis rate 40-fold, while changing it for leaving groups such as (substituted) pyridines or thiophenol made the hydrolysis too slow to observe. Generally, but not always, complexes that readily hydrolyze are cytotoxic. An interesting exception to this rule is [Ru(en)(η6-hmb)(SPh)]+ (18), which hydrolyzes extremely slowly yet shows significant activity. Closer inspection of the reactivity of this and related compounds suggests an ‘activation-by-ligand-oxidation’ mechanism and led to our studies on ruthenium-sulfenate and -sulfinate complexes that will be further discussed below.
The chelating ligand also influences the hydrolysis rate. In general, the effect on substitution reactions depends on the nature of the chelate and the position of the ligand relative to the leaving group. The cis-positioning of the en chelate to the leaving group in [Ru(η6-arene)Cl(en)]+ slows down the aquation and anation reactions due to electronic and steric effects. Replacement of the neutral en ligand by monoanionic acac increases the rate and extent of hydrolysis (80). The presence of a π-acceptor azopyrazole ligand on the other hand decreases the rate of hydrolysis by more than an order of magnitude (76).
The bifunctional amine-tethered ruthenium(II) arene complexes [Ru(η6:η1-C6H5CH2(CH2)nNH2)Cl2] (n = 1,2) (13a,b) show two consecutive hydrolysis steps to yield the mono- and bis-aqua complexes (64). At extracellular chloride concentrations, the majority of the complexes could be expected to be present as the mono-aqua adduct. Equilibrium constants were determined for both steps (for 13b, K1 = 145 mM K2 = 5.4 mM) and found to be considerably higher than those of cisplatin, which also has two reactive sites available.
An important characteristic for the activity of the hydrolysis product, the aqua species, is the pKa of the coordinated water molecule (Figure 16). Acid dissociation gives the hydroxido complex [Ru(η6-arene)(L)(OH)]+ which is less susceptible to substitution reactions than the corresponding aqua complex (81). The pKa values of the aqua complexes can be determined by UV-Vis or NMR spectroscopy. The pKa value of the coordinated water molecule in [Ru(η6-arene)(en)(OH2 )]+ was found to be 7.71, 7.89 and 8.01 for the bip, dha, and tha complexes, respectively (78). This relatively low acidity of the water ligand is important, since under physiological conditions only small amounts (< 10% of the total Ru(II) arene complexes) are predicted to exist as the less reactive hydroxido complexes. Changing the bidentate ligand to monoanionic oxygen chelates such as acac or tropolonate increases the pKa further to 9.12 ([Ru(η6-p-cym)(OH2)(trop)]+) (19H2O) (82) and 9.41 ([Ru(acac)(η6-p-cym)(OH2)]+) (16aH2O) (80), most likely as a consequence of electronic effects exerted by the ligand on the metal center (82). In contrast, the pKa of the aqua adduct in [Ru(azpyz-NMe2)(η6-p-cym)(OH2)]+ (20H2O) (azpyz-NMe2, 4-(1H-pyrazol-3-ylazo)-N,N dimethylaniline) is 4.60, indicative of the low electron density at ruthenium and consistent with the π-acidic nature of the ligand. Indeed, the complex has a low affinity for DNA bases (see below) as it would predominately exist in the more inert hydroxido form at physiological pH (76).
These results clearly show that the hydrolysis rate, and with that drug activation, and the pKa of the aqua adduct can be tuned, a strategy that has previously been successful for Pt anticancer drugs. This opens the possibility of drugs with tailor-made properties. As an aside, it is interesting to note that careful control over the kinetics of exchange or, alternatively, the redox potential of a drug might ultimately lead to personalized anticancer drugs. The characteristics of the drugs could be adapted to fit physical parameters particular to an individual and would allow activation in specific cells. If individual variations in response to a certain drug have a genetic basis, dividing patients into subgroups with a similar genetic profile would allow more efficient treatment, if a series of anticancer drugs with slightly different properties is available.
Loss of Coordinated Arene
We previously stated that the arene ligand in ruthenium(II)-arene complexes is relatively inert towards displacement under physiological conditions. While this is generally true, there are a few exceptions to this rule and this type of reactivity can be used to advantage. Weakly bound arenes, for instance, can be thermally displaced, a property convenient for the synthesis of ruthenium-arene complexes that are not readily available through more common synthetic routes. This way, the reaction of a precursor dimer, [RuCl2(etb)]2 (etb, ethylbenzoate) (68), with either 3-phenyl-1-propylamine or 2-phenylethylamine results in arene replacement and the isolation of the complexes [Ru(η6:η1-C6H5CH2(CH2)nNH2)Cl2] (n = 1,2) (13a,b) in which the amine tether is coordinated to ruthenium as well (64).
The nature of the chelating ligand exerts a strong influence on the lability of the coordinated arene. The presence of strong π-acceptor chelate ligands in [Ru(η6-arene)Cl(L)]+ where L is an azopyridine (17) or azopyrazole (20), can lead to thermal displacement of the arene under mild conditions (low temperatures, aqueous solution) The arene loss is significant, up to 67 % after 24 hours at 310 K, depending on the arene. The competition for π-electron density with the π-acceptor chelate ligands is responsible for the weakening of the Ru(II)-arene bonds and subsequent loss of the arene (76).
Alternatively, the arene displacement can also be photo- rather than thermally-induced. In this respect, we studied the photoactivation of the dinuclear ruthenium-arene complex [{RuCl(η6-indane)}2(μ-2,3-dpp)]2+ (2,3-dpp, 2,3-bis(2-pyridyl)pyrazine) (21). The thermal reactivity of this compound is limited to the stepwise double aquation (which shows biexponential kinetics), but irradiation of the sample results in photoinduced loss of the arene. This photoactivation pathway produces ruthenium species that are more active than their ruthenium-arene precursors (Figure 18). At the same time, free indane fluoresces 40 times more strongly than bound indane, opening up possibilities to use the arene as a fluorescent marker for imaging purposes. The photoactivation pathway is different from those previously discussed for photoactivated Pt(IV) diazido complexes, as it involves photosubstitution rather than photoreduction. Importantly, the photoactivation mechanism is independent of oxygen (see section II on photoactivatable platinum drugs) (83).
Figure 18.

The dinuclear complex [{RuCl(η6-indane)}2(μ-2,3-dpp)]2+ (21) can be photoactivated to yield highly reactive and potentially cytotoxic ruthenium species and the arene indane, which could be used as a fluorescent probe.
Interactions with Nucleobases
Although it has not yet been unequivocally established, the primary cellular target for these organometallic Ru(II) complexes is thought to be DNA, as for many metal-based anticancer drugs including the archetypal drug cisplatin. The study of this interaction and its possible significant effects on DNA structure has therefore been of prime importance in our studies of the ruthenium-arene complexes. Initial studies showed a strong selective binding to N7 of the guanine bases (the most electron-dense site on DNA) on a DNA 14-mer oligonucleotide (59). To gain additional insight into the potential modes of interaction, we have studied in depth the binding of the ruthenium-arene complexes with nucleic acid derivatives as models of DNA. A detailed study of the complexes ([Ru(η6-arene)Cl(en)]+) (arene = bip, dha, tha (10-12)) with the guanine derivatives 9-ethylguanine (9-EtG), guanosine (Guo), and 5′-guanosine monophosphate (5′-GMP) both in the solid state (by X-ray crystallography) and in solution (by NMR studies) elucidated the interactions that give rise to such high specificity (84). Non-covalent interactions turned out to play key roles in the biological molecular recognition process. In addition to direct monofunctional coordination of ruthenium to N7, the crystal structures of [Ru(η6-dha)(en)(9-EtG)]+ and [Ru(en)(9-EtG)(η6-tha)]+ also show strong arene-nucleobase stacking and strong stereospecific intramolecular H-bonding. The hydrogen bonding interaction and the arene-base stacking are illustrated in Figure 19.
Figure 19.

The combination of a) a strong stereospecific hydrogen bonding interaction of the C6O carbonyl of 9-EtG with an en NH in [Ru(η6-dha)(en)(9-EtG)]+ and b) a strong π-π arene-nucleobase stacking interaction is responsible for the high preference of G over A observed for such ruthenium-arene complexes.
The π-π stacking interaction between the outer ring of dha and tha and the guanine base is close to ideal, with interplanar distances and dihedral angles of 3.45 Å/3.3 ° (tha) and 3.31 Å/3.1° (dha). The extensive arene-nucleobase stacking in the absence of arene-arene or base-base stacking in [Ru(η6-arene)(en)(G-N7)]+ represented a new structural feature in adducts of metal-based anticancer drugs with nucleobases. The observed stacking indicates the potential of ruthenium-arenes for intercalation or hydrophobic interactions of the arene ring with duplex DNA (84). Such hydrophobic interactions could provide a major contribution to the driving force for DNA binding. Comparison of the conformations of the G-adducts with the parent chlorido-complexes revealed a significant reorientation and conformational change of the arene, as a result of the arene-nucleobase interaction. The changes involve rotation around the arene-Ru π-bonds, twisting around the Ph–Ph bond (bip), and ring bending (dha, tha). The arene ligands thus possess the flexibility to optimize the geometry for simultaneous covalent binding and arene-base stacking. This may reduce the steric demands of the Ru drug and enhance the DNA affinity.
