

Abstract
Glucocorticoids, such as dexamethasone (Dex), have been used as in vitro inducers of adipogenesis. However, the roles of the glucocorticoid receptor (GR) in adipogenesis have not been well characterized yet. Here we show that inhibition of GR activity using the GR antagonist RU486 prevents human mesenchymal stem cell (hMSC) and mouse embryonic fibroblast (MEF) differentiation into adipocytes. Moreover, in MEFs isolated from GR knockout (GRnull) and GRdim mice deficient in GR DNA-binding activity, adipogenesis was blocked. We identified GRE sites in the first intron of KLF15 by bioinformatical promoter analysis and confirmed their functional relevance by demonstrating GR interaction by chromatin immunoprecipitation. Moreover transfection of MEFs with siRNA for KLF15 significantly attenuated the expressions of adipogenic-marker genes and the lipid accumulation. Our results provide a new mechanism for understanding glucocorticoids dependent adipogenesis and that GR promotes adipogenesis via KLF15 gene expression as a transcriptional direct target.
Keywords: adipogenesis, glucocorticoid receptor, human mesenchymal stem cell, mouse embryonic fibroblast, bioinformatics analysis, Krüppel-like factor 15
Adipose tissue plays a key role in the regulation of energy balance and mobilization as well as the endocrine regulation of nutrient intake. Because of its role in obesity and the metabolic syndrome, there is increasing interest in adipocyte biology, including the differentiation of adipose tissue.
It is well established that fibroblastic cells including 3T3-L1, mesenchymal stem cells and mice embryonic fibroblasts (MEF) can be differentiated into adipocytes under exposure to adipogenic inducers such as fetal bovine serum (FBS), dexamethasone (Dex), 3-isobutyl-1-methylxanthine (IBMX), and insulin (1–4). This adipogenesis process is tightly regulated by the complicated temporal expression of transcription factors (5–7). Among them, the peroxisome proliferator-activated receptor γ (PPARγ) and CCAAT/enhancer-binding protein α (C/EBPα) have been shown to play critical roles in adipogenesis (8, 9). These two proteins act synergistically in the transcriptional activation of various adipocyte-specific genes (10). In addition, two other members of the C/EBP family, C/EBPβ and C/EBPδ, are induced earlier than C/EBPαand PPARγ just after adipogenic stimulation. The ectopic expression of C/EBPβ and C/EBPδ promotes the expression of PPARγ and C/EBPα in fibroblasts and 3T3-L1 (11–13), suggesting that C/EBPβ and C/EBPδare upstream transcriptional regulators for PPARγ and C/EBPα.
Glucocorticoids are potent regulators of adipose differentiation both in vitro and in vivo (14–16). The synthetic glucocorticoid Dex is widely used as a component of the adipogenic induction cocktail for in vitro adipogenesis systems. Cao et al. have shown that treatment of 3T3-L1 preadipocytes with Dex activates C/EBPδ(11), suggesting that the potential mechanism of the role of Dex in adipogenesis is to enhance PPARγ and C/EBPαexpression via promoting C/EBPδ expression (12). In the mesenchymal progenitor cell line, Dex induced C/EBPα, C/EBPβ and C/EBPδ, which lead to an increase in PPARγand aP2 to induce adipocyte differentiation (17).
Glucocorticoids exert their effects through the glucocorticoid receptor (GR), a ligand-activated transcription factor that belongs to the nuclear receptor superfamily (18, 19). Molecular functions of the GR can be divided into two main actions; a transcriptional activator as a dimer and a transcriptional repressor as a monomer. As an activator, the ligand-bound GR binds to palindromic glucocorticoid response elements (GREs) as a dimer, thereby promoting target genes’ transcription (20–23). Dimerization depends on the D loop in the DNA-binding domain (DBD). Transcriptional repression by GR appears to be mediated by GR monomers’ protein-protein interaction with other transcription factors, and neither needs dimerization nor DNA binding. GR dimerizaion activity is abolished by an amino acid substitution in the GR D loop (A458T) (24). In mice carrying this point mutation (GRdim mice), GRE binding-dependent gene expression is impaired, but the monomer GR function i.e. cross-talk with other transcription factors (protein-protein interaction), is maintained. (25). To get insight into the precise molecular mechanism of adipogenesis triggered by Dex, we utilized MEF from two GR mutant mice; GR deficient (GRnull) mice and GRdim mice. These tools allow us to characterize direct GR target genes in adipogenesis.
Among the set of GR targets during adipogenesis screened in this study, we focused on the role of KLF15, a member of the Krüppel-like family of transcription factors. Krüppel-like factors (KLFs) play diverse roles in cell proliferation, cell differentiation, and development. Some members of KLFs, such as KLF2 (26), KLF5 (27) and KLF15 (28) have been implicated in adipogenesis. KLF15 has previously been shown to regulate adipogenesis by inducing GLUT4 (29) and PPARγ (28) expression in 3T3-L1 preadipocytes; KLF15−/− mice exhibit severe hypoglycemia after an overnight fast (30). Previous studies revealed that KLF15 is induced by ectopic expression of C/EBPδ and C/EBPβ, Dex treatment, and cAMP hormonal stimulation in 3T3-L1 cells (28). However, the molecular mechanism for the promotion of KLF15 gene expression is still unknown.
Here we report that glucocorticoid stimulation was required for in vitro adipogenesis and GR activation with glucocorticoids induced KLF15 gene expression in a DNA-dependent manner.
MATERIALS AND METHODS
Reagents and Cells
Dexamethasone (Dex), 3-isobutyl-1-methylxanthine (IBMX), insulin, and RU486 were obtained from Sigma-Aldrich (St. Louice, MO, USA). Pioglitazone was obtained from ALEXIS Biochemicals (Lausen, Switzerland). BMGM culture media for human mesenchymal stem cells (hMSCs) were obtained from Cambrex Bio Science Walkersville (Walkersville, MD, USA). Mouse embryonic fibroblast cells (MEFs) from GRnull mice and GRdim mice were isolated as described (31). The 293T cell line was obtained from American Type Culture Collection. Anti-GR antibody (sc-1002) for chromatin immunoprecipitation was obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA) (32).
Cell culture and Differentiation
HMSCs were maintained in BMGM culture media and cells from passages 3–6 were used for experiments. MEFs were isolated from 13.5-day-old embryos obtained from C57BL/6 mice as described previously (33). Briefly, after dissection of head and visceral organs, embryos were minced and trypsinized for 30 minutes at 37°C. Embryonic fibroblasts were filtered, plated, and maintained in Dulbecco’s modified Eagle medium (DMEM) with 10% fetal calf serum (FCS), 100 U/ml penicillin, and 100 µg/ml streptomycin at 37°C in an atmosphere of 5% CO2. All experiments with wild-type, GRnull, and GRdim MEFs were performed with cells in passages 2–10. For adipocyte differentiation, two-day post-confluent cells were incubated for 3 days in adipogenesis-inducing medium (AIM), consisting of DMEM supplemented with 10% FBS, 500 µM IBMX, 1 µM Dex, 10 µM pioglitazone, 10 µg/mL insulin. Cells were then incubated in adipogenesis maintenance medium (DMEM supplemented with 10% FBS, 10 µg/mL insulin) for 4 days, with a fresh change of medium after 2 days (MEFs), or for 3 days (hMSCs); cells were then returned to AIM medium. Subsequently, lipid accumulation was visualized using Oil Red O stain as described previously (34). Nile Red staining was performed to quantify lipid accumulation. PFA fixed cells were incubated with 1:1000 of a Nile Red stock (0.5 mg/ml in acetone) in 80% glycerol 20% water and 1mg/ml DAPI for nuclear counter staining. Lipid droplets were analysed using Cellomics automated fluorescent microscopy. Cell numbers were counted according to DAPI staining and Nile red stained objects greater than 5.16 µm were considered as lipid droplets and their fluorescent intensity was determined.
Quantitative RT-PCR
Total RNA was extracted using Isogen (Wako, Osaka, Japan) and 2 µg RNA was used for cDNA synthesis with oligo(dT)18 primer by the Ready-to-Go You-First cDNA kit (GE Healthcare Life Sciences, Uppsala, Sweden), according to the manufacturer’s instructions. Quantitative real-time RT-PCR was performed using TaqMan Gene Expression Assay probe for Adiponectin (Hs00605917_m1), C/EBPα (Hs00269972_s1), C/EBP δ (Hs00270931_s1), KLF15 (Hs00362736_m1), PPARγ (Hs01115513_m1), GAPDH (Hs99999905_m1), mAdiponectin (Mm00456425_m1), mC/EBPα (Mm01265914_s1), mC/EBPδ (Mm00786711_s1), mGR (Mm00433832_m1), mHIF3α (Mm00469373_m1), mKLF9 (Mm00495172_m1), mKLF15 (Hs00164004_m1), mPPARγ (Mm01184322_m1), mZbtb (Mm01176868_m1), mZhx3 (Mm00770117_m1) and mGAPDH (Mm99999915_g1)(All Applied Biosystems, Carlsbad, CA, USA). Relative expression was calculated using the ΔΔCt values and results were expressed as 2-ΔΔCt. GAPDH was used as an internal control to normalize differences in each sample. The value of each control sample was set at 1 and was used to calculate the fold change in target genes. For the results in Figure 2c, 4b, 4d and Supplementary Figure 3 quantitative RT-PCR was performed as described (35). Primer information can be provided upon request. Data were analyzed statistically using the Mann-Whitney U test or Steel-Dwass' test (multiple comparison). P values less than 0.05 were considered statistically significant.
