

Abstract
Hydroxyapatite–carbon nanotube (HA–CNT) composites have the potential for improved mechanical properties over HA for use in bone graft applications. Finding an appropriate sintering atmosphere for this composite presents a dilemma, as HA requires water in the sintering atmosphere to remain phase pure and well hydroxylated, yet CNTs oxidize at the high temperatures required for sintering. The purpose of this study was to optimize the atmosphere for sintering these composites. While the reaction between carbon and water to form carbon monoxide and hydrogen at high temperatures (known as the ‘water–gas reaction’) would seem to present a problem for sintering these composites, Le Chatelier's principle suggests this reaction can be suppressed by increasing the concentration of carbon monoxide and hydrogen relative to the concentration of carbon and water, so as to retain the CNTs and keep the HA's structure intact. Eight sintering atmospheres were investigated, including standard atmospheres (such as air and wet Ar), as well as atmospheres based on the water–gas reaction. It was found that sintering in an atmosphere of carbon monoxide and hydrogen, with a small amount of water added, resulted in an optimal combination of phase purity, hydroxylation, CNT retention and density.
Keywords: hydroxyapatite, carbon nanotubes, composites, sintering atmosphere
1. Introduction
Hydroxyapatite (HA; Ca10(PO4)6(OH)2) has been investigated widely as a bone replacement material owing to its bioactivity, which stems from its chemical similarity to the mineral component of bone. While HA promotes bone growth along its surface, its mechanical properties, particularly strength and toughness, are relatively poor (Hench 1991). Carbon nanotubes (CNTs), as one of the strongest and stiffest materials available, have the potential to strengthen and toughen HA, thus expanding the range of clinical uses for the material. However, challenges arise when trying to combine these two materials (White et al. 2007).
Perhaps, the greatest obstacle to the successful production of HA–CNT composites is the choice of appropriate sintering conditions. This is particularly the case for pressureless sintering, when the sintering atmosphere is key, as it must enable the composite to retain the CNTs and maintain HA's stoichiometry and structure at the high temperatures required for full densification. Without water present in the atmosphere, HA tends to lose its hydroxyl (OH) groups, and may decompose to tricalcium phosphate (TCP) and tetracalcium phosphate (TTCP). For use in biological applications, it is important to prevent the dehydroxylation and decomposition of HA, as different calcium phosphate phases have different biological properties. In vitro studies have found that HA is less soluble than other calcium phosphate phases (Klein et al. 1990; Dorozhkin 2009), and α- and β-TCP have been found to resorb faster in vivo than HA (Keller & Triplett 1987). However, since water can react with CNTs to produce H2 and CO, retaining the HA structure may mean oxidizing the CNTs.
While hot pressing or hot isostatic pressing may largely circumvent this problem, by applying enough pressure to prevent oxidizing reactions and dehydroxylation, the processing is more complex and expensive compared with pressureless sintering (deGroot et al. 1990). These techniques have been used with HA–CNT composites in the past with some success (Zhao & Gao 2004; Kealley et al. 2006; Meng et al. 2008). Spark plasma sintering, with its extremely high sintering rates and applied pressure, is another alternative, which has been previously investigated (Xu et al. 2009), yet it is geometrically limited and the equipment is expensive (Nygren & Shen 2004). The goal of this study was to investigate atmospheres for use in pressureless sintering that might allow retention of both CNTs and HA's phase purity and OH groups, while providing enough densification for improved mechanical properties over HA alone.
2. Background
To understand how to design an atmosphere that might allow the retention of both CNTs and OH groups, the competing reactions of CNT oxidation and HA dehydroxylation and decomposition must first be examined.
2.1. CNT oxidation
CNTs may be oxidized by a number of reactions in the type of atmospheres that are standard for HA sintering, such as air and moist Ar or N2, and at the temperatures required for full densification (usually 1200–1300°C). For example:
| 2.1 |
| 2.2 |
| 2.3 |
Our studies using thermogravimetric analysis (TGA) have shown that oxidation in air begins at 550–600°C for the CNTs used in this study (discussed in §4.2). This reaction tends to occur quickly and over a narrow temperature range. The reaction rate between CNTs and water tends to be slower, but depending on the rate of porosity closure of the composite material compared with CNT consumption, all the CNTs in a composite might easily be consumed by water during sintering in a moist atmosphere.
2.2. HA dehydroxylation and decomposition
Dehydroxylation of HA begins at temperatures as low as 900°C in air and 850°C in a water-free atmosphere (Wang & Chaki 1993). When OH groups are removed from the HA structure, two OHs combine to form one molecule of water, leaving one oxygen atom in the lattice:
| 2.4 |
When HA begins to dehydroxylate, it forms oxyhydroxyapatite (OHA), which has a similar crystal structure to HA, but contains vacancies. A completely dehydroxylated HA that has not yet decomposed is called oxyapatite (OA; Ca10(PO4)6O). Trombe & Montel (1978) were the first to demonstrate the existence of OA and OHA and to characterize them. OHA forms according to
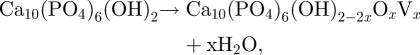 |
2.5 |
where Vx is a hydrogen vacancy. In a moist atmosphere (in which water is explicitly added), it should be possible to sinter HA up to a temperature of 1300°C without dehydroxylation or decomposition (Wang & Chaki 1993). This temperature is based on experimental results by Wang & Chaki and is a typical value reported in the literature when gas is bubbled through water at room temperature (partial pressure of water = 17.5 mm Hg). However, in theory, it should be possible to sinter HA with a Ca/P ratio of precisely 1.67 to temperatures up to 1475°C before decomposition begins, provided that the partial pressure of water in the sintering atmosphere is 500 mm Hg (deGroot et al. 1990). In the case of ‘pressureless’ sintering, assuming atmospheric pressure of 760 mm Hg, achieving this partial pressure of water means bubbling through water very close to boiling.
