

Abstract
Surface Plasmon Resonance (SPR) is a highly sensitive method for the detection of molecular interactions. One interacting partner is immobilized on the sensor chip surface while the other is injected across the sensor surface. This chapter focuses on high affinity immobilization of protein substrates for affinity and kinetic analyses using biotin/streptavidin interaction and GST/anti-GST-antibody interaction.
Keywords: BIAcore 3000, Biotin, Streptavidin, GST, High-Affinity Immobilization, SPR
1. Introduction
By immobilizing a protein on a sensor chip surface, one can take advantage of the optical phenomenon known as Surface Plasmon Resonance (SPR) to observe molecular interactions in real time. Accordingly, SPR can be used to determine rate and affinity constants for interactions involving proteins, nucleic acids, carbohydrates, lipids or small molecules (1–3). This chapter will focus on the high affinity immobilization of protein and peptide ligands to a sensor chip surface. This can be accomplished by a number of different approaches including direct amine coupling, antibody capture, and biotin/streptavidin affinity among others (4–7). The semi-automated nature of SPR biosensor operations, the low consumption of reagents, and the capacity to produce quantitative data about macromolecular interactions make this method preferable to traditional approaches like pulldown assays, analytical chromatography, or isothermal titration calorimetry (ITC) (Note 1).
One approach to high-affinity immobilization is the covalent linkage of an anti-Glutathione S-transferase (GST) antibody to the sensor chip surface (Figure 1). Recombinant proteins fused with GST are readily immobilized on a sensor chip surface through the GST/anti-GST antibody interaction (Figures 2 and 3). Cloning, expression, and purification of GST chimeric proteins are relatively straightforward. Commercial vectors available for the expression of GST-fusion proteins include pGEX (GE Healthcare), pAB-GST (AB Vector), and pAcG (BD Bioscience). The advantage of this approach is that the antibody serves as a universal adaptor, permitting the capture of all GST-fusion proteins on the sensor chip surface. As the antibody is covalently bound to the sensor chip, the anti-GST surface can be regenerated many times and coated with different GST-fusion constructs. Regeneration is accomplished by injecting a glycine-HCl solution at pH 2.2 over the sensor chip surface. As the antibody is covalently bound to the surface, it does not dissociate upon exposure to glycine-HCl pH 2.2 solution, but the GST/anti-GST antibody interaction is disrupted, and thus the chip can be subsequently loaded with a new GST-fusion protein. While the overall output signal strength is lower than other protein immobilization techniques, the anti-GST sensor chip surface can be regenerated upwards of 200 times with different GST-fused proteins, facilitating multiple types of experiments on a single anti-GST antibody-derivatized chip.
Figure 1. A typical single flow cell sensorgram generated during the loading of anti-GST antibody.

The flow cell surface was activated for direct amine coupling of the anti-GST antibody (αGST-Ab) by injecting the coupling solution (40 μl NHS/EDC at 5 μl/min flow rate with EXTRACLEAN command). Anti-GST antibody was then diluted to 30 μg/ml in immobilization buffer and 45 μl were injected at 5 μl/min flow rate, omitting the EXTRACLEAN command. To deactivate esters on the sensor chip surface, ethanolamine was injected (35 μl at 5 μl/min flow rate with EXTRACLEAN command). To test the binding surface, supplied recombinant GST protein was diluted to 5 μg/ml and 100 μl were injected over the sensor chip at 20 μl/min flow rate. To remove GST from the binding surface, 40 μl of glycine-HCl pH 2.2 was injected at 20 μl/min flow rate.
Figure 2. Affinity and kinetic measurements using an anti-GST antibody CM5 sensor chip.

A. Flow cells 1 and 2 were loaded with 1000 RUs of GST and GST-RGS12GoLoco proteins, respectively. Increasing concentrations of Gαi1 were injected using the KINJECT command (300 μl injections with a 200 second dissociation phase at 20 μl/min flow rate). Specific binding was determined by subtracting non-specific binding to a GST control flow cell from the GST-RGS12GoLoco response curve. B. Binding affinities were determined by plotting the maximum response attained at each concentration of Gαi1 versus the concentration of the Gαi1, then by fitting to a Langmuir binding isotherm using GraphPad Prism 5.0 (GraphPad Software, La Jolla, CA) to determine dissociation constant (KD). Kinetic analyses of the association (ka) and dissociation (kd) rates were also used to determine KD using the simultaneous ka/kd (1:1 Langmuir) model (BIAevaluation). Differences in KD determinations between kinetic and equilibrium analyses are likely to reflect mass-transport and/or rebinding limitations to binding assay execution, as indicated by large ka values, nearly linear initial association seen on sensorgrams, and/or greatly slowed dissociation (see Schuck, P. et al. in this Volume for further information).