Figure 2. Induction of Adipocyte Differentiation in MEFs from GRdim and GRnull Mice.
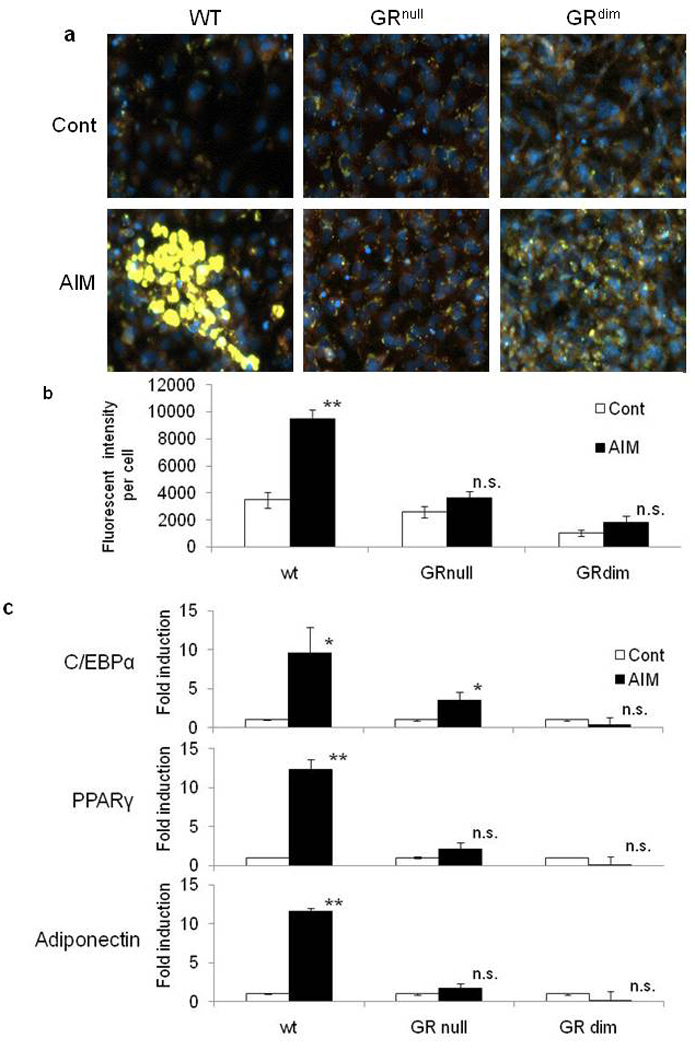
MEFs derived from wild type, GRnull and GRdim mice were differentiated into adipocytes and stained with Nile Red for lipid droplets (yellow) and DAPI (blue) (a). Total fluorescent intensity of lipid droplets per cell were analyzed by Cellomics automated fluorescent microscopy (b). Data were mean ±SEM; n=5. P values were determined by Mann-Whitney U test (** P<0.01 vs control). Analysis of mRNA expression of adipogenesis-specific factors in wild type, GRdim, and GRnull MEFs during adipogenesis using quantitative RT-PCR 1 day after adipogenic differentiation (c). Data are mean ±SEM; n=3. P values were determined by Mann-Whitney U test (** P<0.01 vs control, * P<0.05 vs control).
Figure 4. Expressions of KLF15 and C/EBPδ mRNA in MEFs from Wild Type, GRdim and GRnull Mice.
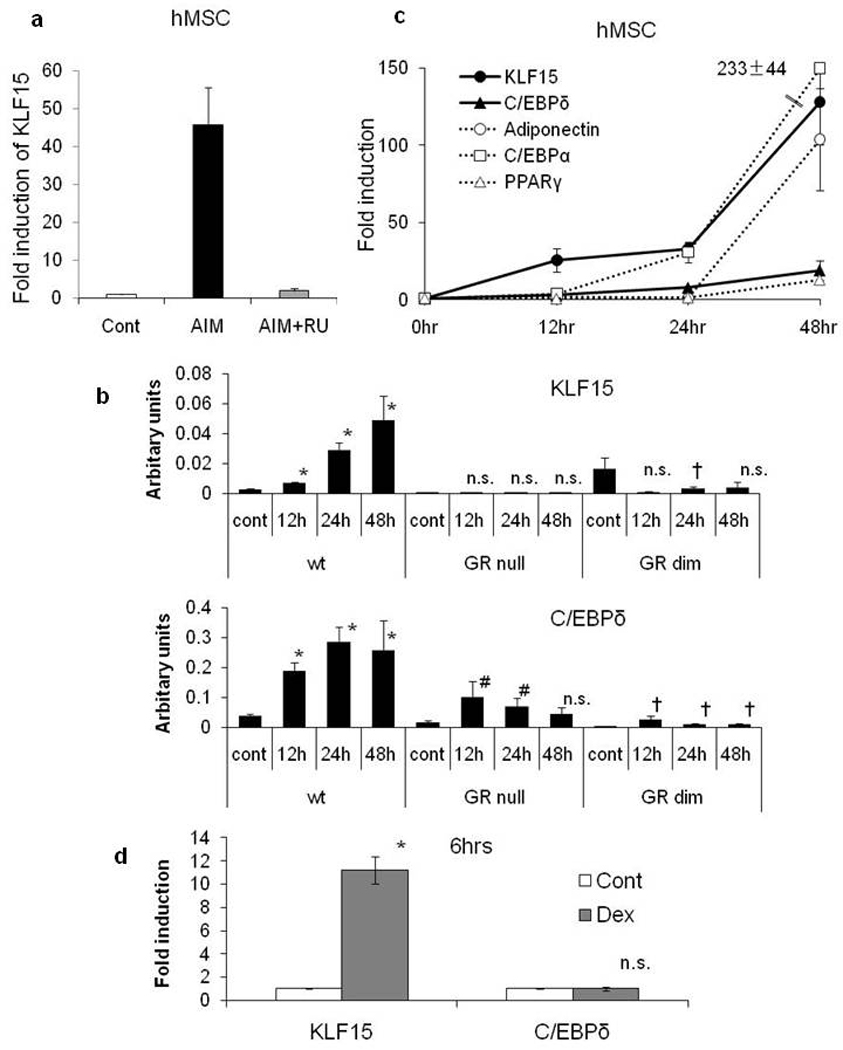
Expression of KLF15 and effect of RU486 for KLF15 in hMSCs 2days after induction (a) and expression of KLF15 and C/EBPδ in wild type, GRdim and GRnull MEFs during adipogenesis (b). Data are mean±SEM; n=3. Data were statistically analyzed with Steel’s test (* P<0.05 vs wild type 0hr, # P<0.05 vs GRnull 0hr, †P<0.05 vs GRdim 0hr). Time courses of expression of KLF15 and adipogenic marker genes in hMSCs(c). Expressions of KLF15 and C/EBPδ in wild type MEFs at 6 hours after Dex alone stimulation(d). Total RNA was analyzed using quantitative RT-PCR. Data are mean±SEM; n=3–4. P values were determined by Mann-Whitney U test (* P<0.05 vs control).
Microarray data analysis
To generate first-strand cDNA, each reaction contained T7-linked oligo(dT) primers and 5 µg total RNA (Affymetrix, Santa Clara, CA, USA). After second-strand synthesis using the SuperScript cDNA system (Invitrogen, Carlsbad, CA, USA), in vitro transcription was performed with biotinylated UTP and CTP (Enzo Life Sciences, New York, NY, USA), resulting in approximately 20-fold amplification of RNA. The biotinylated cRNA targets (20 µg) were then cleaned up, fragmented, and hybridized to GeneChip expression arrays (Mouse Genome 430 2.0 Array, Affymetrix). Arrays were then washed and stained with streptavidin- phycoerythrin before being scanned on an Affymetrix GeneChip scanner. Gene expression was analyzed using the GeneSpring Software 7 (Agilent Technologies, Santa Clara, CA, USA). Genes with signal intensities below 50 were eliminated and normalized, and then genes from cells incubated with AIM medium or Dex treatment that exhibited at least a twofold change relative to the control treatment were selected for analysis. In addition, genes from cells incubated with AIM and RU486 treatment (adipogenesis-RU486 regulated group) that exhibited at least a twofold change compared to cells incubated in AIM medium were also selected. Genes that belong to both the adipogenesis-RU486 treatment group and Dex-treated group were identified as GR-regulated adipogenic genes.
Location Analysis of the Promoter Sequences of KLF15 gene
Promoter regions (from −7500bp to +2000bp) of GR-regulated adipogenic genes conserved between mouse and human genome were analyzed using rVista 2.0 and visualized using the ECR browser (http://ecrbrowser.dcode.org/). MatInspector software searched the highly conserved region for putative GRE sites (Genomatix, Munich, Germany). These GRE sites included androgen receptor (AR) response elements (AREs) and progesterone receptor (PR) response elements (PREs), because GR, AR, and PR bind to a consensus sequence for the superfamily of steroid hormone receptors.