If HA becomes completely dehydroxylated during sintering, forming OA, its crystal structure changes, as OA is not a stable phase. Two outcomes are stoichiometrically possible: OA may decompose to a mixture of TTCP and TCP (β-TCP at temperatures below approx. 1200°C and α-TCP at higher temperatures), or it may decompose to a mixture of TCP and CaO by the following reactions:
| 2.6 |
and
| 2.7 |
Small deviations in the Ca/P ratio of the starting material also can result in the formation of these secondary phases. The phase diagram in figure 1 outlines the possible calcium phosphate phases at various temperatures and compositions for the CaO–P2O5 system. Temperatures T1 and T2 in the phase diagram will depend on the partial pressure of water in the system. If the partial pressure is 500 mm Hg, then T1 and T2 will be 1360°C and 1475°C, respectively. If the partial pressure of water is less, then T1 and T2 will be lowered, potentially substantially. If no water is present, HA cannot form at all, and so the material will be a mixture of TCP + TTCP for all sintering temperatures.
Figure 1.

Phase diagram of the CaO–P2O5 system at high temperatures. T1 and T2 depend on the partial pressure of water. At partial pressure of water = 500 mm Hg, T1 = 1360°C and T2 = 1475°C. Adapted from de Groot et al. 1990.
Experimentally, when considering a compacted tablet that is densifying as these reactions proceed, the material may easily be a mixture of HA, OHA, TCP and TTCP, for example, at a given time. Diffusion of water into and out of the tablet will be slowed by densification and eventually prevented altogether when porosity has been eliminated. Thus, different regions of the tablet may have very localized atmospheres with varying amounts of water. An additional concern with HA–CNT composites is that since CNTs oxidize at temperatures above approximately 600°C, the choice becomes either losing the CNTs or dehydroxylating the HA. Even in dry atmospheres, dehydroxylation produces water (see equation (2.4)), which can then react with the CNTs.
2.3. Role of the water–gas reaction
Faced with the either the loss of CNTs or the decomposition of HA when using standard atmospheres, alternatives should be considered. One possibility lies in the reaction between CNTs and water:
| 2.3 |
This reaction is more commonly known as the ‘water–gas reaction’ from its industrial origins in creating a cooking and heating gas by pouring water over hot coals. While this reaction normally proceeds in the forward direction at high temperatures, Le Chatelier's principle offers the possibility of shifting the equilibrium of the reaction if the concentration of the products is increased, thus potentially retaining carbon (the CNTs) and the water needed to keep HA's OH groups:
To better understand the thermodynamics of the reaction and how it may be possible at a given temperature to preserve the OH groups and the CNTs, the equilibrium constant (Keq) can be plotted against temperature. Figure 2 shows that the equilibrium constant is positive and increases exponentially at temperatures higher than 674°C. Using the value of Keq at 1200°C (a typical sintering temperature for HA), calculation reveals that the partial pressures of CO and H2 at this temperature are 0.4997 atm (379.8 mm Hg) and the partial pressure of water is a very minimal 6 × 10−4 atm (0.456 mm Hg). While the partial pressure of water at this temperature is virtually zero, and thus would not significantly reduce dehydroxylation, it should be kept in mind that these are theoretical values based on the system being in equilibrium, which may be far from reality experimentally. Based on the possibility of using this reaction to reduce CNT oxidation and HA dehydroxylation, some sintering atmospheres were chosen with this reaction in mind.
Figure 2.

Equilibrium constant versus temperature for the water–gas reaction. Values calculated using the software Outokumpu, HSC Chemistry v. 4.1.
3. Material and Methods
3.1. Material synthesis and processing
All materials were synthesized in house. Multi-walled CNTs (MWNTs; diameter range 20–120 nm; davg = 60 nm; aspect ratio = greater than 102) were grown by chemical vapour deposition. A 4-wt% solution of ferrocene (98%–Sigma Aldrich) in toluene (99.5 + %–Sigma Aldrich) was prepared and injected into the first stage of a tube furnace, maintained at 180°C, at a rate of 5.6 ml h−1 for 4 h. The vaporized reactants were carried by an Ar gas flow of 2.4 l min−1 to the second stage of the furnace, maintained at 760°C, where ferrocene decomposed to form the iron catalyst and subsequent MWNT growth occurred directly on the quartz reaction tube.
To encourage interaction between the HA and MWNTs and to improve the dispersion of the MWNTs, they were acid treated to functionalize the ends and wall defects. The MWNTs were ultrasonicated in a 3 : 1 volume ratio of sulphuric (95 + %–Fisher Scientific) to nitric acid (70% assay, Fisher Scientific) for 12 h at approximately 30°C. They were then filtered and washed with deionized water until the pH of the filtered solution was 7. The MWNTs were then dried at 120°C overnight.
Composite materials were made by an in situ precipitation method, wherein the MWNTs were added to the solution as the HA was forming from a precipitation reaction between calcium hydroxide and orthophosphoric acid. The MWNTs were first dispersed in deionized water by a combination of agitation on a vibrating table and ultrasonication before 5 wt% (relative to the expected mass of HA) was added to a 0.5 M aqueous Ca(OH)2 (98.0 + % purity, Acros Organics) solution. Over a period of 4 h at room temperature, a 0.3 M aqueous solution of H3PO4 (85% assay, Acros Organics) was added, drop-wise, to the calcium hydroxide solution containing MWNTs. The resulting precipitate was aged overnight, filtered, then shear mixed for 15 min to achieve a more homogeneous mixture of HA and MWNTs under viscous conditions, before drying overnight at 80°C.