Figure 3. Diverse applications of an anti-GST CM5 sensor chip.

A. A GST-RGS fusion protein was immobilized on the surface of flow cell 1 and GST was immobilized on the surface of flow cell 2 (the latter surface to use to subtract sensorgram changes in resonance due to buffer shifts). Gαi1 pre-incubated with either GDP or GDP+Mg2++AlF4− (AMF), a transition-state mimetic form known to bind avidly to RGS proteins, was injected over the sensor surface. GST-RGS bound to the Gαi1(AMF) but not Gαi1(GDP) as expected. B. GST-H-Ras loaded with a non-hydrolyzable GTP analogue, GTPγS, was loaded in flow cell 1, GST-H-Ras loaded with GDP was loaded in flow cell 2, and GST control was loaded in flow cell 3. An N-terminal construct of c-Raf-1, a known H-Ras binding partner, was injected across the sensor chip surface. The data show the nucleotide state selectivity of the H-Ras/c-Raf-1 interaction. C. GST-RGS14 was loaded in flow cell 1 and GST was loaded in flow cell 2. Gαi1 was injected in GDP+Mg2++AlF4− (AMF) Running Buffer over the sensor chip surface and a Gαi1/RGS14 specific interaction was detected. Specific binding for all injections was determined by subtracting the non-specific binding observed from a GST control flow cell from the experiment-containing flow cells.
The biotin/streptavidin interaction is another high affinity, non-covalent (8, 9) method to immobilize substrate to the sensor chip surface. Because of the low dissociation constant associated with the interaction (approximately 10−15 M (10)), biotinylated substrates are almost irreversibly bound to the sensor chip surface. Protein biotinylation can be performed in vivo or in vitro and can be verified by western blot (Figure 4). Biotinylation can occur concomitantly with protein expression in bacteria by co-transfection of biotin ligase and a vector construct coding for the protein of interest fused to a biotin ligase substrate sequence (G L N D I F E A Q K I E W H E) (11). Alternatively, purified recombinant protein from bacterial, insect, or mammalian expression, with a biotin ligase substrate sequence, can be biotinylated post-purification by incubation with biotin ligase in vitro (Avidity, Aurora, CO) (6, 12). Biotinylated proteins can be directly immobilized on commercially available streptavidin coated sensor chips (Figure 5). To further facilitate the study of molecular interactions, biotinylated peptides can be synthesized (4, 5). Protein/nucleic acid interactions can also be probed using SPR when biotinylated oligonucleotides are immobilized on the sensor chip surface (13) (most oligonucleotide vendors offer biotin as a modification). Advantages to this approach include the assurance that all immobilized molecules will be in the same orientation and that a high substrate density can be achieved using commercially available streptavidin sensor chips. Additionally use of biotin/streptavidin immobilization offers the ability to user harsher regeneration conditions (e.g., 1M urea) than the GST/anti-GST antibody surfaces – so long as the biotinylated moiety (protein, peptide, DNA oligo, etc.) also survives the regeneration without losing its capacity for binding to analyte.
Figure 4. Confirmation of biotinylated protein production.
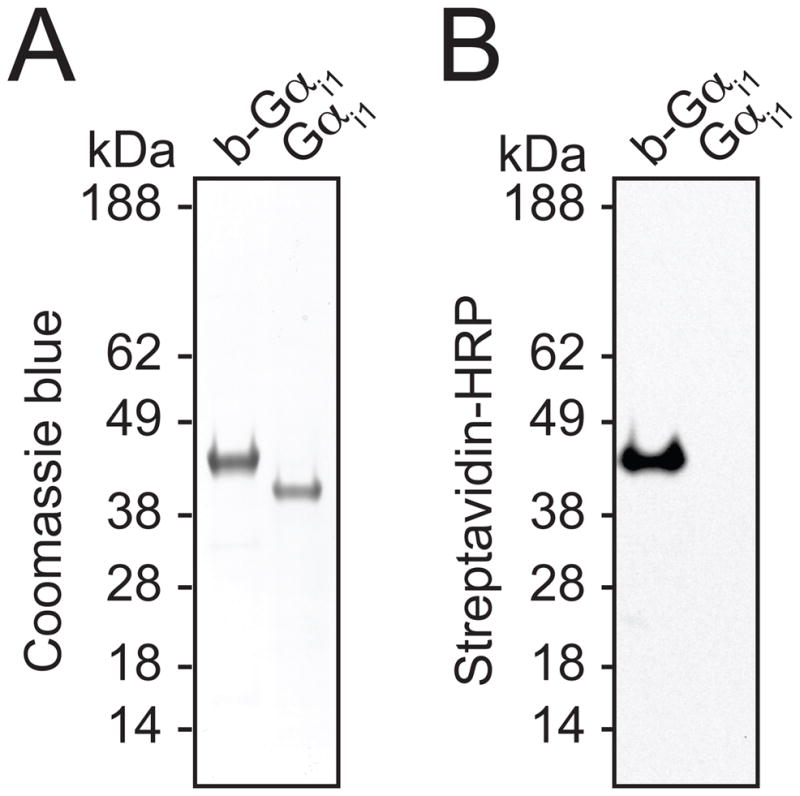
A. SDS-PAGE gel stained with Coomassie Blue dye reveals the migration of biotinylated Gαi1, designated b-Gαi1, and unlabeled Gαi1. B. Immunoblot of SDS-PAGE gel transferred to nitrocellulose using streptavidin-horseradish peroxidase antibody to detect biotinylated protein.