Chromatin immunoprecipitation
Chromatin immunoprecipitation (ChIP) assays were performed as previously described (36, 37). MEFs were washed with PBS and then crosslinked using 1% formaldehyde for 10 min. The crosslinking was quenched with glycine (final concentration, 0.125 M). The samples were then washed twice in PBS and homogenized in ChIP cell lysis buffer (5 mM PIPES [pH 8.0], 85 mM KCl, 0.5% Nonidet P-40, and protease inhibitor cocktail). Nuclei were sedimented and separated from the cellular debris, and then placed into nuclear lysis buffer (50 mM Tris HCl [pH 8.1], 10 mM EDTA, 1% SDS, 10 µM aprotinin, 10 µM leupeptin, and 1 mM phenylmethylsulfonyl fluoride). After 10 min on ice, the lysate was sonicated on ice (Digital Sonifier; BRANSON, Danbury, CT, USA) using three pulses of 2 s each at 4–6 W, and then centrifuged for 20 min at 21,000 × g at 4°C. The supernatant was collected as chromatin samples. At this point, one aliquot was reserved as the input sample. When visualized on an agarose gel, the DNA produced a smear of fragments approximately 200 to 600 bp long. For each immunoprecipitation reaction, 2 µg of chromatin was used. The chromatin was precleared by incubating with protein-G agarose at 4°C for 1 h. Antiserum raised against the GR or pre-immune IgG were added (2 µg each), and the samples rotated at 4°C overnight. Immunoprecipitates were isolated by incubating with blocked protein-G agarose and then washed extensively. Chromatin was eluted from the antibody by incubating for 10 min at room temperature with elution buffer (0.1 M NaHCO3 and 1% SDS). Crosslinking was reversed with NaCl (0.2 M, final concentration) and incubating at 65°C for at least 4 h. Samples were then digested with 40 ng of proteinase K, and DNA was isolated via phenol/chloroform extraction followed by ethanol precipitation. DNA concentrations were calculated by measuring absorbance at 260 nm. PCR primer pairs used were 5’ -GCACGCTGACCCAATGGC- 3’ and 5’ -AGGCCGCTGGACTCCG- 3’ for the GRE1 site (+980/+998), 5’ -CGGAGTCCAGCGGCCT- 3’ and 5’ -GATCCAGGACTTCCTGGACCC- 3’ for the GRE2 site (+1085/+1103) in the first intron of the KLF15 gene.
Luciferase reporter assay
The mouse KLF15 enhancer region (+933/+1216) was amplified by PCR using genomic DNA from C57BL/6 mice with the sense primer 5'- GCACGCTGACCCAATGGC -3' and the antisense primer 5'- GATCCAGGACTTCCTGGACCC -3' and then inserted into the pTA vector (TOYOBO, Osaka, Japan). This plasmid was digested with Nhe I and Xho I restriction enzymes and ligated into the luciferase expression vector pGL3-tk, which had been constructed by insertion of the thymidine kinase (TK) promoter from the herpes simplex virus (HSV) into pGL3-Basic (Promega). The GR-binding site of the KLF15 enhancer region was mutated using the KOD mutagenesis kit according to manufacturer's recommendations (TOYOBO). This mutation changed the wild-type sequence 5' –ATTACATGTTGTTCC- 3' to 5' -ACAACATGTTCATCC- 3' for GRE1 and the wild-type sequence 5' -AGAACAGGGTGCCCC- 3' to 5'- ACAACAGGGTCACCC -3' for GRE2. For the transfection experiment, 293T cells were plated into 96-well plates at 1 × 103 cells/well, and transient transfections were performed the following day using FuGENE HD (Roche Applied Science, Indianapolis, IN, USA). The total amount of plasmid used for the transfection assay per well was 0.1 µg reporter vector/0.01 µg GR expression vector or pcDNA3.1 (Invitrogen)/phRL-TK vector (Promega). On the following day, 100 nM Dex or vehicle (DMSO) were added to cells. After 24 hr, luciferase assays were carried out using Dual-Glo Luciferase Assay kit (Promega). The GR-dependent promoter activity of each construct was expressed as the ratio of activity in Dex/activity in DMSO. All luciferase experiments were performed in triplicate. Statistical significance of the difference between pGL3-intKLF15-tk and mutant reporters was determined by Steel’s-test.
Transfection of siRNA for KLF15 into MEFs
siRNA for KLF15(Silencer® Select siRNAs- s82946, Applied Biosystems) and negative siRNA (Silencer® Select siRNAs Silencer® Negative Control #1, Applied Biosystems) were transfected into MEFs at 80–90% confluence (50nM concentration) with Lipofectamine RNAi Max (Invitrogen) according to the manufacturer’s instructions. After 48hr, transfected MEFs were stimulated with AIM or Control medium.
RESULTS
Inhibition of Adipogenesis by GR Antagonist RU486 in Human Mesenchymal Stem Cells and Mouse Embryonic Fibroblasts
To investigate the role of GR in adipocyte differentiation, hMSCs were induced to differentiate into adipocytes by incubation in adipogenic induction medium (AIM) containing 500 µM IBMX, 1 µM Dex, 10 µM pioglitazone, 10 µg/mL insulin with or without the GR antagonist RU486 (10 µM), followed by insulin treatment every 3 days. As shown in Figure 1a, hMSCs differentiated into adipocytes and accumulated lipid droplets in the presence of AIM, but no obvious lipid accumulation was observed in hMSCs treated with RU486 during adipogenic stimulation. Because the adipogenic process is characterized by expression of specific adipocyte markers such as PPARγ, C/EBPα, and adiponectin, we next examined the expression of these adipocyte-specific genes by quantitave RT-PCR (Figure 1b). The expression of PPARγ, C/EBPα, and adiponectin was increased in hMSCs incubated with adipogenic inducers for 2 days, and these gene expression levels were inhibited completely by RU486. These findings suggest that GR activity is necessary for the adipogenic differentiation of hMSCs.
Figure 1. Effect of GR Antagonist RU486 on Adipogenesis in hMSCs and MEFs.
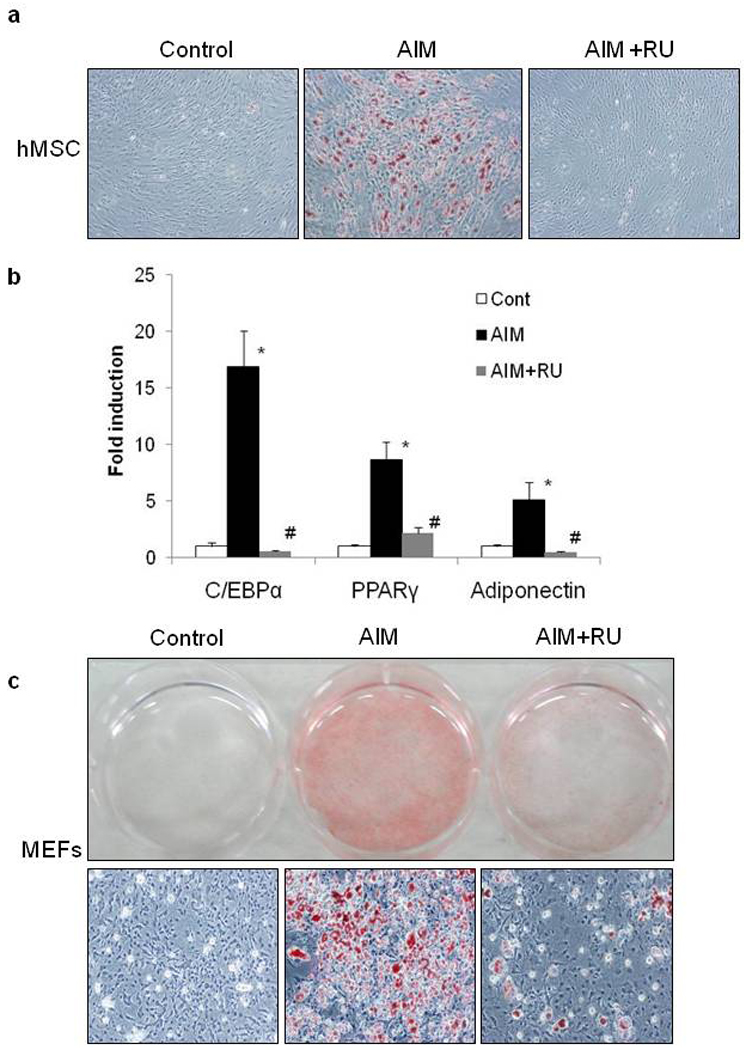
hMSCs were differentiated into adipocytes with AIM medium (containing 500 µM IBMX, 1 µM Dex, 5 µg/mL insulin, 10 µM pioglitazone) with or without 10 µM RU486 for 3 days, followed by treatment with 5 µg/mL insulin alone for 3 days. These treatments were repeated. Cells were stained with Oil Red O at 12 days post-induction (a). Quantitative real time PCR for the determination of expression levels adipogenic genes on day 2 after AIM exposure in the presence or absence of RU486 (b). MEFs were differentiated into adipocytes with AIM for 3 days, followed by treatment with insulin alone for 4 days. These treatments were repeated. Cells were stained with Oil Red O at 14 days post-induction. Macroscopic (upper panel) and microscopic (lower panel) views are shown (c). Data from 3 independent experiments are mean ± SEM; n=6. P values were determined by Steel-Dwass’ test (* P <0.05 vs control, # P<0.05 vs AIM).
We next examined whether the RU486 effect observed in hMSCs is similar in mouse embryonic fibroblasts (MEFs) from wild type mice. Wild type MEFs were differentiated into adipocytes and RU486 inhibited the accumulation of lipid droplets albeit not as efficient as in hMSCs (Figure 1c). Expression of adipocyte-specific marker genes in MEFs from wild type mice were also inhibited by RU486 (Supplementary Figure 1). These data indicated that GR plays a critical role in adipose differentiation from MEFs and hMSCs.
