

Abstract
Patients with diabetes have a much greater risk of developing heart failure than non-diabetic patients, particularly in response to an additional hemodynamic stress such as hypertension or infarction. Previous studies have shown that increased glucose metabolism via the hexosamine biosynthesis pathway (HBP) and associated increase in O-linked-β-N-acetylglucosamine (O-GlcNAc) levels on proteins contributed to the adverse effects of diabetes on the heart. Therefore, in this study we tested the hypothesis that diabetes leads to impaired cardiomyocyte hypertrophic and cell signaling pathways due to increased HBP flux and O-GlcNAc modification on proteins. Cardiomyocytes isolated from type 2 diabetic db/db mice and non-diabetic controls were treated with 1 μM ANG angiotensin II (ANG) and 10 μM phenylephrine (PE) for 24 h. Activation of hypertrophic and cell signaling pathways was determined by assessing protein expression levels of atrial natriuretic peptide (ANP), α-sarcomeric actin, p53, Bax and Bcl-2 and phosphorylation of p38, ERK and Akt. ANG II and PE significantly increased levels of ANP and α-actin and phosphorylation of p38 and ERK in the non-diabetic but not in the diabetic group; phosphorylation of Akt was unchanged irrespective of group or treatment. Constitutive Bcl-2 levels were lower in diabetic hearts, while there was no difference in p53 and Bax. Activation of the HBP and increased protein O-GlcNAcylation in non-diabetic cardiomyocytes exhibited a significantly decreased hypertrophic signaling response to ANG or PE compared to control cells. Inhibition of the HBP partially restored the hypertrophic signaling response of diabetic cardiomyocytes. These results suggest that activation of the HBP and protein O-GlcNAcylation modulates hypertrophic and cell signaling pathways in type 2 diabetes.
Keywords: Hypertrophy, Diabetes, Cardiomyocyte, Cell signaling, O-GlcNAc
Introduction
Diabetes leads to a substantially increased risk for cardiovascular disease, including heart failure (Garcia et al. 1974), which can in part be attributed to an increase in atherosclerosis and an increased incidence of myocardial infarction. However, both clinical (Galderisi et al. 1991) and experimental evidence (Rodrigues and McNeill 1992; Chatham et al. 1996; Ren et al. 1997; Brownlee 2001) also strongly indicate that diabetes leads to changes at the level of the myocardium consistent with the development of a diabetic cardiomyopathy. Furthermore, even accounting for the increase in vascular disease, patients with diabetes have a much greater risk of developing heart failure than non-diabetic patients, particularly in response to an additional hemodynamic stress such as hypertension or infarction (Preis et al. 2009).
While the specific mechanisms leading to development of diabetic cardiomyopathy are not well understood, hyperglycemia is frequently considered to be an important contributing factor. The adverse effects of hyperglycemia on the heart have been attributed to increased oxidative stress (Hayat et al. 2004; Poornima et al. 2006; Boudina and Abel 2007), increased flux through the polyol pathway (Brownlee 2001), increased advanced glycation end products (AGE) (Brownlee 2001; Petrova et al. 2002), PKC activation (Davidoff et al. 2004), and more recently modification of proteins by O-linked-β-N-acetylglucosamine (O-GlcNAc).
The modification of serine and threonine residues on cytosolic and nuclear proteins by O-GlcNAc is increasingly recognized as an important regulatory mechanism involved in signal transduction (Love and Hanover 2005; Slawson et al. 2006; Hart et al. 2007). This atypical glycosylation occurs in the cytosol and the nucleus, rather than in the Golgi or the endoplasmic reticulum, and is regulated by the activities of two key enzymes, O-GlcNAc transferase (OGT) and N-acetylglucosaminidase (O-GlcNAcase) (Love and Hanover 2005; Slawson et al. 2006; Hart et al. 2007). In addition to OGT and O-GlcNAcase, the levels of protein O-GlcNAcylation are also dependent on the metabolism of glucose via the hexosamine biosynthesis pathway (HBP), which leads to the formation of UDP-GlcNAc, the substrate for OGT and the essential sugar donor for O-GlcNAc formation. Indeed, activation of the HBP and the resulting increase in O-GlcNAc have both been implicated in the adverse effects of diabetes on a variety of cells and tissues, including the heart (Brownlee 2001; Clark et al. 2003; Pang et al. 2004; Hu et al. 2005; Fulop et al. 2007a, b, c; Copeland et al. 2008; Laczy et al. 2009).
Hyperglycemia is commonly believed to be the major contributing factor to diabetic complications; paradoxically, however, a number of studies examining the consequences of improved glycemic control in type 2 diabetic patients have reported either no improvements or even an increase in cardiovascular events or deaths (Nissen and Wolski 2007; Gerstein et al. 2008; Duckworth et al. 2009). While the results of these studies could be due to a number of factors, they raise the possibility that hyperglycemia alone may not be the primary mediating factor underlying the adverse effects of diabetes on the heart. Of note, in addition to hyperglycemia (Liu et al. 2000), reactive oxygen species and hyperlipidemia, both of which are characteristics of diabetes, also lead to increased HBP flux and O-GlcNAc levels (Du et al. 2000). Recent studies have also demonstrated the importance of transcriptional regulation of OGT and O-GlcNAcase in modulating cellular levels of O-GlcNAc (Taylor et al. 2008), which could also contribute to the increase in O-GlcNAcylation seen with diabetes. Thus, increases in cardiac O-GlcNAc levels seen in diabetes could occur due to a number of factors other than hyperglycemia.