Figure 5. Loading Streptavidin (SA) Sensor Chip.

A typical sensorgram generated during the loading of biotinylated protein to flow cells 1 and 2. The flow cell surface was stabilized with 3 × 20 μl injections of Regeneration Buffer before loading of biotinylated-Gαi1 (260 nM final concentration, as diluted in Biotin-conjugate Immobilization Buffer), one flow cell at a time, until saturation is observed (note plateau in sensorgram curves).
We have used both of these methods with great success in a number of protein-protein interaction studies (Note 2) (4–7, 12, 14) (Figures 2, 3, and 6).
Figure 6. Affinity measurements using a SA sensor chip.

A. Flow cell 1 was loaded with 450 RUs of Biotin-KB-801 peptide, and flow cell 2 was loaded with 350 RUs mNotch peptide as a control. Increasing concentrations of Gαi1 in GDP+Mg2++AlF4− (AMF) Buffer, the transition state mimetic form, were injected over the sensor surface as indicated, using the KINJECT command (300 μl injections with a 200 second dissociation phase at 20 μl/min flow rate). Binding curves were obtained by subtracting non-specific binding to a non-interacting biotinylated peptide from all experiment-containing flow cells. B. Binding affinities were determined by plotting the maximum response attained at each concentration of Gαi1 versus the concentration of the Gαi1, then by fitting to a rectangular hyperbola to determine KD using GraphPad Prism 5.0 (Graphpad Software, La Jolla, CA). C. Flow cell 1 was loaded with 450 RUs of Biotin-Gαi1, and flow cell 2 was loaded with an equivalent RU signal of denatured Biotin-Gαi1. Varying concentrations of RGS10 in GDP+Mg2++AlF4− (AMF) Buffer were injected over the sensor surface as indicated, using the KINJECT command (300 μl injections with a 200 second dissociation phase at 20 μl/min flow rate). Specific binding was determined by subtracting non-specific binding from a flow cell containing denatured biotin-Gαi1. D. Binding affinities were determined by plotting the maximum response attained at each concentration of RGS10 versus the concentration of the RGS10, then by fitting to a rectangular hyperbola to determine KD using GraphPad Prism 5.0 (GraphPad Software, La Jolla, CA). These final panels were reproduced from Soundararajan et al. 2008 (ref. (7)). Copyright (c) 2008 National Academy of Sciences, U.S.A.
2. Materials
2.1. GST fusion protein production
6 × 1 L Luria Broth prepared and autoclaved in baffled 2800 ml flasks then supplemented with ampicillin (50 μg/ml final concentration).
6 ml 1 M IPTG (isopropyl-beta-D-thiogalactopyranoside) stock solution (Arcos Organics, New Jersey).
200 ml of a BL21(DE3) E. coli overnight culture transformed with desired vector construct (GeneChoice, Frederick, MD).
Beckman J-6 Centrifuge
Beckman LM-8 Ultracentrifuge
High-pressure homogenizer (Emulsiflex, Avestin)
Akta FPLC (GE Healthcare)
HiTrap glutathione Sepharose-4B Fast Flow (GSTrap) column (GE Healthcare).
Sephacryl S200 HR column (GE Healthcare).
Vivaspin 20 ultrafiltration spin column (Sartorius Stedium Biotech, Goettingen Germany) with an appropriate molecular weight cutoff for retention of desired protein during filtration/concentration.
2.2 GST-protein immobilization via antibody
Running Buffer: 10 mM HEPES pH 7.5, 150 mM NaCl, 3 mM EDTA, 0.0005 % (v/v) NP-40 alternative (Calbiochem, La Jolla, CA). Filtered with 0.2 μm polystyrene bottle top filter and degassed by allowing the vacuum to continue for 10 min after filtering is complete. Agitation may be required to release air bubbles from vessel walls.
10 mM Glycine-HCl pH 2.2 (included in GST Capture Kit).
