

Abstract
Protein kinase activation, via autophosphorylation of the activation loop, is a common regulatory mechanism in phosphorylation-dependent signaling cascades. Despite the prevalence of this reaction and its importance in biological regulation, the molecular mechanisms of autophosphorylation are poorly understood. In this study, we developed a kinetic approach to distinguish quantitatively between cis- and trans-pathways in an autocatalytic reaction. Using this method, we have undertaken a detailed kinetic analysis for the autoactivation mechanism of p21-activated protein kinase 2 (PAK2). PAK2 is regulated in vivo and in vitro by small GTP-binding proteins, Cdc42 and Rac. Full activation of PAK2 requires autophosphorylation of the conserved threonine, Thr402, in the activation loop of its catalytic kinase domain. Analyses of the time courses of substrate reaction during PAK2 autoactivation suggest that autophosphorylation of Thr402 in PAK2 obeys a two-step mechanism of cis initiation, followed by trans amplification. The unphosphorylated PAK2 undergoes an intramolecular (cis) autophosphorylation on Thr402 to produce phosphorylated PAK2, and this newly formed active PAK2 then phosphorylates other PAK2 molecules at Thr402 in an intermolecular (trans) manner. Based on the kinetic equation derived, all microscopic kinetic constants for the cis and trans autophosphorylation have been estimated quantitatively. The advantage of the new method is not only its usefulness in the study of fast activation reactions, but its convenience in the study of substrate effects on modification reaction. It would be particularly useful when the regulatory mechanism of the autophosphorylation reaction toward certain enzymes is being assessed.
Keywords: Covalent Regulation, Enzyme Kinetics, Enzyme Mechanisms, Protein Kinases, Protein Phosphorylation, Autoactivation, Intermolecular Reaction, Intramolecular Reaction
Introduction
Protein phosphorylation is one of the most important processes for cellular regulation and signal transduction in eukaryotic cells. The enzymes responsible for catalyzing this reaction are protein kinases, which catalyze phosphate transfer reactions from ATP to serine, threonine, or tyrosine residues in target substrate and provide key mechanisms for control of cellular signaling processes (1). The activities of many protein kinases are themselves regulated by phosphorylation (2). Most protein kinases are activated through phosphorylation of amino acid residue(s) within the activation loop. This can be carried out by an upstream kinase as part of a signaling cascade, or a kinase can autophosphorylate its own activation loop by either an intramolecular (cis) or intermolecular (trans) mechanism (3). Intramolecular autophosphorylation is a first-order reaction and can be described by the single exponential kinetic equation. In contrast, intermolecular autophosphorylation involves in a bimolecular autocatalytic event, and hence there are more variations in overall mechanisms. One important question for trans-autophosphorylation to address is what molecular mechanism might be involved in the initiation of autocatalytic reaction. Uncovering the answers would provide a better understanding of the regulation of a protein kinase and some important clues for development of specific kinase inhibitors that selectively block autoactivation of protein kinases (4, 5).
There are three possible mechanisms through which an unphosphorylated kinase can be activated by trans-autophosphorylation of a critical residue in the activation loop. The first possibility is that the unphosphorylated kinase is completely inactive, and its activation is catalyzed by a small quantity of contamination, the phosphorylated active kinase in Reaction 1,
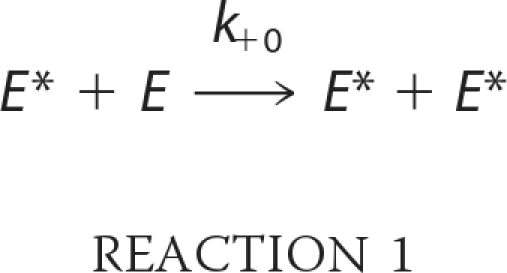 |
where E and E* represent unphosphorylated and phosphorylated enzyme, respectively. In this case, phosphorylation by an upstream kinase is required to initiate kinase autoactivation. A second possible mechanism is one in which unphosphorylated kinase itself has inherent catalytic activity that slowly generates the phosphorylated kinase molecule by an intramolecular reaction, and once the activated kinase is formed, the activation is accelerated by the intermolecular reaction shown in Reaction Scheme 1.
