

Abstract
Structural analogs of the antimalarial Endochin were synthesized and screened for antiplasmodial activity against drug sensitive and multidrug resistant strains of Plasmodium falciparum. Structural features have been identified that are associated with improved potency while other features are associated with equipotency against an atovaquone-resistant clinical isolate. Relative to endochin the most active compound ELQ-121 shows ≈ 100-fold improvement in IC50 for inhibition of P. falciparum in vitro and it also exhibits enhanced metabolic stability. A polyethylene glycol carbonate ester prodrug of ELQ-121 demonstrated in vivo efficacy against P. yoelii in mice. This is the first report of an endochin-like quinolone that is efficacious in treating malaria in a mammalian host.
Introduction
Hans Andersag, who worked for Bayer I.G. Farbenindustrie A.G. laboratories in Elberfeld, Germany, is well known for the discovery of chloroquine (resochin) in the 1930’s. He was also connected with the discovery of “endochin” (Figure 1), a compound that elicited great interest among Bayer scientists because of its efficacy in treatment and prevention of sporozoite-induced malaria in a bird model (P. cathemerium/canary) of the disease (Salzer, et al., 1948). In subsequent work summarized by Kikuth and Mudrow-Reichenow (Kikuth and Mudrow-Reichenow, 1947), Steck (Steck, 1972), and Wiselogle (Wiselogle, 1946), endochin also demonstrated efficacy in treatment and prophylaxis against P. gallinaceum in the chick and P. lophurae in the turkey. Kikuth further reported that endochin exerted gametocidal action against male gametocytes undergoing exflagellation in finches infected with Haemoproteus, a closely related member of the Apicomplexa. Thus, it appeared that endochin could interfere with parasite development at three steps crucial to malaria infection in humans: liver stage, blood stage, and transmission to the mosquito vector. Despite these unique and desirable qualities, endochin’s antimalarial potential was never realized because it failed to cure malaria infections in subsequent experiments in mammalian species ranging from mice to non-human primates (Rhesus monkeys).
Figure 1.

Chloroquine and endochin, two drugs discovered by Hans Andersag and coworkers.
Our prior work with the tricyclic acridone system (Winter, et al., 2006) combined with a desire to minimize the tricyclic pharmacophore led to an interest in antimalarial quinolones and a reexamination of endochin. We recently reported a study of a limited number of endochin analogs, 3-alkyl-4-(1H)quinolone derivatives, together with endochin, and obtained meaningful structure-activity correlations (Winter, et al., 2008). Critical to the antiplasmodial effect was the presence of an extended 3-position alkyl side chain, a methyl group in position 2, and the methoxy group in position 7. Endochin exhibited potent antiplasmodial activity with IC50 values in the low nanomolar range and with slightly diminished activity against an atovaquone-resistant clinical isolate of Plasmodium falciparum. Preliminary mechanistic studies showed that an endochin analog blocked parasite oxygen consumption in parallel with atovaquone. In this study we have prepared additional endochin-like quinolones (ELQ, substituted 4(1H)-quinolones) to broaden our understanding of the structure-activity profile and to begin to optimize this pharmacophore for antimalarial activity in vitro and in vivo. Here we report our findings together with observations that may explain endochin’s poor performance in mammalian systems and which highlight important structural changes that endow the molecule with in vivo efficacy against P. yoelii-infected mice and with equal and potent activity against multidrug-resistant and atovaquone-resistant strains of P. falciparum.
Materials and methods
Chemicals and synthesis procedures
Unless otherwise stated all chemicals and reagents were from Sigma-Aldrich Chemical Company in St. Louis, MO (USA). SYBR Green I dye, used for determination of antiplasmodial IC50 values, was purchased from Invitrogen, Eugene, OR (USA). RPMI-1640, gentamicin, and Albumax II® were purchased from Invitrogen, Carlsbad, CA (USA). Blood, a source for red cells used to culture the parasite, was purchased from Lampire Biologicals, Pipersville, PA (USA). White blood cells were removed by centrifugation followed by removal of the buffy coat and uppermost red blood cells. 3-Heptyl-7-methoxy-2-methyl-4(1H)-quinolone (endochin) was synthesized by the method of Salzer et al. (Salzer, et al., 1948). The synthesis of selected 3-alkyl-2-methyl-4(1H)-quinolones is described below. Each of the quinolone derivatives was characterized by 1H-(500 MHz) nmr and mass spectrometry to ensure identity and purity prior to use in this study.