Inhibition of Adipogenic Differentiation in Mouse Embryonic Fibroblasts from GRnull and GRdim Mice
To further verify the role of GR in adipogenesis at the genetic level, we obtained MEFs from GRnull mice and observed that adipogenesis (Figure 2a and Supplementary Figure 2) and induction of marker gene expression was blocked in GRnull MEFs, as well as RU486 treatment (Figure 2b, c). Furthermore cells with a dimerization impaired GR (GRdim) failed to accumulate lipids and enhancement of adipocyte marker expression during adipogenic stimulation (Figure 2a, b, c). Taken together, these data clearly indicate that ligand dependent GR transcriptional activity with its DNA binding to the target promoter is critical for adipogenesis.
Screening for GR-targets in Adipogenesis by Microarray Expression Analysis
To identify the genes regulated by GR during adipogenesis, total RNA was extracted from MEFs cultured with control medium, AIM medium, AIM with RU486, or Dex alone for 2 days; transcription was analyzed by microarray. As shown in Figure 3a, 1096 genes from AIM-treated cells that displayed at least a twofold expression change relative to control were selected as adipogenesis-specific genes; 203 genes from cells incubated with AIM and RU486 that displayed at least a twofold change compared to cells treated with AIM alone were selected as adipogenesis–RU486-regulated genes; and 1580 genes from Dex-treated cells that displayed at least a twofold change relative to control were selected as Dex-regulated genes. Furthermore, after eliminating overlap and expressed sequence tags (ESTs), 51 genes that belong to both the adipogenesis–RU486-regulated group and the Dex-regulated group were identified as GR-regulated adipogenic genes; a list of these 51 genes is displayed in Table 1. All 51 genes demonstrated an increase or decrease in expression with Dex treatment—45 genes were up-regulated and 6 genes were down-regulated during adipogenesis—and RU486 treatment had the opposite effect on their expression.
Figure 3. Screening for GR-Regulated Adipogenic Induced Genes by Microarray Expression Analysis.
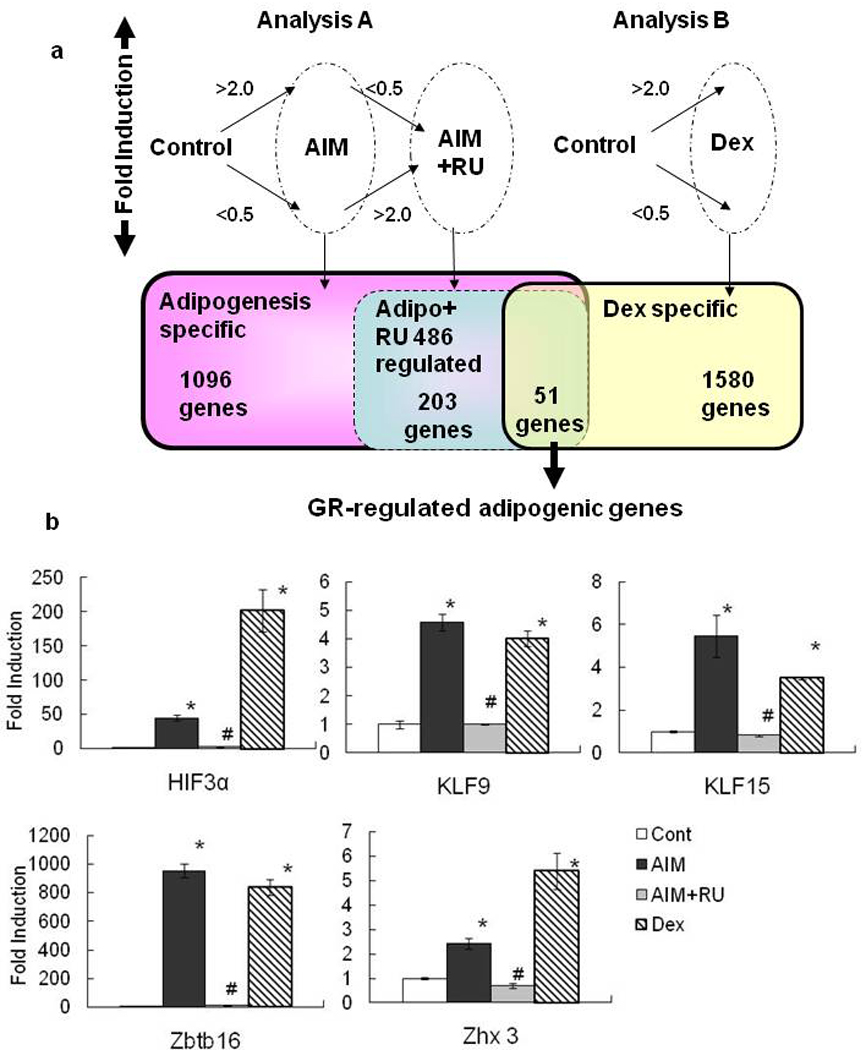
Scheme of microarray data analysis showing genes with increased or decreased expression in each treatment group (a).Genes with signal intensities below 50 were eliminated and normalized, and then genes from cells incubated with AIM medium or Dex treatment that exhibited at least a twofold change relative to the control treatment were selected for analysis. Expression profiles for GR-regulated adipogenic genes in MEF were confirmed with quantitative RT-PCR in Control, AIM, AIM+RU, and Dex treatment groups on Day 2(b).Data are mean ±SEM; n=4. P values were determined by Steel-Dwass’ test (* P<0.05 vs control, # P<0.05 vs AIM).
Table 1.
Microarray analysis of GR-regulated adipogenic genes
| Fold Induction |
Symbol | Description | ||
|---|---|---|---|---|
| AIM | AIM+RU | Dex | ||
| 367.2 | 19.0 | 249.9 | Zbtb16 | zinc finger and BTB domain containing 16 |
| 144.1 | 55.2 | 39.8 | Serpina3c | serine (or cysteine) proteinase inhibitor, clade A, member 3C |
| 78.0 | 15.8 | 39.2 | Hp | haptoglobin |
| 70.5 | 16.0 | 14.8 | Serpina3m | serine (or cysteine) proteinase inhibitor, clade A, member 3M |
| 36.2 | 15.5 | 3.8 | Prg4 | proteoglycan 4 (megakaryocyte stimulating factor, articular superficial zone protein) |
| 23.6 | 0.9 | 11.3 | Hif3a | hypoxia inducible factor 3, alpha subunit |
| 23.4 | 1.6 | 22.6 | BC055107 | cDNA sequence BC055107 |
| 22.8 | 8.1 | 7.3 | Fabp4 | fatty acid binding protein 4, adipocyte |
| 20.1 | 2.3 | 8.6 | Dpep1 | dipeptidase 1 (renal) |
| 19.7 | 2.3 | 9.6 | Fkbp5 | FK506 binding protein 5 |
| 19.6 | 7.3 | 21.6 | Cp | ceruloplasmin |
| 15.6 | 3.3 | 3.7 | 5430433G21Rik | RIKEN cDNA 5430433G21 gene |
| 13.5 | 2.3 | 9.8 | Aldh1a7 | aldehyde dehydrogenase family 1, subfamily A7 |
| 11.7 | 3.6 | 4.4 | B230114P17Rik | RIKEN cDNA B230114P17 gene |
| 11.2 | 4.1 | 10.0 | AI607873 | expressed sequence AI607873 |
| 9.9 | 0.7 | 9.9 | Fmo3 | flavin containing monooxygenase 3 |
| 8.8 | 3.9 | 2.1 | Cd24a | CD24a antigen |
| 8.0 | 0.2 | 6.0 | Fmo2 | flavin containing monooxygenase 2 |
| 7.9 | 2.0 | 3.6 | 1300002F13Rik | RIKEN cDNA 1300002F13 gene |
| 7.6 | 3.4 | 8.1 | Ms4a4d | membrane-spanning 4-domains, subfamily A, member 4D |
| 7.5 | 2.4 | 2.9 | Glul | glutamate-ammonia ligase (glutamine synthase) |
| 6.4 | 1.0 | 3.9 | Tsc22d3 | TSC22 domain family 3 |
| 5.6 | 0.9 | 6.4 | Pdk4 | pyruvate dehydrogenase kinase, isoenzyme 4 |
| 4.6 | 1.5 | 2.1 | Ms4a6b | membrane-spanning 4-domains, subfamily A, member 6B |
| 4.5 | 2.1 | 2.6 | LOC553901 | hypothetical LOC553901 |
| 4.4 | 1.1 | 3.2 | Ndrg2 | N-myc downstream regulated gene 2 |
| 4.3 | 2.1 | 3.0 | Rgs2 | regulator of G-protein signaling 2 |
| 4.3 | 1.1 | 6.4 | Sult1a1 | sulfotransferase family 1A, phenol-preferring, member 1 |
| 4.1 | 1.9 | 3.4 | Gm368 | gene model 368, (NCBI) |
| 3.8 | 1.8 | 2.1 | Zhx3 | zinc fingers and homeoboxes 3 |
| 3.7 | 1.4 | 2.1 | Klf9 | Kruppel-like factor 9 |
| 3.5 | 1.7 | 4.1 | Clca1 /// Clca2 | chloride channel calcium activated 1 /// chloride channel calcium activated 2 |
| 3.5 | 1.6 | 2.2 | Admr | adrenomedullin receptor |
| 3.5 | 1.7 | 3.5 | 2310005P05Rik | RIKEN cDNA 2310005P05 gene |
| 3.4 | 1.1 | 2.8 | Crispld2 | cysteine-rich secretory protein LCCL domain containing 2 |
| 3.1 | 1.5 | 2.2 | Klf15 | Kruppel-like factor 15 |