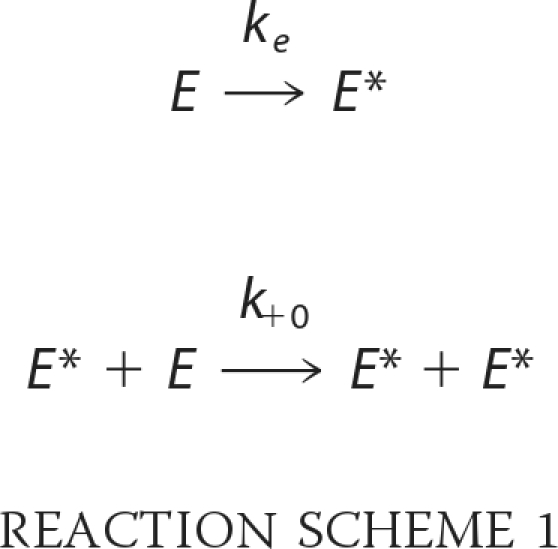 |
The third limiting mechanism is an inter-initiation model in which unphosphorylated kinase molecule, which has residual activity catalyzes the initial phosphorylation in an intermolecular manner shown in Reaction Scheme 2.
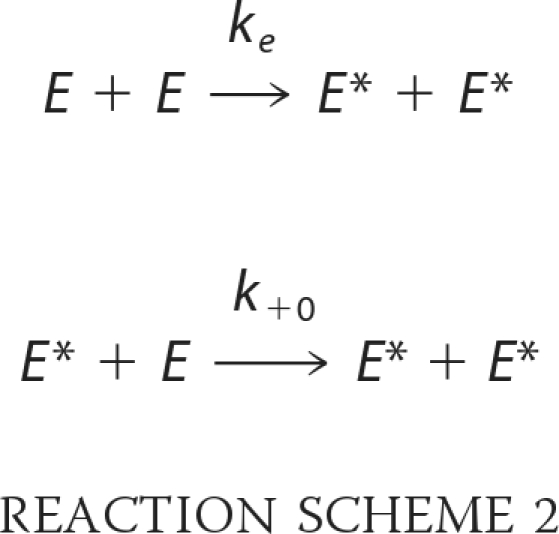 |
A considerable ingenious procedure is therefore required to distinguish between these mechanisms on the basis of various lines of experimental evidence.
The p21-activated protein kinase 2 (PAK2)2 is a serine/threonine kinase regulated by the small GTPases, Cdc42 and Rac, and is involved in the regulation of various cytoskeletal functions, including cell motility and membrane blebbing during apoptosis (6–9). PAK2 consists of a C-terminal kinase domain and an N-terminal regulatory domain containing a GTPase binding domain (GBD) and an autoinhibitory domain (AID). PAK2 is activated by the binding of active Cdc42 and Rac to the GBD, resulting in a conformational change and a subsequent autophosphorylation on several serine and/or threonine residues. Upon activation, PAK2 from human is autophosphorylated at seven sites, six of which have been identified as serine residues in the N-terminal regulatory domain (10, 11). The only phospho-threonine, pThr402, is in the C-terminal kinase domain, and the PAK2 activation was showed to coincide with the phosphorylation of this threonine residue (12). Although autophosphorylation of some N-terminal regulatory sites occurs in cis, phosphorylation of the activation loop threonine (Thr402) is an inter-molecular process (13). The phosphorylation of the activation loop is clearly a critical step in the activation of the PAK kinases, although how the first molecule of active PAK2 is formed remains obscure.
In this study, we developed a new kinetic approach to distinguish effectively between cis- and trans-pathways in an autocatalytic reaction. The mechanism of the autophosphorylation reaction of PAK2 has been further examined using this approach. We demonstrated that the unphosphorylated PAK2 has an activity inherent in itself by which it can self-activate, and that autophosphorylation of Thr402 obeys a two-step mechanism of cis initiation, followed by trans amplification. That is, the unphosphorylated PAK2 undergoes an intramolecular autophosphorylation on Thr402 to produce the first fully activated PAK2, and this newly formed active PAK2 then phosphorylates other PAK2 molecules at Thr402 in an intermolecular manner. On the basis of the kinetic equation of the substrate reaction, all microscopic kinetic constants for the cis and trans autophosphorylation at Thr402 of PAK2 have been estimated quantitatively.