An early response to increased hemodynamic stress, such as hypertension or infarction, is cardiomyocyte hypertrophy, which is typically considered to be a beneficial adaptive response to increased workload. It is only at a later stage that these changes become maladaptive leading to decompensation and heart failure. In neonatal rat ventricular cardiomyocytes (NRVMs), increasing HBP with either hyperglycemia or glucosamine blunted the response to both angiotensin II- (ANG II) and phenylephrine- (PE) induced cellular hypertrophy (Hunton et al. 2002; Pang et al. 2002), which was mediated, at least in part, via increased HBP flux. Therefore, the goal of this study was to test the hypothesis that diabetes leads to blunted hypertrophic and cell signaling responses in cardiomyocytes to ANG II and PE, and that this is mediated via increased HBP and protein O-GlcNAc levels.
Materials and methods
Materials
All chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA) unless otherwise stated.
Animals
Eight-week-old mice C57BL/KsJ-leprdb/leprdb diabetic (db/db) and non-diabetic littermate controls were obtained from the Jackson Laboratories (Bar Harbor, ME, USA) and housed with ad libitum food and water for an additional 4 weeks until the age of 12 weeks. At the time of the experiments, db/db mice weighed 49 ± 3 g compared to 26 ± 1 g for lean controls (P < 0.05), and blood glucose concentrations were 21 ± 1 mM and 7 ± 1 mM, respectively. All animal experiments were approved by the University of Alabama at Birmingham (UAB) Institutional Animal Care and Use Committee and conformed to the Guide for the Care and Use of Laboratory Animals published by the National Institute of Health (NIH publication no. 85–23, 1996).
Isolation of cardiomyocytes
Adult mouse cardiomyocytes were isolated as previously described (Shan et al. 2008). The protocol, with some minor modifications, is based on that originally developed by the Alliance for Cell Signalling (AfCS) for adult mouse cardiomyocyte isolation (O'Connell et al. 2003), which reported that ~70% of cardiomyocytes cultured for 72 h exhibited rod-shaped morphology. Briefly, hearts were rapidly excised from heparinized and anesthetized mice, perfused retrogradely with Ca2+-free perfusion buffer consisting of 0.6 KH2PO4, 0.6 Na2HPO4, 1.2 MgSO4·7H2O, 0.032 phenol red, 12 NaHCO3, 10 KHCO3, 10 HEPES, 30 taurine, 10 2,3-butanedione monoxime and 5.5 glucose, pH 7.46, followed by a buffer containing 12.5 μM CaCl2 and 0.4 mg/ml collagenase type 2 (Worthington). After 15–25 min of perfusion with collagenase containing buffer, the ventricles were removed and finely minced; dispersed myocytes were filtered and allowed to sediment. The supernatant was centrifuged and the pellet resuspended and combined with the original sedimented myocytes in the perfusion buffer containing 5% bovine calf serum and 12.5 μM CaCl2. The calcium concentration was increased gradually from 12.5 μM to 1 mM over ~20 min. Freshly isolated cardiomyocytes were kept at 37°C and all experiments were performed at least 1 h after isolation. As previously reported using this protocol, approximately 80% of cardiomyocytes exhibited predominantly rod-shaped morphology up to at least 18 h following isolation (Shan et al. 2008), which is consistent with report from the Alliance for Cell Signaling (O'Connell et al. 2003). This was slightly lower at ~70% cardiomyocytes isolated from db/db mice (data not shown).
Immunoblot analyses
Protein levels in isolated cardiomyocytes or whole heart lysates were determined using standard immunoblot techniques as previously described (Shan et al. 2008) using antibodies to calsequestrin (Abcam), α-sarcomeric actin (Abcam), atrial natriuretic peptide (ANP, Santa Cruz) and TRPC1 (Alomone Labs), Bcl-2 (Cell Signaling), Bax (Santa Cruz), p53 (Santa Cruz), phosphorylated and total p38 (Santa Cruz), phosphorylated and total ERK (Cell Signaling), phosphorylated Akt (Ser473) and total Akt (Cell Signaling). Protein O-GlcNAc levels were also determined by immunoblot analysis as previously described (Champattanachai et al. 2007; Fulop et al. 2007a, b, c) using the anti-O-GlcNAc antibody CTD110.6. The immunoblots were developed with chemiluminescence (Pierce) and densitometry analyses were performed using Labworks Analysis Software (UVP). Calsequestrin was used as protein loading control throughout, as it was found to have similar levels of expression in both cardiomyocytes from control and db/db mice and was unchanged in response to the various treatment protocols. For evaluation of phosphorylated versus total or comparison of protein levels relative to calsequestrin, membranes were stripped and reprobed with the appropriate antibody as previously described (Fulop et al. 2007a, b, c).