Synthesis of 2-hydroxy-7-methoxy-4-quinolone (ELQ-106)
ELQ-106 is synthesized by a method described by Riegel et al. (Baker, et al., 1946), a modification of the Conrad-Limpach procedure. Diethyl n-heptylmalonate (4.73 g), m-anisidine (2,27 g, 1 equivalent) and 35 ml of Dowtherm A were refluxed for 1 hour, and after cooling, 4,7 g of crude product were filtered off. Recrystallization from aqueous alcohol afforded the pure product as greyish, shiny plates (3.42 g, 64,5 % of theory), m.p. = 182 – 184°C, softening at 179°C. 1H-nmr spectrum (500 MHz, (CD3)2SO, Si(CH3)4 = 0): δNH = 11.11 p.p.m, s, broadened, 0.9H; δOH = 9,84, s, sharp,1H; δ5 = 7.76, d, J = 9.76 Hz, 1H; δ6 = 6.753, m, 2H; δ8 = 6.751 (H6 + H8, overlap), simulated with consecutive change of chemical shifts and J56 = 9.76, J68 = 2,4 Hz; δOCH3 = 3.80, s, 3 H; C7-chain: δCH2(pos.3) = 2.50, m, 2H; δ(CH2)5(middle) = 1.20–1.42, 3 m, 10H; δCH3 = 0,85, t, J = 7.12 Hz, 3H.
Synthesis of 2-Methyl-3-(n-heptyl)-5.7-difluorquinolone (ELQ-121)
The reaction scheme is presented in Figure 2. Ethyl 2-n-heptylacetoacetate (10.0 g, 43.9 mmol), 3.5-difluoroaniline (5.67 g, 43.9mmol), 200 ml benzene and 0.20 g p-toluenesulfonic acid monohydrate are heated in a flask fitted with a water separator for 20 hours; more acid (0.30 g) is added and water removal continued for 3 more days. Solvent is removed (rotary evaporator) and the residue dropped quickly into 15 ml of boiling Dowtherm A, kept at boiling temperature for 5 minutes and allowed to cool. The product crystallizes out upon cooling. After one and one-half hours the mass is broken up and transferred to a suction funnel; soluble components are washed out with a total of about 50 ml of hexane. Re-crystallization from about 100 ml of dimethylformamide leaves 6.43 g of pure product as shiny flakes (50.0 %). Melting point: 294–296°C. 1H-nmr spectrum (400 MHz, (CD3)2SO, Si(CH3)4=0): δCH3(pos.2)=2.33 ppm, s, 3H; C7-chain: δCH2(pos.3)=2.41, dist. t, 2H; δ(CH2)5(middle)= 1.2-1.4, indistinct features, 10H; δ CH3= 0.87, t, J = 6.8 Hz, 3H. δ6= 6.95, d-d-d, J56=12, J67≈ 10, J68≈ 2.5; δ8 ≈ 7.0, d-d-d, J58=1.35 (not resolved in 19F-spectrum), J68=2.5, J78=10.0, H(6) + H(8) = 2H; δNH= 11.4, s(br.), 0.85H. 19F-nmr spectrum (400 MHz, (CD3)2SO, CCl3F=0): δ5= −108,6, t, J = 12.7 Hz,1F; δ7= −106,3, quartett, J =10.6 Hz, 1F. Mass spectrum: M+ = 293, 18 %; (M-C6H13)+ = 208, 100%.
Figure 2.

Chemical synthesis of ELQ-121.
Synthesis of the mixed carbonate ester derivative ELQ-125 of ELQ-121
0.51 g of ELQ-121 was stirred in 10 ml of anhydrous tetrahydrofuran with 75 mg of 60 % NaH (in paraffin, slight excess) in a lightly capped vial for about one half hour, when a pale yellow almost clear solution resulted. To this solution was added 0.54 g of CH3(OCH2CH2)4OCOCl (slight excess) with stirring. After 1 day 3 more drops of the acid chloride was added and stirring continued for one more day. The solution was filtered to remove a white precipitate, evaporated and chromatographed on a short column (Kieselgel, 7 cm i.d. x 5 cm, CH2Cl2). The sample dissolved in methylene chloride was washed onto the column with 50 ml of CH2Cl2, followed by a 1:1-mixture of ethyl acetate and hexane (isomer mixture). The elution was followed by thin-layer chromatography. Later fractions contained a by-product.