EXPERIMENTAL PROCEDURES
Materials
Adenosine 5′-triphosphate (ATP), phospho(enol)pyruvate (PEP), nicotinamide adenine dinucleotide, reduced (NADH), lactate dehydrogenase (LDH), and pyruvate kinase (PK) were purchased from Sigma. Fetal bovine serum, Grace's insect cell culture medium and yeastolate solution were purchased from Invitrogen, MOPS and Tris were purchased from Sigma. Phosphospecific antibodies that react with PAK2 when phosphorylation at Thr402, Ser141, and Ser192/197, respectively, were obtained from Cell Signaling Technology. MLCtide (KKRPQRATSNVFA) was synthesized using standard protocol, purified by reverse-phase preparative HPLC chromatography, and characterized by MALDI-TOF mass spectrometry by Scilight Biotechnology (Beijing) Inc. Other reagents were local products of analytical grade used without further purification. Double-deionized water was used throughout.
Expression and Purification of Proteins
GST-fusion human PAK2 was expressed in Sf9 cells and purified as described previously (13). A constitutively active Cdc42, His-tagged Cdc42L61 was expressed in Escherichia coli and purified by Ni-NTA column (Qiagen), followed by an anion exchange Source 15Q HR 10/10 column (GE Healthcare). The protein purity was over 95% as judged by SDS-PAGE. Protein concentrations were determined spectrophotometrically using theoretical molar extinction coefficients at 280 nm, following the method of Gill and von Hippel (14). The purified proteins were made to 20% glycerol and stored at −80 °C.
Enzyme Assays for PAK2
Enzyme activity of PAK2 was determined with the MLCtide as a substrate using a coupled spectrophotometric assay (15, 16). The standard assay for PAK2 was carried out at 25 °C in a 1.6-ml reaction mixture containing 100 mm MOPS buffer, pH 7.4, 100 mm KCl, 20 mm MgCl2, 1 mm ATP, 200 μm NADH, 1 mm phospho(enol)pyruvate, 20 units/ml lactate dehydrogenase, and 15 units/ml pyruvate kinase, and different concentrations of substrates. Reactions were initiated by the addition of PAK2 to the reaction mixture. Progress of the reaction was monitored continuously by following the formation of NAD+ at 340 nm, on a PerkinElmer Lambda 45 spectrophotometer. The concentrations of ADP formed in PAK2-catalyzed reaction were determined using an extinction coefficient for NADH of 6220 cm−1 m−1 at 340 nm. The concentrations of peptide were determined by turnover with the activated-PAK2 under conditions of limiting peptides.
Western Blotting
The phosphorylation states of PAK2 were assessed by Western blot analysis using specific antibodies. Samples were resolved by SDS-PAGE and electrotransferred onto polyvinylidene fluoride membranes using a Bio-Rad TransBlot system. After incubation of the membranes with anti-phospho-Ser141, Ser192/197, and Thr402 antibodies (Cell Signaling Technology, Inc., 2606, 2605, 2601), specific immunocomplexes were detected by chemiluminescence using ECL reagents(Vigorous Biotechnology Beijing Co., Ltd.). The membrane was finally exposed to x-ray film (KodakInc.).
RESULTS
Effect of Cdc42 on the PAK2-catalyzed Reaction
Recombinant human PAK2 expressed in insect cell was purified as an inactive enzyme. To examine the phosphorylation state of the enzyme preparation used in this study, samples were subjected to Western blot analysis using the phosphospecific antibodies against phospho-Thr402, Ser192/197, and Ser141 of PAK2, respectively (Fig. 1). In the absence of Cdc42L61, no detectable phosphorylation of Thr402 and Ser192/197 were detected, whereas Ser141 exhibited partial phosphorylation. Thus, the first mechanism can be readily excluded since the purified PAK2 contained no phospho-Thr402 contamination. The full-length PAK2 (final concentration of 3 μm) was incubated with 15 μm Cdc42L61 in a reaction mixture containing 100 mm MOPS (pH 7.4), 20 mm MgCl2, 1 mm DTT, and 2 mm ATP at 25 °C, maximal phosphorylation at Thr402, Ser141, and Ser192/197 was achieved within 30 min of Cdc42 stimulation, at which time the PAK2 activation was near 100% complete.
FIGURE 1.

Western blot analysis of PAK2. Lane 1, inactive GST-PAK2. Lane 2, self-activated GST-PAK2 (in the presence of GST-Cdc42L61). Phosphorylation at Ser141, Ser192/197, and Thr402 in PAK2 was monitored with the phosphospecific antibodies (pAb): pPAK2(Ser141) Ab, pPAK2(Ser192/197) Ab, and pPAK2(Thr402) Ab, respectively. Total PAK2 was analyzed by Coomassie Blue staining.