| 3.1 | 0.9 | 4.3 | Ctla2b | cytotoxic T lymphocyte-associated protein 2 beta |
| 3.0 | 0.7 | 2.4 | Stc2 | stanniocalcin 2 |
| 3.0 | 1.1 | 2.8 | Aldh1a1 | aldehyde dehydrogenase family 1, subfamily A1 |
| 2.8 | 1.1 | 2.7 | 1810049K24Rik | Cysteine-rich secretory protein LCCL domain containing 2 |
| 2.4 | 1.1 | 2.3 | Scn7a | sodium channel, voltage-gated, type VII, alpha |
| 2.4 | 1.1 | 2.4 | Rab3d | RAB3D, member RAS oncogene family |
| 2.2 | 0.8 | 2.3 | Prkar2b | protein kinase, cAMP dependent regulatory, type II beta |
| 2.2 | 0.9 | 3.7 | Clec1a | C-type lectin domain family 1, member a |
| 2.1 | 1.0 | 2.3 | Sgk | serum/glucocorticoid regulated kinase |
| 0.4 | 0.9 | 0.4 | 4632408A20Rik | RIKEN cDNA 4632408A20 gene |
| 0.3 | 1.0 | 0.4 | Sec15l1 | SEC15-like 1 (S. cerevisiae) |
| 0.3 | 0.7 | 0.5 | 1810057P16Rik | RIKEN cDNA 1810057P16 gene |
| 0.2 | 0.5 | 0.5 | Slc7a5 | solute carrier family 7 (cationic amino acid transporter, y+ system), member 5 |
| 0.2 | 0.4 | 0.5 | E030004N02Rik | RIKEN cDNA E030004N02 gene |
| 0.1 | 0.3 | 0.5 | Gucy1a3 | guanylate cyclase 1, soluble, alpha 3 |
Candidates for GR Direct Target Genes in Mouse Embryonic Fibroblast during Adipogenesis
To reveal transcriptional network in adipogenesis, we focused on transcription factors which were induced as GR direct target genes in adipogenesis. Among the 45 genes showing adipogenesis and GR dependent upregulation in Table 1, 5 genes are transcriptional factors: Zbtb16 (zinc finger and BTB domain containing 16), HIF3α (hypoxia inducible factor 3, alpha subunit), Zhx3 (zinc fingers and homeoboxes 3), KLF9 (Krüppel-like factor 9), and KLF15.
Expression of these five genes was confirmed by real time RT-PCR using cDNA from MEFs at day 2 after adipogenic induction +/− RU486 or with Dex alone (Figure 3b). Expression of Zbtb16, (also known as ZFP145 or PLZF) was strongly induced by Dex; consistent with a previous report showing that glucocorticoids regulate Zbtb16 gene expression via the promoter region of this gene (38), and so far there is no report on the roles of Zbtb16, HIF3α, Zhx3, or KLF9 in adipogenesis. KLF15 has been reported to be induced by Dex and IBMX (28,39) and act as an adipogenic inducer via PPARγ induction (30,39); however, the molecular relationship between GR dependent transcription and KLF15 expression is largely unrevealed yet.
KLF15 Expression is Dependent on GR Dimerization
Consistent with the above observation, KLF15 expression was increased during adipose differentiation in both hMSCs and wild type MEFs and this induction was inhibited by RU486, suggesting the critical role of GR in KLF15 gene expression in adipogenesis (Figure 3b, 4a). To examine whether induction of KLF15 during adipogenesis is directly regulated by GR or not, we tested the expression of KLF15 using GRnull and GRdim MEFs and found that KLF15 gene expression during adipogenesis was inhibited (Figure 4b). These data suggest that KLF15 expression during adipogenesis needs ligand dependent and DNA binding dependent GR transcriptional activity.
Interestingly, C/EBPδ expression was induced with AIM although basic expression was decreased in both in GRnull and GRdim MEFs (Fold induction: 4.97±0.98 in wild type, 6.38±3.33 in GRnull, 7.91±3.41 in GRdim on 12hrs) (Figure 4b). Kinetics analysis of gene expression showed KLF15 expression is induced earlier than C/EBPδ expression in MSCs and MEFs (Figure 4c and Supplementary Figure 3). Consistent with these findings, Dex treatment strongly and promptly induced KLF15 gene expression, but not C/EBPδ gene expression (Figure 4d). These data suggest that the ligand-bound GR is critical for promoting KLF15 in adipogenesis and that C/EBPδ in adipogenesis can be alternatively induced by other factors (e.g. IBMX) without GR.
GR Directly Binds to GRE Sites in the First Intron of KLF15
Using bioinformatics, we analyzed the promoter regions - KLF15 (7500 to +2000 bp). A conserved region between human and mouse KLF15 promoters was identified in the first intron by rVista 2.0; two putative GRE sites (GRE1 and GRE2) were identified by analysis with MatInspector (Figure 5a, b).
Figure 5. GRE Sites in the KLF15 Gene First Intron.
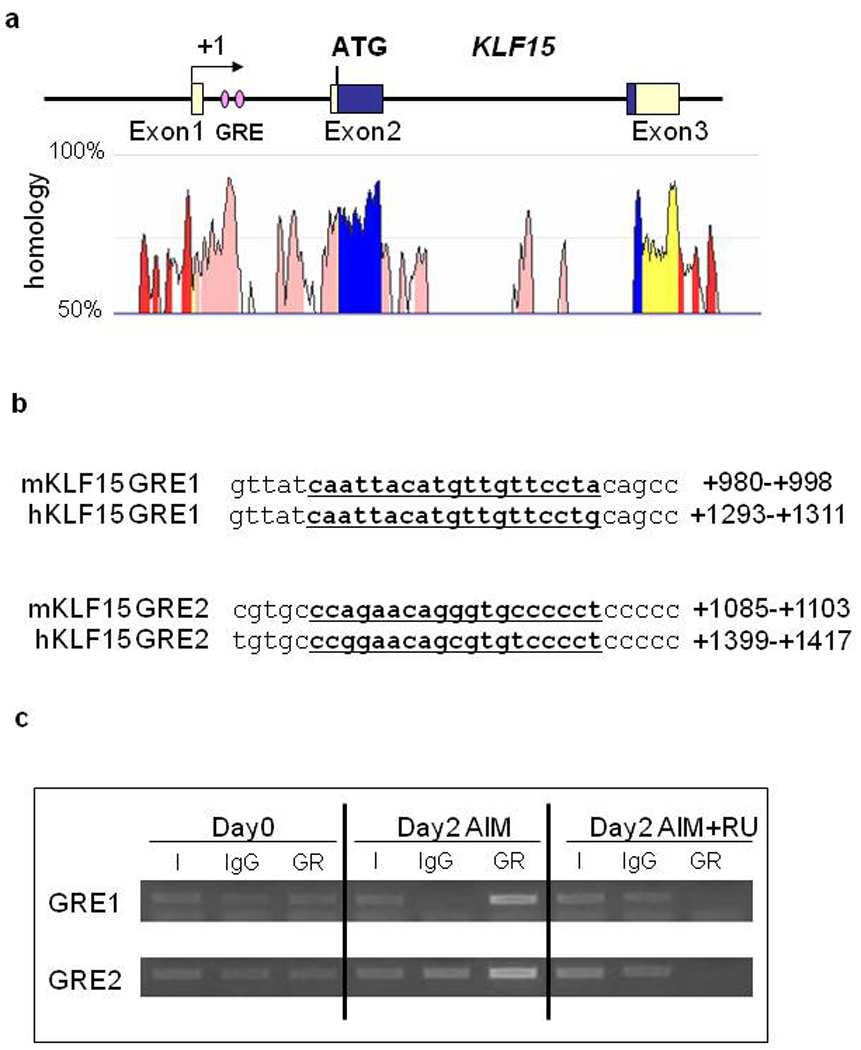
The highly conserved region between human and mouse in the KLF15 first intron (a). GRE site sequence in the first intron of KLF15 (b). The conserved region between human and mouse genomes was identified by rVista 2.0 and the homology graph from the ECR browser, and putative GRE sites were discovered with MatInspector. Chromatin immunoprecipitation was performed in MEFs differentiated with AIM for 2 days. DNA immunoprecipitated with anti-GR antibodies from treated cells was used to amplify the nucleotide sequences located between +933 to +1070 bp (GRE1), and +1055 to +1216 bp (GRE2) in the KLF15 first intron(c).
I: input, IgG: normal rabbit serum IgG.
To confirm these findings, we used the ChIP assay to determine whether GR binds to GRE sites in the first intron of KLF15 during adipocyte differentiation. Chromatin samples were prepared from MEFs undergoing adipogenic differentiation with or without RU486, then immunoprecipitated with a specific antibody against GR. GR was found to be associated with both GRE1 and GRE2 in a conserved region of KLF15 and this association was inhibited by RU486 (Figure 5c). These results confirmed that GR directly binds to GRE sites in the first intron of KLF15 during adipogenesis.
