

Abstract
Neurotransmitter:sodium symporters are crucial for efficient synaptic transmission. The transporter GAT-1 mediates electrogenic cotransport of GABA, sodium, and chloride. The presence of chloride enables the transporter to couple the transport of the neurotransmitter to multiple sodium ions, thereby enabling its accumulation against steep concentration gradients. Here we study the functional impact of mutations of the putative chloride-binding residues on transport by GAT-1, with the emphasis on a conserved glutamine residue. In contrast to another putative chloride coordinating residue, Ser-331, where mutation to glutamate led to chloride-independent GABA transport, the Q291E mutant was devoid of any transport activity, despite substantial expression at the plasma membrane. Low but significant transport activity was observed with substitution mutants with small side chains such as Q291S/A/G. Remarkably, when these mutations were combined with the S331E mutation, transport was increased significantly, even though the activity of the S331E single mutant was only ∼25% of that of wild type GAT-1. Transport by these double mutants was largely chloride-independent. Like mutants of other putative chloride coordinating residues, the apparent affinity of the active Gln-291 single mutants for chloride was markedly reduced along with a change their anion selectivity. In addition to the interaction of the transporter with chloride, Gln-291 is also required at an additional step during transport. Electrophysiological analysis of the Q291N and Q291S mutants, expressed in Xenopus laevis oocytes, is consistent with the idea that this additional step is associated with the gating of the transporter.
Keywords: Chloride Transport, Neurotransmitter Transport, Reconstitution of Membrane Transporters, Sodium Transport, Transport Amino Acids, Chloride-binding Site, GABA, Neurotransmitter Transport, Sodium Coupling, Outward pH Gradient
Introduction
Most neurotransmitters are removed from the synaptic cleft by transporters located in the plasma membrane of surrounding nerve and glial cells. These transporters ensure that the concentrations of the neurotransmitters in the synapse are low, so that the post-synaptic receptors can detect neurotransmitter molecules upon exocytotic release from the presynaptic neurons. With exception of the transporters for glutamate, the transporters for other neurotransmitters, such as GABA, serotonin, dopamine, norepinephrine, and glycine are sodium- and chloride-dependent and belong to the NSS2 family (see Refs. 1 and 2 for reviews). These NSS transporters couple the flux of neurotransmitters not only to that of sodium but also to that of chloride (3–5).
The high resolution crystal structure of the bacterial homologue LeuT (6) appears to be an excellent model for the NSS neurotransmitter transporters (7–10). LeuT consists of 12 TMs with TMs 1–5 related to TMs 6–10 by a pseudo-2-fold axis in the membrane plane. TMs 1 and 6 have breaks in their helical structure approximately halfway across the membrane. These breaks expose main chain carbonyl oxygen and nitrogen atoms for binding of leucine and the two sodium ions, Na1 and Na2. The sodium ions in the binding pocket are both close to the substrate, which is in direct contact, through its carboxyl group, with Na1. Functional characterization of mutants of the GABA transporter GAT-1 suggests that also in this transporter the amino acid substrate participates in the Na1 site and that this site has a higher selectivity and apparent affinity for sodium than the Na2 site (7). In contrast to the NSS neurotransmitter transporters, LeuT and other bacterial homologues do not require chloride for transport (6, 11, 12), and indeed no chloride was observed in the binding pocket of LeuT (6). Recently, the putative chloride-binding site of the NSS neurotransmitter transporters was identified near the Na1 site (4, 5), and it was inferred that the role of chloride is mainly to compensate for the multiple positive charges during the substrate translocation step (5). This site is formed by residues from TMs 2, 6, and 7 (4, 5) (see Fig. 1).
FIGURE 1.

The putative chloride-binding site in GAT-1. The four residues proposed to interact directly with the chloride (orange), Tyr-86 (TM2), Gln-291 and Ser-295 (TM6), and Ser-331 (TM7), are within a distance of 3.5 Å. Also shown are Asn-327 and Ser-328 (both TM7), with the latter depicted in orange. Gln-291 (purple) not only is part of the proposed chloride-binding site but also interacts with Arg-69 (TM1), which in turn interacts with Asp-451 (TM10). This is depicted by green arrows, but Asp-451 itself is not shown. The figure was prepared with Chimera and is an adaptation from Fig. 1 of Ref. 5.
The conformation of the LeuT structure is predominantly outward (extracellular) facing with its binding pocket occluded from the cytoplasm by ∼20 Å of ordered protein (6), and the opposite is true for inward-facing conformations of transporters from different families yet with the same structural fold of LeuT (13–15). In addition to the “thick” cytoplasmic gate, the LeuT structure contains a “thin” extracellular gate where just a few highly conserved residues, a pair of conserved aromatic amino acid residues, and a conserved pair composed of an arginine and an aspartate residue obstruct the binding pocket. Because several of the homologous residues are critical for transport in GAT-1 (16–19), it is likely that the formation of the thin gate is an obligate intermediate during substrate translocation: the transition from the outward-facing conformation to the inward-facing conformation.
A highly conserved glutamine residue, Gln-291 in GAT-1 (Gln-250 in LeuT), has been reported to be crucial for GABA transport by GAT-1 (20). Inspection of the LeuT structure and the putative chloride-binding site suggests two possible reasons why this residue is crucial for transport: 1) hydrogen bond formation with the conserved arginine residue; Arg-69 in GAT-1 (Arg-30 in LeuT), is critical for the formation of the thin gate; and 2) direct interaction with the cotransported chloride predicted by one model (5) (Fig. 1) but not the other (4). In this study we address the question of which of the two postulated roles (or both) this conserved glutamine residue plays in the transport mechanism of the NSS neurotransmitter transporters.