The dried material was ground, dry milled for 48 h, then sieved to eliminate particles larger than 75 µm. Tablets 13 mm in diameter and 0.8 g mass were prepared by uniaxially pressing the powder at 80 MPa to achieve green densities of 43.5 ± 1.5% theoretical density (TD) (relative to a density of 3.10 g cm−3, calculated by rule of mixtures using 3.16 g cm−3 as density of HA and 2.00 g cm−3 as density of MWNTs). A suspension of magnesium stearate in ethanol was used as a lubricant.
3.2. Green material characterization
The green powders were characterized to determine particle size, specific surface area and loading of MWNTs after processing. The distribution of powder particle sizes was measured using a Mastersizer 2000 (Malvern Instruments) and averaging three measurements for each sample. The specific surface area was measured by the BET method using a Micromeritics Tristar 3000. Eight partial pressures were taken and used to calculate the surface area after degassing the samples in N2 at 180°C for at least 2 h. The loading of CNTs post-processing and post-sintering was determined by dissolving the HA from the composite with 2 M hydrochloric acid, leaving just the MWNTs. The HCl solution containing MWNTs was filtered through a PTFE filter (0.2 µm pore size), and the mass of the filter before and after filtration was compared with determine the loading of MWNTs in the composite. The method was tested on HA and on MWNTs alone to ensure the accuracy of the technique.
3.3. Heat treatments
The behaviour of the material sintered in standard atmospheres was investigated in the first instance by TGA using a TA Instruments TGA Q500 (for samples up to 1000°C) and a TA Instruments SDT Q600 (for samples up to 1200°C). The specific atmosphere and the heating and cooling profiles are indicated with the presentation of results for each sample.
For the more in-depth study, eight sintering atmospheres were investigated, including standard atmospheres (air, wet Ar, etc.) and atmospheres based on the water–gas reaction. Four tablets of 0.8 g each were sintered for each experiment at 1200°C for 2 h with a heating and cooling rate of 5°C min−1. Descriptions and detailed methods for each atmosphere are shown in table 1. All experiments were carried out at ambient pressure in a tube furnace (unless otherwise noted).
Table 1.
Detailed methods for each of the 14 heat treatment atmospheres investigated (NF, non-flowing; pH2O (ice), 5 mm Hg; pH2O (room temperature),17.5 mm Hg).
| notation | atmosphere | flow rate (l min−1) | details |
|---|---|---|---|
| Air | Air | NF | no flow, experiment carried out in box furnace. |
| A/W | Ar + H2O | 0.5 | flowing Ar bubbled through water. |
| A | Ar | 0.5 | flowing Ar. |
| H | H2 | 0.8 | flowing H2 between 710°C and 1200°C during both heating and cooling. System flushed with Ar at beginning and end of experiment. |
| CO | CO | 0.4 | same as H, but with CO. |
| CO/H | CO + H2 | 0.8 | same as H, but with flowing CO (0.4 l min−1) and H2 (0.4 l min−1). |
| CO/H/W (ice) | CO + H2 + H2O (ice) | 0.8 | same as CO/H, but gas flowed through water bubbler immersed in ice. |
| CO/H/W | CO + H2 + H2O | 0.8 | same as CO/H/W(ice), but bubbler at room temperature. |
3.4. Sintered material characterization
The sintered tablets were characterized to evaluate their phase purity, CNT loss during heat treatment, per cent TD, level of hydroxylation and appearance. Phase purity was determined by X-ray diffraction (XRD) with a Bruker D8 Gen 10 using CuKα radiation (40 kV, 40 mA, 0.5° divergence slit and 0.5° antiscatter slit). The data were recorded using a step size of 0.01° and a 0.02 s dwell time. Measured diffraction patterns were compared with powder diffraction files from the JCPDS database. The relative amounts of each phase were determined using the semi-quantitative feature of the X'Pert Highscore Plus software (PANalytical, the Netherlands, v.2.2b). The percentage of CNTs lost during the heat treatment was determined using the method outlined in §3.2. The percentage of TD of the samples was determined using the mass and dimensions of the tablets relative to the TD of the tablet (as calculated from the rule of mixtures, using a CNT density of 2 g cm−3 and using a weighted average of the densities of the calcium phosphate phases present). Fourier transform infrared spectroscopy (FTIR) was used to determine the level of hydroxylation in the samples. KBr pellets were prepared with approximately 200 mg of KBr and 2 mg sample, taken from a ground, sintered tablet. A Bruker Optik Tensor 27 spectrometer was used at a resolution of 4 cm−1, covering the wavenumber range of 4000–400 cm−1 in absorbance mode. The appearance of the tablets was also noted before and after sintering for changes in colour and uniformity of colour.
Materials were also examined with electron microscopy. Scanning electron microscopy (SEM) was performed using a JEOL 6340 FEG SEM with an acceleration voltage of 5 kV on platinum-coated samples (in the case of composites). Transmission electron microscopy (TEM) was performed using a JEOL 200CX TEM with an acceleration voltage of 200 kV.
4. Results and discussion
4.1. Green material characteristics
The composite powder was found to have a CNT loading of 3.5 ± 0.3 wt% after processing. This loading was significantly lower than the 5 wt% added to the Ca(OH)2 solution initially, as a higher concentration of CNTs remained in the larger powder particles, which were discarded during processing. The powder particles were found to have a bimodal distribution of sizes, with an average diameter of 25 ± 3 µm. The powder's surface area was 85 ± 4 m2g−1, and it was medium grey in colour. SEM showed that the CNTs were well dispersed in the HA matrix (figure 3).
