

Abstract
The generation of patterns and the diversity of cell types in a multicellular organism require differential gene regulation. At the heart of this process are enhancers or cis-regulatory modules (CRMs), genomic regions that are bound by transcription factors (TFs) that control spatio-temporal gene expression in developmental networks. To date, only a few CRMs have been studied in detail and the underlying cis-regulatory code is not well understood. Here, we review recent progress on the genome-wide identification of CRMs with chromatin immunoprecipitation of TF-DNA complexes followed by microarrays (ChIP-on-chip). We focus on two computational approaches that have succeeded in predicting the expression pattern driven by a CRM either based on TF binding site preferences and their expression levels, or quantitative analysis of CRM occupancy by key TFs. We also discuss the current limits of these methods and highlight some of the key problems that have to be solved to gain a more complete understanding of the structure and function of CRMs.
Keywords: ChIP-on-chip, chromatin immunoprecipitation, cis-regulatory module, enhancer, transcription factor
Introduction
It is widely accepted that enhancer elements or cis-regulatory modules (CRMs) contain the information that mediates spatially and temporally restricted gene expression. CRMs are bound by transcription factors (TFs) that act in a combinatorial and context-dependent manner. The integration of TF input by a CRM eventually promotes or inhibits the expression of the neighboring gene. Yet, TF binding data obtained from in vitro experiments and mutational CRM structure-function analysis have neither been sufficient to predict CRMs, nor have they been able to predict the activity of a given CRM.[1] One of the central aims of current genome biology is to unravel the general principles of TF targeting and information integration on CRMs that determine developmental patterns in vivo. As CRMs do not act in isolation, we also need to understand the complex gene regulatory networks in which key TFs dynamically bind to hundreds of CRMs and cause feedback on other CRMs depending on the developmental context.[2, 3] Therefore, deciphering the CRM binding code, i.e., the grammar that translates bound TF complexes into spatio-temporal expression patterns (at the level of the cis-regulatory code) will eventually allow the identification of all CRMs within a given network to obtain a comprehensive map of their temporal and combinatorial activities (the regulatory network level).
Identifying regulatory regions at the genome level
One way to identify CRMs is by sequence conservation analysis,[4] as functionally relevant TF binding sites are often conserved. Indeed, there is evidence for conservation especially for highly bound and clustered regions.[5–8] A recent study has demonstrated that functional regulatory motifs can be extracted using this type of phylogenetic footprinting.[9] However, it appears that CRMs that lead to the same embryonic expression patterns in different Drosophila species often lack conservation and differ dramatically in sequence and spacing of TF binding sites.[10] This means that conservation analysis can be an indicator of functionality of a regulatory motif and, therefore, a simple tool for identifying CRMs; however, lack of conservation does not necessarily mean absence of function.
Another strategy is chromatin immunoprecipitation with antibodies against key regulator TFs followed by microarray hybridization (“ChIP-on-chip”) or deep DNA sequencing (“ChIP-seq”) that has recently allowed high-throughput genome-wide localization of CRMs.[5, 6, 8, 11, 12] ChIP is an accurate technique[6] for unbiased, high-resolution in vivo assessment of protein–DNA interactions that gives a quantitative measure of TF occupancy without requiring previous knowledge about TF concentration, diffusion rate, expression pattern, target affinity or interaction with other factors. The identification of TF binding peak clusters allowed the mapping of a remarkably large number of novel candidate CRMs that had not been found in genetic screens, through conservation analyses, or with DNA binding assays.[5, 8] Moreover, large-scale projects such as the model organism Encyclopedia of DNA Elements (modENCODE11) will provide us with unprecedented, comprehensive information about the global and dynamic activity of CRMs by mapping and comparing functional elements in the Caenorhabditis elegans and Drosophila genomes.