EXPERIMENTAL PROCEDURES
Generation and Subcloning of Mutants
Mutations were made by site-directed mutagenesis of GAT-1-WT or GAT-1-S331E in the vector pBluescript SK− (Stratagene) using single-stranded uracil-containing DNA as described previously (21, 22). Briefly, the GAT-1-containing plasmid was used to transform Escherichia coli CJ236 (dut−, ung−). From one of the transformants, single-stranded uracil-containing DNA was isolated upon growth in uridine-containing medium according to the standard protocol from Stratagene, using helper phage R408. This yields the sense strand, and consequently, mutagenic primers were designed to be antisense. The mutants were subcloned into GAT-1-WT residing in the vector pBluescript SK−, using the unique restriction enzymes NheI and AgeI. The coding and noncoding strands were sequenced between these two restriction sites.
Cell Growth and Expression
HeLa cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum, 200 units/ml penicillin, 200 μg/ml streptomycin, and 2 mm glutamine. Infection with recombinant vaccinia/T7 virus vTF7–3 (23) and subsequent transfection with plasmid DNA, as well as GABA transport, were done as published previously (24). Transport was terminated after 3 (initial rate) or 10 min (close to maximal transport) as indicated in the figure legends. The values for the mutants were normalized to those of GAT-1-WT. At 150 mm NaCl, the latter values ranged from 51 to 61 and from 92 to 112 pmol/mg of protein for the 3- and 10-min time points, respectively. In experiments determining the dependence of transport on the external chloride concentration, the indicated chloride concentrations were achieved by equimolar replacement of NaCl in the transport solution (150 mm NaCl, 0.5 mm MgSO4, and 5 mm KPi, pH 7.4) with sodium gluconate. In all of the experiments described, the expression vector was pBluescript SK−.
Cell Surface Biotinylation
Labeling of wild type and mutant transporters at the cell surface, using Sulfo-NHS-SS-Biotin (Pierce), quenching the reaction, cell lysis, and isolation of the biotinylated proteins by streptavidin-agarose beads (Pierce) were done as described (25). After SDS-PAGE (10% gel) and transfer to nitrocellulose, the GAT-1 protein was detected with an affinity-purified antibody, directed against an epitope from the cytoplasmic C-terminal tail of GAT-1, at a 1:1,000 dilution, with horseradish peroxidase-conjugated secondary antibody at a 1:40,000 dilution, and with ECL. 1% of goat serum was present in all antibody, blocking, and washing solutions to minimize the appearance of nonspecific bands. The films were scanned using the Minibis Pro from DNR Bio Imaging Systems Ltd. Using GelCapture version 5.3 software. Quantitative densitometry was done using TINA version 2.09 software.
Reconstitution
Reconstitution of the transporters into liposomes using spin columns was done as described (25) and for each transport reaction 10 μl of reconstituted proteoliposomes, containing either 120 mm KPi, pH 7.4, or 150 mm KMES, pH 6.0 (as indicated), was added to 360 μl of external medium containing 75 mm NaCl plus 60 mm NaPi, pH 7.4, supplemented with 2.8 μm of valinomycin and 1 μCi of [3H]GABA (89.5 Ci/mmol). In those cases where “out” media of different compositions were used, this is indicated in the text. Transport reactions were terminated by filtration, and radioactivity, retained after washing the filters, was determined by liquid scintillation counting as described (26).
Expression in Oocytes and Electrophysiology
cRNA was transcribed using mMESSAGE-mMACHINE (Ambion) and injected into Xenopus laevis oocytes, as described (17). Oocytes were placed in the recording chamber, penetrated with two agarose-cushioned micropipettes (1%/2 m KCl, resistance varied between 0.5 and 3 mΩ), voltage clamped using GeneClamp 500 (Axon Instruments) and digitized using Digidata 1322 (Axon Instruments both controlled by the pClamp9.0 suite (Axon Instruments). Voltage jumping was performed using a conventional two-electrode voltage clamp as described previously (27). The standard buffer, termed ND96, was composed of 96 mm NaCl, 2 mm KCl, 1.8 mm CaCl2, 1 mm MgCl2, 5 mm Na-HEPES, pH 7.5). When nominal sodium-free medium was used, NaCl was replaced by equimolar concentrations of choline Cl, and Na-HEPES was replaced by Tris-HEPES. Current-voltage relations represent steady-state substrate-elicited net currents ((Ibuffer+substrate) − (Ibuffer)) and were analyzed by Clampfit version 8.2 or 9.0 (Axon Instruments). Because of the variability in expression level within and between different oocyte batches, the data shown in Fig. 9 have been normalized as indicated in the figure legend.
FIGURE 9.

Voltage dependence of GABA-induced steady-state currents. The GABA-induced currents (defined in the Legend to Fig. 8) from 420 to 480 ms at each potential were averaged and normalized to the GABA-induced current of GAT-1-WT at −140 mV. These currents were then plotted against the corresponding potential (V (mV)). The data are the means ± S.E. of at least three repeats. The GABA-induced currents by GAT-1-WT ranged from −380 to −650 nA.