The second strong non-covalent interaction observed was the stereospecific H-bonding of an amine NH to the C6O carbonyl of G (average N⋯O distance 2.8 Å, N–H⋯O angle 163°). This strong H-bonding interaction partly accounts for the high preference for binding of {Ru(η6-arene)(en)}+ to G versus A. Indeed, subsequent studies on the thermodynamics and kinetics of binding of ([Ru(η6-arene)Cl(en)]+) (arene = bip, dha, tha) to mononucleotides and mononucleosides (G, A, C, and T) confirmed the importance of these specific interactions. For the mononucleosides, selective binding to N7 of guanosine (100%), moderate binding to N3 of thymidine (35%), weak binding to N3 of cytidine (14%), and hardly any binding to adenosine (< 5% to N1/N7) was observed (81). Similar affinities for Ru were found for the corresponding mononucleotides, except for an additional, significant amount (40-60%) of 5′-phosphate binding with 5′-AMP, 5′-CMP, and 5′-TMP, but not with 5′-GMP (although initially phosphate bound intermediates were detected for 5′-GMP, bound phosphate was displaced by N7). No binding to the phosphodiester groups of 3′,5′-cyclic guanosine monophosphate (3′,5′-cGMP) or 3′,5′-cAMP was observed, suggesting that the complexes do not bind to the phosphodiester backbone of DNA. Kinetic experiments also indicated that the binding of Ru(II) arene complexes with arenes that can take part in π-π stacking (dha, tha, bip) are up to an order of magnitude faster than those containing arenes which cannot (p-cym (22), bz). This demonstrates the significant role arene-purine base-stacking plays in stabilizing the transition state in the associative substitution reactions.
The differences in the extent of binding to Ru(II) can be explained by analyzing the possible H-bonding and nonbonding repulsive interactions, in addition to the electronic properties of the nucleobase binding sites themselves. We previously saw the contribution of non-covalent interactions of amino Ru-arenes with the exocyclic carbonyl group of G. In contrast, adenosine has a C6 NH2 group and as a consequence binding at either N7 or N1 is weakened by repulsive interactions. The reactivity of the various binding sites of the nucleobases decreases in the order G(N7) > T(N3) > C(N3) > A(N1, N7). This strong preference for G was confirmed by competition studies with 5′-GMP versus either 5′-AMP or 5′-CMP or 5′-TMP; in each case, essentially only the 5′-GMP adduct forms. The ability of the NH proton of en to act as an H-bond donor toward an exocyclic oxo group but not toward an amino group therefore plays an important role in the selective site recognition of these ruthenium-arene anticancer complexes (81). Extensive NMR studies mirrored the observations made for the solid state structures of the ruthenium-arene nucleobase adducts. 1H 2D NOESY NMR experiments, for instance, confirm that the pendant phenyl ring in [Ru(η6-bip)(en)(9EtG-N7)]+ adopts a syn conformation with respect to the G base. It is therefore not surprising that replacement of en by acac results in changes in the nucleobase selectivity. The complex [Ru(acac)Cl(η6-p-cym)] (16a) binds equally well to guanosine and adenosine and no binding to either thymidine or cytidine was observed (80). The N7/N1 coordination ratio for adenosine binding was about 4:1 at pH 5.8. Molecular models demonstrate that adenine coordination can be stabilized by hydrogen bonding between N6H2 as a donor and an acac oxygen as hydrogen-bond acceptor. This hydrogen bonding interaction was later observed in the crystal structure of [Ru(η6-p-cym)(9-EtA)(Ph2acac)]+ (16b) (Ph2acac, diphenylacetylacetonate) (85). In contrast, the crystal structure of [Ru(η6-p-cym)(9-EtG)(Ph2acac)]+ reveals a repulsive interaction between the exocyclic C6O carbonyl and the acac oxygen, which further explains the weaker affinity for G compared with the en complexes (85). Indeed, competitive binding of Guo and Ado showed a slightly higher affinity for Ado, but the adducts were found to be kinetically labile.
Interactions with Oligonucleotides
Binding studies of ruthenium-arene complexes with oligonucleotides have provided insight into their modes of interaction with duplex DNA. The reaction of [Ru(η6-bip)Cl(en)]+ (10) with the single-strand DNA 14-mer d(ATACATGGTACATA) or its complementary strand d(TATGTACCATGTAT) gave rise to mono-ruthenated and/or di-ruthenated species depending on the Ru:oligonucleotide ratio, consistent with selective binding at G (86). Surprisingly, when a ruthenated single strand was annealed with its complementary strand, the product was not simply a double strand ruthenated at a single site. 2D NOESY NMR data showed that all four guanine residues were ruthenated at N7. Hence, we concluded that ruthenation is indeed highly specific, but also that {Ru(η6-bip)(en)}2+ is mobile at elevated temperatures (353 K) and migration between guanine residues can be facile. This contrasts with observations for DNA with bound Pt(II) am(m)ines for which migration is rare.
Intramolecular NOEs and other NMR data were consistent with arene ring intercalation between DNA bases. Molecular models suggested two different interactions (Figure 20). One in which the ruthenium complex was bound to G and its pendant arene ring is intercalated between a G and T residue, and a second in which the ruthenium arene is stacked on a flipped-out T on the surface of the major groove, i.e. is non-intercalated, forcing a T base to stack underneath it by tilting. Furthermore the data suggest that arene intercalation is dynamic: equilibria can exist between the intercalated and non-intercalated conformers. We also reported on the ruthenation of single- and double-stranded self-complementary hexameric DNA (d(CGGCCG)) (87). The complexes [Ru(η6-arene)Cl(en)]+, arene = p-cym (22) and bip (10), in this study were chosen to compare potential intercalators (bip) with non-intercalators (p-cym). For ss-DNA, all three G’s were readily ruthenated, but for duplex DNA a preferential ruthenation of G3 and G6, and no binding to G2 was detected. This was attributable to unfavorable steric interactions between the duplex and arene for binding at G2. The differences between the two arenes manifested themselves in different strengths of hydrogen bonding between the amine NH to the C6O carbonyl of G. The intercalation of the pendant phenyl ring in the bip adduct resulted in weakening of the stereospecific hydrogen bond compared to the p-cymene adduct. The arene ligand plays a major role in distorting the duplex either through steric interaction (p-cymene) or through intercalation (biphenyl) (87).
Figure 20.

Molecular models of two conformers of 14-mer duplex d(ATACATGGTACATA) ruthenated at N7 of one of the guanine residues with [Ru(η6-bip)Cl(en)]+ (10). Conformer a) shows the nonintercalated phenyl ring of the arene stacked on a thymine residue.
This work provides important evidence for elucidating the cytotoxic effect of the ruthenium-arene complexes and the influence of the arene thereon, for instance with respect to excision repair of DNA lesions and DNA destabilization. It also established two different classes of Ru(II) arene anticancer drugs, i.e. those bearing an arene that has the possibility to intercalate and those that do not. This distinction is important as we will see further differences in DNA binding interactions for these two classes (vide infra).
Interactions with DNA
In collaboration with the Brabec group, we studied the DNA interactions of several organometallic [RuCl(η6-arene)(en)]+ complexes in cell-free media. Binding studies using calf thymus (CT) double-helical DNA (ri = 0.1) showed that the bip, dha, and tha complexes 10-12 bind about an order of magnitude faster than cisplatin, with t½ values of 10, 15, and 10 min, respectively (88). The influence of the arene is remarkable as for p-cymene (22) a t½ of 3.5 h was obtained. These results correlate well with the ability of the arene for intercalation/π-π stacking. DNA binding was almost quantitative and irreversible and transcription mapping experiments identified G as the preferential binding site, consistent with our previous studies on the DNA models. DNA binding of the achiral Ru(II) complexes with bip, dha, or tha as the arenes induced a sizeable circular dichroism (CD) spectrum for Ru(II)-arene absorption bands, whereas p-cym binding did not result in an induced CD band. Linear dichroism (LD) data showed that the p-cym complex stiffened DNA, while the other complexes bend it. Differential pulse polarography measurements demonstrated nondenaturational alterations in DNA with bip, dha, and tha and denaturational alterations with p-cym (88). All these observations point to differences in binding interactions between ruthenium-arene complexes that can intercalate and those that cannot. Interestingly, the adducts of the p-cym complex distort the conformation and thermally destabilize DNA distinctly more that the other three adducts (88). This is remarkable since an intercalating arene enhances cytotoxicity in a number of tumor cell lines (59, 89). It is known that biological activity is modulated by the ‘downstream’ effects of damaged DNA, such as recognition of damaged DNA by specific proteins and/or repair. The different distortions might lead to different ‘downstream’ effects, which could eventually explain the differences in observed cytotoxicity.