Statistical analysis
All data are presented as mean ± SE. Statistical analysis was performed by either an unpaired t tests or one-way analysis of variance (ANOVA) followed by Dunnett's multiple comparisons test as appropriate. Statistically significant differences between groups were defined as P < 0.05.
Results
Diabetes blunts hypertrophic response to ANG II and PE
Cardiomyocytes from control and diabetic (db/db) mice were incubated for 24 h in the presence or absence of either ANG II (1 μM) or PE (10 μM) as previously described (Hunton et al. 2002; Pang et al. 2002; Hunton et al. 2004). In the control group, in response to both ANG II and PE, there was a significant increase in ANP expression, an early marker of cardiomyocyte hypertrophy (Fig. 1a). In contrast, in diabetic cardiomyocytes ANP expression was unaffected by either agonist. As expected, ANG II and PE also induced a robust increase in α-actin expression in the control group, whereas there was no effect in the diabetic group (Fig. 1b).
Fig. 1.

Expression of a ANP, b α-actin and c TRPC1 protein in cardiomyocytes isolated from non-diabetic control (control) and diabetic (db/db) hearts following 24 h treatment with ANG (1 μM) or PE (10 μM). Upper panels are representative immunoblots and the lower panels are mean densitometric data from three individual experiments normalized to calsequestrin. *P < 0.05 versus control untreated group
Transient receptor potential channel 1 (TRPC1) was reported to mediate agonist-induced Ca2+ signaling, and increased expression has been implicated as playing a causal role in the development of cardiac hypertrophy (Ohba et al. 2007). Similar to ANP and α-actin, there was a significant increase in TRPC1 protein in control cardiomyocytes with both ANG II and PE treatments (Fig. 1c); in contrast, there were no significant changes in TRPC1 expression in diabetic cardiomyocytes with either treatment.
To provide additional insight into the effect of diabetes on hypertrophic signaling pathways, the effect of PE on phosphorylation of p38, ERK and Akt were also assessed (Fig. 2). PE treatment significantly increased p38 and ERK phosphorylation in control cardiomyocytes, but similar to ANP, α-actin and TRPC1, this response was significantly blunted in the diabetic cardiomyocytes. However, PE had no effect on Akt phosphorylation in either control or diabetic cardiomyocytes.
Fig. 2.

Expression of phosphorylated (P) and total (T) a p38, b ERK and c Akt protein in cardiomyocytes isolated from non-diabetic control (control) and diabetic (db/db) hearts following 24 h treatment with ANG (1 μM) or PE (10 μM). Upper panels are representative immunoblots and the lower panels are mean densitometric data from three individual experiments of phosphorylated proteins normalized to their respective total protein. *P < 0.05 versus control untreated group
Inhibition of the hexosamine biosynthesis pathway restores the hypertrophic response to ANG II and PE in diabetic cardiomyocytes
Cardiomyocytes from diabetic mice were treated with azaserine (5 μM) (Champattanachai et al. 2007) or 6-diazo-5-oxo-L-norleucine (Don, 20 μM) (Kang et al. 2009) inhibitors of glutamine-fructose 6-phosphate amidotransferase (GFAT) to determine whether increased HBP flux contributed to the blunted hypertrophic response to ANG II and PE. In the absence of ANG II or PE, azaserine and Don had no effect on ANP or α-actin expression in diabetic cardiomyocytes; however, both azaserine and Don significantly increased the ANG II- and PE-induced increase in ANP and α-actin expression, to levels similar to those seen in the control group (Fig. 3).
Fig. 3.

a Immunoblots for ANP, α-actin and calsequestrin in untreated cardiomyocytes and cardiomyocytes following 24 h treatment with ANG (1 μM) or PE (10 μM). Cardiomyocytes isolated from db/db animals (d) were also treated with the GFAT inhibitors azaserine (5 μM) or Don (20 μM). Mean densitometric data for b ANP and c α-actin expression normalized to calsequestrin from three individual experiments. Data presented as mean ± SE of five individual experiments. *P < 0.05 versus db/db group induced with ANG or PE
Increased HBP and O-GlcNAc levels mimic the effects of diabetes
One of the consequences of increased HBP flux is an increase in protein O-GlcNAc levels; therefore, we assessed protein O-GlcNAc levels in control and diabetic hearts and found that they were significantly increased (Fig. 4a). In earlier studies, we found that diabetes led to increases in specific O-GlcNAc-positive protein bands (Fulop et al. 2007a, b, c); consequently, densitometric analysis was performed on five individual bands in the immunoblots. The results, normalized to calsequestrin, are summarized in Fig. 4b and demonstrate a significant increase in O-GlcNAc levels in each of the bands evaluated.
Fig. 4.