RESULTS
Transport by Gln-291 Mutants
When the conserved glutamine residue located in TM6 of GAT-1 was mutated such as to either maintain the same functional group of the side chain (Q291N) or to keep its length constant (Q291E), [3H]GABA transport activity was basically eliminated (Fig. 2, gray bars), in full agreement with an earlier study (20). This loss of activity is not due to decreased steady-state levels of the mutant transporters on the plasma membrane relative to WT, because the intensity of the bands in the biotinylated fraction of these and almost all of the other mutants studied is similar to that of WT as shown by surface biotinylation (Table 1 and supplemental Fig. S1). The transporters in this biotinylated fraction represent only a small proportion of the total (supplemental Fig. S2).
FIGURE 2.

Transport activity of Gln-291 mutants in whole cells. GAT-1 WT and Gln-291 mutants in the background of either WT (light gray bars) or S331E (dark gray bars) were transiently expressed in HeLa cells, and [3H]GABA transport was measured for 10 min, as described under “Experimental Procedures.” The data are given as percentages of the activity of WT-GAT-1 and are the means ± S.E. (error bars) of at least three separate experiments performed in quadruplicate.
TABLE 1.
Cell surface expression of GAT-1 mutants
The bands were scanned after exposure of the films for 30 s. Scanning and quantification of the bands on the developed films was done as described under “Experimental Procedures.” The response for the bands with medium intensity was linear up to 2 min. The sum of the intensities of the monomer and the dimer bands were normalized against the corresponding value for GAT-1-WT (percentage of GAT-1-WT ± S.E.).
| Intensity | n | |
|---|---|---|
| GAT-1-WT | 100 | |
| S331E | 110 ± 8 | 3 |
| Q291E | 89 ± 23 | 8 |
| Q291E/S331E | 74 ± 17 | 6 |
| Q291N | 100 ± 10 | 9 |
| Q291N/S331E | 110 ± 11 | 7 |
| Q291S | 71 ± 8 | 8 |
| Q291S/S331E | 143 ± 21 | 8 |
| Q291G | 111 ± 16 | 8 |
| Q291G/S331E | 121 ± 10 | 6 |
| Q291C | 94 ± 19 | 6 |
| Q291C/S331E | 85 ± 20 | 4 |
| Q291A | 20 ± 9 | 7 |
| Q291A/S331E | 80 ± 18 | 6 |
| Y86F | 133 ± 16 | 4 |
| S295C | 157 ± 28 | 4 |
| N327Q | 138 ± 26 | 4 |
| S331A | 113 ± 30 | 5 |
| S331G | 82 ± 15 | 5 |
Remarkably, significant albeit low transport activity was observed when the glutamine was mutated to smaller amino acid residues, and this was particularly true for the Q291G mutant (Fig. 2). Even though there was a large variability in the ratio of the intensities of the biotinylated bands of the mutants relative to those of WT, it is obvious that, with the exception of Q291A, also for these Gln-291 substitution mutants and the other mutants studied in this paper, the low activity relative to WT cannot be accounted for by a corresponding decrease of steady-state transporter levels on the plasma membrane. (Table 1 and supplemental Fig. S1). Because transport in intact cells expressing Q291A is comparable with that of the other Gln-291 mutants despite its reduced levels in the biotinylated samples, the intrinsic activity of this mutant may actually be higher than the other Gln-291 mutants.
Because the amide group on the side chain of Gln-291 was implicated as a coordinating group for chloride (5) (Fig. 1), we reasoned that the low activity by the Gln-291 mutants could be due to a lowered apparent affinity for the cotransported chloride anion. The physiologically relevant chloride concentration of 150 mm is almost saturating in the case of GAT-1-WT, but in the Gln-291 mutants, the rate of [3H]GABA transport as a function of the chloride concentration was still in the linear range at this concentration (Fig. 3A). If we assume that the decreased transport rate by the mutants is entirely due to impaired chloride affinity, so that at saturating concentrations the rate would be similar to that of GAT-1-WT, the apparent affinity for the mutants is decreased by 7–20-fold. As a control for the data with the Gln-291 mutants, we analyzed the chloride dependence of the rate of [3H]GABA transport with mutants of Ser-328, where the distance of its hydroxyl group to the chloride predicted by the model is almost doubled as compared with that of the amide N-H of Gln-291 (Fig. 1) (5). Clearly the apparent chloride affinity of the S328C and S328N mutants is very similar to that of GAT-1-WT (Fig. 3B).
FIGURE 3.

Chloride dependence of Gln-291 and Ser-328 mutants. HeLa cells expressing WT and mutants of GAT-1 were assayed for [3H]GABA transport as described under “Experimental Procedures.” Transport was carried out for 3 min at the indicated chloride concentrations with gluconate as the substituting ion. The data shown at the indicated chloride concentrations are normalized to those of WT GAT-1 at 150 mm chloride (no gluconate substitution) and are the means ± S.E. of at least three separate experiments performed in quadruplicate. A, dependence of WT (□), Q291A (♦), Q291G (●), Q291S (▴), and Q291T (■). B, chloride dependence of transport by WT (□), S328C (▴), and S328N (○).