Further experiments focused therefore on [RuCl(en)(η6-tha)]+ (12) and [RuCl(η6-p-cym)(en)]+ (22), which represent the two different classes, and their conformational distortion of short oligonucleotide duplexes. Chemical probes demonstrated that the induced distortion extended over at least 7 basepairs for [RuCl(η6-p-cym)(en)]+ (22), whereas the distortion was less extensive for [RuCl(en)(η6-tha)]+ (12). Isothermal titration calorimetry also showed that the thermodynamic destabilization of duplex was more pronounced for [RuCl(η6-p-cym)(en)]+ (22) (89). DNA polymerization was markedly more strongly inhibited by the monofunctional Ru(II) adducts than by monofunctional Pt(II) compounds. The lack of recognition of the DNA monofunctional adducts by HMGB1, an interaction that shields cisplatin-DNA adducts from repair, points to a different mechanism of antitumor activity for the ruthenium-arenes. DNA repair activity by a repair-proficient HeLa cell-free extract (CFE) showed a considerably lower level of damage-induced DNA repair synthesis (about 6 times) for [RuCl(en)(η6-tha)]+ compared to cisplatin. This enhanced persistence of the adduct is consistent with the higher cytotoxicity of this compound (89).
DNA binding studies on the series of isomeric [Ru(η6-arene)Cl(en)]+ compounds with o-, m- or p-terphenyl as the arene further illustrate the importance of the hydrophobicity and intercalation potential of the arene. The more extended p-terphenyl isomer 14 showed faster binding, a larger unwinding angle and a more significant distortion of the duplex compared to the m-terphenyl isomer (74).
The tethered bifunctional complexes [Ru(η6:η1-C6H5CH2(CH2)nNH2)Cl2] (n = 1,2) (13a,b) readily bound to CT DNA but failed to produce stop sites on the pSP73KB plasmid DNA for RNA synthesis and very low amounts of cross-linking were observed, indicating the formation mainly of monofunctional adducts on DNA. This, together with a small observed unwinding angle, may explain why these complexes exhibit low cytotoxicities (64).
Dinuclear ruthenium-arene complexes can also bind rapidly to CT DNA, again preferentially to G bases. DNA-directed RNA synthesis was inhibited more effectively than by the corresponding mononuclear complex (70). A large unwinding angle of 31° induced by binding of the dinuclear complex [{RuCl(η6-bip)}2(en)2-(CH2)6]2+ (23) to pSP3KB DNA, twice that of the mononuclear complex (14°), attributable to DNA crosslinking and structure perturbation by the two pendant phenyl rings (70). These results illustrate the synergistic effects of tethering the complexes together.
Interactions with Biomolecules
Candidate metallodrugs encounter a plethora of biomolecules upon administration, be it in, for instance, the blood plasma, the cell membrane, or the cytosol, before they reach the proposed target site (e.g. DNA in the nucleus or mitochondrion). The study of the interactions between the metal-based anticancer agent and proteins, antioxidants and other cellular components is therefore of key importance in understanding both the biological activity both in vitro and in vivo. Such interactions can be responsible for drug inactivation (related to resistance) or activation (e.g. in the case of prodrugs) and drug delivery. Indeed, the biomolecule can even be the actual target of the metallodrug. Whereas the interaction of ruthenium-arene complexes with (models of) DNA has been extensively investigated, protein interactions are much less well characterized. Advances in particular in mass spectrometric methods, in combination with NMR and single crystal X-ray diffraction studies, now provide an opportunity to map the interactions of ruthenium-arene anticancer drugs with the proteome and identify key protein targets. This area is currently attracting much interest (53).
We reported the first crystal structure of a half-sandwich arene ruthenium(II)-enzyme complex (Figure 21) (90). The crystal structure showed a {Ru(η6-p-cym)X2} half-sandwich fragment bound selectively to Nε of the imidazole ring of the only histidine residue (His15) in egg-white lysozyme, a small 14 kDa single-chain protein. The electron density for the additional two ligands on ruthenium was modeled as chloride, although partial occupancy by water cannot be ruled out. The selective binding of the Ru-fragment places the complex in an environment that is asymmetric by nature. Since there is much current interest in ruthenium-catalyzed enantioselective synthesis, these results suggest that such sites can provide a basis for the design of novel catalytic centers (90).
Figure 21.
X-ray crystal structure of a half-sandwich arene ruthenium(II)-enzyme complex: [RuCl2(η6-p-cym)(lysozyme)]
Horse heart cytochrome c also has two solvent-accessible histidine residues, His26 (interior residue) and His33 (surface exposed), but our NMR and ESI-MS binding studies of [Ru(η6-bip)Cl(en)]+ (10) show no binding to the histidine residues even with an 10-fold excess of ruthenium. Monoruthenated enzymes were detected both in water (pH 8.7) and buffer (pH 7.6) and ICP-AES revealed that 50 % of cytochrome c was ruthenated. Surprisingly, 2D [1H, 15N] HSQC NMR data suggest that ruthenium is bound to the N-terminal amino group or to a carboxylate group (Glu, Asp or C-terminus), rather than to the histidines (91).
Histidine residues are, however, generally regarded as major possible binding sites for ruthenium-arene complexes in proteins. To model this interaction, we also studied the reaction of [RuCl(en)(η6-bip)]+ (10) with l-histidine at 310 K in aqueous solution (91). The reaction was quite sluggish and did not reach equilibrium until 24 h at 310 K, by which only about 22% of the complex had reacted. Two isomeric imidazole-bound histidine adducts could be discerned, with more or less equal binding through Nε and Nδ to ruthenium. Competitive binding experiments of [Ru(η6-bip)Cl(en)]+ with an oligonucleotide and either l-histidine or cytochrome c showed almost selective binding to the oligonucleotide and neither the amino acid nor the protein can compete effectively with the guanine site in the oligonucleotide (91).
Reaction with sulfur-containing amino acids l-cysteine and l-methionine appear to play a major role in the biological chemistry of both Pt(II), Pt(IV) and Ru(III) anticancer agents, making it important to study these interactions with Ru(II)-arenes as well. [Ru(η6-bip)Cl(en)]+ (10) reacts slowly with l-cysteine with only 50% of the complex reacting after 48 h (92). Initially, S- or O-bound 1:1 [Ru(η6-bip)(l-Cys)(en)]+ adducts are formed after hydrolysis of the chlorido complex. The mononuclear adducts then convert to the final products, which could be identified as unusual dinuclear ruthenium complexes, in which the one or two en chelates have been displaced to form singly or doubly S-bridged adducts. Methionine binding was found to be equally slow, with only 23% of the ruthenium complex reacted after 48 h to give S-bound [Ru(η6-bip)(en)(S-l-Met)]2+.
These amino acid and protein binding results might account for the low toxic side effects of this class of anticancer agents (72). On the other hand, the relatively weak binding to amino acids and proteins could perhaps aid in transport and delivery of active species to cancer cells prior to binding to DNA or RNA (75).
Of special interest is the interaction of the family of ruthenium-arenes with the tripeptide glutathione (γ-l-Glu-l-Cys-Gly; GSH). This major intracellular antioxidant, present at millimolar concentrations in the cell, is known for its ability to detoxify heavier transition metal ions, including some platinum and ruthenium anticancer complexes (93). Given the abundance of GSH in the cell, competition experiments with GSH and cGMP were undertaken to obtain more insight into binding selectivity of the ruthenium-arene anticancer complexes and to determine whether a large molar excess of GSH could prevent nucleobase binding. Our initial studies on the reaction of [Ru(η6-bip)Cl(en)]+ and GSH gave some surprising results (Figure 22) (94). Under physiological and anaerobic conditions (20 μM [Ru], 5 mM GSH, pH = 7 buffered solution, 310 K), the reaction yielded the thiolato adduct [Ru(η6-bip)(en)(S-GS)]+ as the major product. Unexpectedly, under an O2 atmosphere, the thiolato adduct fully converted to a sulfenato complex in 48 h, strongly suggesting that the sulfenato originates from oxidation of the thiolato complex. The sulfenate is likely to be coordinated through S rather than O, as indicated by the observed S=O stretching frequency. Sulfenates are generally too reactive to be isolated, but can be stabilized by coordination to a transition metal.
Figure 22.

Remarkable ‘activation-by-ligand-oxidation’ pathways for the reaction of ruthenium-arenes with thiolates. a) reaction of [Ru(η6-bip)(en)(OH2)]+ with GSH; b) direct synthesis of ruthenium-arene sulfenato complexes; c) the air-stable thiolato complexes are oxidized in the presence of the antioxidant GSH.
Competition experiments with 250 mol equiv GSH and 25 mol equiv cGMP subsequently showed that although initially both glutathione (thiolato and sulfenato) and cGMP adducts were formed, after 72 hours the cGMP adduct was identified as the dominant product of the reaction. Our results indicated that the sulfenato ligand is readily displaced by cGMP, whereas the thiolato ligand is not (Figure 22). The oxidation to sulfenate (perhaps followed by protonation, vide infra) could possibly weaken the ruthenium-sulfur bond and in this way introduce a good leaving group. X-ray absorption studies that provide insight into the covalency of the metal-sulfur bonds of the thiolato, sulfenato and sulfinato complexes are currently under way (95). This ‘activation-by-ligand-oxidation’ is remarkable and clearly distinguishes these complexes from, for instance, cisplatin. It provides a facile route for DNA and RNA ruthenation even in the presence of a large excess of GSH (94). These observations also shed more light on the activity of [Ru(en)(η6-hmb)(SPh)]+ (18), a complex of significant, but also initially somewhat unexpected cytotoxicity, given that the complex does not hydrolyze (75).