a Representative anti-O-GlcNAc immunoblots of whole heart homogenates from non-diabetic mice (control; n = 3) and three diabetic (db/db; n = 3) mice; b mean intensity of O-GlcNAc proteins determined by densitometric analysis with levels normalized to calsequestrin for bands 1–5 as indicated; c representative anti-O-GlcNAc immunoblots from cardiomyocytes treated with high glucose (25 mM; +HG), glucosamine (5 mM; +GlcN) or PUGNAc (100 μM; +PUGNAc) for 24 h; d mean intensity of O-GlcNAc proteins determined by densitometric analysis with levels normalized to calsequestrin for all bands and bands 1–5 as indicated. *P < 0.05 versus control or untreated
Cardiomyocytes from non-diabetic control mice were incubated with either high glucose (25 mM) or glucosamine (5 mM) to increase HBP flux, or 100 μM PUGNAc, an inhibitor of O-GlcNAcase which increases O-GlcNAc levels in cardiomyocytes independent of the HBP (Nagy et al. 2006). Each of these interventions led to a marked increase in O-GlcNAc levels compared to untreated cardiomyocytes (Fig. 4c). Densitometric analysis of five individual bands, as indicated, demonstrated that for four out of the five bands O-GlcNAc levels were significantly increased in each treatment group (Fig. 4d).
As high glucose, glucosamine and PUGNAc all increased O-GlcNAc levels in normal cardiomyocytes in a similar manner to that seen with diabetic cardiomyocytes, we examined the effect of each intervention on ANP and α-actin levels following ANG II and PE treatment. Increasing O-GlcNAc levels had no effect on ANP or α-actin expression in the absence of either ANG II or PE and, consistent with the data in Fig. 1, there was a robust increase in both ANP and α-actin levels in untreated normal cardiomyocytes (Fig. 5); however, treatment with high glucose, glucosamine or PUGNAc completely blocked this response (Fig. 5) in a manner similar to that seen in diabetic cardiomyocytes (Fig. 1). To confirm that the effects seen with high glucose were not due to changes in osmolarity, additional control experiments were conducted comparing the response of normal control cardiomyocytes to Ang II and PE in the presence of the osmotic control, 25 mM mannitol, or 5 or 25 mM glucose (Fig. 6). As shown in Fig. 6, mannitol-treated cells exhibited the same response as cells treated with 5 mM glucose confirming that the changes reported in Fig. 5 were a consequence of elevated glucose concentration and not a result of increased osmolarity.
Fig. 5.

a Immunoblots for ANP, α-actin and calsequestrin in control non-diabetic cardiomyocytes following 24 h treatment with ANG (1 μM) or PE (10 μM) in the presence of high glucose (25 mM; +HG), glucosamine (5 mM; +GlcN) or PUGNAc (100 μM; +PUGNAc). Mean densitometric data for b ANP and c α-actin expression normalized to calsequestrin from three individual experiments. *P < 0.05 versus 5 mM glucose
Fig. 6.

a Immunoblots for ANP, α-actin and calsequestrin in control non-diabetic cardiomyocytes following 24 h treatment with ANG (1 μM) or PE (10 μM) in the presence of normal glucose (5 mM), high glucose (25 mM; HG) or D-mannitol (25 mM). Mean densitometric data for b ANP and c α-actin expression normalized to calsequestrin from three individual experiments. *P < 0.05 versus 5 mM glucose
Effect of diabetes on p53, Bax and Bcl-2 in the heart
As cell survival pathways contribute to the regulation of cellular hypertrophy, we analyzed the expression levels of key modulators of apoptosis, p53, Bax and Bcl-2, in the hearts from control and db/db mice (Fig. 7). There was no difference in either p53 or Bax between groups; however, the anti-apoptotic protein Bcl-2 was significantly decreased in the hearts from diabetic mice (Fig. 7c).
Fig. 7.
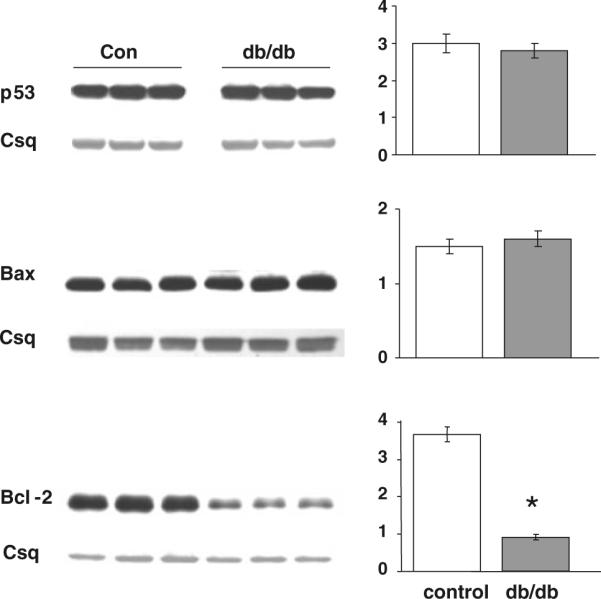
Expression levels and mean densitometric data of pro-apoptotic proteins p53 and Bax, and the anti-apoptotic protein Bcl-2 in the hearts of non-diabetic (Con) and diabetic (db/db) mouse whole heart homogenates. *P < 0.05 versus Con
Discussion
It is well established that diabetes is an important risk factor for cardiovascular disease and is associated with a significantly increased risk of heart failure independent of the increased incidence of vascular disease and other risk factors. Diabetes has also been attributed to specific defects at the level of the cardiomyocyte that could contribute to contractile dysfunction and increased risk for heart failure; however, there is little consensus regarding the specific mechanisms underlying these defects. Clinical studies have shown left ventricular hypertrophy in diabetic hearts (Devereux et al. 2000), although animal studies have not consistently demonstrated this association (Marsh et al. 2009). This has likely been complicated by the numerous animal models of diabetes that have been studied, as well as the presence or absence of concomitant pathologies, such as hypertension. For example, in untreated STZ-induced type 1 diabetic rat hearts, hypertrophic signaling is higher compared to non-diabetic hearts (Rosenkranz et al. 2003), whereas in the type 2 diabetic db/db mouse heart under basal conditions we observed no differences compared to wild-type hearts (Figs. 1, 5, 6). Despite differences in baseline indices, however, both our study and that of Rosenkranz et al. consistently demonstrated a blunted hypertrophic response of diabetic hearts to pro-hypertrophic stimuli. Taken together, these data support the notion that diabetic hearts are at increased risk of pathological hypertrophic signaling and remodeling in the presence of concomitant stress.