To test the idea that the decreased transport rate by the Gln-291 mutants is largely due to a defect in chloride binding, we reasoned that the impact of these mutations would be reduced when introduced into the background of S331E, a chloride-independent mutant of GAT-1 (5). In contrast to GAT-1-WT, the transport activity of GAT-1-S331E was almost independent on the presence of chloride with transport levels of ∼80% in the absence of chloride of those in the presence of chloride (Fig. 4). This is apparently because the introduced glutamate residue may provide a negative charge in TM7, which can mimic the negative charge provided by the cotransported chloride anion (5). As observed previously (5), the rate of [3H]GABA transport by S331E is lower than that of GAT-1-WT (Fig. 2). Introduction of the Gln-291 mutants into the background of GAT-1-S331E would be expected to reduce the transport rates even further if the mutations are functionally independent. However, the activity of the Q291X/S331E double mutants was actually higher than that of the Q291X single mutants, with the exception of the totally inactive Q291E mutant (Fig. 2, black versus gray bars). Again, the expression levels of the Q291X/S331E double mutants at the plasma membrane were similar to those of GAT-1-S331E and GAT-1-WT (Table 1 and supplemental Fig. S1). Thus, except for Q291A, where expression at the plasma membrane was lower than that of Q291A/S331E (Table 1 and supplemental Fig. S1), the increased activity of the Q291X/S331E double mutants is not due to improved surface expression by the S331E mutation.
FIGURE 4.
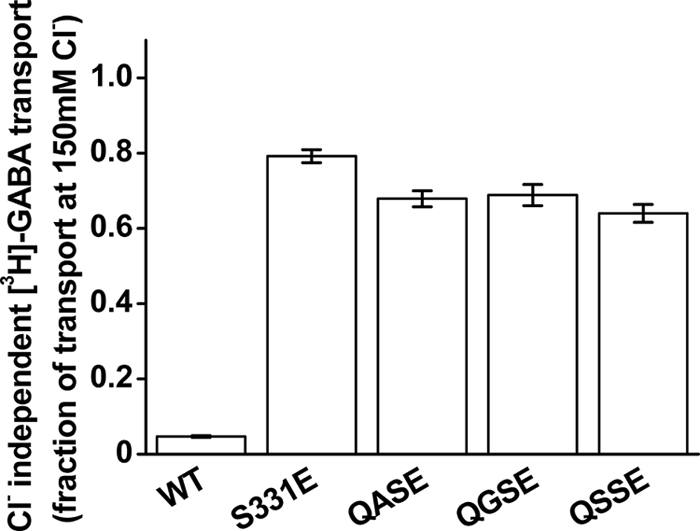
Chloride independence of the Q291X/S331E mutants. The mutants Q291A, Q291G, and Q291S in the background of S331E (QASE, QGSE, and QSSE, respectively), as well as WT and S331E GAT-1, were transiently expressed, and [3H]GABA transport was measured for 3 min, as described under “Experimental Procedures,” in the presence or absence of chloride (150 mm NaCl or sodium gluconate in the external medium, respectively). The data are given as the ratio of transport in the chloride-free medium to that in chloride-containing medium, and the values are the means ± S.E. of at least three separate experiments performed in quadruplicate.
Because of the low rates of transport of the single mutants, it was difficult to determine their kinetic parameters of [3H]GABA transport. However, because of the increased transport, we were able to do so in the case of the double mutants. The averaged Km of S331E (n = 3) was 0.92 ± 0.30 μm, which is comparable with the Km of GAT-1-WT (1.35 ± 0.18 μm) (5). On the other hand, the vmax of S331E was only 5.8 ± 1.7% of that of GAT-1-WT (5) (6.9 ± 1.4 pmol/min/well; see also supplemental Fig. S3). The vmax values for Q291A/S331E, Q291G/S331E, and Q291S/S331E were 2.5 ± 0.1, 1.7 ± 0.2, and 2.0 ± 0.3 pmol/min/well, respectively (n = 3) (see also supplemental Fig. 3). The decreased vmax of the double mutants was partially offset by an increased apparent affinity with Km values of 0.66 ± 0.13, 0.30 ± 0.04, and 0.39 ± 0.05 μm for Q291A/S331E, Q291G/S331E, and Q291S/S331E, respectively (n = 3) (see also supplemental Fig. S3). As in GAT-1-S331E, the activity of the Q291X/S331E double mutants was dramatically less chloride-dependent than that of GAT-1-WT (Fig. 4).
Reconstitution of Gln-291 Mutants in Liposomes
In contrast to GAT-1-WT, GABA uptake by the chloride-independent S331E mutant is dramatically stimulated by the imposition of an outwardly directed proton gradient apparently because the mutation renders the chloride cotransporter into a proton antiporter (5). If the increase in transport activity of Q291X mutants by the introduction of the S331E mutation is indeed because transport is rendered independent of chloride, one would predict that this effect will be optimally observed when an outwardly directed proton gradient is imposed. This idea was tested with proteoliposomes inlaid with the Q291N/S331E and the Q291S/S331E transporters. In this system an artificial sodium gradient can be imposed, using potassium as an internal cation and sodium in the external medium, and an interior negative membrane can be imposed by the addition of the potassium-specific ionophore valinomycin. Indeed, lowering the internal pH from 7.4 to 6.0 and keeping the external pH at 7.4 gave rise to a marked stimulation of [3H]GABA transport not only by S331E but also by Q291N/S331E and the Q291S/S331E (Fig. 5). In contrast with GAT-1-WT, the imposition of the outwardly directed proton gradient actually caused some inhibition (Fig. 5). On the other hand, transport by the reconstituted single mutants Q291N and Q291S was very low, regardless of the internal pH (Fig. 5). Transport by Q291S/S331E in the presence of an outwardly directed proton gradient was similar to that by the single GAT-1-S331E mutant, whereas it was significantly lower in the case of the Q291N/S331E mutant (Fig. 5).
FIGURE 5.