We further explored the oxidation of a ruthenium-coordinated thiolate and the chemistry of the resulting sulfenato-complexes (Figure 22) (96). Sulfenato complexes could be directly obtained from the oxidation of [Ru(η6-arene)(en)(SR)]+ with hydrogen peroxide. The resulting sulfenato complexes are stable towards hydrolysis at pH 7 at ambient temperature. The ruthenium-sulfenato complex is rather basic (pKa of 3.37 for [Ru(en)(η6-hmb)(S(O)iPr)]+) (24b) and protonation at lower pH leads to hydrolysis (96). The thiolato complexes are not sensitive to air, in contrast to the analogous GSH adducts. Surprisingly, however, we found that [Ru(en)(η6-hmb)(SiPr)]+ (24a) could be fully oxidized to [Ru(en)(η6-hmb)(S(O)iPr)]+ (24b) by dioxygen in the presence of GSH, with concomitant conversion of the latter to GSSG. The oxygen atom transfer from an intermediate of GSH autooxidation to the organometallic ruthenium arene thiolato complex was remarkably efficient (97).
Our work on thiolato oxidation is of broader relevance, as there is much current interest in the function of protein cysteinyl sulfenates in signal transduction, oxygen metabolism, oxidative stress, and their role in the activity of nitrile hydratase (98-100).
Catalytic Activity of Ruthenium-Arene Complexes
Although a different mechanism of action than cisplatin is assumed for the family of cytotoxic ruthenium-arene compounds, the general reactivity assumed for most of the cytotoxic ruthenium-arene complexes is consistent with the classic cisplatin paradigm of aquation followed by DNA binding, which in turn leads to ‘downstream’ effects that ultimately trigger apoptosis. The reactivity of the cytotoxic agent is therefore limited to a one-off event. It would be desirable to develop metallo-anticancer agents that exert their cytotoxic effect in a catalytic manner, rather than the common single reactivity. The possibility of multiple turnovers would potentially increase the potency of a drug and thus allow lower doses to be administered to the patient.
The exceptional ability of bioorganometallics to acts as catalysts has not yet been widely explored (101). Half-sandwich ruthenium complexes, for instance, are highly active catalysts in a wide variety of chemical transformations, most notably transfer hydrogenations, amongst many others. With this in mind, we have studied the regioselective reduction of NAD+ to NADH by [Ru(η6-arene)Cl(en)]+ complexes using formate as the hydrogen donor under physiological conditions (102). NAD(P)+/NAD(P)H is one of the major redox couples found in the cell involved in many electron transfer reactions. Off-setting the NAD(P)+/NAD(P)H equilibrium by catalytic conversion might be a way of exerting a cytotoxic effect through disturbing the cell’s redox state. The reactivity was again found to be highly dependent on the arene, with [RuCl(en)(η6-hmb)]+ (25) giving the best results. Turnover frequencies were modest (1.46 h−1 at saturation, pH 7.4 and 37 °C), however, and formation of the ruthenium-hydride species proved rate-limiting. Lung cancer cells were found to be remarkably tolerant to formate even at millimolar concentrations, opening up the possibility of in vivo biocatalysis if more active organometallic complexes would become available (102).
One point of concern for in vivo activity is that metal-based catalysts might readily be inactivated by poisoning. The myriad of cellular components such as thiols that are present in millimolar concentrations, e.g. GSH, provide a challenge for organometallic complexes. It is therefore intruiging that [Ru(η6-arene)(azpy)I]+ complexes can act as catalysts for the oxidation of the major intracellular reducing agent glutathione to glutathione disulfide, in a cycle which appears to involve ligand-centered redox reactions (Figure 23) (77). The influence of the halide is remarkable in this case as the chloride complex is rather unstable and not cytotoxic/catalytically active. The [Ru(η6-arene)(azpy)I]+ complexes do not hydrolyse, but do deplete millimolar amounts of GSH over 24 h by catalytic oxidation to GSSG under physiological conditions. Concomitant hydrogenation of dissolved dioxygen leads to the formation of hydrogen peroxide and, a reactive oxygen species (ROS), which which are thought to be ultimately responsible for the cell death. The catalytic ruthenium anticancer agents thus lead to oxidative stress by simultaneously producing ROS and consuming its antioxidant defenses in the cell (77). It is important to note that the azpy ligands themselves do not show this type of reactivity and the ligands are catalytically active only upon coordination to the metal center, i.e. controlling ligand reactivity by metal-ion coordination (77).
We started this section by stating that the advent of bioorganometallics provides the medicinal chemist with access to new types of reactivity and therefore with new opportunities for anticancer drug design. Our studies on the ruthenium-arene anticancer drugs and results by others show that ruthenium-based compounds in particular do live up to this expectation. The general structure of the ruthenium-arenes provides a versatile platform for structural modification and activity optimization. Correlating structure and reactivity with cytotoxicity is complicated, but some general trends can be discerned. This way, control can be exerted over some kinetic (e.g. hydrolysis rate) and thermodynamic (pKa, specific binding interactions) parameters that govern both activation of the drug and the strength of the interaction with cellular targets. It is interesting to note here that not only the complexes on the (relatively) fast side of the scale of ligand-exchange exhibited good cytotoxicity. Some kinetically inert complexes proved equally cytotoxic and presumably operate through completely different, non-classical mechanisms of action. A good example of the latter is the simultaneous depletion of GSH with concomitant generation of ROS by a catalytically active Ru(II)-arene anticancer agent (77). These results show the scope for design of ruthenium-arene anticancer drugs, and in particular their ability to operate in ways beyond the classical cisplatin-paradigm of selective DNA-binding. A major aim that needs to be addressed in future work is the targeted delivery of organometallic anticancer drugs to cancer cells only. Also, additional features that generate cytotoxic activity other than disruption of DNA replication, such as the inclusion of molecular fragments that can interfere in cancer cell-specific cellular pathways, could be explored (11, 103).
IV. Osmium(II)-Arenes: A cytotoxic family of the heavier congener
The application of organometallic complexes of the other group 8 elements, iron and osmium, in anticancer drug design has until recently been almost exclusively focused on iron, with the ferrocenyl derivative of tamoxifen (ferrocifen) being the most prominent example (104). Organometallic osmium compounds have been little explored in this respect.
We have recently extended our interest to the analogous half-sandwich osmium-arene complexes and are exploring the chemical and biological properties of [Os(η6-arene)(XY)Z]n+ complexes (Figure 25) (105). Both the aqueous chemistry and the biological activity of osmium complexes have been little studied. Third row transition metals are usually considered to be more inert than those of the first and second rows. Similar to the five orders of magnitude decrease in substitution rates of Pt(II) complexes compared to Pd(II), the [Os(η6-arene)(L)X]n+ complexes were expected to display rather different kinetics than their Ru(II)-arene analogues. A few other reports on the anticancer activity of osmium-arene complexes have also appeared recently (106-108).
Figure 25.

a) General aqueous reactivity of osmium(II)-arenes: the formation of a hydroxido-bridged dimer can play a major role in the aqueous chemistry of osmium(II)-arenes, especially if the chelate XY is an O,O-bidentate ligand; b) molecular structures of the osmium-arene anticancer agents 26-30.
Structure and Reactivity
In our initial studies (105), we looked at the structure and reactivity of [OsCl(L)(η6-arene)]n+ with N,N-chelating ligand en (26) and O,O-chelating ligand acac (27a,b) (Figure 25). X-ray crystal structure determinations showed the complexes to be isostructural with their ruthenium analogues, with the same M–Cl bond lengths, even though the ligand exchange rates are different (vide infra) (Figure 24). The hydrolysis of [Os(η6-bip)Cl(en)]+ (26) is ca. 40 times slower (t½ 6.4 h) than that of its Ru(II) analogue 10, consistent with often-observed slower exchange rates on osmium compared to ruthenium.
Figure 24.

Comparison between the osmium- and ruthenium-arenes, exemplified by the respective [M(η6-bip)Cl(en)]+ complexes. Although the crystal structures show the complexes to be isostructural with similar M–Cl bond lengths (a), the properties of the complexes are quite different, illustrated by the differences in hydrolysis rate (t1/2), pKa and 5′-GMP binding (the black box denotes the amount of OPO3-bound 5′-GMP), (b).