The fraction containing ELQ-125 was brought to dryness, leaving 0.46 g of a very pale yellow oil (50 % of theory). 1H-nmr spectrum (400 MHz, CDCl3, Si(CH3)4=0): TMCH3(pos.2)=2.74 ppm, s, 3H; C7-chain: TMCH2(pos.3)=2.72, dist.t, overlap with TMCH3(pos.2), together 5H; TMCH2(middle)=1.2-1.6, indistinct features, 10H; TMCH3=0.88, t, J=6.88 Hz, 3H; polyether chain of carbonate: TM = 3.55–3.72, 2 m, 12H; TM=2,82, m, 2H; TM=4.46, m, 2H; TMCH3=3.36, s, 3H. TM6=6.97, d-d-d; J56=8.9, J67 =9.6, J68=2.5, 1H; TM8= 7.48, d-d-d, J58=1.3 (not res. in 19F-spectrum), J68 = 2.5, J78=9.6, 1H. 19F-nmr spectrum (400 MHz, CDCl3, Si(CH3)4=0): TM5= −108.6, quartett, Javerage = 8.9, 1F; TM7= −114.2, d-d or t, JH9.7 Hz,1F. Mass spectrum: M+ = 527, < 1%; CH3OCH2CH2+ = 59, 100 %.
Synthesis of 6-chloro-2-methyl-3-(n-heptyl)-quinolone (ELQ-130)
Ethyl 2-n-heptylacetoacetate, was prepared from 5.15 g of ethylacetoacetate, 1 equivalent of sodium ethoxide and 1 equivalent 1-bromoheptane in alcohol. When the reaction was completed (1 day of boiling), the solvent was removed under vacuum, the residue stirred with 150 ml of hexane, filtered and the solvent removed from the filtrate. The crude ethyl 2-n-heptylacetoacetate obtained was mixed with 5.0 g p-chloroaniline (1 equivalent, calculated for ethyl acetoacetate used to pepare n-heptylacetoacetate), 90 ml of benzene and 20 drops of concentrated HCl and heated. The water formed in the condensation was continuously removed with a water separator. Since the reaction was not completed after 20 hours, another 20 drops of conc. HCl were added and heating continued. After another 24 hours, little starting materials could be detected by g.c.-m.s.
After removal of the benzene under vacuum, the residue was added within 4 minutes to 65 ml of boiling Dowtherm A and held at boiling temperature for 10 minutes. After 90 minutes of cooling, the product was filtered off, washed with hexane and recrystallized from aqueous alcohol. A total 2.13 g of product was obtained (18.6 % of theory). G.c.-m.s. (DB5, 30 M, 150°C: 2 min., then 11°C/min.→ 280°C). Rt = 14,76 min, M+ = 291(11 %), 293 (4%); 207 (100%), 209 (30 %) = (M-C6H13)+. M.p. = 265-269°C, darkening at 210°C. 1H-nmr spectrum (400 MHz, (CD3)6SO, Si(CH3)4 = 0): δNH = 11.54 p.p.m., s, 1H; δ5 = 7,97, d, J = 2.43 Hz, 1H; δ7 = 7.60, d-d, J = 8.84, J = 2.49, 1H; δ8 = 7.50, d, J = 8.81, 1H; δCH3(pos.2) = 2.38, s, 3H; C7-cahin: δCH2(pos.3) = 2.48, dist. t (overlap with solvent signal); δ(CH2)5(middle) = 1.17–1.45, m, 10H; δCH3 = 0.86, t, J = 6.90, 3H.
Parasites
Plasmodium falciparum strains D6 and Dd2 were obtained from the MR4 (ATCC, Manassas, VA, USA). D6 is sensitive to chloroquine but mildly resistant to mefloquine (Oduola, et al., 1987) while Dd2 is resistant to multiple quinoline and antifolate antimalarial agents as summarized by Singh (Singh and Rosenthal, 2001). Tm90.C2B (provided by Dr. Dennis Kyle, WRAIR, Silver Spring, MD, USA) is resistant to atovaquone, chloroquine, mefloquine, and quinine (Suswam, et al., 2001).