Buffer A: 50 mM Tris-HCl pH 7.5, 100 mM NaCl, 1 mM dithiothreitol, 5 % (weight/volume) glycerol.
Buffer B: 50 mM Tris-HCl pH 7.5, 100 mM NaCl, 10 mM reduced glutathione, 1 mM dithiothreitol, 5 % (weight/volume) glycerol.
S200 buffer: 50 mM Tris-HCl pH 8.0, 250 mM NaCl, 5 mM dithiothreitol, 2.5 % (weight/volume) glycerol.
GST Capture Kit (GE Healthcare; ref. (15)): goat anti-GST antibody 0.8 mg/ml in 0.15 M NaCl, recombinant GST 0.2 mg/ml in 10 mM HEPES pH 7.4, 0.15 M NaCl, 3 mM EDTA, 0.005 % Surfactant P20, immobilization buffer (10 mM sodium acetate pH 5.0), regeneration solution (10 mM glycine-HCl pH 2.2).
Sensor Chip CM5 (GE Heathcare).
Amine Coupling Kit (GE Healthcare): 750 mg 1-ethyl-3-(3 dimethyl-aminopropyl)carbodiimide hydrochloride (EDC) resuspended in water to 0.2 M concentration, 115 mg N-hydroxysuccinimide (NHS) resuspended in water to 0.05 M, 10.5 ml 1.0 M ethanolamine-HCl pH 8.5.
2.3 Binding assay
GST-fusion protein (see 2.1)
10 mM Glycine-HCl pH 2.2 (included in GST Capture Kit).
2.4 Biotin-conjugated protein production
6 × 1 L Luria Broth prepared and autoclaved in baffled 2800 ml flasks then supplemented with ampicillin (50 μg/ml) and chloramphenicol (10 μg/ml final concentration).
6 ml 1 M IPTG (isopropyl-beta-D-thiogalactopyranoside) stock solution (Arcos Organics, New Jersey).
50 mM D-Biotin (Sigma) dissolved in ethanol. Add 1 ml to each flask for a 50 μM final concentration.
B strain (hsdR, lon11, sulA1) E. coli transformed with chloramphenicol-resistance plasmid expressing biotin ligase BirA (Avidity, Aurora, Colorado).
Beckman J-6 Centrifuge
Beckman LM-8 Ultracentrifuge
High-pressure homogenizer (Emulsiflex, Avestin)
Akta FPLC (GE Healthcare)
Buffer N1: 50 mM HEPES pH 7.5, 400 mM NaCl, 30 mM imidazole, 5 % (weight/volume) glycerol.
Buffer N2: 50 mM HEPES pH 7.5, 400 mM NaCl, 1 M imidazole, 5 % (weight/volume) glycerol.
S200 Buffer: 10 mM HEPES pH 7.5, 300 mM NaCl, 5 mM dithiothreitol, 5 % (weight/volume) glycerol.
HisTrap Fast Flow column (GE Healthcare).
Sephacryl S200 HR column (GE Healthcare).
Vivaspin 20 ultrafiltration spin column (Sartorius Stedium Biotech, Goettingen Germany) with an appropriate molecular weight cutoff for desired protein.
2.5 Biotinylation verification
Laemmli Loading Buffer (2x) (Sigma).
TBS-T Buffer - Tris Buffered Saline (Sigma) with 0.1% v/v Tween 20 (Fisher).
Blocking Buffer – 5% w/v powdered non-fat milk in TBS-T.
1:10,000 dilution (in Blocking Buffer) of stock Streptavidin Horseradish Peroxidase (SA-HRP; GE Healthcare).
Polyacrylamide denaturing gel. We routinely use NuPAGE Bis-Tris 4–12% gradient gels (Invitrogen).
Polyacrylamide gel electrophoresis apparatus and Coomassie Stain.
MES Buffer for electrophoresis: (50 mM Tris base, 50 mM 3–(N–Morpholino) propanesulfonic acid, 1 mM EDTA, 0.01% SDS at pH 7.3).
SeeBlue molecular weight marker (Invitrogen).
Western blot transfer apparatus (homemade).
ECL Plus Chemiluminescence Detection Kit (GE Healthcare).
2.6 Streptavidin immobilization and binding assay
Biotin-conjugate Immobilization Buffer: 10 mM HEPES pH 7.5, 150 mM NaCl, 3 mM EDTA, 0.0005 % (v/v) NP-40 alternative (Calbiochem La Jolla, CA). Filtered with 0.2 μm polystyrene bottle top filter and degassed by allowing the vacuum to continue for 10 min after filtering is complete. Agitation may be required to release air bubbles from vessel walls.
Regeneration Buffer: 1 M NaCl, 50 mM NaOH.
Sensor Chip SA (GE Healthcare).