Two computational approaches for predicting expression patterns
Although it is currently not clear how many of the candidate regulatory elements are functional, ChIP technology, and conservation analyses have allowed tremendous progress in the identification of CRMs. An emerging goal and major challenge of the field is now to predict the activity of an uncharacterized CRM based on its organization. This has been recently attempted by two computational modeling approaches that differ in the input parameters (Fig. 1): One made use of TF expression levels as well as the arrangement and quality of their binding sites to predict the expression profile of an arbitrary DNA sequence. Segal et al.[13] achieved this by generating a model based on the biochemical properties and binding site preferences of eight key TFs (Bicoid, Hunchback, Caudal, Kruppel, Giant, TorRE, Knirps, and Tailless) of the early Drosophila segmentation network (Fig. 1A). Its CRMs are complex, as they do not act as simple switches based on the binding of selector TFs, but they are able to read gradients of TF activity.[14] The model nonetheless accurately predicted the activity of relatively broadly active gap gene CRMs (e.g., for knirps, giant, and hunchback) even across species. This means that knowing the TF concentration as well as the arrangement and quality of TF binding sites can be sufficient to explain complex segmentation patterns. Moreover, these parameters appear to be crucial for CRM output in this developmental context. The algorithm reached its limits for more refined pair-rule modules (e.g., even skipped); however, such failure could still give useful hints at missing components, e.g., the lack of activators or repressors.
Figure 1.
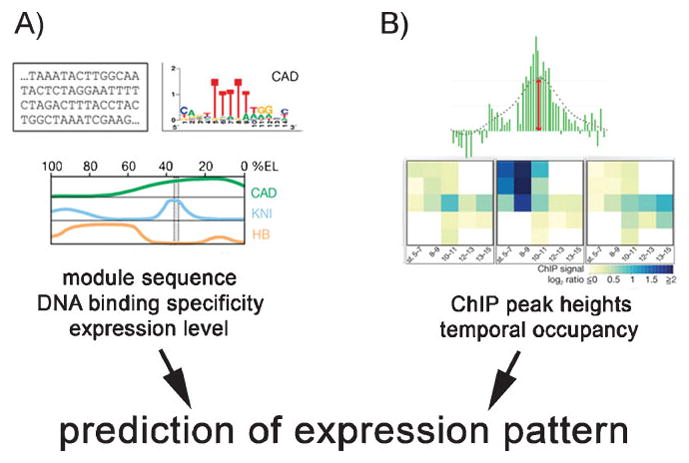
Two computational approaches to predict gene expression patterns differ in their input parameters. A: This approach uses DNA sequence data, TF binding specificity and expression levels. B: This approach is based on the binding intensity of peaks obtained by ChIP-on-chip (see text for details) and temporal occupancy of the regulatory sequence (adapted by permission from Macmillan Publishers Ltd: Nature,[8, 13] copyright 2008, 2009).
As detailed knowledge of biochemical TF properties is often not available, a second approach based on the ChIP-on-chip technology was developed by Zinzen et al.,[8] who generated a comprehensive catalogue of CRMs involved in Drosophila mesoderm development that are bound by five key TFs (Twist, Myocyte enhancer factor 2, Tinman, Bagpipe, Biniou). The authors identified clusters of TF binding peaks that were grouped into more than 8,000 candidate CRMs. By addressing 15 consecutive developmental time points, the authors were also able to assess the temporal occupancy of these regions. They then tested the activities of 36 CRMs with an in vivo reporter assay and found that 35 were sufficient to drive expression in mesodermal tissue. Finally, to ask whether combinatorial TF information obtained from the ChIP experiments (Fig. 1B) was able to serve as predictor of mesodermal CRM activity, the authors defined five broad and partially overlapping categories [meso muscle, visceral (gut) muscle, somatic muscle, meso and somatic muscle, visceral, and somatic muscle] and trained a machine-learning algorithm (a support vector machine) with the respective CRM activity information. Remarkably, the algorithm correctly predicted spatio-temporal expression patterns for 25 out of 35 CRMs (71%), simply based on the spatio-temporal CRM occupancy and intensity of the TF binding peaks obtained by ChIP-on-chip.