It is interesting, however, that similar water exchange rates have been reported for the Os(II) and Ru(II) complexes [M(η6-benzene)(OH2)3]2+ (109), indicative of a major influence of the chelating ligand. Indeed, changing the chelating ligand to acac resulted in a significant increase in rate and extent of aquation. DFT calculations show that aquation of [Os(η6-arene)Cl(L)] is indeed more facile for L = acac compared to L = en, and the mechanism is more dissociative in nature (110). However, hydroxido-bridged dimers [Os2(η6-arene)2(μ-OH)3]+ play a dominant role in the aqueous chemistry of [Os(acac)(η6-arene)Cl] (27a,b), whereas they are only a minor component, and then only at high pH, in the ruthenium systems (Figure 25). The hydroxido-bridged dimer is the only species present on dissolution of [Os(acac)Cl(η6-p-cym)] (27a) at micromolar concentrations, conditions relevant for biological testing. The pKa values of the aqua complexes are all significantly lower (ca. 1.5 units) than those for the corresponding ruthenium-arene complexes, e.g. 6.34 for [Os(η6-bip)(en)(OH2)]2+ and 7.12 for [Os(acac)(η6-bip)(OH2)]+. Therefore, at physiological pH almost all of the hydrolyzed en complex would be present in the hydroxide form, [Os(η6-bip)(en)(OH)]+. The higher acidity of osmium arene aqua complexes proved to be general and can be attributed to increased mixing of the δσ* (Os) → σ (OH−) orbitals (105).
The aqueous chemistry of N,O-chelated complexes appeared to be intermediate between that of the neutral N,N-chelates and anionic O,O-chelates, with significant effects of the chosen N- and O-group (111). The aminoacidate complexes hydrolyzed rapidly, but replacement of the primary amine by the π-acceptor pyridine slowed down the rate of hydrolysis. Important was the observation that the inactive aminoacidate complexes again proved unstable with respect to hydroxido-bridged dimer formation, whereas no such dimer formation was observed with the active pico (28) and oxine (30) complexes (vide infra for cytotoxicity). The strength of the Os–N(pyridine) bond seems crucial for the stability of the complex. At high chloride concentrations typical of blood plasma (ca. 100 mM) the complex [OsCl(η6-p-cym)(pico)] (28a) is likely to be mainly present as the less reactive intact chlorido species. At lower [Cl−] concentration (4 mM, typical of cell nucleus), the complex is activated by hydrolysis. As previously stated for the ruthenium-arenes this presents an interesting prodrug activation strategy for DNA binding.
Nucleobase Binding
Nucleobase interactions showed slow interaction of [Os(η6-bip)Cl(en)]+ (26) with 9-EtG and then only to a limited extent (45% after 22 h). No binding to Ado, Cyt or Thy was observed. [Os(acac)(η6-bip)Cl] (27b) on the other hand binds rapidly to both 9-EtG and Ado (< 10 min), but not to Cyt or Thy. Notably, the hydroxo-bridged dimer was inert towards nucleobases (105). Two major adducts were observed in the reaction between [Os(η6-bip)Cl(en)]+ (26) and 5′-GMP attributable to N7 and phosphate adducts, which formed in a 2:1 N7/phosphate ratio (112) (Figure 24). Only about 40% of 5′-GMP is bound after 30 h. This contrasts with the 5′-GMP binding of [Ru(η6-bip)Cl(en)]+ (10), for which quantitative and almost selective binding to N7 is observed after 1 d (81).
Nucleobase competition binding experiments showed binding of [OsCl(η6-p-cym)(pico)] (28a) to both G and A, but with a strong preference for G (111). Intriguingly, a modest binding constant for G binding was found (log K 3.95), but the slow dissociation of 9-EtG at micromolar concentrations, makes it likely for G adducts on DNA or RNA to persist once formed. Little and no binding to the pyrimidine bases Cyt and Thy was observed.
Cytotoxicity
[OsCl(acac)(η6-arene)] (27a,b) showed no cytotoxicity towards A2780 and A549 cell lines, probably attributable to formation of the inert hydroxido-bridged dimer. Replacement of acac for the potentially more stable five-membered chelate ring of maltolato did provide some stabilization towards dimer formation compared to the acac complex, but under biologically relevant conditions the hydroxido-bridged dimer remained the dominant species (110). Although the complexes were not active, the results did show that kinetics of ligand substitution on osmium can be controlled by variation of the ligand and thus proved very valuable for our further studies.
Initial experiments showed that [Os(η6-bip)Cl(en)]+ (26) was not cytotoxic towards cancer cells (105), but a later reassessment of the cytotoxic activity of this compound showed that it indeed was active at micromolar concentrations (IC50 values of 7.6 (A2780) and 10 μM (A549)) (112). A possible explanation for the initial lack of activity may be the partial decomposition of the complex in stock test solutions prepared in DMSO, as was evidenced in subsequent studies (112). The cytotoxicity data are now more in line with the chemical properties of the complex, i.e. observed hydrolysis rate and guanine binding.
The use of an extended arene (tetrahydroanthracene) in [OsCl(en)(η6-tha)]+ (29) gave rise to a similar potency (112). This contrasts the data for ruthenium-arenes, where the same substitution gave rise to a 10-fold increase in activity. Further work therefore needs to determine if the extended Os-arenes can intercalate into DNA in a manner similar to Ru-arenes. Replacement of the N,N-chelating ligand en for other N,N-bidentates with pyridine, aliphatic amine or azopyridine donor atoms lead to loss of activity, probably because of slower hydrolysis and higher acidity of the coordinated water (112).
Further ligand variation led to the isolation of other osmium-arenes with moderate to high cytotoxicity towards cancer cells (111). We found that the aqueous chemistry of the organometallic osmium complexes could be fine-tuned so as to achieve cancer cell cytotoxicity by using mixed N,O-chelate ligands. Several aminoacidates, picoline (pico) and 8-hydroxyquinoline (oxine) were tested, but inclusion of pyridine as the N donor rather than a primary amine proved key for cytotoxic activity. The complexes [Os(η6-bip)Cl(pico)] (28b), [OsCl(η6-p-cym)(pico)] (28a), and [OsCl(η6-p-cym)(oxine)] (30) exhibit cytotoxic activity against human ovarian (A2780) and lung (A549) cancer cells, with IC50 values of 5-60 μM. Notable is the high activity of [Os(η6-bip)Cl(pico)] (28b) with IC50 values of 4.2 (A2780) and 8 μM (A549) (111). The four active complexes [Os(η6-bip)Cl(en)]+ (26), [Os(η6-bip)Cl(pico)] (28b), [OsCl(η6-p-cym)(pico)] (28a), and [OsCl(η6-p-cym)(oxine)] (30) all show similar potency towards cisplatin-sensitive and resistant A2780 cell lines, indicating a different detoxification mechanism than cisplatin. Intriguingly, [Os(η6-bip)Cl(en)]+ (26) shows an even greater activity in the resistant cell line (resistance factor 0.55) (113).
DNA binding
An in-depth study of osmium-arene anticancer drug binding to DNA was carried out in collaboration with the group of Brabec (113). It is notable that the tested complexes all bind polymeric DNA. The complexes [Os(η6-bip)Cl(pico)] (28b) and [OsCl(η6-p-cym)(oxine)] (30) bind rapidly to CT DNA (t½ ca. 2 h. at ri = 0.1), whereas [Os(η6-bip)Cl(en)]+ (26) and [OsCl(η6-p-cym)(pico)] (28a) bind up to two and four times more slowly with t½ values of 4.6 and 8.3 h, respectively (113). For comparison, the t½ of DNA binding of the ruthenium-arene [Ru(η6-bip)Cl(en)]+ is ca. 10 min, approximately 28 times faster than its osmium analogue, which might allow more of the latter to reach its target site (88). The extent of DNA binding ranges from ca. 72% for [OsCl(η6-p-cym)(pico)] (28a) to 95% for [Os(η6-p-bip)Cl(pico)] (28b), illustrating the importance of an extended arene ring for DNA binding. RNA synthesis transcription mapping showed the major stop sites to be guanine residues (with some minor ones as adenine), similar to cisplatin and the ruthenium analogues. Electrophoretic mobility experiments on osmium-adducts of DNA duplex oligonucleotides (up to 21 bp) showed that no DNA bending is induced upon osmium binding. The binding of the osmium-arene complex leads to a large degree of unwinding (21-27°) (except for the oxine complex) (113), much larger than that observed for ruthenium(II) complexes (88), This large unwinding angle might be explained by an additional interaction of the arene ligand with the duplex upon strong binding of osmium. The complex [OsCl(η6-p-cym)(oxine)] (30) on the other hand shows hardly any significant unwinding of DNA (< 2.5 °). This suggests that this complex interacts with DNA in a different manner, which could correlate with its markedly lower cytotoxic activity (113).
In line with expectations of kinetic inertness of third-row transition metals, little interest has been vested in the development of osmium anticancer drugs, as ligand-exchange rates did not seem favorable on the timescale of cellular processes. Our work, however, shows that the kinetic lability of such complexes can be tuned to such extent that anticancer activity comes within range. We have demonstrated how rational chemical design can thus be applied to osmium-arene complexes resulting in specific windows of reactivity, stability, and cancer cell cytotoxicity (111). This has allowed us to design complexes with cytotoxicities comparable to their ruthenium analogues, but with reactivities that are 100 times less. Such a range of kinetic effects can be useful for balancing cytotoxicity and unwanted side-effects of anticancer drugs, as illustrated previously by the clinical profiles of cisplatin and the less-labile second generation drug carboplatin (112). The osmium complexes are therefore interesting candidates for further investigation.