There is evidence that increased flux through the HBP and increased protein O-GlcNAc levels contribute to diabetic complications in a number of different tissues (Laczy et al. 2009); for example, in the heart increased O-GlcNAc levels have been associated with impaired EC coupling and slower cardiomyocyte relaxation (Hu et al. 2005; Fulop et al. 2007a, b, c). Here we show for the first time that from adult type 2 diabetic db/db mice, impaired agonist-induced hypertrophic and cell signaling responses were elicited compared to cardiomyocytes from non-diabetic animals. Inhibition of the HBP restored the sensitivity of diabetic cardiomyocytes to activation of hypertrophic signaling pathways; furthermore, we found that acute activation of the HBP and increased O-GlcNAc levels in normal cardiomyocytes blunted the hypertrophic signaling responses in a manner similar to that seen in the diabetic cardiomyocytes. Taken together, these data support the notion that activation of the HBP and the resulting increase in protein O-GlcNAcylation lead to impaired activation of hypertrophic signaling in cardiomyocytes, which may be an important contributing factor to increased incidence of heart failure associated with diabetes.
Although it has been established that diabetes increases HBP and O-GlcNAc levels in the heart (Hu et al. 2005; Fulop et al. 2007a, b, c), the impact of these changes on the response of cardiomyocytes to stimuli, such as hypertrophic agonists, had not previously been studied. Pang et al. (2002) reported that in neonatal cardiomyocytes, hyperglycemia blunted ANG II-induced cardiomyocyte hypertrophy and that HBP inhibition with azaserine partially prevented the effects of hyperglycemia. In contrast to studies in neonatal cardiomyocytes (Hunton et al. 2002; Pang et al. 2002), we observed no significant increase in cell size with either ANG II or PE (data not shown), likely due to the differences between neonatal and adult cardiomyocytes, as well as the fact that the treatment period in the earlier studies was 48 h rather than 24 h used here. Nevertheless, in control cardiomyocytes treated with ANG II or PE for 24 h, there were significantly increased levels of ANP and α-actin, both well-characterized molecular markers of cardiac hypertrophy (Fig. 1a, b). We also showed that TRPC1, another protein that has been implicated in mediating cardiomyocyte hypertrophy (Ohba et al. 2007), was also increased by both ANG II and PE (Fig. 1c). Thus, treatment of control cardiomyocytes with either ANG II or PE clearly activated hypertrophic signaling pathways. In contrast, treatment of diabetic cardiomyocytes with either agonist resulted in a complete lack of change in expression of any of these markers of cardiomyocyte hypertrophy (Fig. 1).
To determine whether diabetes modulated the response of other signaling pathways, we also examined the effects of PE treatment on phosphorylation levels of p38, ERK MAPK and Akt. We found no difference in the total level of proteins between control and diabetic groups in either untreated or PE treated conditions; however, there was a significant increase in both p38 and ERK phosphorylation in the control group following PE treatment, which was completely absent in the diabetic group (Fig. 2a, b). PE had no effect on Akt phosphorylation in either control or diabetic cardiomyocytes, which is consistent with the notion that PE more closely mimics pathophysiological hypertrophic signaling, whereas Akt activation was typically associated with physiological hypertrophy (Shiojima and Walsh 2006).
To determine whether the blunted response of diabetic cardiomyocytes to ANG II and PE was due to increased HBP flux, diabetic cardiomyocytes were treated with either azaserine or DON, both inhibitors of GFAT. We found that both GFAT inhibitors significantly increased ANP and α-actin levels following ANG II and PE treatment (Fig. 3). We also found that increasing HBP flux in normal cardiomyocytes, either by incubating with high glucose or with glucosamine, also attenuated the ANG II and PE-induced increase in ANP and α-actin (Fig. 5). One consequence of increased HBP flux is higher protein O-GlcNAc levels and, as previously reported (Champattanachai et al. 2007; Fulop et al. 2007a, b, c), we found that diabetes as well as acute treatment with high glucose or glucosamine increased O-GlcNAc levels (Fig. 4). We also found that increasing O-GlcNAc levels in normal cardiomyocytes independent of the HBP, by inhibiting O-GlcNAcase with PUGNAc, had similar effects on both high glucose and glucosamine (Fig. 5). These data provide strong support for the notion that activation of the HBP leading to increased protein O-GlcNAc levels contributes to the blunted response of diabetic cardiomyocytes to ANG II and PE.