Figure 3.

SEM micrograph showing homogeneous distribution of CNTs in HA matrix. Scale bar, 1 µm.
4.2. Behaviour in standard atmospheres as shown by TGA
The vast majority of publications involving HA sintering have used either air or an inert gas (such as nitrogen or argon) bubbled through water as the sintering atmosphere. Despite evidence that HA dehydroxylates and decomposes without the presence of water, some studies have used dry inert atmospheres to sinter HA composites in order to prevent reactions with the secondary phases (Li et al. 2007).
The mass–temperature profiles of HA and HA–CNT composite are shown for a dry Ar atmosphere in figure 4. Generally, the shape of the two curves was similar, with two distinct regions of mass loss during heating—one between room temperature and about 500°C and the second from about 700–1200°C. The first mass loss can be attributed to adsorbed water, while the second can be attributed to dehydroxylation in the case of HA (Liao et al. 1999; Wang et al. 2004). For the composite, the second region of mass loss is due to both dehydroxylation and possibly some degree of CNT oxidation from water produced by the dehydroxylation process. During cooling, a slight increase in mass was observed. Despite the experiment being carried in dry Ar, this may still be due to rehydroxylation, as weight gain owing to rehydroxylation during cooling has been observed even in vacuum (Trombe & Montel 1978). These phenomena were observed in the comprehensive atmosphere study discussed in the next section, and will be described in more detail.
Figure 4.

Mass–temperature profile of HA and composite in dry Ar following the sintering profile outlined in §2.3. Heating and cooling rate = 5°C min−1. Arrows indicate directions of heating/cooling. The text suggests reasons for mass loss/gain. Solid thick line, HA; black dotted line, composite.
Figure 5 compares the behaviour of HA and composite materials in several atmospheres up to 1000°C. The oxidation of CNTs alone is also shown as a reference to demonstrate the temperature range over which CNTs oxidize in air. All of the samples showed a nearly identical rate of weight loss up to 500°C. Above this temperature, the different atmospheres resulted in markedly different behaviour. The profiles for HA in air and dry nitrogen were nearly identical until 800°C. Above this temperature, the HA in air gained a slight amount of mass, whereas the HA in nitrogen lost a small amount of mass. It is not clear, why the HA in air would have gained mass, as the air contained negligible water, but the HA in dry nitrogen may have lost weight owing to some dehydroxylation.
Figure 5.

Mass–temperature profile of HA and composite in air, dry nitrogen and wet nitrogen, with CNTs alone (dotted line and right-hand axis) for comparison of oxidation temperature. Heating rate = 10°C min−1.
The graph also compares two atmospheres for the composites—air and nitrogen bubbled through water at room temperature. Between 520°C and 710°C the composite in air lost 5.5 per cent of its mass, which is approximately equal to the loading of CNTs (unprocessed powder, expected to contain approx. 5 wt% CNTs was used). While this mass loss began at the same temperature as the mass loss in the CNT sample, the mass loss ended at a temperature approximately 35°C higher than the CNTs alone, possibly because the addition of HA slows the rate of oxidation. Above 710°C, the composite material in air experienced a gradual, additional weight loss of 0.4 per cent, possibly owing to partial dehydroxylation. The profile of the composite in wet nitrogen was quite different. This sample did not experience any significant weight loss past 500°C until nearly 700°C, and, most importantly, it should be noted that oxidation of CNTs occurred at a temperature nearly 150°C higher in wet nitrogen than in air. The total mass loss between 600°C and 1000°C was 4.3 per cent. Thus, a smaller percentage of CNTs was oxidized by wet nitrogen than by air.
4.3. Comprehensive atmosphere study
Table 2 shows the results of the characterization of composite material sintered in the eight atmospheres described in table 1. Three atmospheres (A, H, CO) resulted in decomposition of the material, as shown in XRD (see electronic supplementary material). According to the phase diagram shown in figure 1, TTCP + α-TCP can result either when the sintering temperature is extremely high, with or without water in the atmosphere, or at lower sintering temperatures, such as 1200°C, when very little water is present. The combination of HA + TTCP + CaO would then seem to result from either localized differences in the partial pressure of water or an incomplete phase transition with a material which has a Ca/P ratio of less than 1.67. By examining FTIR data, the level of hydroxylation can be determined. In table 2, the results for each atmosphere are listed as either ‘decomposition’, to indicate a significant amount of secondary phases, or some degree of hydroxylation is listed. The electronic supplementary material provides full details of FTIR results.
Table 2.
Characterization results for eight sintering atmospheres. (T, TTCP; α, α-TCP; O, CaO. ‘Layer’ indicates region of greater thickness compared with ‘coating’.)