Running Buffer: 10 mM HEPES pH 7.4, 150 mm NaCl, 50 μM EDTA, 100 μM GDP, 0.0005 % (v/v) NP-40 alternative (Calbiochem). Filtered with 0.2 μm polystyrene bottle top filter and degassed by allowing the vacuum to continue for 10 min after filtering is complete. Agitation may be required to release air bubbles from vessel walls (See Note 3).
3. Methods
The carboxymethylated dextran surface is highly negatively charged, and it is therefore difficult to immobilize proteins with a significant negative charge. CM4 sensor chips are available with a lower degree of carboxymethylation, and are better suited for the immobilization of such proteins. Quantitative analysis of binding data using saturation binding or kinetic methods requires specific binding to be measured. To determine specific binding, we usually perform background subtraction with an appropriate control for non-specific binding. For biotin-streptavidin experiments, we usually use denatured protein or an irrelevant biotinylated peptide. For GST experiments, we use GST alone or an irrelevant GST-fusion protein. As controls for protein-protein interaction specificity, we often employ the use of proteins with loss-of-function mutations. This can provide definitive supporting evidence as to the specificity of molecular interactions being tested (16, 17) (Note 4).
3.1. GST-fusion Protein Production
Inoculate 6 × 1 L Luria Broth (containing antibiotic) with E. coli transformed with protein expression construct.
Culture at 37 °C while shaking at 210 rpm.
Reduce temperature to 20 °C after the optical density of the culture reaches OD600 is 1.0.
Induce protein expression with IPTG (500μM final concentration) and culture 14 hours at 20 °C while shaking at 210 rpm.
Harvest cells by centrifugation at 10,000 × g for 10 min and resuspend in 25 ml Buffer A.
Lyse cells using a high-pressure homogenizer at 10,000 psi.
Clarify lysate by ultra-centrifugation for 45 min at 160,000 × g.
Purify by GST affinity chromatography. We use an Akta FPLC. Load clarified lysate onto 5 ml GSTrap column. Unbound protein is washed through the column with five column volumes of Buffer A. GST-fusion protein is eluted from the column with five column volumes of Buffer B. Fractions containing GST-fusion protein are pooled and subjected to size exclusion chromatography using a Sephacryl S200 HR column. Monodisperse protein is eluted in S200 buffer, concentrated using a VivaSpin 20 ultrafiltration spin column at 4 °C 3,000 × g, and stored at −80 °C.
3.2 GST Immobilization via antibody (based on BIAcore GST Capture Kit Instructions for Use (15))
DOCK CM5 sensor chip
PRIME system with Running Buffer.
Start sensorgram flow cells 1–4.
Mix 100 μl 0.05 M N-hydroxysuccinimide (NHS) from Amine Coupling Kit with 100 μl 0.2 M 1-Ethyl-3-[3-dimethylaminopropyl]carbodiimide hydrochloride (EDC) from Amine Coupling Kit and INJECT 40 μl of resultant mixture with EXTRACLEAN at 5 μl/min flow rate.
Dilute anti-GST antibody (from GST Capture Kit) (Note 5) in immobilization buffer, also from kit, to 30 μg/ml. We dilute 3 μl of antibody in 70 μl immobilization buffer. INJECT 45 μl at 5 μl/min, as this is sufficient antibody to derive all four flow cells. Do not select the EXTRACLEAN command (see Note 6).
INJECT (with EXTRACLEAN) 35 μl ethanolamine from Amine Coupling Kit at 5 μl/min flow rate.
-
Flow Running Buffer at 20 μl/min.
To check the capacity of the newly-created anti-GST antibody chip to bind GST, perform the following steps:
Dilute recombinant GST (from the GST Capture Kit) to 5 μg/ml in Running Buffer.
INJECT 100 μl of diluted GST over the anti-GST antibody surface at 20 μl/min flow rate across all flow cells. The equilibrium saturation value provides a measure of surface capacity of individual flow cells chip. A freshly prepared sensor chip surface will load ~2000 RUs of GST (Figure 1) (Note 7).
Regenerate surface with a 40 μl glycine-HCl pH 2.2 injection at a flow rate of 20 μl/min across all flow cells.
3.3 Immobilized GST-fusion protein-based binding assay
Load sensor chip surface by using MANUAL INJECT command to inject GST-fusion protein over desired flow cell(s). 500 nM is a good starting concentration for the gradual loading of protein to the sensor chip surface; using the MANUAL INJECT command will allow for repeating stopping and re-starting of protein injection until desired level of binding is achieved.
If loading flow cells independently, load the same molar amounts of protein to the surface of each flow cell; if the proteins are of a similar molecular weight, this will result in loading the same number of RUs to each flow cell.