Parasite culture and drug sensitivity
Three different laboratory strains of P. falciparum were cultured in human erythrocytes by standard methods under a low oxygen atmosphere (5% O2, 5% CO2, 90% N2) in an environmental chamber (Trager and Jensen, 1976). The culture medium was RPMI-1640, supplemented with 25 mM HEPES buffer, 25 mg/L gentamicin sulfate, 45 mg/L hypoxanthine, 10 mM glucose, 2 mM glutamine, and 0.5% Albumax II (complete medium). The parasites were maintained in fresh human erythrocytes suspended at a 2% hematocrit in complete medium at 37°C. Stock cultures were sub-passaged every 3 to 4 days by transfer of infected red cells to a flask containing complete medium and uninfected erythrocytes.
In vitro antimalarial activity of the ELQ derivatives was assessed by the SYBR Green I fluorescence-based method (the “MSF assay”) described previously by us (Smilkstein, et al., 2004) with minor modifications (Winter, et al., 2006). Experiments were set up in triplicate in 96 well plates (Costar, Corning) with two-fold dilutions of each drug across the plate in a total volume of 100 microliters and at a final red blood cell concentration of 2% (v/v). Stock solutions of each drug were prepared by dissolving in ethanol or dimethylsulfoxide (as appropriate) at 10mM. The dilution series was initiated at a concentration of 1μM and the experiment was repeated beginning with a lower initial concentration for those compounds in which the IC50 value was below 10nM. Automated pipeting and dilution was carried out with the aid of a programmable Precision 2000 robotic station (BioTek, Winooski, VT). An initial parasitemia of 0.2% was attained by addition of normal uninfected red cells to a stock culture of asynchronous parasite infected red cells (PRBC). The plates were incubated for 72 hrs at 37°C in an atmosphere of 5% CO2, 5% O2, and 90% N2. After this period the SYBR Green I dye-detergent mixture (100μl) was added and the plates were incubated at room temperature for an hour in the dark and then placed in a 96-well fluorescence plate reader (Spectramax Gemini-EM, Molecular Diagnostics) for analysis with excitation and emission wavelength bands centered at 497 and 520 nm, respectively. The fluorescence readings were plotted against the logarithm of the drug concentration and curve fitting by nonlinear regression analysis (GraphPad Prism software) yielded the drug concentration that produced 50% of the observed decline relative to the maximum readings in drug-free control wells (IC50).
In vivo efficacy in a murine malaria model of patent infection with P. yoelii
The activity of the prototype ester, ELQ-125, against the blood stages was assessed using a modified Thompson procedure (Ager, 1984). Mice (female, CF1, Charles River Labs) were infected intravenously with about 500,000 P. yoelii parasitized erythrocytes from a donor animal. Drug administration was initiated once the parasitemia had risen to between 3 to 5% as determined microscopically by examination of Giemsa-stained blood smears. The test compound, ELQ-125, was taken into NeoBee® M-5 (a mixture of glycerol esters of caprylic and capric fatty acids derived from coconut oil, Stephan Company, Northfield, Illinois, USA) and used without dilution. The drug was administered by oral gavage once daily for 3 days. On the 4th day blood films were prepared and the extent of parasitemia was determined microscopically. ED50 and ED90 values (mg/kg/day) were derived from the dose required to reduce the parasite burden by 50% and 90%, respectively, relative to drug-free controls. The procedures involved, together with all matters relating to the care and housing of the animals used in this study, were approved by the Portland VA Medical Center Institutional Animal Care and Use Committee.
In vitro metabolic stability of endochin
An initial assessment of the stability and metabolic fate of endochin (ELQ-100) was performed. Hepatic murine microsomal fractions (1 mg/ml) were incubated with 1 and 10 μM endochin (added in 5 μl of methanol) in the presence of NADPH (1 mM) for 0, 10, 20 and 40 min. The reactions were terminated by addition of 2 volumes of ice-cold methanol. Precipitated protein was removed by centrifugation and the supernatant was transferred directly to an autosampler vial for LC/MS analysis. Aliquots (10 μl) were injected onto a BetaBasic C18 (100 x 2.1 mm, 5microns) column and analytes were separated with a Surveryor HPLC system using a linear gradient of water containing 0.1% acetic acid (solvent A) and methanol with 0.1% acetic acid (solvent B) from 50% B to 98% B over 5 min after a 0.1 min delay and held at 98% B for 7 additional min before returning to the starting conditions. The flow rate was 0.3 ml/min. The HPLC was interfaced to an LCQ Advantage (ThermoElectron) ion trap mass spectrometer equipped with an electrospray ionziation (ESI) source to enable monitoring in the positive mode of selected ions corresponding to unmetabolized endochin (m/z=288.2), the desmethyl endochin metabolite (m/z=274.2), as well as other possible mono (m/z=290.2 and m/z=304.2) and dihydroxylated metabolites (m/z=320.2).