V. Concluding Remarks
In general transition metal complexes provide enormously versatile platforms for drug design. Many variations in the metal itself, the types and numbers of coordinated ligands and hence in the strengths of coordination bonds and in the kinetics of ligands substitution processes are available. In this review, we have highlighted our efforts towards the development of photoactive platinum anticancer agents and the ruthenium- and osmium-arene families of anticancer agents. The key to successful design of clinically useful drugs is effective activity whilst minimizing side effects. Targeting is important to ensure that sufficient amounts of the active drug reach the target site and that unwanted reactions do not occur along the way. Hence both the thermodynamics and kinetics of ligand exchange and redox processes must be carefully controlled. This presents a major challenge for the transition metal medicinal chemist- an exciting one too. We have shown that such control is indeed possible by systematic ligand variation. This way we were able to design photoactive platinum(IV) complexes that are stable and non-toxic in the dark. The strategy of activation by light to yield highly cytotoxic species at irradiated spots only allows for local, targeted treatment and holds the promise of less invasive chemotherapy. Similarly, our work on the ruthenium- and osmium-arenes showed that thermodynamic and kinetic parameters, such as those associated with aquation, can vary over several orders of magnitude, thus allowing the chemist to fine-tune the properties of the agent for increased cytotoxicity. Not only is direct coordination to the metal important in biological recognition processes but so are second coordination sphere interactions, e.g. H-bonding, hydrophobic interactions, as exemplified by the stereospecific hydrogen bonding interactions of the ruthenium-arenes and arene intercalation upon coordination of the complex to DNA.
While for the photoactive Pt(IV) complexes both metal- and ligand-centered redox reactions can be involved in biological mechanisms of action, it is interesting to note that ligand-centered redox processes can also dominate reactions of arene complexes. The catalytic depletion of GSH by azopyridine complexes with concomitant generation of ROS, for instance, is a ligand-based process, which is nonetheless modulated and made possible by coordination of the ligand to ruthenium. Such reactions are often not possible for the ligands alone in purely organic drugs. Additionally, metal complexes themselves are renowned as catalysts and the possibility of designing catalytically-active metallo-drugs which are not prematurely poisoned in the biological system represents a significant challenge.
Figure 26.

Bar charts relate the influence of different chelates in [Os(η6-arene)Cl(XY)]n+ (XY = N,N- N,O- or O,O-) on cytotoxicity, stability with respect to hydroxido-dimer formation, hydrolysis rates, and pKa of the aqua adduct for osmium-arene complexes. Shading indicates the range in observed values. Adapted from ref. 111.
VI. Acknowledgments
PCAB thanks the Netherlands Organization for Scientific Research (NWO) for financial support through a Rubicon Scholarship. We thank the EPSRC, BBSRC, Royal Society, Wellcome Trust, EC (Marie Curie and COST), Scottish Enterprise, and Oncosense Ltd for support of our recent research on therapeutic metal complexes. The University of Edinburgh (former employer of PJS) has filed patent applications relating to the ruthenium-arene, and platinum diazido complexes and University of Warwick for osmium-arene complexes under study in the PJS laboratory.
VII. References
- 1.Krogsgaard-Larsen P, Liljefors T, Madsen U, editors. Textbook of Drug Design and Discovery. Tayler & Francis; London: 2002. [Google Scholar]
- 2.Guo Z, Sadler PJ. Angew. Chem. Int. Ed. 1999;38:1513–1531. doi: 10.1002/(SICI)1521-3773(19990601)38:11<1512::AID-ANIE1512>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 3.Hambley TW. Dalton Trans. 2007:4929–4937. doi: 10.1039/b706075k. [DOI] [PubMed] [Google Scholar]
- 4.Mukherjee A, Sadler PJ. Wiley Encyclopedia of Chemical Biology. 2008. Metals in Medicine: An Introductory Review. in press. [Google Scholar]
- 5.Ronconi L, Sadler PJ. Coord. Chem. Rev. 2007;251:1633–1648. doi: 10.1016/j.ccr.2008.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Storr T, Thompson KH, Orvig C. Chem. Soc. Rev. 2006;35:534–544. doi: 10.1039/b514859f. [DOI] [PubMed] [Google Scholar]
- 7.Hall MD, Mellor HR, Callaghan R, Hambley TW. J. Med. Chem. 2007;50:3403–3411. doi: 10.1021/jm070280u. [DOI] [PubMed] [Google Scholar]
- 8.Jung Y, Lippard SJ. Chem. Rev. 2007;107:1387–1407. doi: 10.1021/cr068207j. [DOI] [PubMed] [Google Scholar]
- 9.Van Zutphen S, Reedijk J. Coord. Chem. Rev. 2005;249:2845–2853. [Google Scholar]
- 10.Kelland L. Nat. Rev. Cancer. 2007;7:573–584. doi: 10.1038/nrc2167. [DOI] [PubMed] [Google Scholar]
- 11.Bruijnincx PCA, Sadler PJ. Curr. Opin. Chem. Biol. 2008;12:197–206. doi: 10.1016/j.cbpa.2007.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rautio J, Kumpulainen H, Heimbach T, Oliyai R, Oh D, Jarvinen T, Savolainen J. Nat. Rev. Drug. Discov. 2008;7:255–270. doi: 10.1038/nrd2468. [DOI] [PubMed] [Google Scholar]
- 13.Rose MJ, Mascharak PK. Curr. Opin. Chem. Biol. 2008;12:238–244. doi: 10.1016/j.cbpa.2008.02.009. [DOI] [PubMed] [Google Scholar]
- 14.Johnson TR, Mann BE, Clark JE, Foresti R, Green CJ, Motterlini R. Angew. Chem. Int. Ed. 2003;42:3722–3729. doi: 10.1002/anie.200301634. [DOI] [PubMed] [Google Scholar]
- 15.Reedijk J. Platinum Metals Rev. 2008;52:2–11. [Google Scholar]
- 16.Helm L, Nicolle GM, Merbach AE. Adv. Inorg. Chem. 2005;57:327–379. [Google Scholar]
- 17.Hall MD, Failes TW, Yamamoto N, Hambley TW. Dalton Trans. 2007:3983–3990. doi: 10.1039/b707121c. [DOI] [PubMed] [Google Scholar]
- 18.Kratz F, Muller IA, Ryppa C, Warnecke A. ChemMedChem. 2008;3:20–53. doi: 10.1002/cmdc.200700159. [DOI] [PubMed] [Google Scholar]
- 19.Farrer NJ, Sadler PJ. Austr. J. Chem. 2008;61:669–674. [Google Scholar]
- 20.Szacilowski K, Macyk W, Drzewiecka-Matuszek A, Brindell M, Stochel G. Chem. Rev. 2005;105:2647–2694. doi: 10.1021/cr030707e. [DOI] [PubMed] [Google Scholar]
- 21.Bednarski PJ, Mackay FS, Sadler PJ. Anti-Cancer Agents Med. Chem. 2007;7:75–93. doi: 10.2174/187152007779314053. [DOI] [PubMed] [Google Scholar]
- 22.Choy H, Park C, Yao M. Clin. Cancer Res. 2008;14:1633–1638. doi: 10.1158/1078-0432.CCR-07-2176. [DOI] [PubMed] [Google Scholar]
- 23.Kratochwil NA, Zabel M, Range K-J, Bednarski PJ. J. Med. Chem. 1996;39:2499–2507. doi: 10.1021/jm9509105. [DOI] [PubMed] [Google Scholar]