The surface is now ready for the injection of analyte. This should be done using the KINJECT command. Begin with an injection of buffer as a control, then proceed with increasingly more concentrated injections of analyte. (See Note 8 for considerations about when regeneration might be required) (Figure 2,3).
To dissociate GST-fusion proteins from the antibody surface, inject 40 μl glycine-HCl pH 2.2 at flow rate of 20 μl/min across all flow cells. As the anti-GST antibody is covalently attached, this regeneration procedure does not negatively affect the ability to bind another round of injected GST-fusion proteins.
3.4 Biotin-conjugated Protein Production
Inoculate 6 × 1 L of sterile Luria Broth containing ampicillin (50 μg/ml final concentration) and chloramphenicol (10 μg/ml final concentration) with E. coli transformed with BirA and protein expression construct sub-cloned with a biotin ligase targeting sequence.
Culture at 37 °C while shaking at 210 rpm.
Reduce temperature to 20 °C when the culture attains an optical density OD600 of 1.0.
Induce protein expression with IPTG (500μM final concentration), add D-biotin solution (50 μM final concentration) and culture 14 hours at 20 °C while shaking at 210 rpm.
Harvest cells by centrifugation at 10,000 × g for 10 min and resuspend in 25 ml buffer N1.
Lyse cells using a high-pressure homogenizer at 10,000 psi.
Clarify lysate by ultra-centrifugation for 45 min at 160,000 × g.
Purify by affinity chromatography. Our expression vectors for biotinylating proteins also contain an N-terminal hexahistidine tag, which demonstrates affinity for charged nickel, and thus the biotinylated proteins are purified by a nickel chelating column (HisTrap). We use an Akta FPLC (GE Healthcare). Load lysate onto a 5 ml HisTrap column. Unbound protein is washed through the column with five column volumes of Buffer N1. His-fusion protein is eluted from the column with sequential steps of 3 % then 30 % Buffer N2 in five column volumes each. Fractions containing His-fusion protein are pooled and subjected to size exclusion chromatography using a Sephacryl S200 HR column. Monodisperse protein is eluted in S200 buffer concentrated using a VivaSpin 20 ultrafiltration spin column at 4 °C and 3,000 × g, and stored in aliquots at −80 °C.
3.5 Biotinylation Verification
In separate tubes, add 2 μg of biotinylated protein and 2 μg of a non-biotinylated control protein to 40 μl of 2X Laemelli Loading Buffer and boil for 5 minutes.
Load a 12 well denaturing gel as follows: Lane 1 - markers, Lane 2 - biotin-protein, Lane 3 - control protein, Lanes 4–5 blank, Lane 6 - markers, Lane 7 -biotin-protein, Lane 8 - control protein, Lanes 9–12 blank.
Following completion of gel-electrophoresis, cut the gel vertically along Lane 5.
Stain lanes 1–5 with Coomassie Brilliant Blue dye. This is a positive control to verify equivalent protein loading.
Transfer the lanes 5–12 onto a nitrocellulose membrane using a standard electroblotting transfer technique.
After the completion of the transfer, wash the nitrocellulose membrane 3 times in TBS-T.
At room temperature, soak the membrane in Blocking Buffer with gentle rocking for 1 hour.
Wash 3X with TBS-T.
Dilute enough SA-HRP in TBS-T/milk solution (1 μl SA-HRP to 2.5 ml TBS-T/milk solution) to cover the entire blot and incubate with rocking for 1 hour at room temperature.
Wash 3X with TBS-T
Detect the presence of SA-HRP using chemiluminescence (Figure 4).
3.6 Streptavidin Immobilization and binding assay
Stabilize the sensor chip surface with multiple injections (usually three or more) of Regeneration Buffer at a flow rate of 20 μl/min (Note 9).
Maintain a 5 μl/min flow rate and, one flow cell at a time using the MANUAL INJECT command, inject biotinylated protein (Figure 5, Note 10).
The negative control used to subtract buffer shifts should be a biotinylated protein/peptide relevant to the experiment. For Gα subunits, we often use a biotinylated Gα that is denatured on the surface by performing 4 × 20 μl injections of Regeneration Buffer post-immobilization.