Uptake of [3H]GABA in proteoliposomes. Proteoliposomes were prepared from HeLa cells expressing the indicated GAT-1 mutants as described under “Experimental Procedures” using 10 μl of proteoliposomes containing 15–30 μg of protein, which were diluted into 360 μl of 75 mm NaCl plus 60 mm NaPi, pH 7.4, supplemented with 1 μCi of [3H]GABA and 2.8 μm valinomycin for each triplicate time point (10 min). The “in” medium contained either 120 mm KPi, pH 7.4 (gray bars) or 150 mm KMES, pH 6.0 (black bars). The data are given as percentages of activity of GAT-1-WT at pH 7.4, which ranged from 15.2–28.0 pmol/mg·protein, and are the means ± S.E. of at least four separate experiments.
Transport of [3H]GABA by the two reconstituted double mutants and S331E was completely dependent on the presence of sodium in the external medium, just like GAT-1-WT, with undetectable activity when the sodium was replaced by choline; <0.5% (n = 5) of the values in the presence of sodium. On the other hand, transport by the S331E single mutant and the double mutants Q291N/S331E and Q291S/S331E was basically chloride-independent with 96.0 ± 4.2, 98.4 ± 9.4, and 89.4 ± 5.2% activity of that in chloride-containing medium, respectively, when gluconate replaced chloride (n = 5). Transport by GAT-1-WT was undetectable under these conditions: <0.5% (n = 5) of the value in the presence of chloride.
Chloride Dependence of Transport by Other Putative Chloride-binding Site Mutants
The side chains of Tyr-86 of TM2, Ser-295 of TM6, and Ser-331 of TM7 have also been implicated as chloride coordinating groups (4, 5) (Fig. 1). Moreover, in one model Asn-327 was assigned as a binding residue as well (4), whereas in the other model it is at slightly larger distance than the 3.5 Å required for a direct interaction (5) (Fig. 1). In the case of the conservatively substituted Y86F, the rate of [3H]GABA transport at the physiologically relevant chloride concentration of 150 mm was 50–60% of that of the rate of GAT-1-WT (Fig. 6). However, in contrast to GAT-1-WT, the transport rate by this mutant was basically a linear function of the chloride concentration just as observed with the Gln-291 mutants (Fig. 3A). In the case of Asn-327, the equivalent residue in LeuT participates in the Na1 site (6), and functional evidence with the N327C and N327A mutants is consistent with the idea that this also is true for GAT-1 (7). However, because of the low transport activity observed with these two mutants (7), we made the conserved replacement mutant N327Q. Indeed, the rate of [3H]GABA transport of the mutant at 150 mm chloride was significantly higher than that of the other Asn-327 mutants, namely ∼25% of that of the rate of GAT-1-WT. Also for this mutant, this chloride concentration was far from saturation (Fig. 6). The equivalent residue of Ser-295 in LeuT, Thr-254, also participates in the Na1 site (6). We reported previously that S295A was defective in [3H]GABA transport (7) but found that S295C had low but significant activity (19). The rate of [3H]GABA transport of S295C at 150 mm chloride was only ∼10% of that of the rate of GAT-1-WT, and also for this mutant this chloride concentration was far from saturation (Fig. 6). A similar result was also observed with the S331G and S331A mutants (Fig. 6).
FIGURE 6.

Chloride dependence of mutants of the other putative chloride site residues. HeLa cells expressing WT and the indicated mutants of GAT-1 were assayed for [3H]GABA transport as described under “Experimental Procedures” and in Fig. 3. The data shown at the indicated chloride concentrations are normalized to those of GAT-1-WT at 150 mm chloride (no gluconate substitution) and are the means ± S.E. of at least three separate experiments performed in quadruplicate.
Anion Selectivity of Transport by Other Putative Chloride-binding Site Mutants
We showed previously that when GAT-1-Ser-331 was mutated to a smaller residue, such as alanine or glycine, larger anions, such as iodide or thiocyanate, became much more effective in their ability to stimulate transport. This is consistent with the idea that the truncation of the side chain of Ser-331 creates more space in the chloride-binding site (5). Increased iodide/chloride and thiocyanate/chloride selectivities were also observed with the S295C and Y86F mutants but not with mutants at Ser-328 (Fig. 7), presumably because this residue is too far away to coordinate chloride directly (Fig. 1). In the case of N327Q, we observed a decrease in the above selectivity ratios, and the same was true for the Gln-291 mutants, with a larger effect by the Q291G than by the Q291S mutation. Possible interpretations of these results will be discussed below.
FIGURE 7.

Anion selectivity of the putative chloride site mutants. [3H]GABA transport by HeLa cells transiently expressing GAT-1-WT and the indicated mutants was measured for 10 min at room temperature as described under “Experimental Procedures,” in a medium containing 150 mm chloride (NaCl, black bars), iodide (NaI, white bars), or thiocyanate (NaSCN, gray bars). The data are normalized to the activity of each mutant in the presence of chloride and are the means ± S.E. of at least three separate experiments performed in quadruplicate.
Electrophysiological Analysis of Gln-291 Mutants
Our observations thus far (Figs. 2 and 5 and supplemental Fig. S1) suggest that Gln-291 not only participates in the chloride-binding site but fulfills an additional role in transport. To obtain evidence for the idea that this role may involve hydrogen bond formation with Arg-69, we analyzed the transporter-mediated currents of the Q291N and Q291S mutants expressed in X. laevis oocytes.