| atmosphere | tablet appearance | CNT loss (%) | XRD | FTIR | sintered density (% TD) |
|---|---|---|---|---|---|
| Air: Air | uniformly pale blue | 97 ± 0.3 | HA | excellent OH retention | 96.2 ± 0.5 |
| A/W: Ar + H2O | white layer, black inside | 50 ± 2 | HA | significant OH loss | 92.3 ± 2.0 |
| A: Ar | uniformly black | 40 ± 3 | T (65%), α (35%) | decomposition | 90.6 ± 0.4 |
| H: H2 | uniformly black | 37 ± 3 | T (75%), α (25%) | decomposition | 89.7 ± 0.5 |
| CO: CO | uniformly black | 58 ± 2 | HA (61%), T (37%), O (2%) | decomposition | 87.1 ± 0.1 |
| CO/H: CO + H2 | medium grey coating, dark grey inside | 54 ± 2 | HA | good OH retention | 82.8 ± 0.9 |
| CO/H/W(ice): CO + H2 + H2O (ice) | light grey coating, dark grey inside | 62 ± 2 | HA | some OH loss | 86.3 ± 0.7 |
| CO/H/W: CO + H2 + H2O | pale grey coating, medium grey inside | 88 ± 1 | HA | significant OH loss | 95.4 ± 0.7 |
The sample densities varied widely, from 82.8 per cent in CO/H to 96.2 per cent in air. The values were calculated as percentage of TD, with the TD being calculated based on the phase percentages determined by XRD and using the rule of mixtures. The TDs of the phases were taken as: HA = 3.16 g cm−3, α-TCP = 2.86 g cm−3, TTCP = 3.05 g cm−3 and CaO = 3.42 g cm−3. What this calculation did not take into account was the amount of OHA that might have been in a sample. It is difficult to determine a theoretical value for each sample, as a quantitative extent of dehydroxylation was not determined, nor were adjusted lattice parameters for each sample measured. It is presumed that the % TD for dehydroxylated samples should be marginally higher than the reported value. An additional compounding factor in calculating the % TD is that the XRD measurements were taken near the surface of the sample; however, the phase composition may change in the interior of the sample.
CNT loss varied considerably, from 40 per cent in Ar to 97 per cent in air. The presence of CNTs after sintering was verified by examining in SEM and TEM material left on the filter when the CNT loading measurements were made on a CO/H/W(ice) sample. Figure 6 shows an (b) SEM and (c) TEM micrograph of the material, with CNTs which have not undergone heat treatment (a) for comparison. While the material still retained the basic CNT shape, the CNTs appeared to be damaged, relatively short, and many were unrolled.
Figure 6.

(a) SEM image of CNTs before sintering, (b) SEM and (c) TEM images of CNTs from a CO/H/W(ice) sample after sintering showing damage to the CNTs. Comparison of (a) with (b) and (c) shows that CNT sidewalls and ends were more rough after sintering and some CNTs have unrolled. Scale bars, (a,b) 1 µm, (c) 100 nm.
The data were also analysed to determine whether there was any correlation between the final CNT loading and the final density of the samples. In previous studies, CNTs have been shown to prevent full densification of ceramics, particularly when pressure is not used to sinter (Peigney 2003). Figure 7 shows a graph of CNT loading versus final density, using data points from table 2. The data show a loose trend, with higher density values tending to correspond to low CNT loadings and lower density values tending to correspond to higher CNT loadings.
Figure 7.

Final density versus CNT loading for sintered samples.
The appearance of the tablets was also found to depend on the atmosphere in which they were sintered. While all samples began as very dark grey in colour before sintering, the colour of the sintered materials ranged from black to very light in colour. As expected, the samples sintered in air, which contained virtually no CNTs after sintering, were a very light colour, although rather than white they were a very pale blue, which can be attributed to manganese impurities in the starting materials (Yubao et al. 1993). To a certain extent, the degree of greyness of the samples correlated with the loading of CNTs. Samples described as ‘black’ or ‘dark grey’ had relatively high loadings, whereas samples described as ‘medium grey’ or ‘pale grey’ had relatively low loadings. However, it was misleading that most of the samples seemed relatively dark in colour compared with their unsintered counterparts. A sample which contained only about 1 wt% CNTs following sintering might still appear as dark in colour as a unsintered sample with 3.5 wt% CNTs, at first suggesting that the CNT loading should be much higher than the measured value of 0.5 wt%. However, an apparent darkening in colour is likely also attributable to a higher density.
In addition to the general colour of the tablet, it was noted that some tablets had coatings or layers on the outside that were lighter in colour than the interior of the tablets. This would suggest that the outer layers had a lower loading of CNTs than the interior. In table 2, a ‘coating’ refers to a very small amount of a lighter colour (less than 0.1 mm), whereas a ‘layer’ refers to a thicker amount (greater than 0.1 mm). In the case of A/W, the layer was particularly pronounced and white in colour, suggesting virtually no CNTs were present in those areas. Figure 8 shows a tablet of uniform colour and one with a white layer. As the CNT content and porosity varied along the thickness of the tablets with lighter layers, these samples might also present mechanical deficiencies.
Figure 8.

Sintered tablets of (a) uniform colour and (b) with a white layer.
SEM micrographs of the surface and cross section of a sintered tablet revealed further information. Figure 9a shows long, thin holes on the top surface of the composite tablet, which are about the right size and shape to be artefacts from oxidized CNTs. No CNTs were observed on the surface. The cross-section image (figure 9b) shows the interface of the black and white regions. In the black region, CNTs can still be seen, while in the white region, there appear to be no CNTs. Also of note is that the black region seems much more porous than the white region. In the white region, necking between particles is observed, while in the black region, it appears that sintering has not yet begun. This supports earlier observations that composites with more CNTs tend to be more porous.
Figure 9.

SEM micrographs of (a) a white surface of a sintered tablet with a layer and (b) the white–black interface sintered at 1000°C. The surface image shows holes remaining after CNT oxidation, while the interface image shows CNTs remaining in the black region and no CNTs present in the white region. The black region also shows less sintering than the white region. Scale bars, (a,b) 1 µm.