- Once the binding surface has been established, multiple rounds of injections of analyte can be performed (Figure 6, Note 11). These injections can be automated as follows (GST-fusion protein is used as an example):
Command Explanation DEFINE APROG galpha //a unique name for this block PARAM %position //defines ‘position’ is the only variable FLOW 20 //sets the flow rate (units μl/min) FLOWPATH 1,2,3,4 //can be modified to inject only two flow cells KINJECT %position 200 300 //performs a stabilized injection, 200 μl volume and 300 sec dissociation time END DEFINE APROG loadGST //specifies what to load to sensor chip surface FLOW 5 FLOWPATH 1,2 //specifies flow cells to load INJECT R2F3 15 //specifies position to retrieve GST from stock tube and amount (in μl) EXTRACLEAN END DEFINE APROG loadGSTfusion //specifies what to load to sensor chip FLOW 5 FLOWPATH 3,4 INJECT R2F4 20 //specifies position to retrieve GST-fusion protein and amount (in μl) EXTRACLEAN END DEFINE APROG glycine //specifies removal of GST/GST-fusion from antibody surface FLOW 20 FLOWPATH 1,2,3,4 INJECT R2F6 40 EXTRACLEAN END MAIN RACK 1 THERMO_C //specifies which thermal block is to be found in the rack positions RACK 2 THERMO_A RACK R Reag_A DETECTION 1,2,3,4 APROG loadGST //loads a GST control APROG loadGSTfusion //loads the experiment APROG galpha R2A1 //injection of 200 μl of the sample in rack position R2A1 APROG glycine //regenerates the binding surface APPEND Standby //puts the machine in standby mode END
Footnotes
We have frequently observed that protein-protein interactions described in the literature using affinity-pulldowns with purified proteins cannot be reproduced using SPR. A major reason for this discrepancy is that artifactual binding interactions are observed in affinity-pulldown experiments when impure and aggregated protein preparations are used, and non-quantitative analysis is performed. Thus it is most effective to do experiments with homogenous purified protein samples. Moreover, investigators should not be overly discouraged at their inability to replicate published literature based on affinity-pulldown experiments.
From our experience, the BIAcore 3000 does not provide reproducible data in the presence of moderate to high concentrations of DMSO (> 0.1%). Therefore, this system does not provide a robust approach to determine the effects on protein-protein interactions of low affinity (Kd > 1 μM) small molecule inhibitors that are often dissolved in DMSO.
The addition of GDP is specific to studying heterotrimeric Gα subunits and may not be needed for studying other protein families.
A large component of observed resonance changes upon analyte injection can be due to “buffer shifts.” This is due to altering the refractive index of the liquid at the sensor surface (18) and should not be misconstrued as specific binding. If identical running and analyte buffers are used, no “buffer shift” is observed. Buffer shift is typically removed by background subtraction as described in Methods. To avoid buffer shift, we advise preparing analytes directly in the running buffer for the experiment, or at high enough stock concentrations that dilution removes the majority of “buffer shift”.
We have attempted to use other GST-antibodies from other vendors and have found them to not be amenable to multiple regenerations.
For high quality data, do not use the EXTRACLEAN feature when using the KINJECT command. This often results in additional noise immediately before the injection begins.
When generating anti-GST sensor surfaces, we typically observe differential immobilization of antibody on the four flow cells. This is due to the serial nature of the sensor chip fluidic system. Injected solutions flow from Fc1 to Fc2 then to Fc3 and finally to Fc4. Thus Fc4 can have significantly lower amounts immobilized ligand than Fc1 due to depletion of ligand in the flowing injection stream. To overcome this problem, differential flow cell loading by multiple, independent injections can be performed to equalize immobilization levels across all four flow-cells. Alternatively, to account for this phenomenon, we routinely switch the flow cells on which ligands are immobilized.
The binding surface of the anti-GST-coated sensor chip requires regeneration under two circumstances:
- One wishes to change the immobilized GST-fusion protein prior to the next analyte injection.
- One observes a slow dissociation of the binding partner, such that the sensorgram does not return to baseline in a given timed interval after the injection of analyte is complete. For accurate binding data in this case, it is necessary to re-load the GST-fusion protein after each round of analyte binding.
Regeneration Buffer is injected in repeated, sequential 20 μl amounts until the sensorgram baseline returns to less than 10 RUs from the pre-injection baseline.
When using biotinylated protein, we usually saturate the surface. However, when using biotinylated peptides, we usually try not to saturate the surface and load approximately 100–500 RU’s to the surface. If mass transport is a concern, we load multiple flow cells with varying amounts of biotinylated peptides to make sure our measurements are not affected by mass transport effects.
Full-length biotinylated proteins immobilized on sensor chips are often not stable when undocked; these sensor chips are likely not to function if reinserted into the BIAcore apparatus. In contrast, streptavidin-bound biotinylated peptide and anti-GST antibody sensor chips are more stable and can be undocked, stored at 4 °C inside a 50 ml polypropylene capped tube filled with running buffer for months at a time, then redocked, and additional binding experiments conducted.