It has been reported that hyperglycemia-induced cardiomyocyte apoptosis was associated with an increase in p53 expression, due at least in part to increased HBP flux and O-GlcNAcylation (Fiordaliso et al. 2001); however, in the diabetic heart we found no significant changes in p53 protein levels between hearts from control and diabetic hearts (Fig. 7), despite significant increases in tissue O-GlcNAc levels (Fig. 4). There was also no change in the level of the pro-apoptotic factor Bax; however, there was a greater than threefold decrease in the anti-apoptotic protein Bcl-2 in diabetic hearts compared to controls. While these data do not provide a causal link between the previously reported increase in apoptosis seen in diabetic hearts either under basal conditions or following agonist stimulation, they do suggest that decreased basal Bcl-2 levels, combined with impaired response to hypertrophic agonists, may be contributing factors to the adverse effects of diabetes on the heart.
Activation of the HBP and increased O-GlcNAc levels are often associated with the adverse effects of hyperglycemia and diabetes; however, there is also growing evidence that acute activation of these same pathways increase the tolerance to stress, including ischemia/reperfusion (Champattanachai et al. 2007; Fulop et al. 2007a, b, c; Liu et al. 2007; Jones et al. 2008; Chatham and Marchase 2010). Indeed, we have previously shown that hearts from STZ-induced diabetic animals and control hearts pretreated with glucosamine exhibited increased tolerance to Ca2+-overload stress induced by the calcium paradox protocol (Liu et al. 2006) and that the onset of diabetes in the type-2 diabetic Zucker diabetic fatty rat was associated with decreased injury following ischemia/reperfusion (Wang and Chatham 2004) as well as an increase in protein O-GlcNAcylation (Fulop et al. 2007). It may appear somewhat paradoxical to suggest that on the one hand the adverse effects of diabetes on the heart is a consequence of attenuated hypertrophic and pro-survival signaling mediated by increased HBP and O-GlcNAc levels, while also demonstrating that acute increases in O-GlcNAc result in increased tolerance to ischemia/reperfusion injury. Clearly, additional studies are required to better elucidate the mechanisms underlying the beneficial and deleterious effects of increased O-GlcNAc on the heart.
One limitation of the studies is that they were conducted in isolated cardiomyocytes; however, we have previously shown that the inotropic response to PE in the intact heart was blunted following diabetes and this could be partially restored by inhibition of HBP (Pang et al. 2004). Thus, it would seem likely that the observations seen here in isolated cardiomyocytes would be reproduced in the intact heart. While the results from this study link increased O-GlcNAc levels to the impaired hypertrophic response seen in cardiomyocytes from db/db mice, we have not demonstrated the specific mechanism(s) by which this occurs. Earlier studies in neonatal cardiomyocytes implicated a role for O-GlcNAc in attenuating the increase in intracellular Ca2+ induced by ANG II and PE and required for subsequent NFAT nuclear translocation (Hunton et al. 2002; Pang et al. 2002; Nagy et al. 2006). However, we cannot rule out the possibility that the impaired hypertrophic responses seen here are a result of decreased receptor density or receptor cycling. Indeed alpha-adrenergic receptor density is reduced in the diabetic heart (Heyliger et al. 1982; Tanaka et al. 1992). In Fig. 4, it can be seen that multiple bands in the O-GlcNAc immunoblot are increased in diabetes as well as with treatment with glucosamine and PUGNAc; however, the specific O-GlcNAcylated protein or proteins, which contribute to the attenuated response to ANG II and PE, are yet to be identified. Given that relatively large numbers of proteins are targets for O-GlcNAcylation, the use of O-GlcNAc immunopurification techniques followed by shotgun proteomics, as recently described by Teo et al. (2010), will likely be needed to identify the proteins that are involved.
While further studies are clearly warranted to better elucidate the mechanisms underlying the relationship between the HBP, O-GlcNAc and cardiac hypertrophic signaling, these data provide further support for the notion that elevated protein O-GlcNAc levels contribute to the adverse effects of diabetes on the heart. It should be noted that even in the absence of any other hemodynamic stress, diabetes leads to impaired cardiomyocyte contractility and ventricular function (Davidoff et al. 2004; Davidoff 2006; Ren et al. 2010), which has also been attributed, at least in part, to increased O-GlcNAcylation (Hu et al. 2005; Fulop et al. 2007a, b, c). The ability of the heart to respond appropriately to adverse stimuli, such as hypertension, is dependent upon the “normal” hypertrophic response. Thus, the impaired hypertrophic signaling seen here could accelerate the progression to heart failure in diabetic patients following acute MI or with concomitant hypertension. Consequently, we postulate that a diabetes-induced increase in protein O-GlcNAc levels not only contributes to contractile dysfunction, but also to impaired hypertrophic signaling, which combined with the decreased levels of the anti-apoptotic protein Bcl-2 could led to increased cardiomyocyte death, thereby contributing to the increased incidence of heart failure associated with type 2 diabetes.