Arg-69 of GAT-1 is absolutely essential for transport (16), but sodium-dependent transient currents could be observed only in the most conservative substitution mutant at this position, namely R69K (17). Interestingly, the voltage dependence of these transient currents indicated that the apparent affinity of the empty R69K transporter for sodium was increased (17). Remarkably, this was also the case for the Q291N mutant (Fig. 8). In contrast to the corresponding currents by GAT-1-WT, Q291N exhibited only outward transient currents in the “on” phase when jumping from a holding potential of −25 mV to more positive potentials, and no transients were observed with jumps to more negative potentials (Fig. 8). These transient currents were capacitative, because transient currents of the same magnitude but opposite direction were seen in the “off” phase, jumping back to the holding potential (Fig. 8). Like with R69K (17), the voltage dependence of the transients suggest that at a holding potential of −25 mV, all of the Q291N transporters are already in the sodium-bound state. This bound sodium is released by jumps to positive potentials, thereby giving rise to the outward transient currents seen in the on phase in Fig. 8. Indeed, when the external sodium concentration was progressively decreased, inward transient currents became apparent (data not shown). The similarity of Q291N and R69K (17) with respect to their increased apparent sodium affinity is consistent with the idea that Arg-69 and Gln-291 interact during the transport cycle, so that mutation in either residue may have a similar impact.
FIGURE 8.

Sodium-dependent transient currents and GABA-induced steady-state currents. The membrane voltage of oocytes expressing GAT-1-WT or the indicated mutants was stepped from a holding potential of −25 mV to voltages between −140 to +60 mV in 25-mV increments. Each potential was held clamped for 500 ms, followed by 500 ms of a potential clamped at −25 mV. For each condition, the traces shown are from the same oocytes that are representative of at least three repeats: NaCl, absolute (unsubtracted) currents in ND96; NaCl-ChCl, currents in nominal sodium-free medium (choline substitution) were subtracted from those in ND96; and GABA-induced, currents in ND96 were subtracted from those in the same medium supplemented with 1 mm (GAT-1-WT) or 10 mm (Q291N and Q291S) of GABA. The dashed lines indicate no current.
In contrast to Q291N, the voltage dependence of the transient currents by Q291S was similar to that of GAT-1-WT (Fig. 8), suggesting that the apparent sodium affinity of the empty Q291S mutant transporters was not increased. Whereas the removal of chloride reduces the apparent sodium affinity of GAT-1-WT (28), there was little effect of removal of chloride (gluconate substitution) on the voltage dependence of the sodium-dependent transients by Q291N and Q291S (data not shown). In contrast to Q291N, GABA induced transport currents in oocytes expressing Q291S (Fig. 8). However, the half-maximal concentration of GABA for the activation of the currents by Q291S was 1.25 ± 0.1 mm (n = 3), ∼20–50-fold that for GAT-1-WT. Moreover, the voltage dependence of these currents was different from those of GAT-1-WT (Fig. 9).
DISCUSSION
Our study shows that the conserved glutamine residue Gln-291 of GAT-1 is important for the function of this transporter. However, this residue is not irreplaceable, because low but significant transport is observed when this residue is substituted by amino acid residues with a small side chain (Fig. 2). These mutations have a major impact on the apparent affinity for chloride, which is cotransported with sodium and GABA (3) (Fig. 3). This is in harmony with the idea that the side chain of Gln-291 participates directly in the binding of chloride (Fig. 1) (5). Indeed in the background of the S331E mutation, which is chloride-independent, the functional impact of many of the Gln-291 mutations is greatly reduced when compared with that of the single Gln-291 mutants (Fig. 2). With the exception of Q291A, the increased transport activity in the S331E background was not due to increased levels of the double mutant transporters on the plasma membrane plasma membrane (Table 1). In the case of the Q291S mutation, there is a small stimulatory effect on surface expression (Table 1), but the experiments where all transporters are solubilized and reconstituted into proteoliposomes (Fig. 5) clearly show that the stimulation by the S331E mutation is due to an intrinsic effect on function. Because the introduced glutamate could undergo reversible protonation/deprotonation, the S331E mutation apparently converts GABA transport from chloride cotransport to proton countertransport (5). The latter has been directly demonstrated with the chloride-independent bacterial homologue Tyt1 (29). Indeed, the elimination of the impact of the Q291S mutation by the S331E mutation was seen only when an outwardly directed proton gradient was present (Fig. 5).
It is counterintuitive that conservative replacement of Gln-291, namely Q291N and Q291E, impacts GABA transport more than its replacement by small amino acid residues (Fig. 2). In the case of Q291E, one would expect chloride-independent transport just as with S331E (Fig. 4) (5). The fact that this does not happen (Fig. 2) is probably due to a perturbation of a step different from chloride binding by the Q291E mutation. One possibility is that the Q291E and Q291N mutations also interfere with the functioning of the hydrogen bonding network associated with the thin extracellular gate, involving the interactions of Arg-69 of TM1 with Asp-451 of TM10 and with Gln-291 (6). In the case of Q291N, a significant restoration of the activity by the S331E mutation is observed in intact cells and in the reconstituted system (Figs. 2 and 5). Nevertheless the activity of the Q291N/S331E mutant is lower than that of the Q291S/S331E mutant, which has similar activity to that of the S331E single mutant, especially in the reconstituted system (Fig. 5). It is possible that as long as small substitutions are introduced at Gln-291, this has a relatively minor effect on the functioning of the hydrogen bonding network associated with the thin extracellular gate. On the other hand, the perturbation by the Q291N mutation may be larger, and therefore lower levels of transport are observed in the background of a chloride-independent GAT-1. This idea is compatible with the electrophysiological experiments showing that Q291S is more similar to WT than Q291N (Figs. 8 and 9).