GR Induces Promoter Activity via KLF15 First Intron
To define the mechanism by which GR induces KLF15 expression, we performed transient transfection studies in 293T cells using reporter constructs of the KLF15 intron (+933 to +1216), including 2 GRE sites under the HSV TK promoter (Figure 6a). Dex treatment resulted in an approximate 10-fold induction of expression of the KLF15 promoter and this induction was inhibited by RU486 treatment (Figure 6b).
Figure 6. Promoter Activity of Reporter Constructs Containing +933bp to +1216bp of the KLF15 First Intron.
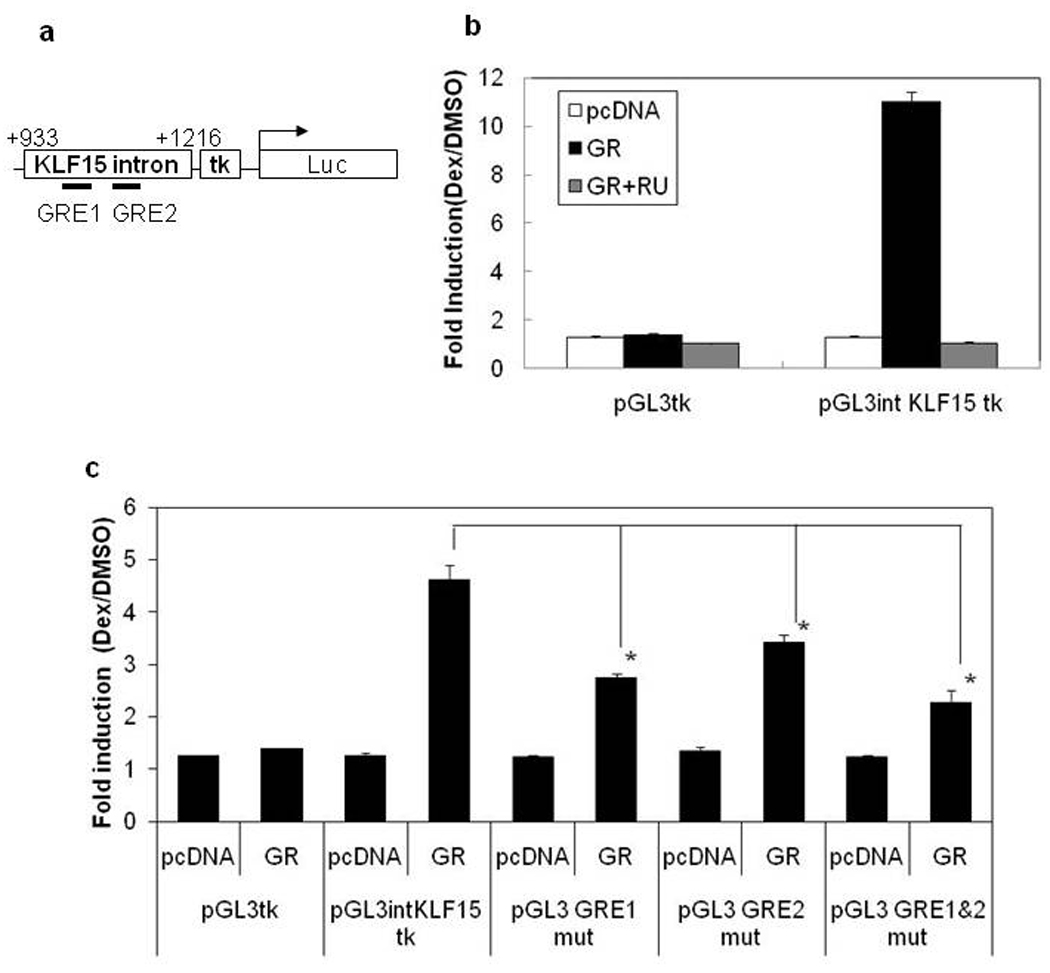
Map of pGL3 intKLF15 tk reporter vector (a). 293T cells were transiently transfected with the reporter construct pGL3-intKLF15-tk (b) or deletion constructs pGL3-GRE1mut (GRE1 mutation), pGL3-GRE2mut (GRE2 mutation), pGL3-GRE1&2mut (GRE1 and GRE2 mutations) (c), and empty vector (pcDNA) or GR expression vector. After 24 hr, cells were incubated in Dex (100 nM) or DMSO (vehicle) with or without 10 µM RU486 for 24 hr before the luciferase assay. Results were expressed as fold induction compared to cells treated with DMSO alone. Data are expressed as the mean ± SD of triplicates from representative experiments. Results were evaluated by Steel’s-test (* P< 0.05 vs pGL3-intKLF15-tk with Dex).
To determine which potential GR-binding site in the first intron of KLF15 was involved in transactivation, we introduced point mutations at the GRE 1 site (ATTACATGTTGTTCC → ACAACATGTTCATCC) and at the GRE2 site (AGAACAGGGTGCCCC → ACAACAGGGTCACCC). Both mutations in the GRE1 or GRE2 resulted in a loss of transactivation (Figure 6c). Moreover, introduction of mutations at both GRE1 and GRE2 caused an even greater reduction in transactivation. These data suggest that GR can transactivate the KLF15 promoter activity by binding these GRE consensus sites.
Knockdown of KLF15 Inhibited Adipogenesis
Finally, to investigate the function of KLF15 on adipogenesis, we examined the effect of siRNA for KLF15 during adipogenic differentiation in MEFs. As shown in Figure 7a, MEFs transfected with nonspecific siRNA (siNega) differentiated into adipocytes and accumulated lipid droplets, but less lipid accumulation was observed in MEFs transfected with siRNA for KLF15 (siKLF15) during adipogenic stimulation. We examined the expression of adipocyte-specific genes by quantitative RT-PCR 2days after stimulation (Figure 7b). KLF15 gene expression was inhibited by siRNA for KLF15 (siKLF15) and adipocyte-marker genes, C/EBPα and PPARγ were reduced whereas adiponectin was not affected. Thus, KLF15 induction contributes to adipocyte differentiation.
Figure 7. Effect of KLF15 Knockdown on Adipogenesis in MEFs.
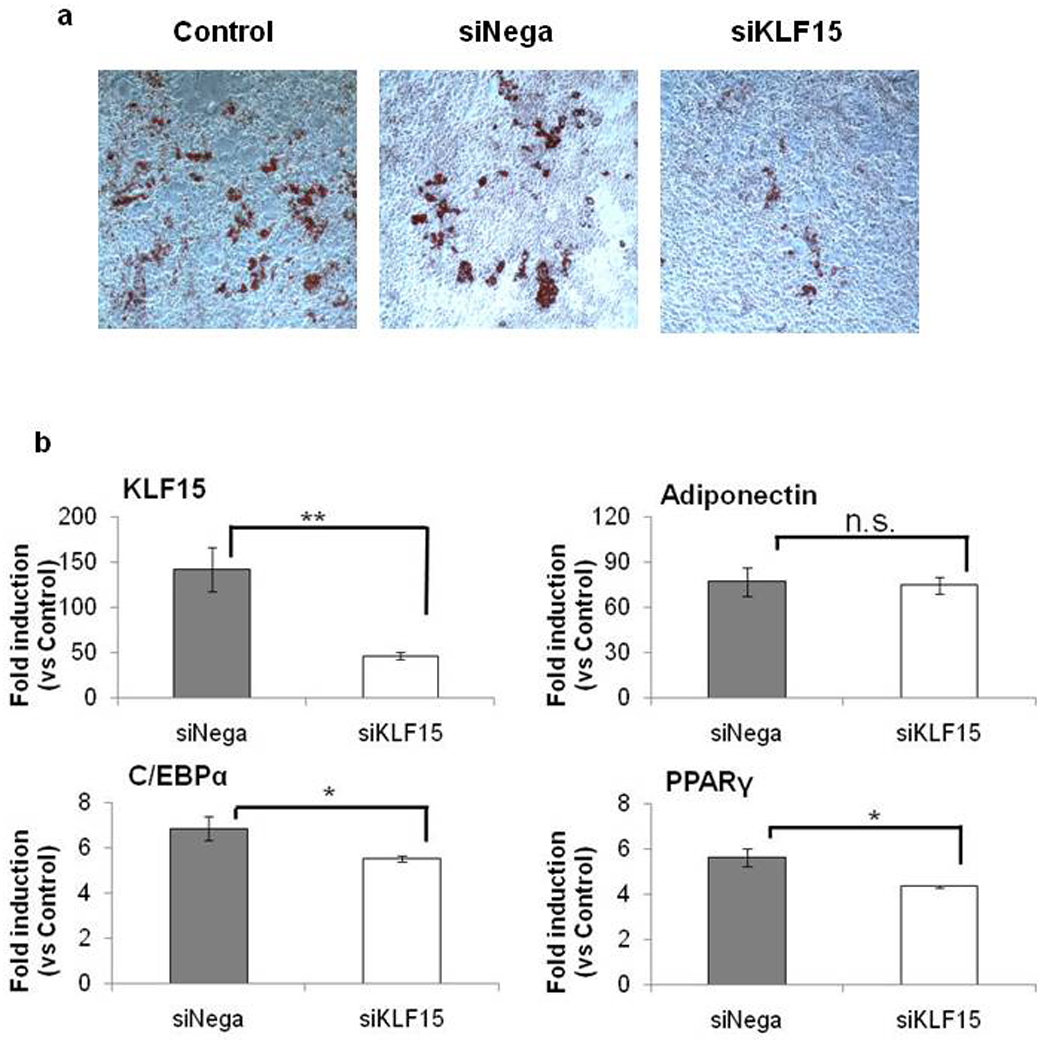
50nM siRNA for KLF15 (siKLF15) and 50nM nonspecific siRNA(siNega) were transfected into MEFs. After 48hr, cells were differentiated into adipocytes in the same way as wild type MEFs Figure 1 and were stained with Oil Red O at 14 days post-induction (a). Expression of KLF15 and adipogenic-specific marker genes in siRNA-transfected MEFs(b). Total RNA was extracted from cells 2 days after initiating adipose differentiation and analyzed using quantitative RT-PCR. Data are mean ±SEM; n=6–10. P values were determined by Mann-Whitney U test (** P<0.01 vs siNega, * P<0.05 vs siNega).