References
- 1.Malmqvist M, Karlsson R. Biomolecular interaction analysis: affinity biosensor technologies for functional analysis of proteins. Curr Opin Chem Biol. 1997;1:378–383. doi: 10.1016/s1367-5931(97)80077-4. [DOI] [PubMed] [Google Scholar]
- 2.Jason-Moller L, Murphy M, Bruno J. Curr Protoc Protein Sci. Unit 19. Chapter 19. 2006. Overview of Biacore systems and their applications; p. 13. [DOI] [PubMed] [Google Scholar]
- 3.Majka J, Speck C. Analysis of protein-DNA interactions using surface plasmon resonance. Adv Biochem Eng Biotechnol. 2007;104:13–36. [PubMed] [Google Scholar]
- 4.Johnston CA, Afshar K, Snyder JT, Tall GG, Gonczy P, Siderovski DP, Willard FS. Structural determinants underlying the temperature-sensitive nature of a Galpha mutant in asymmetric cell division of Caenorhabditis elegans. J Biol Chem. 2008;283:21550–21558. doi: 10.1074/jbc.M803023200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Johnston CA, Lobanova ES, Shavkunov AS, Low J, Ramer JK, Blaesius R, Fredericks Z, Willard FS, Kuhlman B, Arshavsky VY, Siderovski DP. Minimal determinants for binding activated G alpha from the structure of a G alpha(i1)-peptide dimer. Biochemistry. 2006;45:11390–11400. doi: 10.1021/bi0613832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kimple AJ, Willard FS, Giguere PM, Johnston CA, Mocanu V, Siderovski DP. The RGS protein inhibitor CCG-4986 is a covalent modifier of the RGS4 Galpha-interaction face. Biochim Biophys Acta. 2007;1774:1213–1220. doi: 10.1016/j.bbapap.2007.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Soundararajan M, Willard FS, Kimple AJ, Turnbull AP, Ball LJ, Schoch GA, Gileadi C, Fedorov OY, Dowler EF, Higman VA, Hutsell SQ, Sundstrom M, Doyle DA, Siderovski DP. Structural diversity in the RGS domain and its interaction with heterotrimeric G protein alpha-subunits. Proc Natl Acad Sci U S A. 2008;105:6457–6462. doi: 10.1073/pnas.0801508105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Green NM. Avidin. 1. The Use of (14-C)Biotin for Kinetic Studies and for Assay. Biochem J. 1963;89:585–591. doi: 10.1042/bj0890585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Melamed MD, Green NM. Avidin. 2. Purification and Composition. Biochem J. 1963;89:591–599. doi: 10.1042/bj0890591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haun M, Wasi S. Biotinylated antibodies bound to streptavidin beads: a versatile solid matrix for immunoassays. Anal Biochem. 1990;191:337–342. doi: 10.1016/0003-2697(90)90228-2. [DOI] [PubMed] [Google Scholar]
- 11.Beckett D, Kovaleva E, Schatz PJ. A minimal peptide substrate in biotin holoenzyme synthetase-catalyzed biotinylation. Protein Sci. 1999;8:921–929. doi: 10.1110/ps.8.4.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Willard FS, Low AB, McCudden CR, Siderovski DP. Differential G-alpha interaction capacities of the GoLoco motifs in Rap GTPase activating proteins. Cell Signal. 2007;19:428–438. doi: 10.1016/j.cellsig.2006.07.013. [DOI] [PubMed] [Google Scholar]
- 13.Webster CI, Cooper MA, Packman LC, Williams DH, Gray JC. Kinetic analysis of high-mobility-group proteins HMG-1 and HMG-I/Y binding to cholesterol-tagged DNA on a supported lipid monolayer. Nucleic Acids Res. 2000;28:1618–1624. doi: 10.1093/nar/28.7.1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Willard FS, Siderovski DP. Covalent immobilization of histidine-tagged proteins for surface plasmon resonance. Anal Biochem. 2006;353:147–149. doi: 10.1016/j.ab.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 15.BIAcore. GST Capture Kit Instructions for Use GE Healthcare. 2005. [Google Scholar]
- 16.Willard FS, Kimple AJ, Johnston CA, Siderovski DP. A direct fluorescence-based assay for RGS domain GTPase accelerating activity. Anal Biochem. 2005;340:341–351. doi: 10.1016/j.ab.2005.02.015. [DOI] [PubMed] [Google Scholar]
- 17.Willard FS, Zheng Z, Guo J, Digby GJ, Kimple AJ, Conley JM, Johnston CA, Bosch D, Willard MD, Watts VJ, Lambert NA, Ikeda SR, Du Q, Siderovski DP. A point mutation to Galpha i selectively blocks GoLoco motif binding: direct evidence for Galpha/GoLoco complexes in mitotic spindle dynamics. J Biol Chem. 2008;283:36698–36710. doi: 10.1074/jbc.M804936200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Decker J, Reischl U. Molecular diagnosis of infectious diseases. Humana Press; Totowa, N.J: 2004. [Google Scholar]