It is impressive that this learning/prediction experiment shows that one can, to some extent, predict CRM activity in the early Drosophila embryo without having fully deciphered the cis-regulatory code.[8] However, the categories for scoring the CRM expression patterns were quite broad (e.g., “somatic muscle,” “visceral muscle”) and the origin of more refined expression in subsets of mesodermal tissue was not addressed. The algorithm was less efficient when knowledge of key regulators was sparse, as evidenced by the class of somatic muscle tissue where only the general muscle differentiation factor Mef2 is known. In this case, only two out of seven known CRMs were correctly predicted. It could be that the algorithm had learned rather simple rules, as correct predictions seemed to often match the expression domains of the key TFs. For instance, binding of the Twist selector gene is a strong predictor of mesoderm expression, while binding of the specification factor Biniou clearly determines CRM activity in visceral muscles. Nevertheless, the success of the method is fascinating and testing the algorithm on larger datasets and in different developmental contexts will tell us how powerful this approach is.
Taken together, these two novel strategies for predicting CRMs underline the importance of strong experimental data as a basis for computational approaches to understand gene regulation. The first one (predicting expression patterns based on TF concentration and binding affinity) turned out to be a powerful tool for predicting complex segmentation patterns without knowing the whole map of CRMs involved, and will be useful in regulatory networks where detailed knowledge about the biochemical properties of key regulators is available.[13] The second one (predicting expression patterns based on binding intensity and temporal CRM occupancy) does not require this information and instead succeeds by using in vivo TF binding and CRM activity data.[8] This more general approach can be readily applied to any CRM network for which ChIP-on-chip datasets are available.
Variability of the cis-regulatory code and CRM architecture
Although predicting CRM activity based on few input parameters is possible and hints at general regulatory rules, the assumption that there would be only one general cis-regulatory code is overly simplistic. In line with this hypothesis, Zinzen et al.[8] made the interesting observation that, in some instances, TF occupancy on CRMs was variable and that flexible TF binding profiles (in terms of TF identity, binding duration and ChIP peak heights) could converge toward similar spatio-temporal activity in the mesoderm. An example of this unexpected plasticity is a CRM that belongs to the visceral muscle class despite showing additional binding of the early factors Twist and Tinman. The lack of stringency and the discovery of redundant versions of the code are in line with studies of embryonic patterning with the even skipped enhancer as a model CRM that reported that the arrangement and evolutionary conservation of TF binding sites can be flexible and still result in the same pattern across species.[10, 15, 16] Variability of motif sequence, spacing, order, relative orientation, and motif composition of a given CRM confirms that the rules governing regulatory output can be variable.[17]
The early-acting and variable CRMs mentioned above resemble one of two extreme types of CRMs, the “billboard” type:[18] such CRMs are flexible in motif arrangement and processing of information, as they are able to translate multiple interactions into defined outputs. Contrary to this variability, the second CRM type is more restrictive as it requires highly cooperative and coordinate action of TFs on rather precisely arranged binding sites like the interferon-beta enhancer[19] (“enhanceosomes”-type CRMs). Interesting candidates for this CRM type are rhodopsins that require fewer than 300 base pairs to control subtype-specific spatial and temporal pattern.[20] They share motifs perfectly conserved over 60 million years, such as the RCSI/P3 motif located 15–30 base pairs upstream of the TATA box that is thought to be recognized in vivo by a Pax6 protein, the master regulator of eye development.[21, 22] Additionally, the distinction of photoreceptor subtypes also involves highly conserved home-odomain binding sites that are only present in the CRMs of the appropriate rhodopsins and not in the others.[23] An explanation for the more restricted architecture of these late-acting CRMs is that they act to elaborate a differentiated state rather than establishing differences in cell fate between adjacent cells as the examples for the billboard type mentioned above. Such differences between CRMs should be given more attention in the future and we need the characterization of a larger number of individual CRMs to determine how representative these two categories are or whether most CRMs are in between them.