- 24.Kratochwil NA, Guo Z, Del Socorro Murdoch P, Parkinson JA, Bednarski PJ, Sadler PJ. J. Am. Chem. Soc. 1998;120:8253–8254. [Google Scholar]
- 25.Kratochwil NA, Parkinson JA, Bednarski PJ, Sadler PJ. Angew. Chem. Int. Ed. 1999;38:1460–1463. doi: 10.1002/(SICI)1521-3773(19990517)38:10<1460::AID-ANIE1460>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 26.Vogler A, Hlavatsch J. Angew. Chem. Int. Ed. 1983;22:154–155. [Google Scholar]
- 27.Vogler A, Kern A, Huttermann J. Angew. Chem. Int. Ed. 1978;17:524–525. [Google Scholar]
- 28.Muller P, Schroder B, Parkinson JA, Kratochwil NA, Coxall RA, Parkin A, Parsons S, Sadler PJ. Angew. Chem. Int. Ed. 2003;42:335–339. doi: 10.1002/anie.200390110. [DOI] [PubMed] [Google Scholar]
- 29.Kasparkova J, Mackay FS, Brabec V, Sadler PJ. J. Biol. Inorg. Chem. 2003;8:741–745. doi: 10.1007/s00775-003-0474-3. [DOI] [PubMed] [Google Scholar]
- 30.Bednarski PJ, Grunert R, Zielzki M, Wellner A, Mackay FS, Sadler PJ. Chem. Biol. 2006;13:61–67. doi: 10.1016/j.chembiol.2005.10.011. [DOI] [PubMed] [Google Scholar]
- 31.Blisard KS, Harrington DA, Long DA, Jackson JE. J. Comp. Pathol. 1991;105:367–375. doi: 10.1016/s0021-9975(08)80106-5. [DOI] [PubMed] [Google Scholar]
- 32.Mackay FS, Woods JA, Moseley H, Ferguson J, Dawson A, Parsons S, Sadler PJ. Chem. Eur. J. 2006;12:3155–3161. doi: 10.1002/chem.200501601. [DOI] [PubMed] [Google Scholar]
- 33.Mackay FS. PhD Thesis. University of Edinburgh; Edinburgh: 2006. [Google Scholar]
- 34.Mackay FS, Moggach SA, Collins A, Parsons S, Sadler PJ. Inorg. Chim. Acta. DOI:10.1016/j.ica.2008.02.039. [Google Scholar]
- 35.Mackay FS, Woods JA, Heringova P, Kasparkova J, Pizarro AM, Moggach SA, Parsons S, Brabec V, Sadler PJ. Proc. Natl. Acad. Sci. U. S. A. 2007;104:20743–20748. doi: 10.1073/pnas.0707742105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ronconi L, Sadler PJ. Chem. Commun. 2008:235–237. doi: 10.1039/b714216a. [DOI] [PubMed] [Google Scholar]
- 37.Albota M, Beljonne D, Bredas J-L, Ehrlich JE, Fu J-Y, Heikal AA, Hess SE, Kogej T, Levin MD, Marder SR, McCord-Maughon D, Perry JW, Rockel H, Rumi M, Subramaniam G, Webb WW, Wu X-L, Xu C. Science. 1998;281:1653–1656. doi: 10.1126/science.281.5383.1653. [DOI] [PubMed] [Google Scholar]
- 38.Fish RH, Jaouen G. Organometallics. 2003;22:2166–2177. [Google Scholar]
- 39.Jaouen G, editor. Bioorganometallics. Wiley-VSH Verlag GmbH & Co; Weinheim: 2006. [Google Scholar]
- 40.Halpern J. Pure Appl. Chem. 2001;73:209–220. [Google Scholar]
- 41.Vessieres A, Top S, Beck W, Hillard E, Jaouen G. Dalton Trans. 2006:529–541. doi: 10.1039/b509984f. [DOI] [PubMed] [Google Scholar]
- 42.Alberto R, Schibli R, Waibel R, Abram U, Schubiger AP. Coord. Chem. Rev. 1999;190-192:901–919. [Google Scholar]
- 43.Dougan SJ, Sadler PJ. Chimia. 2007;61:704–715. [Google Scholar]
- 44.Yan YK, Melchart M, Habtemariam A, Sadler PJ. Chem. Commun. 2005:4764–4776. doi: 10.1039/b508531b. [DOI] [PubMed] [Google Scholar]
- 45.Rosenberg B, VanCamp L, Trosko JE, Mansour VH. Nature. 1969;222:385–386. doi: 10.1038/222385a0. [DOI] [PubMed] [Google Scholar]
- 46.Ang WH, Dyson PJ. Eur. J. Inorg. Chem. 2006:4003–4018. [Google Scholar]
- 47.Clarke MJ. Coord. Chem. Rev. 2002;232:69–93. [Google Scholar]
- 48.Jakupec MA, Galanski M, Arion VB, Hartinger CG, Keppler BK. Dalton Trans. 2008:183–194. doi: 10.1039/b712656p. [DOI] [PubMed] [Google Scholar]
- 49.Durig JR, Danneman J, Behnke WD, Mercer EE. Chem.-Biol. Interact. 1976;13:287–294. doi: 10.1016/0009-2797(76)90081-8. [DOI] [PubMed] [Google Scholar]
- 50.Rademaker-Lakhai JM, Van Den Bongard D, Pluim D, Beijnen JH, Schellens JHM. Clin. Cancer Res. 2004;10:3717–3727. doi: 10.1158/1078-0432.CCR-03-0746. [DOI] [PubMed] [Google Scholar]
- 51.Hartinger CG, Zorbas-Seifried S, Jakupec MA, Kynast B, Zorbas H, Keppler BK. J. Inorg. Biochem. 2006;100:891–904. doi: 10.1016/j.jinorgbio.2006.02.013. [DOI] [PubMed] [Google Scholar]
- 52.Bergamo A, Sava G. Dalton Trans. 2007:1267–1272. doi: 10.1039/b617769g. [DOI] [PubMed] [Google Scholar]
- 53.Timerbaev AR, Hartinger CG, Aleksenko SS, Keppler BK. Chem. Rev. 2006;106:2224–2248. doi: 10.1021/cr040704h. [DOI] [PubMed] [Google Scholar]
- 54.Reisner E, Arion VB, Keppler BK, Pombeiro AJL. Inorg. Chim. Acta. 2008;361:1569–1583. [Google Scholar]
- 55.Brindell M, Stawoska I, Supel J, Skoczowski A, Stochel G, van Eldik R. J. Biol. Inorg. Chem. 2008;13:909–918. doi: 10.1007/s00775-008-0378-3. [DOI] [PubMed] [Google Scholar]
- 56.Habtemariam A, Kelland L, Sadler PJ. unpublished results.
- 57.Morris R, Habtemariam A, Guo Z, Parsons S, Sadler PJ. Inorg. Chim. Acta. 2002;339:551–559. [Google Scholar]
- 58.Guo Z, Habtemariam A, Sadler PJ, James BR. Inorg. Chim. Acta. 1998;273:1–7. [Google Scholar]
- 59.Morris RE, Aird RE, Del Socorro Murdoch P, Chen H, Cummings J, Hughes ND, Parsons S, Parkin A, Boyd G, Jodrell DI, Sadler PJ. J. Med. Chem. 2001;44:3616–3621. doi: 10.1021/jm010051m. [DOI] [PubMed] [Google Scholar]
- 60.Allardyce CS, Dyson PJ, Ellis DJ, Heath SL. Chem. Commun. 2001:1396–1397. [Google Scholar]
- 61.Meggers E, Atilla-Gokcumen GE, Bregman H, Maksimoska J, Mulcahy SP, Pagano N, Williams DS. Synlett. 2007:1177–1189. [Google Scholar]
- 62.Smalley KSM, Contractor R, Haass NK, Kulp AN, Atilla-Gokcumen GE, Williams DS, Bregman H, Flaherty KT, Soengas MS, Meggers E, Herlyn M. Cancer Res. 2007;67:209–217. doi: 10.1158/0008-5472.CAN-06-1538. [DOI] [PubMed] [Google Scholar]
- 63.Muetterties EL, Bleeke JR, Wucherer EJ, Albright T. Chem. Rev. 1982;82:499–525. [Google Scholar]
- 64.Melchart M, Habtemariam A, Novakova O, Moggach SA, Fabbiani FPA, Parsons S, Brabec V, Sadler PJ. Inorg. Chem. 2007;46:8950–8962. doi: 10.1021/ic700799w. [DOI] [PubMed] [Google Scholar]
- 65.Pizarro AM, Melchart M, Sadler PJ. unpublished results.
- 66.Gray JC, Habtemariam A, Winnig M, Meyerhof W, Sadler PJ. J. Biol. Inorg. Chem. 2008;13:1111–1120. doi: 10.1007/s00775-008-0396-1. [DOI] [PubMed] [Google Scholar]
- 67.Habtemariam A, Melchart M, Fernandez R, Parsons S, Oswald IDH, Parkin A, Fabbiani FPA, Davidson JE, Dawson A, Aird RE, Jodrell DI, Sadler PJ. J. Med. Chem. 2006;49:6858–6868. doi: 10.1021/jm060596m. [DOI] [PubMed] [Google Scholar]
- 68.Therrien B, Ward TR, Pilkington M, Hoffmann C, Gilardoni F, Weber J. Organometallics. 1998;17:330–337. [Google Scholar]
- 69.Hoeschele JD, Habtemariam A, Muir J, Sadler PJ. Dalton Trans. 2007:4974–4979. doi: 10.1039/b706246j. [DOI] [PubMed] [Google Scholar]
- 70.Chen H, Parkinson JA, Novakova O, Bella J, Wang F, Dawson A, Gould R, Parsons S, Brabec V, Sadler PJ. Proc. Natl. Acad. Sci. U. S. A. 2003;100:14623–14628. doi: 10.1073/pnas.2434016100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang F, Aird RE, Habtemariam A, Jodrell DI, Sadler PJ, Guichard SM. unpublished.