To elucidate the mechanism of the autoactivation of full-length PAK2, we first performed kinase assay to measure the rate of PAK2-catalyzed reaction. Enzyme activity of PAK2 was determined using a continuous spectrophotometric assay that couples production of ADP to oxidation of NADH, measured as a decrease in absorbance at 340 nm. A synthetic peptide derived from myosin light chain, MLCtide (residues 11–23), was used as an exogenous substrate (17). As shown in Fig. 2A, the activated PAK2 is highly efficient toward MLCtide substrate, even in the absence of Cdc42 (curve 1). In contrast, when the unphosphorylated PAK2 was incubated with MgATP and MLCtide, no significant kinase activity (less than 1%) was detected (curve 2 in Fig. 2A). The addition of the activated PAK2 to the reaction mixture resulted in a linear increase of the absorbance at 340 nm (curve 3 in Fig. 2A) with a slope identical to that of the control experiment in the absence of unphosphorylated PAK2 (curve 1 in Fig. 2A). If the unphosphorylated, inactive PAK2 molecule alone can be trans-phosphorylated by other activated PAK2, then the total active PAK2 concentration would be increased during the assay reaction, and the rates of PAK2-catalyzed MLCtide phosphorylation should be significantly greater than that observed in the control experiment. Therefore, our results demonstrated that the unphosphorylated PAK2 alone is not a substrate of the activated PAK2 in the absence of Cdc42. We next investigated the effect of Cdc42 on the activated PAK2 activity. The initial rates of the activated PAK2-catalyzed MLCtide phosphorylation in the presence of various concentrations of Cdc42 were measured (Fig. 2B). It can be seen from this figure, the progressive addition of Cdc42 had no effect on the initial velocities of PAK2-catalyzed reactions, suggesting that once PAK2 is activated, the continued binding of Cdc42 is not necessary for PAK2 kinase activity, and even following dissociation of the GTPase, the kinase would remain in an “open” state allowing the catalyzed-reaction to occur.
FIGURE 2.

Effect of interaction between Cdc42L61 and PAK2 on PAK2-catalyzed reaction. A, time courses of PAK2-catalyzed reactions. The reaction mixture contained standard assay buffer mixture and 200 μm MLCtide. The reactions were initiated by adding 3 nm phosphorylated PAK2 (trace 1) and 7.5 nm unphosphorylated PAK2 (trace 2). No significant enzyme activity was observed upon addition of unphosphorylated PAK2 in the absence of Cdc42L61. Following addition of 3 nm phosporylated PAK2 (as the arrow indicated), a linear increase in absorbance at 340 nm occurred (trace 3). B, dependence of the initial rate of the PAK2-catalyzed reaction on Cdc42L61 concentration. The reaction mixture contained standard assay buffer mixture, 200 μm MLCtide and different concentrations of Cdc42L61. The reaction was initiated by addition of 3 nm phosphorylated PAK2 into the assay system.
Theoretical Analysis
The general mechanism for PAK2 autoactivation can be written as Scheme 3 (see supplemental information), where E and E* represent unphosphorylated and phosphorylated enzyme, L represent activator, and S and P represent exogenous substrate and its corresponding product, respectively. According to the results given above, it is reasonable to assume that the binding of Cdc42 to the activated PAK2 does not affect its kinase activity for both autophosphorylation of PAK2 and phosphorylation of exogenous substrate MLCtide (that is, KS = K′S, k2 = k′2, and k+0 = k″+0, and k′+0 = k‴+0). Therefore, the kinetic equation for describing the time-dependent behavior of enzyme-catalyzed reaction during the autocatalytic activation is given by Equation 1 (see supplemental information),
 |
where vs = k2[T]0[S]/(KS + [S]) is the steady state velocity of the substrate reaction catalyzed by PAK2, and the expressions of the observed rate constant kobs and β are given by Equation 2 for the intra-initiation model,
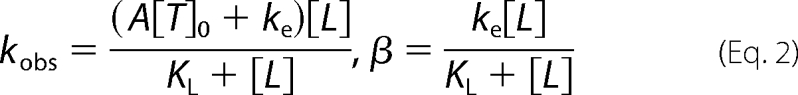 |
or Equation 3 for the inter-initiation model.