Results and Discussion
In vitro activity and pharmaco-resistance pattern of endochin-like quinolones against a panel of P. falciparum parasites
We synthesized over 30 endochin analogs (ELQs) by the Conrad and Limpach method (Conrad and Limpach, 1891) and screened them for antiplasmodial activity in vitro against chloroquine (CQ) sensitive (D6), multidrug resistant (Dd2), and chloroquine/quinine/atovaquone (ATV)-resistant (Tm90.C2B) strains of P. falciparum. This information is provided in Table 1. As reported previously it may be observed that endochin (ELQ-100) exhibits potent activity with IC50 values of ≈3-4nM vs. D6 and Dd2, and 11.4nM vs. the ATV-resistant Tm90.C2B clinical isolate, i.e., a modest level of ATV cross-resistance. Additional exploration of the structure-activity relationships in this study revealed that the potency of the endochin molecule can be greatly influenced by chemical modification. The following observations on the structure-activity relationships (SAR) can be made:
Table 1.
Antiplasmodial activity of ELQs (IC50 ’s) vs. Plasmodium falciparum strains in vitro (nM).*
| Code | MW | Chemical Structure | LogP | D6 | Dd2 | C2B |
|---|---|---|---|---|---|---|
| ELQ-100 | 287.4 | 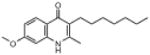 |
3.35 | 4 | 3 | 11 |
| ELQ-102 | 341.4 | 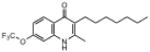 |
5.00 | 86 | 86 | 102 |
| ELQ-103 | 397.2 | 5.31 | 1 | 1 | 5 | |
| ELQ-106 | 289.4 | 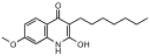 |
3.55 | >2,500 | >2,500 | >2,500 |
| ELQ-107 | 412.5 | 3.90 | 1,800 | 1,500 | 1,300 | |
| ELQ-109 | 291.8 | 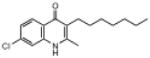 |
4.03 | 6 | 2 | 64 |
| ELQ-110 | 302.4 | 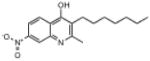 |
4.11 | 18 | 21 | 41 |
| ELQ-114 | 259.3 | 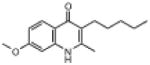 |
2.52 | 8 | 12 | 52 |
| ELQ-115 | 245.3 | 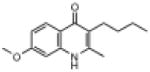 |
2.10 | 29 | 29 | 92 |
| ELQ-117 | 273.4 | 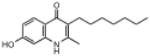 |
3.09 | 759 | 805 | 824 |
| ELQ-118 | 282.4 | 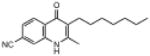 |
3.51 | 18 | 45 | ND |
| ELQ-119 | 311.8 | 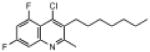 |
6.7 | 567 | 455 | 2,625 |
| ELQ-120 | 275.4 | 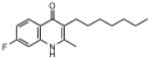 |
3.63 | 5 | 5 | 24 |
| ELQ-121 | 293.3 | 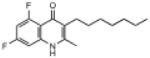 |
3.79 | 0.1 | 0.1 | 81 |
| ELQ-122 | 313.8 | 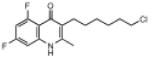 |
3.4 | 1 | 1 | 24 |
| ELQ-124 | 326.3 | 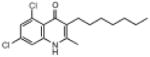 |
4.59 | 112 | 101 | 1,700 |
| ELQ-125 | 527 | 0.4 | 0.4 | 146 | ||
| ELQ-127 | 257 | 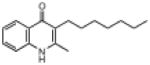 |
3.5 | 35 | 34 | 43 |
| ELQ-129 | 325 | 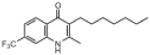 |
4.4 | 217 | 180 | 7,900 |
| ELQ-130 | 291.8 | 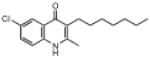 |
4.03 | 18 | 12 | 16 |
| ELQ-131 | 275.4 | 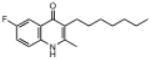 |
3.63 | 40 | 40 | 37 |
| ELQ-132 | 293.3 | 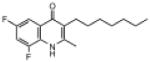 |
3.79 | 115 | 134 | 110 |
| ELQ-133 | 303.2 | 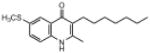 |
3.92 | 206 | 257 | 461 |
| ELQ-134 | 307.4 | 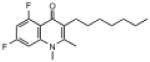 |
4.58 | 25 | 20 | >2,500 |
Results are the average of at least 3 independent determinations, each carried out in triplicate. ND = not determined
The length of the 3-position side chain influences the antiplasmodial effect. Our data show that the 7 carbon chain length (endochin) is superior to C6 > C5 > C4 with values ranging from ≈3nM (ELQ-100) to ≈30nM (C4, ELQ-115). ELQ-103 with a trifluoroundecyl side chain exhibits IC50 values in the low nanomolar range for all 3 tested strains.