DISCUSSION
In this study we used the GR antagonist RU486 and GR-mutant mice to provide evidence for an essential role for GR in adipogenesis. The GR and in particular its dimerization function are essential for in vitro adipogenesis, since cells with a GR impaired in dimerized DNA binding derived from GRdim mice fail to differentiate into adipocytes. Thus genes under the control of GR-DNA interaction, which comprise a subset of GR regulated genes are involved in adipogenesis (40). The Krüppel-like factor 15 (KLF15) gene was identified as a direct GR target in MEF during adipogenesis and KLF15 plays the critical role in adipogenic differentiation. Previous studies showed that KLF15 is induced by ectopic expression of C/EBPδ and C/EBPβ, and Dex and IBMX stimulation in 3T3-L1 cells (28). Our data showed that KLF15 is more rapidly induced then C/EBPδ mRNA, suggesting a C/EBPδ- independent pathway for GR mediated KLF15 induction (Figure 8).
Figure 8. Schematic Model of the Pathways Controlling Differentiation.
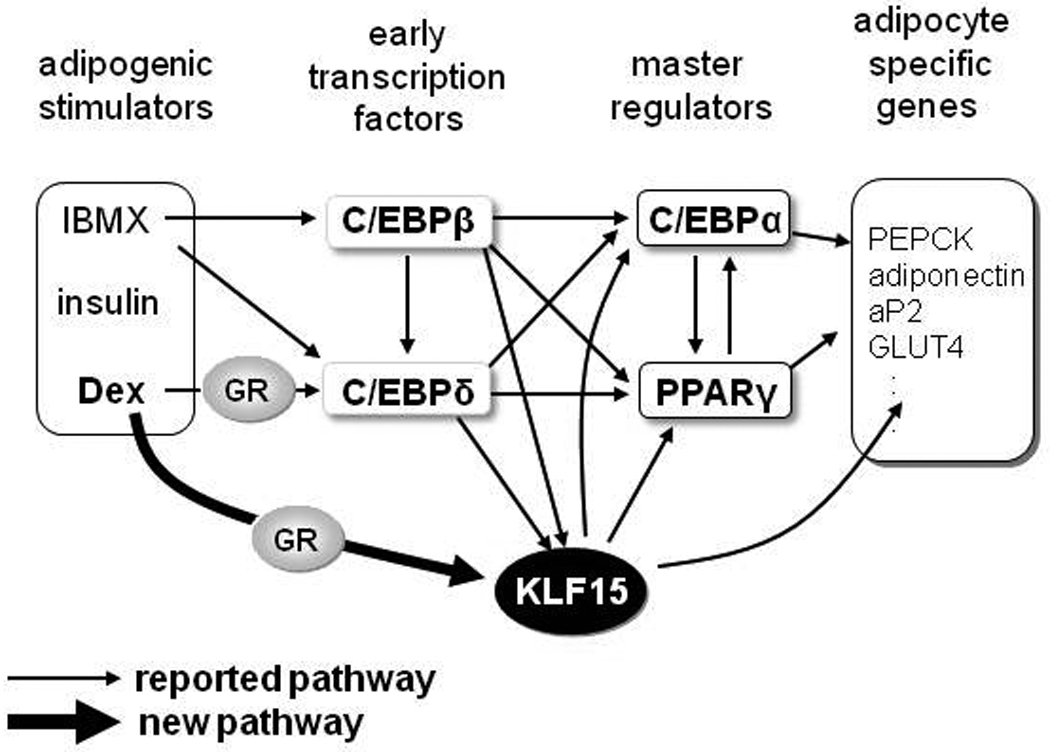
GR directly regulates KLF15 expression, which acts as an adipocyte differentiation activator at the transcriptional level in hMSCs and MEFs.
KLF15 is expressed in major tissues such as liver, kidney, heart, skeletal muscle, and adipose tissue. KLF15−/− mice display severe hypoglycemia with a deficiency in amino acid metabolic enzymes in the liver and muscle (30). These findings raise the question of the function of adipose tissue in these mice. GR plays an important role in glucose and amino acid homeostasis in liver, muscle, and adipose tissue during gluconeogenesis under fasting conditions (30). Fasting increases endogenous glucocorticoids, and induces gluconeogenesis-associated genes, such as PEPCK and G6Pase in the liver (41); liver-specific GR-deficient mice displayed hypoglycemia while fasting, similar to KLF15−/− mice. Expression of KLF15 is increased in the liver (42), muscle (43), and adipocytes in fasting conditions (data not shown). These findings suggest that the glucocorticoid effects may be partially mediated by KLF15 in vivo.
In GRnull mice or GRdim mice, developments of white adipose tissues in embryos are normal comparative to wild type under normal conditions (data not shown); however, various stresses, such as a high fat diet, may reveal the functions of GR in adipogenesis in vivo.
Further in vitro and in vivo studies to investigate the expression of the adipogenic regulators and GR target genes (KLF15 etc.) using GRnull embryos and GRdim mice, will generate a clearer picture of the roles of GR during adipocyte differentiation. Excessive glucocorticoids cause ectopic adipose distribution (central obesity, buffalo hump etc.) and contribute to metabolic disease through the facilitation of adipose tissue expansion. Our findings in this study will allow a more focused approach towards studying the mechanisms underlying the effect of glucocorticoids on adipose tissue in vivo.
Supplementary Material
ACKNOWLEDGEMENTS
This study was supported by NIH grants AR-50631, AR-56120, the Arthritis National Research Foundation, Grants from the Genome Network Project (MEXT), Grants-in Aid for Scientific Research (MEXT), Labour Sciences Research Grants, Research on Child Health and Development, Grant ID10-43 from the National Institute of Biomedical Innovation, The Japan Health Sciences Foundation and the Boehringer Ingelheim Stiftung (to J.T.).
Footnotes
Supplementary information is available at Laboratory Investigation's website.