Acknowledgments
This work was supported by National Institute of Health grants (HL-77100, HL-67464, HL-101192). We are grateful for the technical support provided by Charlye A. Brocks and Dan Shan.
Footnotes
Present Address: S. A. Marsh Program in Nutrition and Exercise Physiology, Washington State University, Spokane, WA, USA
References
- Boudina S, Abel ED. Diabetic cardiomyopathy revisited. Circulation. 2007;115:3213–3223. doi: 10.1161/CIRCULATIONAHA.106.679597. [DOI] [PubMed] [Google Scholar]
- Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813–820. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- Champattanachai V, Marchase RB, Chatham JC. Glucosamine protects neonatal cardiomyocytes from ischemia–reperfusion injury via increased protein-associated O-GlcNAc. Am J Physiol Cell Physiol. 2007;292:C178–C187. doi: 10.1152/ajpcell.00162.2006. [DOI] [PubMed] [Google Scholar]
- Chatham JC, Marchase RB. The role of protein O-linked beta-N-acetylglucosamine in mediating cardiac stress responses. Biochim Biophys Acta. 2010;1800:57–66. doi: 10.1016/j.bbagen.2009.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatham JC, Forder JR, McNeill JH, editors. The Heart in Diabetes. Kluwer Academic Publishers; Norwell: 1996. [Google Scholar]
- Clark RJ, McDonough PM, Swanson E, Trost SU, Suzuki M, Fukuda M, Dillmann WH. Diabetes and the accompanying hyperglycemia impairs cardiomyocyte cycling through increased nuclear O-GlcNAcylation. J Biol Chem. 2003;278:44230–44237. doi: 10.1074/jbc.M303810200. [DOI] [PubMed] [Google Scholar]
- Copeland RJ, Bullen JW, Hart GW. Cross-talk between GlcNAcylation and phosphorylation: roles in insulin resistance and glucose toxicity. Am J Physiol Endocrinol Metab. 2008;295:E17–E28. doi: 10.1152/ajpendo.90281.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidoff AJ. Convergence of glucose- and fatty acid-induced abnormal myocardial excitation–contraction coupling and insulin signalling. Clin Exp Pharmacol Physiol. 2006;33:152–158. doi: 10.1111/j.1440-1681.2006.04343.x. [DOI] [PubMed] [Google Scholar]
- Davidoff AJ, Davidson MB, Carmody MW, Davis ME, Ren J. Diabetic cardiomyocyte dysfunction and myocyte insulin resistance: role of glucose-induced PKC activity. Mol Cell Biochem. 2004;262:155–163. doi: 10.1023/b:mcbi.0000038231.68078.4b. [DOI] [PubMed] [Google Scholar]
- Devereux RB, Roman MJ, Paranicas M, O'Grady MJ, Lee ET, Welty TK, Fabsitz RR, Robbins D, Rhoades ER, Howard BV. Impact of diabetes on cardiac structure and function: the strong heart study. Circulation. 2000;101:2271–2276. doi: 10.1161/01.cir.101.19.2271. [DOI] [PubMed] [Google Scholar]
- Du XL, Edelstein D, Rossetti L, Fantus IG, Goldberg H, Ziyadeh F, Wu J, Brownlee M. Hyperglycemia-induced mitochondrial superoxide overproduction activates the hexosamine pathway and induces plasminogen activator inhibitor-1 expression by increasing Sp1 glycosylation. Proc Natl Acad Sci USA. 2000;97:12222–12226. doi: 10.1073/pnas.97.22.12222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duckworth W, Abraira C, Moritz T, Reda D, Emanuele N, Reaven PD, Zieve FJ, Marks J, Davis SN, Hayward R, Warren SR, Goldman S, McCarren M, Vitek ME, Henderson WG, Huang GD. Glucose control and vascular complications in veterans with type 2 diabetes. N Engl J Med. 2009;360:129–139. doi: 10.1056/NEJMoa0808431. [DOI] [PubMed] [Google Scholar]
- Fiordaliso F, Leri A, Cesselli D, Limana F, Safai B, Nadal-Ginard B, Anversa P, Kajstura J. Hyperglycemia activates p53 and p53-regulated genes leading to myocyte cell death. Diabetes. 2001;50:2363–2375. doi: 10.2337/diabetes.50.10.2363. [DOI] [PubMed] [Google Scholar]
- Fulop N, Marchase RB, Chatham JC. Role of protein O-linked N-acetyl-glucosamine in mediating cell function and survival in the cardiovascular system. Cardiovasc Res. 2007a;73:288–297. doi: 10.1016/j.cardiores.2006.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulop N, Mason MM, Dutta K, Wang P, Davidoff AJ, Marchase RB, Chatham JC. The impact of type-2 diabetes and aging on cardiomyocyte function and O-Linked N-acetylglucosamine levels in the heart. Am J Physiol Cell Physiol. 2007b;292:C1370–C1378. doi: 10.1152/ajpcell.00422.2006. [DOI] [PubMed] [Google Scholar]