Replacement of the 7-OCH3 group by hydroxy (ELQ-117) greatly diminishes antiplasmodial activity whereas replacement by either Cl (ELQ-109) or F (ELQ-120) results in only a modest reduction in in vitro potency. Derivatives bearing other electronegative substituents at the 7-position (e.g., CN, CF3, OCF3, and NO2) proved inferior, and all of these molecules exhibited modest to significant cross-resistance against the Tm90.C2B strain. It is interesting that the in vitro activity of the 7-H analog (ELQ-127) is weakened by roughly 5-fold relative to that of endochin however it remains equally active against all three parasite strains. Electron-donating substituents (i.e., other than OCH3) placed at the 7-position caused diminished antiplasmodial activity.
The 2-CH3 group appears to be important because replacement of it with hydroxy is accompanied by a dramatic loss of effectiveness, e.g., compare ELQ-100 to ELQ-106 vs. the D6 strain with IC50 values of 3.8nM and >2,500nM, respectively.
Moving the chlorine atom from the 7-position (e.g., ELQ-109) to the 6-position (e.g., ELQ-130) results in a modest reduction in antimalarial response (strain D6 IC50 values of 5.8nM and 22.2nM, respectively) however equal sensitivity is observed against the atovaquone-resistant Tm90.C2B clinical isolate only for ELQ-130. Similar results were observed for the congener with a fluorine atom at position 6 (ELQ-131). Taken together with results from the 7-H derivative, ELQ-127, these observations combine to suggest that the mutation appearing in the cytochrome b gene of this clinical isolate (which is linked to a high level of atovaquone resistance) introduces steric hindrance to bulky substituents occupying the 7-position of quinolone ring system.
Placement of 2 halogens on the benzenoid ring had a mixed effect. The 5,7-dichloro endochin analog (ELQ-124) exhibited weak in vitro activity while the corresponding 5,7-difluoro construct (ELQ-121) proved to be one of the most potent compounds in the tested series with IC50 values of ≈0.05nM against D6 and Dd2 and about 300 times higher against Tm90.C2B. By contrast, the 6,8-difluoro positional isomer exhibited diminished but respectable antimalarial potency together with an equal response toward all three tested P. falciparum strains (with IC50 values ranging from ≈110nM to 134nM for all 3 strains).
The results of testing ELQ-134 and ELQ-119, structural analogs of our most potent quinolone construct, are particularly revealing. ELQ-134 is the N-methyl derivative of ELQ-121 and it shows greatly diminished potency (roughly 300-fold reduced). ELQ-119 contains a chlorine atom at the 4-position and it is over a 1000 times less potent than the parent drug based on in vitro testing. Taken together, this information suggests that the ability to tautomerize between the quinolone and quinolinol forms is important for antiplasmodial potency.