REFERENCES
- 1.Green H, Kehinde O. An established preadipose cell line and its differentiation in culture. II. Factors affecting the adipose conversion. Cell. 1975;5:19–27. doi: 10.1016/0092-8674(75)90087-2. [DOI] [PubMed] [Google Scholar]
- 2.Green H, Kehinde O. Spontaneous heritable changes leading to increased adipose conversion in 3T3 cells. Cell. 1976;7:105–113. doi: 10.1016/0092-8674(76)90260-9. [DOI] [PubMed] [Google Scholar]
- 3.Rosen OM, Smith CJ, Fung C, et al. Development of hormone receptors and hormone responsiveness in vitro. Effect of prolonged insulin treatment on hexose uptake in 3T3-L1 adipocytes. J Biol Chem. 1978;253:7579–7583. [PubMed] [Google Scholar]
- 4.Rubin CS, Hirsch A, Fung C, et al. Development of hormone receptors and hormonal responsiveness in vitro. Insulin receptors and insulin sensitivity in the preadipocyte and adipocyte forms of 3T3-L1 cells. J Biol Chem. 1978;253:7570–7578. [PubMed] [Google Scholar]
- 5.Gregoire FM, Smas CM, Sul HS. Understanding adipocyte differentiation. Physiol Rev. 1998;78:783–809. doi: 10.1152/physrev.1998.78.3.783. [DOI] [PubMed] [Google Scholar]
- 6.Rosen ED, Walkey CJ, Puigserver P, et al. Transcriptional regulation of adipogenesis. Genes Dev. 2000;14:1293–1307. [PubMed] [Google Scholar]
- 7.Farmer SR. Transcriptional control of adipocyte formation. Cell Metab. 2006;4:263–273. doi: 10.1016/j.cmet.2006.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rosen ED, Hsu CH, Wang X, et al. C/EBPalpha induces adipogenesis through PPARgamma: a unified pathway. Genes Dev. 2002;16:22–26. doi: 10.1101/gad.948702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rosen ED, Sarraf P, Troy AE, et al. PPAR gamma is required for the differentiation of adipose tissue in vivo and in vitro. Mol Cell. 1999;4:611–617. doi: 10.1016/s1097-2765(00)80211-7. [DOI] [PubMed] [Google Scholar]
- 10.Tontonoz P, Hu E, Spiegelman BM. Stimulation of adipogenesis in fibroblasts by PPAR gamma 2, a lipid-activated transcription factor. Cell. 1994;79:1147–1156. doi: 10.1016/0092-8674(94)90006-x. [DOI] [PubMed] [Google Scholar]
- 11.Cao Z, Umek RM, McKnight SL. Regulated expression of three C/EBP isoforms during adipose conversion of 3T3-L1 cells. Genes Dev. 1991;5:1538–1552. doi: 10.1101/gad.5.9.1538. [DOI] [PubMed] [Google Scholar]
- 12.Wu Z, Bucher NL, Farmer SR. Induction of peroxisome proliferator-activated receptor gamma during the conversion of 3T3 fibroblasts into adipocytes is mediated by C/EBPbeta, C/EBPdelta, and glucocorticoids. Mol Cell Biol. 1996;16:4128–4136. doi: 10.1128/mcb.16.8.4128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yeh WC, Cao Z, Classon M, et al. Cascade regulation of terminal adipocyte differentiation by three members of the C/EBP family of leucine zipper proteins. Genes Dev. 1995;9:168–181. doi: 10.1101/gad.9.2.168. [DOI] [PubMed] [Google Scholar]
- 14.Masuzaki H, Paterson J, Shinyama H, et al. A transgenic model of visceral obesity and the metabolic syndrome. Science. 2001;294:2166–2170. doi: 10.1126/science.1066285. [DOI] [PubMed] [Google Scholar]
- 15.Newell-Price J, Bertagna X, Grossman AB, et al. Cushing's syndrome. Lancet. 2006;367:1605–1617. doi: 10.1016/S0140-6736(06)68699-6. [DOI] [PubMed] [Google Scholar]
- 16.Hauner H, Schmid P, Pfeiffer EF. Glucocorticoids and insulin promote the differentiation of human adipocyte precursor cells into fat cells. J Clin Endocrinol Metab. 1987;64:832–835. doi: 10.1210/jcem-64-4-832. [DOI] [PubMed] [Google Scholar]
- 17.Ito S, Suzuki N, Kato S, et al. Glucocorticoids induce the differentiation of a mesenchymal progenitor cell line, ROB-C26 into adipocytes and osteoblasts, but fail to induce terminal osteoblast differentiation. Bone. 2007;40:84–92. doi: 10.1016/j.bone.2006.07.012. [DOI] [PubMed] [Google Scholar]
- 18.Yamamoto KR. Steroid receptor regulated transcription of specific genes and gene networks. Annu Rev Genet. 1985;19:209–252. doi: 10.1146/annurev.ge.19.120185.001233. [DOI] [PubMed] [Google Scholar]
- 19.Beato M, Herrlich P, Schutz G. Steroid hormone receptors: many actors in search of a plot. Cell. 1995;83:851–857. doi: 10.1016/0092-8674(95)90201-5. [DOI] [PubMed] [Google Scholar]
- 20.Karin M. New twists in gene regulation by glucocorticoid receptor: is DNA binding dispensable? Cell. 1998;93:487–490. doi: 10.1016/s0092-8674(00)81177-0. [DOI] [PubMed] [Google Scholar]
- 21.McKay LI, Cidlowski JA. Molecular control of immune/inflammatory responses: interactions between nuclear factor-kappa B and steroid receptor-signaling pathways. Endocr Rev. 1999;20:435–459. doi: 10.1210/edrv.20.4.0375. [DOI] [PubMed] [Google Scholar]
- 22.De Bosscher K, Vanden Berghe W, Haegeman G. The interplay between the glucocorticoid receptor and nuclear factor-kappaB or activator protein-1: molecular mechanisms for gene repression. Endocr Rev. 2003;24:488–522. doi: 10.1210/er.2002-0006. [DOI] [PubMed] [Google Scholar]
- 23.Barnes PJ. Corticosteroid effects on cell signalling. Eur Respir J. 2006;27:413–426. doi: 10.1183/09031936.06.00125404. [DOI] [PubMed] [Google Scholar]
- 24.Heck S, Kullmann M, Gast A, et al. A distinct modulating domain in glucocorticoid receptor monomers in the repression of activity of the transcription factor AP-1. Embo J. 1994;13:4087–4095. doi: 10.1002/j.1460-2075.1994.tb06726.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reichardt HM, Kaestner KH, Tuckermann J, et al. DNA binding of the glucocorticoid receptor is not essential for survival. Cell. 1998;93:531–541. doi: 10.1016/s0092-8674(00)81183-6. [DOI] [PubMed] [Google Scholar]
- 26.Banerjee SS, Feinberg MW, Watanabe M, et al. The Kruppel-like factor KLF2 inhibits peroxisome proliferator-activated receptor-gamma expression and adipogenesis. J Biol Chem. 2003;278:2581–2584. doi: 10.1074/jbc.M210859200. [DOI] [PubMed] [Google Scholar]
- 27.Oishi Y, Manabe I, Tobe K, et al. Kruppel-like transcription factor KLF5 is a key regulator of adipocyte differentiation. Cell Metab. 2005;1:27–39. doi: 10.1016/j.cmet.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 28.Mori T, Sakaue H, Iguchi H, et al. Role of Kruppel-like factor 15 (KLF15) in transcriptional regulation of adipogenesis. J Biol Chem. 2005;280:12867–12875. doi: 10.1074/jbc.M410515200. [DOI] [PubMed] [Google Scholar]
- 29.Gray S, Feinberg MW, Hull S, et al. The Kruppel-like factor KLF15 regulates the insulin-sensitive glucose transporter GLUT4. J Biol Chem. 2002;277:34322–34328. doi: 10.1074/jbc.M201304200. [DOI] [PubMed] [Google Scholar]
- 30.Gray S, Wang B, Orihuela Y, et al. Regulation of gluconeogenesis by Kruppel-like factor 15. Cell Metab. 2007;5:305–312. doi: 10.1016/j.cmet.2007.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reichardt HM, Kaestner KH, Tuckermann J, et al. DNA binding of the glucocorticoid receptor is not essential for survival. Cell. 1998;93:531–541. doi: 10.1016/s0092-8674(00)81183-6. [DOI] [PubMed] [Google Scholar]
- 32.Pascussi JM, Busson-Le Coniat M, Maurel P, et al. Transcriptional analysis of the orphan nuclear receptor constitutive androstane receptor (NR1I3) gene promoter: identification of a distal glucocorticoid response element. Mol Endocrinol. 2003;17:42–55. doi: 10.1210/me.2002-0244. [DOI] [PubMed] [Google Scholar]
- 33.Todaro GJ, Green H. Quantitative studies of the growth of mouse embryo cells in culture and their development into established lines. J Cell Biol. 1963;17:299–313. doi: 10.1083/jcb.17.2.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ramirez-Zacarias JL, Castro-Munozledo F, Kuri-Harcuch W. Quantitation of adipose conversion and triglycerides by staining intracytoplasmic lipids with Oil red O. Histochemistry. 1992;97:493–497. doi: 10.1007/BF00316069. [DOI] [PubMed] [Google Scholar]
- 35.Tuckermann JP, Kleiman A, Moriggl R, et al. Macrophages and neutrophils are the targets for immune suppression by glucocorticoids in contact allergy. J Clin Invest. 2007;117:1381–1390. doi: 10.1172/JCI28034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Phuc Le P, Friedman JR, Schug J, et al. Glucocorticoid receptor-dependent gene regulatory networks. PLoS Genet. 2005;1:e16. doi: 10.1371/journal.pgen.0010016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kassel O, Schneider S, Heilbock C, et al. A nuclear isoform of the focal adhesion LIM-domain protein Trip6 integrates activating and repressing signals at AP-1- and NF-kappaB-regulated promoters. Genes Dev. 2004;18:2518–2528. doi: 10.1101/gad.322404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fahnenstich J, Nandy A, Milde-Langosch K, et al. Promyelocytic leukaemia zinc finger protein (PLZF) is a glucocorticoid- and progesterone-induced transcription factor in human endometrial stromal cells and myometrial smooth muscle cells. Mol Hum Reprod. 2003;9:611–623. doi: 10.1093/molehr/gag080. [DOI] [PubMed] [Google Scholar]
- 39.Baudry A, Yang ZZ, Hemmings BA. PKBalpha is required for adipose differentiation of mouse embryonic fibroblasts. J Cell Sci. 2006;119:889–897. doi: 10.1242/jcs.02792. [DOI] [PubMed] [Google Scholar]
- 40.Frijters R, Fleuren W, Toonen EJ, et al. Prednisolone-induced differential gene expression in mouse liver carrying wild type or a dimerization-defective glucocorticoid receptor. BMC Genomics. 2010;11:359. doi: 10.1186/1471-2164-11-359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Opherk C, Tronche F, Kellendonk C, et al. Inactivation of the glucocorticoid receptor in hepatocytes leads to fasting hypoglycemia and ameliorates hyperglycemia in streptozotocin-induced diabetes mellitus. Mol Endocrinol. 2004;18:1346–1353. doi: 10.1210/me.2003-0283. [DOI] [PubMed] [Google Scholar]
- 42.Teshigawara K, Ogawa W, Mori T, et al. Role of Kruppel-like factor 15 in PEPCK gene expression in the liver. Biochem Biophys Res Commun. 2005;327:920–926. doi: 10.1016/j.bbrc.2004.12.096. [DOI] [PubMed] [Google Scholar]
- 43.Yamamoto J, Ikeda Y, Iguchi H, et al. A Kruppel-like factor KLF15 contributes fasting-induced transcriptional activation of mitochondrial acetyl-CoA synthetase gene AceCS2. J Biol Chem. 2004;279:16954–16962. doi: 10.1074/jbc.M312079200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.