The problem of TF binding specificity
In addition to the problem of variability in binding to the CRM and its architecture, the mechanisms underlying TF binding specificity are not fully understood. It has long been known that TFs recognize short, degenerate sites of five to ten base pairs.[24] However, many of these sites occur by chance and may be bound by TFs,[12] but only a subset is believed to be functional. On the other hand, how can TFs direct specific expression, when for instance many homeodomain TFs that have different functions in vivo bind to very similar sequences in vitro? In this respect, considerable progress has been made in the classification of most fly and mouse homeodomain TFs,[25–27] but there are still some gaps in our understanding of what distinguishes the members of a TF group in terms of specific DNA binding.[28] It appears now that the amino acids contacting base pairs in the major groove are not the only ones to be important; minor groove contacts and even the primary sequence of the DNA target that dictates three-dimensional structure of the DNA also matter considerably.[28, 29] In addition, interactions with cofactors affect specificity.[30]
The problem of “nonspecific” versus “specific” binding is also apparent in some of the recent ChIP-on-chip studies that have unexpectedly reported significant and reproducible low-level TF binding to up to thousands of sites for 21 TFs of different binding domain types in the Drosophila blastoderm.[7, 9, 12] These low-level bound regions were either in protein-coding or non-conserved non-coding regions of housekeeping genes and/or genes that have no detectable expression in the blastoderm.[7] Simple explanations for this puzzling result could be a methodological problem (noise in the chip experiments), heterogeneity of the blastoderm tissue or non-functionality of these sites. The latter assumption would also explain why there was no apparent selection against this low-level binding, and could mean that some of these neutral elements are material for the evolution of new CRMs. However, some of these regions that are not sufficient for driving expression could have a more subtle function in regulating neighboring genes that are not detected in reporter assays that only address broad expression patterns.[31] Alternatively, they may be relevant for interactions with other CRMs, for regulating the number of available TFs in the nucleus, or they only become meaningful at a later time point in development, for instance when the chromatin state changes. These options have to be addressed in future experiments, as they are of critical importance for our understanding of TF-CRM interactions.
Conclusion
There has been tremendous progress in the mapping and analysis of CRMs in recent years. To understand a cis-regulatory network in its entirety, it is essential to identify (i) the expression patterns of key TFs, (ii) CRMs and their temporal TF occupancy, and (iii) the logic of individual CRMs. For requirement (i) and (ii), ChIP, either combined with microarrays or deep sequencing,[6] complemented by conservation analysis, has proven to be a powerful tool that will provide global insights into combinatorial TF binding. The study by Zinzen et al.[8] shows that the relative level of TF occupancy is relevant and that the temporal dynamics should be taken into account in future studies. Moreover, larger datasets, for instance from the modENCODE project,[11] will tell us how robust and reliable machine learning algorithms are in predicting CRM activity, and it will be interesting to test whether some of the results found for embryonic CRMs can be applied to other developmental contexts.
Concerning requirement (iii), one should keep in mind that cis-regulatory logic cannot be fully understood by simply knowing key regulators, their binding affinities and the localization of their binding sites, as shown by the failure to reconstruct CRMs.[1] This means that we have to expand our knowledge about CRM architecture by thoroughly dissecting a larger number of individual CRMs. Taken together, the increasing knowledge obtained from the detailed dissection of individual CRMs, in combination with genome-wide ChIP-on-chip datasets, will allow us to make further progress toward understanding the cis-regulatory code.
Acknowledgments
We thank Stephen Small, Robert Johnston, Dominic Didiano, and Tilman Triphan for discussion and suggestions on the paper. This work was supported by NIH EY13010-11 to C. D. and an EMBO long-term fellowship (ALTF 462-2008) to J. R.