[T]0 and [L] refer to the total concentrations of PAK2 and Cdc42, and A is the apparent second-order rate constant in Equation 4,
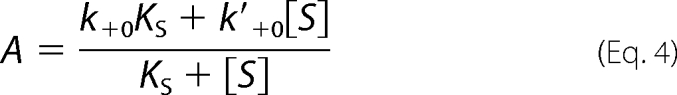 |
where k+0 and k′+0 are microscopic rate constants for the free enzyme and enzyme-substrate complex, respectively. A comparison of the kobs expressions for the second and third mechanisms suggests that a plot of kobs as a function of [T]0 should yield a straight line with a slope and a positive y intercept in the intra-initiation case, but a straight line with a y intercept of zero in the inter-initiation case. Thus, these two mechanisms can be distinguished by determining the effects of varying enzyme concentration on the value of kobs at fixed concentrations of substrate and Cdc42.
SCHEME 3.

Similar to irreversible inhibitor (18), covalent modification can also be classified as competitive, noncompetitive, and uncompetitive on the basis of the interaction mode of the exogenous (S) and isogenous (EL) substrates with the enzyme. Experimentally, the type of modification can be ascertained by studying the effect of the substrate concentration [S] on the apparent second-order rate constant A.
Competitive:
 |
Noncompetitive:
Uncompetitive:
 |
Therefore, the three types of substrate competition in covalent modification can be distinguished by suitable plots of A against [S]. For the competitive binding, a plot of 1/A against [S] will give a straight line. For an uncompetitive binding, the plot of 1/A against 1/[S] will be a straight line, whereas A will be independent of [S] for a noncompetitive binding.
Kinetics of PAK2 Autoactivation
We applied the kinetic theory of the substrate reaction during modification of enzyme activity (18, 19) to the study of the autoactivation process of PAK2. Fig. 3 shows the time courses of product formation at different PAK2 concentrations. The progress curves start off the lag phases, and go up with increasing time due to the activation of PAK2. To distinguish between the second and third mechanisms, the progress curves were analyzed with Equation 1, and the values of kobs, vs, and β were determined by nonlinear curve-fitting analysis. As shown in the inset of Fig. 3, a plot of kobs against enzyme concentration gives a straight line with a positive intercept at the ordinate. The nonzero intercept indicates that the autoactivation proceeds through the second mechanism: an initial intramolecular step followed by an intermolecular activation event.
FIGURE 3.

Time courses of substrate reaction in the presence of different concentrations of the full-length PAK2. The concentrations of Cdc42L61 and MLCtide in the assay system were 0.8 μm and 200 μm respectively. The full-length PAK2 was added to the reaction mixture to start the reaction. Final concentrations of PAK2 were 7.5, 11.25, 15, and 22.5 nm for curves 1–4, respectively. Other conditions were the same as in Fig. 2. The data were fitted to Equation 1 to determine the kinetic parameters, vs, kobs, and β. The solid black lines are the best fitting results according to Equation 1. Inset: plots of the observed rate constant, kobs, against enzyme concentration, [T]0.
To determine the kinetic parameters for the Cdc42-mediated PAK2 autoactivation, two sets of experiments were performed. First, the time courses of product formation at different Cdc42 concentrations, but fixed MLCtide and PAK2 concentrations, were monitored (Fig. 4). From the progress curves of product formation, kobs, vs, and β were determined by fitting the experimental data to Equation 1 as described before. The inset of Fig. 4 shows that the observed rate constant kobs gives rise to the hyperbolic dependence on the Cdc42 concentration, and the data fit to the expression of kobs with a KL of 6.3 ± 0.9 μm and an upper limit of 0.066 ± 0.004 s−1. The dissociation constant for the binding of Cdc42 to the full-length PAK2 (KL = 6.3 μm) is 2 orders of magnitude higher than those between Cdc42 and the isolated GBD fragments (20–22). A possible explanation for this discrepancy might be that there are contacts between the kinase domain and its regulatory domain in full-length PAK2, which modify the interaction of Cdc42 with the regulatory domain (23, 24).
FIGURE 4.