Because of the altered voltage dependence of the GABA-induced currents by Q291S (Fig. 9), they are unlikely to reflect coupled GABA transport. This is also consistent with the very low apparent affinity for GABA observed with the currents, not seen with radioactive uptake, as judged by the impact of the Q291S mutation in the S331E background (supplemental Fig. S3). In contrast to radioactive uptake, the GABA-induced currents may reflect not only electrogenic sodium- and chloride-coupled GABA translocation, but also GABA-induced sodium leak currents (30). It is therefore possible that the GABA-induced inward-rectifying currents by Q291S reflect uncoupled leak currents rather than coupled GABA transport.
In contrast to Q291N, no restoration of activity by the S331E mutation was observed with Q291E (Fig. 2). It is possible that the Q291E mutation is more disruptive to the hydrogen bonding network than the Q291N mutation. Alternatively in the Q291E/S331E double mutant, it is possible that the presence of two potentially negative charged residues in the binding pocket at a short distance from each other might be energetically nonfavorable and could lead to profound changes in the binding pocket that could abolish transport.
Analysis of the chloride dependence of transport by mutants at residues proposed to coordinate the bound chloride (4, 5) showed that the apparent affinity for the anion was reduced in all cases (Figs. 3 and 6). This was not the case for the farther away Ser-328 (Fig. 1), which served as a negative control. Even though Ser-328 appears to be too far away to directly coordinate the chloride ion, mutation of the residue to aspartate or glutamate gave rise to largely chloride-independent transport (5). Although these mutations extend the length of the side chain, the introduced carboxyl group would still not be in the same position as that of the S331E mutation. These observations suggest that a negative charge introduced at a somewhat larger distance from Na1 than the chloride ion may nevertheless still be able to compensate for the excess positive charge of the sodium ions during substrate translocation.
Not all mutations had an equally strong impact on function. The strongest impact on transport at 150 mm of chloride was observed with the Gln-291 mutants (Fig. 2A) and with S295C and the Ser-331 mutants (Fig. 6). Mutations at the latter position are easier to understand than at the two others because Ser-295 and Gln-291 appear to influence additional functions. Because of the proximity of the Na1 and Cl sites, it is expected that sodium facilitates the binding of chloride and vice versa. Indeed, the latter has been documented in GAT-1 (28). It is therefore possible that the reduction of the apparent chloride affinity of S295C and N327Q (Fig. 6) is partly due to effects of these mutations on sodium binding. In the case of Gln-291, the situation is different. This position is not predicted to interact directly with the sodium sites (6), and the removal of chloride did not affect the voltage dependence of the sodium-dependent transient currents.
It is possible that the side chain hydroxyl of Ser-331 is perhaps coordinating chloride at the closest distance. Tyr-86 also has no known additional function, yet the impact of the Y86F mutation on transport was smaller than that of S331G and S331A (Fig. 6). One interpretation of these results is that the hydroxyl group of Tyr-86 is farther away from the bound chloride than that of Ser-331. An alternative, more likely, explanation is that mutation of different putative chloride coordinating residues is expected to have different outcomes dependent on their interactions with neighboring residues.
The strong impact on the chloride dependence of GABA transport by N327Q (Fig. 6) could be due to the fact that the mutation extends the length of the side chain, perhaps bringing the amide group closer to the bound chloride (and sodium) than in GAT-1-WT. This longer side chain could also be the reason that larger anions, such as iodide or thiocyanate, are less efficient to substitute for chloride than in GAT-1-WT (Fig. 7), as if the binding site for chloride became more compacted. Indeed, iodide and thiocyanate became markedly more effective than chloride to stimulate transport by S331A and even more so in S331G where the side chain becomes even shorter (5). Similar, albeit less dramatic effects on anion selectivity were observed with Y86F (Fig. 7). In the case of S295C, the side chain maintains a similar size. Nevertheless iodide and thiocyanate became more efficient in stimulating transport than in GAT-1-WT (Fig. 7). This suggests that the selectivity change may not only be a function of size but also of other factors such as effects of the mutated residue on the position of other chloride coordinating residues. The decreased ability of iodide and thiocyanate to support transport in the Q291G and Q291S mutants (Fig. 7) may be due to the altered position of the mutant side chains as a result of the absence of the interaction between Arg-69 and Gln-291.
The two different models of the chloride-binding site are very similar, and in both the chloride ion is close to the Na1 site (4, 5), in accordance with the observation that either chloride or the endogenous negative charge are required during the translocation of positive charge associated with sodium and substrate translocation (5). All of the residues are highly conserved, suggesting that the same site is used for all of the NSS neurotransmitter transporters. Both models include (GAT-1 numbering) the side chains of Ser-331, Tyr-86, and Ser-295. The difference between the models is that the side chain of Asn-327 (4) provides the fourth coordinating group rather than Gln-291 (5). Our present study provides experimental evidence, supporting the idea that Gln-291 is a chloride coordinating residue, which also fulfills an additional role during transport. To unravel the details of this additional role is an important goal for future studies.
Supplementary Material
Acknowledgments
We thank Niels Danbolt (Oslo University) for a generous gift of the affinity purified antibody against GAT-1. We greatly appreciate Gary Rudnick sharing his unpublished manuscript (now published together with this article (31)).