Abbreviations
- CRM
cis-regulatory module
- TF
transcription factor
- ChIP
chromatin immunoprecipitation
References
- 1.Johnson LA, Zhao Y, Golden K, et al. Reverse-engineering a transcriptional enhancer: a case study in Drosophila. Tissue Eng Part A. 2008;14:1549–59. doi: 10.1089/ten.tea.2008.0074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Levine M, Davidson EH. Gene regulatory networks for development. Proc Natl Acad Sci USA. 2005;102:4936–42. doi: 10.1073/pnas.0408031102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bonn S, Furlong EE. Cis-regulatory networks during development: a view of Drosophila. Curr Opin Genet Dev. 2008;18:513–20. doi: 10.1016/j.gde.2008.09.005. [DOI] [PubMed] [Google Scholar]
- 4.Mereiles-Filho ACA, Stark A. Comparative genomics of gene regulation – conservation and divergence of cis-regulatory information. Curr Opin Genet Dev. 2009;19:565–70. doi: 10.1016/j.gde.2009.10.006. [DOI] [PubMed] [Google Scholar]
- 5.Zeitlinger J, Zinzen RP, Stark A, et al. Whole-genome ChIP-chip analysis of Dorsal, Twist, and Snail suggests integration of diverse patterning processes in the Drosophila embryo. Genes Dev. 2007;21:385–90. doi: 10.1101/gad.1509607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Visel A, Blow MJ, Li Z, et al. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature. 2009;457:854–8. doi: 10.1038/nature07730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.MacArthur S, Li XY, Li J, et al. Developmental roles of 21 Drosophila transcription factors are determined by quantitative differences in binding to an overlapping set of thousands of genomic regions. Genome Biol. 2009;10:R80. doi: 10.1186/gb-2009-10-7-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zinzen RP, Girardot C, Gagneur J, et al. Combinatorial binding predicts spatio-temporal cis-regulatory activity. Nature. 2009;462:65–70. doi: 10.1038/nature08531. [DOI] [PubMed] [Google Scholar]
- 9.Kheradpour P, Stark A, Roy S, et al. Reliable prediction of regulator targets using 12 Drosophila genomes. Genome Res. 2007;17:1919–31. doi: 10.1101/gr.7090407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ludwig MZ, Bergman C, Patel NH, et al. Evidence for stabilizing selection in a eukaryotic enhancer element. Nature. 2000;403:564–7. doi: 10.1038/35000615. [DOI] [PubMed] [Google Scholar]
- 11.Celniker SE, Dillon LAL, Gernstein MB, et al. Unlocking the secrets of the genome. Nature. 2009;459:927–30. doi: 10.1038/459927a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li XY, MacArthur S, Bourgon R, et al. Transcription factors bind thousands of active and inactive regions in the Drosophila blastoderm. PLoS Biol. 2008;6:e27. doi: 10.1371/journal.pbio.0060027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Segal E, Raveh-Sadka T, Schroeder M, et al. Predicting expression patterns from regulatory sequence in Drosophila segmentation. Nature. 2008;451:535–40. doi: 10.1038/nature06496. [DOI] [PubMed] [Google Scholar]
- 14.Chopra VS, Levine M. Combinatorial patterning mechanisms in the Drosophila embryo. Brief Funct Genomic Proteomic. 2009;8:243–9. doi: 10.1093/bfgp/elp026. [DOI] [PubMed] [Google Scholar]
- 15.Clyde DE, Corado MS, Wu X, et al. A self-organizing system of repressor gradients establishes segmental complexity in Drosophila. Nature. 2003;426:849–53. doi: 10.1038/nature02189. [DOI] [PubMed] [Google Scholar]