Time courses of substrate reaction in the presence of different concentrations of Cdc42L61. The reaction was started by the addition of the full-length PAK2 to the reaction mixture. The final concentration of enzyme and MLCtide was 7.5 nm and 200 μm, respectively. Concentrations of Cdc42L61 were 1.6, 3.2, 4.8, 8, and 12.6 μm for curves 1–5, respectively. Other conditions were the same as in Fig. 2. The data were fitted to Equation 1 to determine the kinetic parameters, vs, kobs, and β. Inset: plot of the observed rate constant, kobs against Cdc42L61 concentration, [L]. The solid line represents the best fitting result according to kobs = (A[T0] + ke)[L]/(KL + [L]) with KL = 6.3 μm and A[T]0 + ke = 0.066 s−1.
To further assess the effect of exogenous substrates on the autophosphorylation of PAK2, we next carried out the autoactivation assays in the presence of varied concentrations of MLCtide, but at a fixed concentrations of Cdc42 and PAK2 (Fig. 5). For a two-step mechanism of cis initiation followed by trans amplification, Equation 1 can be rewritten as Equation 8,
 |
where θ = [L]/(KL + [L]). With the fixed values of θ and [T]0, the best-fit curves were obtained by nonlinear regression analysis with Equation 8, from which the kinetic parameters, vs, A, and ke were determined. The steady-state velocity vs gives rise to the hyperbolic dependence on the MLCtide concentration (Fig. 6A), which fits the Michaelis-Menten equation with a KS of 292 ± 17 μm and k2 of 34.3 ± 1.0 s−1. In contrast, the apparent second-order rate constant A decreases on increasing the concentration of MLCtide (Fig. 6B). Knowing the value of KS, the values of k+0 and k′+0 were determined to be 14.0 ± 0.5 μm−1 s−1 and 0.78 ± 0.68 μm−1 s−1 by fitting the experimental data to the analytical expression of A (Equation 4). It can be seen that the parameter k′+0, as inferred from these data, had a very large standard deviation. The large magnitude of the standard deviation in the estimate of k′+0 could signify that the Equation 4 does not contain this parameter. When k′+0 = 0, Equation 4 can be simplified to Equation 5. In this case, a plot of 1/A against [S] will give a straight line, as indeed the case for the autoactivation of PAK2 (Fig. 6C). Re-fitting the experimental data with Equation 5 gives a remarkable correspondence, and the continuous lines in Fig. 6B represent the best fit with k+0 = 14.4 ± 0.3 μm−1 s−1 and a fixed KS = 292 μm. This result indicates that trans-autophosphorylation of PAK2 is competitively inhibited by binding of the exogenous substrate, MLCtide, at the active site. A plot of ke against MLCtide concentration shows the value of ke is not affected by the exogenous substrate (Fig. 6D), consistent with our model in which MLCtide cannot bind to EL to form ELS ternary complex. An average of these experiments yielded ke = 0.0254 ± 0.0007 s−1. All kinetic parameters for the autoactivation of the intact PAK2 are summarized in Table 1.
FIGURE 5.

Time courses of substrate reaction in the presence of different concentrations of MLCtide. The reaction was started by the addition of PAK2 to the reaction mixture. The final concentration of enzyme and Cdc42L61 ware 7.5 nm and 1.6 μm, respectively. Concentrations of MLCtide were 100, 200, 300, and 400 μm for curves 1–4, respectively. Other conditions were the same as in Fig. 2. The data were fitted to Equation 8 to determine the kinetic parameters, vs, A, and ke.
FIGURE 6.

Effect of MLCtide concentration on PAK2 autoactivation. A, plot of the steady-state velocities vs against substrate concentration, [S]. The solid line represents the best fitting result according to Michealis-Menten equation with KS = 292 μm, k2 = 34 s−1. B, plot of the apparent second-order rate constant A against substrate concentration, [S]. The continuous line represents the best fitting result according to Equation 5 with k+0 = 14.4 μm−1 s−1, and a fixed KS = 292 μm. C, plot of the reciprocal of the apparent second-order rate constant, A, against substrate concentration, [S]. D, plot of the intra-initiation rate constant ke against substrate concentration, [S]. The solid line represents the best fitting result with ke of 0.0254 ± 0.0007 s−1.
TABLE 1.