4.4. Explanation of CNT loss
At high temperatures, CNT loss or damage in an HA–CNT composite can originate from two sources—oxidation by reactive species in the sintering atmosphere and oxidation by water formed during dehydroxylation of the HA. When considering the atmospheres tested in this study, reactive species were present in the sintering atmosphere owing to impurities in the gases (with the obvious exception of air) and water added to the atmosphere. The amount that each of these components may contribute to CNT loss can be calculated, although there are many assumptions made and sources of error in the calculations that may not result in an entirely accurate explanation of CNT loss. Nonetheless, the calculations can provide useful insight, which can partially explain CNT loss for different atmospheres. Full details of these calculations are available in the electronic supplementary material.
To give an idea of how much CNT oxidation owing to gas impurities and dehydroxylation could be explained by calculations, table 3 lists the predicted values of CNT loss for each heat treatment experiment. The right-most column lists the difference between the observed CNT loss and the calculation, where a negative value indicates an overestimate of CNT loss. Note that the oxidation from added water vapour is not included in the table calculations.
Table 3.
Calculation of oxidation of CNTs in various sintering atmospheres as a result of gas impurities and dehydroxylation of HA. Note that water added explicitly to the atmosphere is not taken into account (NF, non-flowing; impur., impurities; DH, dehydroxylation).
| atmosphere | flow rate (l min−1) | CNT loss (%) | oxidation (%) |
CNT loss, un-accounted (%) | ||
|---|---|---|---|---|---|---|
| impur. | DH | impur. + DH | ||||
| Air: Air (NF) | NF | 97 ± 0.3 | 100 | 36 | >100 | −3 ± 0.3 |
| A/W: Ar + H2O | 0.5 | 50 ± 2 | 1.3 | 36 | 37 | 13 ± 2 |
| A: Ar | 0.5 | 40 ± 3 | 1.3 | 36 | 37 | 3 ± 3 |
| H: H2 | 0.8 | 37 ± 3 | 0.3 | 36 | 36 | 1 ± 3 |
| CO: CO | 0.4 | 58 ± 2 | 39 | 36 | 75 | −17 ± 2 |
| CO/H: CO + H2, flowing | 0.8 | 54 ± 2 | 35 | 36 | 71 | −17 ± 2 |
| CO/H/W(ice): CO + H2 + H2O | 0.8 | 62 ± 2 | 35 | 36 | 71 | −9 ± 2 |
| CO/H/W: CO + H2 + H2O | 0.8 | 88 ± 1 | 35 | 36 | 71 | 17 ± 1 |
As the right-most column shows, these calculations only partially account for CNT loss. A few atmospheres are more straightforward than others, in this respect. The air atmosphere is the most basic, as the CO2 and O2 in the air in the furnace are easily enough to oxidize all the CNTs in the samples. Ar should be the next most straightforward, as it does not involve any added water and contains only inert gas and its impurities. As the material sintered in Ar was found to have decomposed and, thus, to have dehydroxylated, the full oxidation contribution from dehydroxylation can be expected to be fairly accurate, and, as the table shows, adding the contribution from gas impurities and dehydroxylation explains the CNT loss within a reasonable amount of error.
The explanation of results for the other dry atmospheres (H, CO and CO/H) may be slightly more complicated, as they involve CO and/or H2, which play a role in the water–gas reaction. The calculation for H matches the experimental value very well. The CO calculation, on the other hand, overestimated the CNT loss compared with the experimental value. As the sintered material had decomposed, it seems likely that the dehydroxylation component fully contributed to oxidation. The impurity component could be an overestimate, however, as at some point some degree of closed porosity should have formed (despite the somewhat low sintered density of the composites), making access to the CNTs more difficult. The amount by which the CNT loss is overestimated by calculation is equal to approximately half the value for the impurity calculation, suggesting that gas access to the CNTs could have become much more difficult during the dwell at 1200°C, which is when porosity closure should be at its peak. Indeed, it may be that the impurity component is overestimated in all cases, but Ar and H2 have such small amounts of impurities (0.002% and 0.0004%, respectively) relative to CO (0.2%) that the overestimate is not noticeable.
For CO/H, again the calculation overestimates the CNT loss, and by approximately the same amount as for CO. Thus, it could be that the impurity value is once again an overestimation. However, XRD and FTIR showed that the end material was well-hydroxylated HA, thus bringing into question if any oxidation occurred owing to dehydroxylation. If the system was in thermodynamic equilibrium, then a small amount of water would have been present at all temperatures, thus suppressing dehydroxylation. However, the partial pressure of water in this system at equilibrium would be extremely low by 1200°C (0.11 mm Hg), suggesting a large amount of dehydroxylation would occur.
CO/H differs from H and CO in that it contains both CO and H2. While Le Chatelier's principle suggests that an increase in either or both species on one side of an equation will shift the equilibrium towards the opposite side (in this case, either CO or H2 or the combination might suppress dehydroxylation and the reaction of CNTs with water), figure 10 shows that the thermodynamic situation is a bit more complex.
Figure 10.

Change in partial pressures of gases with temperature for the water–gas reaction.
Figure 10 plots the variation of the partial pressures of the gases involved in the water–gas reaction with temperature. As temperature increases, the partial pressure of water tends to decrease. As the temperature decreases during the cooling stage, more water will be present in the system. If the material is dehydroxylated by the end of the dwell at 1200°C, then CO and H2 will tend to react to provide the necessary amount of water to fulfil thermodynamic equilibrium, and this water could serve to rehydroxylate the material during the cooling stage. However, in the case of an atmosphere of only CO or H2, while the heating stage may be similar to an atmosphere of CO +H2, the cooling stage will be different. If the material dehydroxylated and the resulting water either reacted with the CNTs or was carried away by the flowing gas, then during cooling, only one of the two components necessary to form more water would be present, and therefore the material cannot be rehydroxylated. This would result in either a dehydroxylated or decomposed material, as was the result for H and CO. With CO/H, on the other hand, as both components necessary to make additional water were present, then the resulting material was more hydroxylated. It is unclear whether this new water could reach the interior of the tablet, just as it seems possible that gas impurities may no longer be able to reach the interior of the tablet owing to closed porosity. It should be noted, however, that the CO/H samples had the lowest density of any samples (approx. 82%), and thus may not have had a large degree of closed porosity, making rehydroxylation more possible than for denser materials.