- 72.Aird RE, Cummings J, Ritchie AA, Muir M, Morris RE, Chen H, Sadler PJ, Jodrell DI. Brit. J. Cancer. 2002;86:1652–1657. doi: 10.1038/sj.bjc.6600290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Guichard SM, Else R, Reid E, Zeitlin B, Aird R, Muir M, Dodds M, Fiebig H, Sadler PJ, Jodrell DI. Biochem. Pharmacol. 2006;71:408–415. doi: 10.1016/j.bcp.2005.10.053. [DOI] [PubMed] [Google Scholar]
- 74.Bugarcic T, Novakova O, Halamikova A, Zerzankova L, Vrana O, Kasparkova J, Habtemariam A, Parsons S, Sadler PJ, Brabec V. J. Med. Chem. 2008;51:5310–5319. doi: 10.1021/jm8003043. [DOI] [PubMed] [Google Scholar]
- 75.Wang F, Habtemariam A, Van Der Geer EPL, Fernandez R, Melchart M, Deeth RJ, Aird R, Guichard S, Fabbiani FPA, Lozano-Casal P, Oswald IDH, Jodrell DI, Parsons S, Sadler PJ. Proc. Natl. Acad. Sci. U. S. A. 2005;102:18269–18274. doi: 10.1073/pnas.0505798102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dougan SJ, Melchart M, Habtemariam A, Parsons S, Sadler PJ. Inorg. Chem. 2006;45:10882–10894. doi: 10.1021/ic061460h. [DOI] [PubMed] [Google Scholar]
- 77.Dougan SJ, Habtemariam A, McHale SE, Parsons S, Sadler PJ. Proc. Natl. Acad. Sci. U. S. A. 2008;105:11628–11633. doi: 10.1073/pnas.0800076105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wang F, Chen H, Parsons S, Oswald IDH, Davidson JE, Sadler PJ. Chem. Eur. J. 2003;9:5810–5820. doi: 10.1002/chem.200304724. [DOI] [PubMed] [Google Scholar]
- 79.Fairhurst MT, Swaddle TW. Inorg. Chem. 1979;18:3241–3244. [Google Scholar]
- 80.Fernandez R, Melchart M, Habtemariam A, Parsons S, Sadler PJ. Chem. Eur. J. 2004;10:5173–5179. doi: 10.1002/chem.200400640. [DOI] [PubMed] [Google Scholar]
- 81.Chen H, Parkinson JA, Morris RE, Sadler PJ. J. Am. Chem. Soc. 2003;125:173–186. doi: 10.1021/ja027719m. [DOI] [PubMed] [Google Scholar]
- 82.Melchart M, Habtemariam A, Parsons S, Moggach SA, Sadler PJ. Inorg. Chim. Acta. 2006;359:3020–3028. [Google Scholar]
- 83.Magennis SW, Habtemariam A, Novakova O, Henry JB, Meier S, Parsons S, Oswald IDH, Brabec V, Sadler PJ. Inorg. Chem. 2007;46:5059–5068. doi: 10.1021/ic062111q. [DOI] [PubMed] [Google Scholar]
- 84.Chen H, Parkinson JA, Parsons S, Coxall RA, Gould RO, Sadler PJ. J. Am. Chem. Soc. 2002;124:3064–3082. doi: 10.1021/ja017482e. [DOI] [PubMed] [Google Scholar]
- 85.Melchart M, Habtemariam A, Parsons S, Sadler PJ. J. Inorg. Biochem. 2007;101:1903–1912. doi: 10.1016/j.jinorgbio.2007.04.018. [DOI] [PubMed] [Google Scholar]
- 86.Liu HK, Berners-Price SJ, Wang F, Parkinson JA, Xu J, Bella J, Sadler PJ. Angew. Chem. Int. Ed. 2006;45:8153–8156. doi: 10.1002/anie.200602873. [DOI] [PubMed] [Google Scholar]
- 87.Liu HK, Wang F, Parkinson JA, Bella J, Sadler PJ. Chem. Eur. J. 2006;12:6151–6165. doi: 10.1002/chem.200600110. [DOI] [PubMed] [Google Scholar]
- 88.Novakova O, Chen H, Vrana O, Rodger A, Sadler PJ, Brabec V. Biochemistry. 2003;42:11544–11554. doi: 10.1021/bi034933u. [DOI] [PubMed] [Google Scholar]
- 89.Novakova O, Kasparkova J, Bursova V, Hofr C, Vojtiskova M, Chen H, Sadler PJ, Brabec V. Chem. Biol. 2005;12:121–129. doi: 10.1016/j.chembiol.2004.11.008. [DOI] [PubMed] [Google Scholar]
- 90.McNae IW, Fishburne K, Habtemariam A, Hunter TM, Melchart M, Wang F, Walkinshaw MD, Sadler PJ. Chem. Commun. 2004;10:1786–1787. doi: 10.1039/b408141b. [DOI] [PubMed] [Google Scholar]
- 91.Wang F, Bella J, Parkinson JA, Sadler PJ. J. Biol. Inorg. Chem. 2005;10:147–155. doi: 10.1007/s00775-004-0621-5. [DOI] [PubMed] [Google Scholar]
- 92.Wang F, Chen H, Parkinson JA, Murdoch PDS, Sadler PJ. Inorg. Chem. 2002;41:4509–4523. doi: 10.1021/ic025538f. [DOI] [PubMed] [Google Scholar]
- 93.Reedijk J. Chem. Rev. 1999;99:2499–2510. doi: 10.1021/cr980422f. [DOI] [PubMed] [Google Scholar]
- 94.Wang F, Xu J, Habtemariam A, Bella J, Sadler PJ. J. Am. Chem. Soc. 2005;127:17734–17743. doi: 10.1021/ja053387k. [DOI] [PubMed] [Google Scholar]
- 95.Sriskandakumar T, Kennepohl P, Petzold H, Sadler PJ. unpublished results.
- 96.Petzold H, Xu J, Sadler PJ. Angew. Chem. Int. Ed. 2008;47:3008–3011. doi: 10.1002/anie.200705342. [DOI] [PubMed] [Google Scholar]
- 97.Petzold H, Sadler PJ. Chem. Commun. 2008 doi: 10.1039/b805358h. [DOI] [PubMed] [Google Scholar]
- 98.Harrop TC, Mascharak PK. Acc. Chem. Res. 2004;37:253–260. doi: 10.1021/ar0301532. [DOI] [PubMed] [Google Scholar]
- 99.Claiborne A, Conn Mallett T, Yeh JI, Luba J, Parsonage D, Klinman JP, Dove JE. Adv. Protein Chem. 2001;58:215–236. doi: 10.1016/s0065-3233(01)58006-7. [DOI] [PubMed] [Google Scholar]
- 100.Claiborne A, Yeh JI, Mallett TC, Luba J, Crane EJ, Charrier V, Parsonage D. Biochemistry. 1999;38:15407–15416. doi: 10.1021/bi992025k. [DOI] [PubMed] [Google Scholar]
- 101.Streu C, Meggers E. Angew. Chem. Int. Ed. 2006;45:5645–5648. doi: 10.1002/anie.200601752. [DOI] [PubMed] [Google Scholar]
- 102.Yan YK, Melchart M, Habtemariam A, Peacock AFA, Sadler PJ. J. Biol. Inorg. Chem. 2006;11:483–488. doi: 10.1007/s00775-006-0098-5. [DOI] [PubMed] [Google Scholar]
- 103.Wang D, Lippard SJ. Nat. Rev. Drug. Discov. 2005;4:307–320. doi: 10.1038/nrd1691. [DOI] [PubMed] [Google Scholar]
- 104.Hillard E, Vessieres A, Thouin L, Jaouen G, Amatore C. Angew. Chem. Int. Ed. 2006;45:285–290. doi: 10.1002/anie.200502925. [DOI] [PubMed] [Google Scholar]
- 105.Peacock AFA, Habtemariam A, Fernandez R, Walland V, Fabbiani FPA, Parsons S, Aird RE, Jodrell DI, Sadler PJ. J. Am. Chem. Soc. 2006;128:1739–1748. doi: 10.1021/ja055886r. [DOI] [PubMed] [Google Scholar]
- 106.Dorcier A, Ang WH, Bolano S, Gonsalvi L, Juillerat-Jeannerat L, Laurenczy G, Peruzzini M, Phillips AD, Zanobini F, Dyson PJ. Organometallics. 2006;25:4090–4096. [Google Scholar]
- 107.Dorcier A, Dyson PJ, Gossens C, Rothlisberger U, Scopelliti R, Tavernelli I. Organometallics. 2005;24:2114–2123. [Google Scholar]
- 108.Schmid WF, John RO, Arion VB, Jakupec MA, Keppler BK. Organometallics. 2007;26:6643–6652. [Google Scholar]
- 109.Stebler-Roethlisberger M, Hummel W, Pittet PA, Buergi HB, Ludi A, Merbach AE. Inorg. Chem. 1988;27:1358–1363. [Google Scholar]
- 110.Peacock AFA, Melchart M, Deeth RJ, Habtemariam A, Parsons S, Sadler PJ. Chem. Eur. J. 2007;13:2601–2613. doi: 10.1002/chem.200601152. [DOI] [PubMed] [Google Scholar]
- 111.Peacock AFA, Parsons S, Sadler PJ. J. Am. Chem. Soc. 2007;129:3348–3357. doi: 10.1021/ja068335p. [DOI] [PubMed] [Google Scholar]
- 112.Peacock AFA, Habtemariam A, Moggach SA, Prescimone A, Parsons S, Sadler PJ. Inorg. Chem. 2007;46:4049–4059. doi: 10.1021/ic062350d. [DOI] [PubMed] [Google Scholar]
- 113.Kostrhunova H, Florian J, Novakova O, Peacock AFA, Sadler PJ, Brabec V. J. Med. Chem. 2008;51:3635–3643. doi: 10.1021/jm701538w. [DOI] [PubMed] [Google Scholar]