Kinetic parameters for autoactivation of PAK2
Kinetic parameters for autoactivation of PAK2 were determined by the progress curve method and conventional method at 25 °C and pH 7.4 in 100 mm MOPS buffer. The standard deviation for individual parameters was obtained from the nonlinear regression fit of the data to the corresponding equations using the software Sigmaplot 9.
| Kinetic parameter | Value |
|---|---|
| k2 (s−1) | 34.32 ± 1.05 |
| Ks (μm) | 292 ± 17 |
| k2/Ks (μm−1 s−1) | 0.117 ± 0.010 |
| k+0 (μm−1 s−1) | 14.44 ± 0.26 |
| k′+0 (μm−1 s−1) | 0 |
| ke (s−1) | 0.0254 ± 0.0007 |
According to our definition, the parameter k+0 determined from the autoactivation kinetics is equal to kcat/Km for the isogenous substrate (unphosphorylated PAK2 in complex with Cdc42). The second order rate constant kcat/Km is a physiologically relevant parameter for the reaction of free enzyme with free substrate and reflects both binding affinity and catalytic efficiency. As shown in Table 1, the k+0 value for trans-autophosphorylation of PAK2 is more than 100-fold higher than k2/KS for the exogenous substrate, MLCtide. Therefore, the PAK2-Cdc42 complex is a more efficient substrate for the active enzyme, suggesting that there are additional contacts outside the immediate vicinity of the phosphorylation site that mediate the recognition of one PAK2 molecule by another.
DISCUSSION
The autophosphorylation of the activation loop is a critical step in the activation of PAK2 kinase, but the molecular details of this process remain poorly understood. To further this understanding, we have undertaken a detailed kinetic study of PAK2 activation mechanism. Analyses of the time courses of substrate reaction during PAK2 autoactivation suggest that autophosphorylation of Thr402 in PAK2 obeys a two-step mechanism of cis initiation, followed by trans amplification, and binding of the exogenous substrate MLCtide at the active site of PAK2 completely abolish the PAK2 autophosphorylation at site Thr402. Compared with the conventional method of taking aliquots at time intervals during the activation process and assaying for the enzyme activity or modification extend, the advantage of the present method is not only its continuous nature and usefulness in the study of fast modification reactions, but its convenience in the study of substrate effects on modification reaction. This new method would be particularly useful when the regulatory mechanism of the reversible phosphorylation reaction toward certain enzymes is being assessed.
Ligand-induced autophosphorylation of protein kinase is considered to be a trigger of intracellular signal transduction (2, 3). Autophosphorylation can be achieved via two basic routes: intramolecular (in cis) and intermolecular (in trans) reactions. There is an abundance of structural data for protein kinases, which have provided insight into how the kinase transforms from an inactive state into an active state by trans-autophosphorylation. However, how a protein kinase autophosphorylates its activation loop in cis remains obscure. In the inactive state, the nonphosphorylated activation loop often folds back into the active site and sterically blocks substrate binding. This raises the question of how protein kinases that become self-activated by autophosphorylation of their activation loops in cis overcoming these steric constraints (3). Most of the kinases that autophosphorylate their activation loops in cis require protein binding partners, which temporally regulates the conformation of protein kinases and promote their activation (25–28). For example, autophosphorylation of the critical activation loop tyrosine of DYRK is intramolecular and mediated by the nascent kinase passing through a transitory intermediate form (27). In contrast, GSK-3β is not activated during protein translation but requires the binding and activity of the molecular chaperone, heat shock protein 90 kDa (Hsp90), which mediates a cis autophosphorylation event in the activation loop of GSK-3β (at Tyr216) in response to growth factors (25). Therefore, we propose that binding of Cdc42 to the N-terminal regulatory domain of PAK2 does more than just relieve inhibitory constraints imposed on the catalytic domain by the inhibitory switch, and may also provide an essential chaperone-like function that transiently coverts the intermediate form of PAK2 into an intramolecular kinase capable of autophosphorylating the activation loop threonine in the protein kinase domain.
Supplementary Material
This work was supported by the RRDCC basic research grant of Roche R&D Center (China) Ltd., MOST Grants 2006CB503900 and 2007CB914400, and NSFC Grant 30770476.

The on-line version of this article (available at http://www.jbc.org) contains supplemental information.
- PAK2
- p21-activated kinase 2
- MESG
- 7-methyl-6-thioguanosine
- PK
- pyruvate kinase
- LDH
- lactate dehydrogenase
- MLCtide
- synthetic peptide, KKRPQRATSNVFA
- Ab
- antibody.