CNT loss in atmospheres with water explicitly added is more difficult to account for with certainty. It is unclear how much of the CNT loss was due to water and how much was due to impurities, as at some stage closed porosity probably prevented some oxidation by the impurities and water. Comparing first CO/H/W(ice) and CO/H/W (CO + H2 + H2O bubbled through ice water and room temperature water, respectively), one would expect more CNT loss from CO/H/W, as the partial pressure of water is higher. As predicted, CO/H/W resulted in an 88 per cent loss of CNTs, whereas CO/H/W(ice) resulted in just 62 per cent. A/W (Ar + H2O bubbled through at room temperature) resulted in just 50 per cent loss of CNTs compared with 88 per cent for CO/H/W, which can be at least partly explained by the higher amount of impurities in CO compared with Ar. The unaccounted-for CNT loss for A/W and CO/H/W is similar (13% and 17%, respectively), which is to be expected as both should have incorporated similar amounts of water into the atmosphere relative to their flow rates.
4.5. Change in properties throughout tablet thickness
All atmospheres which contained either added water or the possibility of producing added water (i.e. contained both CO and H2) resulted in samples with some degree of coating or layer. All ‘dry’ atmospheres, on the other hand, resulted in a uniform colour throughout. These observations led to a more in-depth study of these layers, the results of which can more fully explain the interaction between the material and the atmosphere.
To examine if the properties of the tablet changed with depth, one 1 g tablet of HA and one 1 g tablet of composite were sintered in wet Ar according to the conditions described for the A/W atmosphere. This atmosphere was chosen as it produced a noticeable white layer but still a relatively high loading of CNTs in the interior. After sintering, the tablets, which were approximately 4 mm in thickness, were examined on the top surface and at 1, 2, 3 and 4 mm from the top surface with XRD and FTIR to create a thickness profile. The XRD and FTIR results revealed that the HA tablet remained phase pure and fairly well hydroxylated throughout its thickness. The composite tablet, on the other hand, remained phase pure and well hydroxylated on the outer surfaces, but completely dehydroxylated and decomposed in the interior.
To investigate if the thickness of the white layer changed with sample thickness, three composite tablets of different masses (0.5 g, 1.0 g and 1.5 g) were sintered at 1200°C. Cross-sectional fracture of the tablets revealed that the white layer thickness did not depend on sample thickness, suggesting that the white layer is caused by oxidizing gas trying to pass into the tablet, and that it is a time-dependent reaction unrelated to material mass or geometry.
From all this information, it appears that at some stage the oxidation from water added to the atmosphere becomes more significant than oxidation owing to water formed during dehydroxylation, and CNTs more easily oxidize on the outer layer of the tablet. As CNTs tend to inhibit densification, eventually the outer layer becomes CNT free and nearly all open porosity is eliminated, effectively trapping the interior of the tablet from reaction with the sintering atmosphere. One of two things may happen at this point to cause a variation in phase composition and hydroxylation. Firstly, the entire sample may have already dehydroxylated, and only very slowly (owing to slow diffusion from the very low open porosity) are the outside layers rehydroxylated by the water in the sintering atmosphere. Secondly, it may be that the material has not completely dehydroxylated when the white layer forms. In this case, dehydroxylation may continue on the interior of the tablet and the resulting water would then oxidize CNTs. As it is very difficult for water to rehydroxylate the interior from the outside, the interior may fully dehydroxylate and then begin to decompose, while the exterior remains hydroxylated.
5. Conclusions
Choosing a sintering atmosphere for HA–CNT composites presents a dilemma, as CNTs oxidize at high temperatures, yet water is necessary to prevent dehydroxylation and decomposition of HA. Thus, the purpose of this study was to investigate a wide range of atmospheres that might enable both CNTs and OH groups to be retained while resulting in a material with high density. One of the considerations when selecting potential atmospheres was using carbon monoxide and/or hydrogen in the atmosphere to shift the equilibrium of the water–gas reaction to retain CNTs and water.
Dry atmospheres were found to retain more CNTs than those containing water, but also resulted in decomposed and dehydroxylated materials. Materials sintered in wet atmospheres (including CO/H—CO + H2, which is expected to produce its own water) decomposed less than in dry atmospheres, or not at all, and were hydroxylated to some extent. However, wet atmospheres, particularly A/W (Ar + H2O), resulted in a lighter-coloured coating or layer, which was found to be undesirable owing to variations in the degree of sintering and composition throughout the tablet, which could also result in mechanical property deficiencies. CNT loss was found to result from reactive species in the atmosphere (impurities and water) as well as water produced from the dehydroxylation of HA, which then reacted with the CNTs. A tendency was found that samples with more CNTs had lower densities.
As CO/H/W(ice) (CO + H2 + H2O bubbled through ice water) remained phase pure and hydroxylated, did not have a thick white layer, and had a good balance of CNT loading and density, it was determined to be the optimal atmosphere in which to sinter HA–CNT composites. Thus, applying Le Chatelier's principle to the water–gas reaction was successful. With further adjustments to the sintering parameters, improvements in the properties of composites sintered in this atmosphere should be possible.