- 16.Ochoa-Espinosa A, Small S. Developmental mechanisms and cis-regulatory codes. Curr Opin Genet Dev. 2006;16:165–70. doi: 10.1016/j.gde.2006.02.014. [DOI] [PubMed] [Google Scholar]
- 17.Brown CD, Johnson DS, Sidow A. Functional architecture and evolution of transcriptional elements that drive gene coexpression. Science. 2007;317:1557–60. doi: 10.1126/science.1145893. [DOI] [PubMed] [Google Scholar]
- 18.Arnosti DN, Kulkarni MM. Transcriptional enhancers: Intelligent enhanceosomes or flexible billboards? J Cell Biochem. 2005;94:890–98. doi: 10.1002/jcb.20352. [DOI] [PubMed] [Google Scholar]
- 19.Panne D, Maniatis T, Harrison SC. An atomic model of the interferon-beta enhanceosome. Cell. 2007;129:1111–23. doi: 10.1016/j.cell.2007.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pichaud F, Desplan C. A new visualization approach for identifying mutations that affect differentiation and organization of the Drosophila ommatidia. Development. 2001;128:815–26. doi: 10.1242/dev.128.6.815. [DOI] [PubMed] [Google Scholar]
- 21.Sheng G, Thouvenot E, Schmucker D, et al. Direct Regulation of rhodopsin1 by Pax-6/eyeless in Drosophila: Evidence for the evolutio-narily conserved function of Pax-6 in photoreceptors. Genes Dev. 1997;11:1122–31. doi: 10.1101/gad.11.9.1122. [DOI] [PubMed] [Google Scholar]
- 22.Papatsenko D, Nazina A, Desplan C. A conserved regulatory element present in all Drosophila rhodopsin genes mediates Pax6 functions and participates in the fine-tuning of cell-specific expression. Mech Dev. 2001;101:143–53. doi: 10.1016/s0925-4773(00)00581-5. [DOI] [PubMed] [Google Scholar]
- 23.Tahayato A, Sonneville R, Pichaud F, et al. Otd/Crx, a dual regulator for the specification of ommatidia subtypes in the Drosophila retina. Dev Cell. 2003;5:391–402. doi: 10.1016/s1534-5807(03)00239-9. [DOI] [PubMed] [Google Scholar]
- 24.Biggin MD. To bind or not to bind. Nat Genet. 2001;28:303–4. doi: 10.1038/91045. [DOI] [PubMed] [Google Scholar]
- 25.Berger MF, Badis G, Gehrke AR, et al. Variation in homeodomain DNA binding revealed by high-resolution analysis of sequence preferences. Cell. 2008;133:1266–76. doi: 10.1016/j.cell.2008.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Noyes MB, Christensen RG, Wakabayashi A, et al. Analysis of homeodomain specificities allows the family-wide prediction of preferred recognition sites. Cell. 2008;133:1277–89. doi: 10.1016/j.cell.2008.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Affolter M, Slattery M, Mann RS. A lexicon for homeodomain-DNA recognition. Cell. 2008;133:1133–5. doi: 10.1016/j.cell.2008.06.008. [DOI] [PubMed] [Google Scholar]
- 28.Parker SC, Hansen L, Abaan HO, et al. Local DNA topography correlates with functional noncoding regions of the human genome. Science. 2009;324:389–92. doi: 10.1126/science.1169050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rohs R, West SM, Sosinsky A, et al. The role of DNA shape in protein-DNA recognition. Nature. 2009;461:1248–53. doi: 10.1038/nature08473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Joshi R, Passner J, Rohs R, et al. Functional specificity of a Hox protein mediated by the recognition of minor groove structure. Cell. 2007;131:530–43. doi: 10.1016/j.cell.2007.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Keränen SV, Fowlkes CC, Luengo Hendriks CL, et al. Three-dimensional morphology and gene expression in the Drosophila blastoderm at cellular resolution. II: dynamics. Genome Biol. 2006;7:R124. doi: 10.1186/gb-2006-7-12-r124. [DOI] [PMC free article] [PubMed] [Google Scholar]