REFERENCES
- 1. Adams J. A. (2001) Chem. Rev. 101, 2271–2290 [DOI] [PubMed] [Google Scholar]
- 2. Nolen B., Taylor S., Ghosh G. (2004) Mol. Cell 15, 661–675 [DOI] [PubMed] [Google Scholar]
- 3. Lochhead P. A. (2009) Sci. Signal 2, pe4. [DOI] [PubMed] [Google Scholar]
- 4. Deacon S. W., Beeser A., Fukui J. A., Rennefahrt U. E., Myers C., Chernoff J., Peterson J. R. (2008) Chem. Biol. 15, 322–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Oliver A. W., Knapp S., Pearl L. H. (2007) Trends Biochem. Sci. 32, 351–356 [DOI] [PubMed] [Google Scholar]
- 6. Molli P. R., Li D. Q., Murray B. W., Rayala S. K., Kumar R. (2009) Oncogene 28, 2545–2555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Arias-Romero L. E., Chernoff J. (2008) Biol. Cell 100, 97–108 [DOI] [PubMed] [Google Scholar]
- 8. Kumar R., Gururaj A. E., Barnes C. J. (2006) Nat. Rev. Cancer 6, 459–471 [DOI] [PubMed] [Google Scholar]
- 9. Bokoch G. M. (2003) Annu. Rev. Biochem. 72, 743–781 [DOI] [PubMed] [Google Scholar]
- 10. Gatti A., Huang Z., Tuazon P. T., Traugh J. A. (1999) J. Biol. Chem. 274, 8022–8028 [DOI] [PubMed] [Google Scholar]
- 11. Manser E., Huang H. Y., Loo T. H., Chen X. Q., Dong J. M., Leung T., Lim L. (1997) Mol. Cell. Biol. 17, 1129–1143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yu J. S., Chen W. J., Ni M. H., Chan W. H., Yang S. D. (1998) Biochem. J. 334, 121–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wu H., Wang Z. X. (2003) J. Biol. Chem. 278, 41768–41778 [DOI] [PubMed] [Google Scholar]
- 14. Gill S. C., von Hippel P. H. (1989) Anal. Biochem. 182, 319–326 [DOI] [PubMed] [Google Scholar]
- 15. Roskoski R., Jr. (1983) Methods Enzymol. 99, 3–6 [DOI] [PubMed] [Google Scholar]
- 16. McClure W. R. (1969) Biochemistry 8, 2782–2786 [DOI] [PubMed] [Google Scholar]
- 17. Chew T. L., Masaracchia R. A., Goeckeler Z. M., Wysolmerski R. B. (1998) J. Muscle Res. Cell Motil 19, 839–854 [DOI] [PubMed] [Google Scholar]
- 18. Tsou C. L. (1988) Adv. Enzymol Relat Areas Mol. Biol. 61, 381–436 [DOI] [PubMed] [Google Scholar]
- 19. Tian W. X., Tsou C. L. (1982) Biochemistry 21, 1028–1032 [DOI] [PubMed] [Google Scholar]
- 20. Stevens W. K., Vranken W., Goudreau N., Xiang H., Xu P., Ni F. (1999) Biochemistry 38, 5968–5975 [DOI] [PubMed] [Google Scholar]
- 21. Nomanbhoy T., Cerione R. A. (1999) Biochemistry 38, 15878–15884 [DOI] [PubMed] [Google Scholar]
- 22. Thompson G., Owen D., Chalk P. A., Lowe P. N. (1998) Biochemistry 37, 7885–7891 [DOI] [PubMed] [Google Scholar]
- 23. Buck M., Xu W., Rosen M. K. (2004) J. Mol. Biol. 338, 271–285 [DOI] [PubMed] [Google Scholar]
- 24. Buchwald G., Hostinova E., Rudolph M. G., Kraemer A., Sickmann A., Meyer H. E., Scheffzek K., Wittinghofer A. (2001) Mol. Cell. Biol. 21, 5179–5189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lochhead P. A., Kinstrie R., Sibbet G., Rawjee T., Morrice N., Cleghon V. (2006) Mol. Cell 24, 627–633 [DOI] [PubMed] [Google Scholar]
- 26. Bhattacharyya R. P., Reményi A., Good M. C., Bashor C. J., Falick A. M., Lim W. A. (2006) Science 311, 822–826 [DOI] [PubMed] [Google Scholar]
- 27. Lochhead P. A., Sibbet G., Morrice N., Cleghon V. (2005) Cell 121, 925–936 [DOI] [PubMed] [Google Scholar]
- 28. Ge B., Gram H., Di Padova F., Huang B., New L., Ulevitch R. J., Luo Y., Han J. (2002) Science 295, 1291–1294 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.