Acknowledgements
A.A.W. would like to acknowledge funding from the Marshall Aid Commemoration Commission and the National Science Foundation Graduate Research Fellowship programme. The authors would also like to thank S. Pattinson for providing the TEM image.
Footnotes
One contribution to a Theme Supplement ‘Scaling the heights–challenges in medical materials: an issue in honour of William Bonfield, Part II. Bone and tissue engineering’.
References
- deGroot K., Klein C. P. A. T., Wolke J. G. C., deBlieck-Hogervorst J. M. A. 1990. Chemistry of calcium phosphate bioceramics. In CRC handbook of bioactive ceramics, Vol. II: calcium phosphate and hydroxylapatite ceramics (eds Yamamuro T., Hench L. L., Wilson J.), pp. 3–16. Boca Raton, FL: CRC Press. [Google Scholar]
- Dorozhkin S. V. 2009. Calcium orthophosphates in nature, biology and medicine. Materials 2, 399–498. ( 10.3390/ma2020399) [DOI] [Google Scholar]
- Hench L. L. 1991. Bioceramics: from concept to clinic. J. Am. Ceram. Soc. 74, 1487–1510. ( 10.1111/j.1151-2916.1991.tb07132.x) [DOI] [Google Scholar]
- Kealley C., Elcombe M., van Riessen A., Ben-Nissan B. 2006. Development of carbon nanotube-reinforced hydroxyapatite bioceramics. Physica B 385, 496–498. ( 10.1016/j.physb.2006.05.254) [DOI] [Google Scholar]
- Keller E. E., Triplett W. W. 1987. Iliac bone grafting: review of 160 consecutive cases. J. Oral Max. Surg. 45, 11–24. ( 10.1016/0278-2391(87)90079-6) [DOI] [PubMed] [Google Scholar]
- Klein C. P. A. T., Deblieckhogervorst J. M. A., Wolke J. G. C., De Groot K. 1990. Studies of the solubility of different calcium-phosphate ceramic particles in vitro. Biomaterials 11, 509–512. ( 10.1016/0142-9612(90)90067-Z) [DOI] [PubMed] [Google Scholar]
- Li A. M., Sun K. N., Dong W. F., Zhao D. M. 2007. Mechanical properties, microstructure and histocompatibility of MWCNTs/HAp biocomposites. Mater. Lett. 61, 1839–1844. ( 10.1016/j.matlet.2006.07.159) [DOI] [Google Scholar]
- Liao C. J., Lin F. H., Chen K. S., Sun J. S. 1999. Thermal decomposition and reconstitution of hydroxyapatite in air atmosphere. Biomaterials 20, 1807–1813. ( 10.1016/S0142-9612(99)00076-9) [DOI] [PubMed] [Google Scholar]
- Meng Y. H., Tang C. Y., Tsui C. P., Chen D. Z. 2008. Fabrication and characterization of needle-like nano-HA and HA/MWNT composites. J. Mater. Sci.-Mater. Med. 19, 75–81. ( 10.1007/s10856-007-3107-5) [DOI] [PubMed] [Google Scholar]
- Nygren M., Shen Z. 2004. Spark plasma sintering: possibilities and limitations. Key Eng. Mater. 264–268, 719–724. ( 10.4028/www.scientific.net/KEM.264-268.719) [DOI] [Google Scholar]
- Peigney A. 2003. Tougher ceramics with nanotubes. Nat. Mater. 2, 15–16. ( 10.1038/nmat794) [DOI] [PubMed] [Google Scholar]
- Trombe J. C., Montel G. 1978. Some features of the incorporation of oxygen in different oxidation states in the apatitic lattice, I: on the existence of calcium and strontium oxyapatites. J. Inorg. Nucl. Chem. 40, 15–21. ( 10.1016/0022-1902(78)80298-X) [DOI] [Google Scholar]
- Wang P. E., Chaki T. K. 1993. Sintering behavior and mechanical properties of hydroxyapatite and dicalcium phosphate. J. Mater. Sci.-Mater. Med. 4, 150–158. ( 10.1007/BF00120384) [DOI] [Google Scholar]
- Wang T., Dorner-Reisel A., Muller E. 2004. Thermogravimetric and thermokinetic investigation of the dehydroxylation of a hydroxyapatite powder. J. Eur. Ceram. Soc. 24, 693–698. ( 10.1016/S0955-2219(03)00248-6) [DOI] [Google Scholar]
- White A. A., Best S. M., Kinloch I. A. 2007. Hydroxyapatite-carbon nanotube composites for biomedical applications: a review. Int. J. Appl. Ceram. Technol. 4, 1–13. ( 10.1111/j.1744-7402.2007.02113.x) [DOI] [Google Scholar]
- Xu J. L., Khor K. A., Sui J. J., Chen W. N. 2009. Preparation and characterization of a novel hydroxyapatite/carbon nanotubes composite and its interaction with osteoblast-like cells. Mater. Sci. Eng. C-Biomimetic Supramol. Syst. 29, 44–49. ( 10.1016/j.msec.2008.05.009) [DOI] [Google Scholar]
- Yubao L., Klein C. P. A. T., Xingdong Z., deGroot K. 1993. Relationship between the colour change of hydroxyapatite and the trace element manganese. Biomaterials 14, 969–972. ( 10.1016/0142-9612(93)90187-7) [DOI] [PubMed] [Google Scholar]
- Zhao L. P., Gao L. 2004. Novel in situ synthesis of MWNTs-hydroxyapatite composites. Carbon 42, 423–426. ( 10.1016/j.carbon.2003.10.024) [DOI] [Google Scholar]