In vivo efficacy of an ELQ-polyethylene glycol carboxylate ester against patent infections of P. yoelii mice
While many of our ELQ derivatives exhibit potent antiplasmodial activity in vitro their relatively poor aqueous solubility makes it difficult to administer them to animals. We therefore designed and synthesized ELQ prodrugs containing a water-solubilizing pro-moiety that could be metabolically released after drug administration. We synthesized a prodrug ester of ELQ-121 and have found that the prodrug formulation (ELQ-125, Figure 3) exhibits improved water solubility, miscibility with NeoBee M-5, a pharmaceutical delivery vehicle, and established in vivo efficacy. In a test of drug efficacy against a patent P. yoelii infection in mice, presenting with a >1% parasitemia (5 mice/group) at the beginning of a 3-day (once daily) treatment regimen, doses of 100mg/kg/day and 50mg/kg/day completely cleared parasites from the bloodstream. These treatment doses were without evident toxicity based on weight loss, grooming and locomotion of treated mice. In each case, ELQ-125, a clear and colorless syrup, was administered orally with NeoBee M-5 (vol. = 100μl). At 25mg/kg/day, parasitemia was suppressed by >99% relative to controls (assessed on the day following the last dose). Follow-up experiments performed in the same manner established ED90 (22mg/kg/day) and ED50 (11mg/kg/day) values for ELQ-125 against P. yoelii infections in CF1 mice.
Figure 3.

Chemical structure of the polyethylene glycol (PEG) ester prodrug, ELQ-125.
We believe that these results have great significance. Even though we have not fully optimized the pro-moiety or the nature of the critical 3-position substituent, this “first-of-a-kind” construct proved to be highly efficacious by oral dosing. Including other enhancements that we’ve incorporated into the pharmacophore (i.e., halogens in the benzenoid ring system), we have overcome the primary obstacle (oral bioavailability) that has blocked the therapeutic advancement of endochin for over 60 years. Additional studies are needed to confirm that oral administration of ELQ-125 increases exposure to ELQ-121 in drug treated animals; a detailed in vivo pharmacokinetic study is planned.
In vitro microsomal metabolism of endochin
We performed an initial assessment of the stability and metabolic fate of endochin (ELQ-100) in the presence of mammalian microsomal elements. Hepatic murine microsomal fractions were incubated with endochin in the presence of NADPH for 0, 10, 20 and 40 min. Samples were processed and injected into an LC-MS monitoring in the positive mode of selected ions corresponding to unmetabolized endochin, the O-demethylated endochin metabolite, as well as other possible mono and dihydroxylated metabolites. In summary of our findings, the O-demethylated metabolite, ELQ-117, was detected at the earliest time point in abundance (Figure 4). In addition to the O-demethylated product we also observed formation of monohydroxy metabolites of endochin that are believed to result from both side chain alkyl hydroxylation and N-hydroxylation events. Incubations were also conducted with human and rat hepatic microsomes under identical conditions and we observed a similar metabolic profile compared to the murine system. Taken together these findings show that while endochin exerts potent antiparasitic activity in vitro, it undergoes extensive metabolism in vivo which may account for its poor performance against malaria and toxoplasmosis in mice; it is possible that these metabolic transformations do not occur in the avian systems used by malaria researchers in the 1940’s.
Figure 4.

LC/MS metabolite profiles of 10 μM endochin obtained after t=0 (Panel A) and t=10 min of incubation at 37°C with murine hepatic microsomes in the presence of NADPH (Panel B). The top panels represent the total ion chromatograms of all ions monitored and the extracted ion chromatogram for the indicated ions are shown below. Endochin elutes at 9.2 min (m/z=288.2) and the 7-desmethyl metabolite elutes at 8.7 min (m/z=274.2) as confirmed by cochromatography with standards. The other ions monitor potential hydroxylated (m/z=290.2 and m/z=304.2) and dihydroxylated (m/z=320.2) metabolites.
Microsomal stability studies were also carried out with ELQ-121. These experiments indicate that ELQ-121 exhibits enhanced metabolic stability compared to endochin (murine microsomes + NADPH t1/2 = 2.5 min for endochin and 14.7 min for ELQ-121) although extensive hydroxylation of the alkyl side chain was evident.
Concluding remarks
Our ongoing study of endochin suggests that the primary cause of its poor performance in mammals is due to metabolic instability. We have modified the quinolone nucleus to enhance metabolic stability (to a modest extent) and effected additional structural changes that endow our current lead compounds with potent intrinsic activity against multidrug-resistant parasites (IC50’s in the low to sub-nanomolar range) and with the therapeutic power to clear a patent P. yoelii infection in mice by the oral route of administration. ELQs are attractive therapeutics because they exhibit many desirable characteristics of drug-like molecules and have the potential for targeting multiple developmental stages of the parasite life cycle. Efforts to optimize this chemotype to enhance metabolic stability, antiplasmodial activity, and oral bioavailability are ongoing and we will continue to publish our findings on this very intriguing series of compounds.