This work was supported by grants from the Israel Science Foundation and the European Union Consortium European Drug Initiative on Channels and Transporters.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S3.
- NSS
- neurotransmitter:sodium symporter
- TM
- transmembrane domain.
REFERENCES
- 1. Kanner B. I. (1994) J. Exp. Biol. 196, 237–249 [DOI] [PubMed] [Google Scholar]
- 2. Nelson N. (1998) J. Neurochem. 71, 1785–1803 [DOI] [PubMed] [Google Scholar]
- 3. Keynan S., Kanner B. I. (1988) Biochemistry 27, 12–17 [DOI] [PubMed] [Google Scholar]
- 4. Forrest L. R., Tavoulari S., Zhang Y. W., Rudnick G., Honig B. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 12761–12766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zomot E., Bendahan A., Quick M., Zhao Y., Javitch J. A., Kanner B. I. (2007) Nature 449, 726–730 [DOI] [PubMed] [Google Scholar]
- 6. Yamashita A., Singh S. K., Kawate T., Jin Y., Gouaux E. (2005) Nature 437, 215–223 [DOI] [PubMed] [Google Scholar]
- 7. Zhou Y., Zomot E., Kanner B. I. (2006) J. Biol. Chem. 281, 22092–22099 [DOI] [PubMed] [Google Scholar]
- 8. Zhang Y. W., Rudnick G. (2006) J. Biol. Chem. 281, 36213–36220 [DOI] [PubMed] [Google Scholar]
- 9. Vandenberg R. J., Shaddick K., Ju P. (2007) J. Biol. Chem. 282, 14447–14453 [DOI] [PubMed] [Google Scholar]
- 10. Dodd J. R., Christie D. L. (2007) J. Biol. Chem. 282, 15528–15533 [DOI] [PubMed] [Google Scholar]
- 11. Androutsellis-Theotokis A., Goldberg N. R., Ueda K., Beppu T., Beckman M. L., Das S., Javitch J. A., Rudnick G. (2003) J. Biol. Chem. 278, 12703–12709 [DOI] [PubMed] [Google Scholar]
- 12. Quick M., Yano H., Goldberg N. R., Duan L., Beuming T., Shi L., Weinstein H., Javitch J. A. (2006) J. Biol. Chem. 281, 26444–26454 [DOI] [PubMed] [Google Scholar]
- 13. Faham S., Watanabe A., Besserer G. M., Cascio D., Specht A., Hirayama B. A., Wright E. M., Abramson J. (2008) Science 321, 810–814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Shimamura T., Weyand S., Beckstein O., Rutherford N. G., Hadden J. M., Sharples D., Sansom M. S., Iwata S., Henderson P. J., Cameron A. D. (2010) Science 328, 470–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shaffer P. L., Goehring A., Shankaranarayanan A., Gouaux E. (2009) Science 325, 1010–1014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pantanowitz S., Bendahan A., Kanner B. I. (1993) J. Biol. Chem. 268, 3222–3225 [PubMed] [Google Scholar]
- 17. Kanner B. I. (2003) J. Biol. Chem. 278, 3705–3712 [DOI] [PubMed] [Google Scholar]
- 18. Bismuth Y., Kavanaugh M. P., Kanner B. I. (1997) J. Biol. Chem. 272, 16096–16102 [DOI] [PubMed] [Google Scholar]
- 19. Rosenberg A., Kanner B. I. (2008) J. Biol. Chem. 283, 14376–14383 [DOI] [PubMed] [Google Scholar]
- 20. Mari S. A., Soragna A., Castagna M., Santacroce M., Perego C., Bossi E., Peres A., Sacchi V. F. (2006) Cell Mol. Life Sci. 63, 100–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kunkel T. A., Roberts J. D., Zakour R. A. (1987) Methods Enzymol. 154, 367–382 [DOI] [PubMed] [Google Scholar]
- 22. Kleinberger-Doron N., Kanner B. I. (1994) J. Biol. Chem. 269, 3063–3067 [PubMed] [Google Scholar]
- 23. Fuerst T. R., Niles E. G., Studier F. W., Moss B. (1986) Proc. Natl. Acad. Sci. U.S.A. 83, 8122–8126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Keynan S., Suh Y. J., Kanner B. I., Rudnick G. (1992) Biochemistry 31, 1974–1979 [DOI] [PubMed] [Google Scholar]
- 25. Bennett E. R., Su H., Kanner B. I. (2000) J. Biol. Chem. 275, 34106–34113 [DOI] [PubMed] [Google Scholar]
- 26. Radian R., Bendahan A., Kanner B. I. (1986) J. Biol. Chem. 261, 15437–15441 [PubMed] [Google Scholar]
- 27. Borre L., Kanner B. I. (2004) J. Biol. Chem. 279, 2513–2519 [DOI] [PubMed] [Google Scholar]
- 28. Mager S., Kleinberger-Doron N., Keshet G. I., Davidson N., Kanner B. I., Lester H. A. (1996) J. Neurosci. 16, 5405–5414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhao Y., Quick M., Shi L., Mehler E. L., Weinstein H., Javitch J. A. (2010) Nat. Chem. Biol. 6, 109–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Krause S., Schwarz W. (2005) Mol Pharmacol 68, 1728–1735 [DOI] [PubMed] [Google Scholar]
- 31. Tavoulari S., Rizwan A. N., Forrest L. R., Rudnick G. (2011) J. Biol. Chem. 286, 2834–2842 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.