Article Highlights.
Synthesis of endochin analogs with enhanced antiparasitic activity.
Identification of endochin analogs that are equipotent against multidrug resistant parasites.
Identification of endochin analogs that are equipotent against atovaquone resistant parasites.
Prodrug of endochin analog ELQ-121 cures murine malaria by oral route.
Acknowledgments
We want to acknowledge financial support from the Stanley Medical Research Institute of Chevy Chase, Maryland (USA), from the Merit Review Program of the Department of Veterans Affairs, and the US National Institutes of Health RO1 program (Grant #.R01 AI079182). An international PCT application has been filed by the US Department of Veterans Affairs and Oregon Health Sciences University to protect the intellectual property described in this report.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Cited Literature
- Ager AJ. Rodent malaria models. Springer-Verlag; Berlin: 1984. [Google Scholar]
- Baker R, Lappinn G, Riegel B. Some 3-alkyl-2,4-quinolinediols. Journal of the American Chemical Society. 1946;68:1284–1285. doi: 10.1021/ja01211a048. [DOI] [PubMed] [Google Scholar]
- Conrad M, Limpach L. Synthese von Chinolinderivaten mittelst Acetessigester. Berichte der deutschen chemischen Gesellschaft. 1891;24:2990–2992. [Google Scholar]
- Kikuth W, Mudrow-Reichenow L. Über kausalprophylaktisch bei Vogelmalaria wirksame Substanzen. Zeitschrift für Hygiene und Infektionskrankheiten, medizinische Mikrobiologie, Immunologie und Virologie. 1947;127:151–165. [PubMed] [Google Scholar]
- Oduola AM, Milhous WK, Salako LA, Walker O, Desjardins RE. Reduced in-vitro susceptibility to mefloquine in West African isolates of Plasmodium falciparum. Lancet. 1987;2:1304–1305. doi: 10.1016/s0140-6736(87)91195-0. [DOI] [PubMed] [Google Scholar]
- Salzer W, Timmler H, Andersag H. Über einen neuen, gegen Vogelmalaria wirksamen Verbindungstypus. Chemische Berichte. 1948;81:12–19. [Google Scholar]
- Singh A, Rosenthal PJ. Comparison of efficacies of cysteine protease inhibitors against five strains of Plasmodium falciparum. Antimicrobial Agents and Chemotherapy. 2001;45:949–951. doi: 10.1128/AAC.45.3.949-951.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smilkstein M, Sriwilaijaroen N, Kelly JX, Wilairat P, Riscoe M. Simple and inexpensive fluorescence-based technique. Antimicrobial Agents and Chemotherapy. 2004;48:1803–1806. doi: 10.1128/AAC.48.5.1803-1806.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steck EA. Chemotherapy of malaria. Division of Medicinal Chemistry. Walter Reed Army Institute of Research, U.S. Government Printing Office; Washington DC: 1972. [Google Scholar]
- Suswam E, Kyle D, Lang-Unnasch N. Plasmodium falciparum: the effects of atovaquone resistance on respiration. Experimental Parasitology. 2001;98:180–187. doi: 10.1006/expr.2001.4639. [DOI] [PubMed] [Google Scholar]
- Trager W, Jensen JB. Human malaria parasites in continuous culture. Science. 1976;193:673–675. doi: 10.1126/science.781840. [DOI] [PubMed] [Google Scholar]
- Winter RW, Kelly JX, Smilkstein MJ, Dodean R, Bagby GC, Rathbun RK, Levin JI, Hinrichs D, Riscoe MK. Evaluation and lead optimization of anti-malarial acridones. Experimental Parasitology. 2006;114:47–56. doi: 10.1016/j.exppara.2006.03.014. [DOI] [PubMed] [Google Scholar]
- Winter RW, Kelly JX, Smilkstein MJ, Dodean R, Hinrichs D, Riscoe MK. Antimalarial quinolones: Synthesis, potency, and mechanistic studies. Experimental Parasitology. 2008;118:487–497. doi: 10.1016/j.exppara.2007.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiselogle FY. A Survey of Antimalarial Drugs, 1941-1945. J. W. Edwards; Ann Arbor, Michigan: 1946. pp. xi–1921. [Google Scholar]