- Fulop N, Zhang Z, Marchase RB, Chatham JC. Glucosamine cardioprotection in perfused rat heart associated with increased O-Linked N-acetylglucosamine protein modification and altered p38 activation. Am J Physiol Heart Circ Physiol. 2007c;292:H2227–H2236. doi: 10.1152/ajpheart.01091.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galderisi M, Anderson KM, Wilson PWF, Levy D. Echocardiographic evidence for the existence of a distinct diabetic cardiomyopathy (The Framingham Heart Study) Am J Cardiol. 1991;68:85–89. doi: 10.1016/0002-9149(91)90716-x. [DOI] [PubMed] [Google Scholar]
- Garcia MJ, McNamara PM, Gordon T, Kannell WB. Morbidity and mortality in diabetics in the Framingham population. Diabetes. 1974;23:105–111. doi: 10.2337/diab.23.2.105. [DOI] [PubMed] [Google Scholar]
- Gerstein HC, Miller ME, Byington RP, Goff DC, Jr, Bigger JT, Buse JB, Cushman WC, Genuth S, Ismail-Beigi F, Grimm RH, Jr, Probstfield JL, Simons-Morton DG, Friedewald WT. Effects of intensive glucose lowering in type 2 diabetes. N Engl J Med. 2008;358:2545–2559. doi: 10.1056/NEJMoa0802743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart GW, Housley MP, Slawson C. Cycling of O-linked beta-N-acetylglucosamine on nucleocytoplasmic proteins. Nature. 2007;446:1017–1022. doi: 10.1038/nature05815. [DOI] [PubMed] [Google Scholar]
- Hayat SA, Patel B, Khattar RS, Malik RA. Diabetic cardiomyopathy: mechanisms, diagnosis and treatment. Clin Sci (Lond) 2004;107:539–557. doi: 10.1042/CS20040057. [DOI] [PubMed] [Google Scholar]
- Heyliger CE, Pierce GN, Singal PK, Beamish RE, Dhalla NS. Cardiac alpha- and beta-adrenergic receptor alterations in diabetic cardiomyopathy. Basic Res Cardiol. 1982;77:610–618. doi: 10.1007/BF01908314. [DOI] [PubMed] [Google Scholar]
- Hu Y, Belke D, Suarez J, Swanson E, Clark R, Hoshijima M, Dillmann WH. Adenovirus-mediated overexpression of O-GlcNAcase improves contractile function in the diabetic heart. Circ Res. 2005;96:1006–1013. doi: 10.1161/01.RES.0000165478.06813.58. [DOI] [PubMed] [Google Scholar]
- Hunton DL, Lucchesi PA, Pang Y, Cheng X, Dell'Italia LJ, Marchase RB. Capacitative calcium entry contributes to nuclear factor of activated T-cells nuclear translocation and hypertrophy in cardiomyocytes. J Biol Chem. 2002;277:14266–14273. doi: 10.1074/jbc.M107167200. [DOI] [PubMed] [Google Scholar]
- Hunton DL, Zou LY, Pang Y, Marchase RB. Adult rat cardiomyocytes exhibit capacitative calcium entry. Am J Physiol Heart Circ Physiol. 2004;286:H1124–H1132. doi: 10.1152/ajpheart.00162.2003. [DOI] [PubMed] [Google Scholar]
- Jones SP, Zachara NE, Ngoh GA, Hill BG, Teshima Y, Bhatnagar A, Hart GW, Marban E. Cardioprotection by N-acetylglucosamine linkage to cellular proteins. Circulation. 2008;117:1172–1182. doi: 10.1161/CIRCULATIONAHA.107.730515. [DOI] [PubMed] [Google Scholar]
- Kang JG, Park SY, Ji S, Jang I, Park S, Kim HS, Kim SM, Yook JI, Park YI, Roth J, Cho JW. O-GlcNAc protein modification in cancer cells increases in response to glucose deprivation through glycogen degradation. J Biol Chem. 2009;284:34777–34784. doi: 10.1074/jbc.M109.026351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laczy B, Hill BG, Wang K, Paterson AJ, White CR, Xing D, Chen YF, Darley-Usmar V, Oparil S, Chatham JC. Protein O-GlcNAcylation: a new signaling paradigm for the cardiovascular system. Am J Physiol Heart Circ Physiol. 2009;296:H13–H28. doi: 10.1152/ajpheart.01056.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K, Paterson AJ, Chin E, Kudlow JE. Glucose stimulates protein modification by O-linked GlcNAc in pancreatic beta cells: linkage of O-linked GlcNAc to beta cell death. Proc Natl Acad Sci USA. 2000;97:2820–2825. doi: 10.1073/pnas.97.6.2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Pang Y, Chang T, Bounelis P, Chatham JC, Marchase RB. Increased hexosamine biosynthesis and protein O-GlcNAc levels associated with myocardial protection against calcium paradox and ischemia. J Mol Cell Cardiol. 2006;40:303–312. doi: 10.1016/j.yjmcc.2005.11.003. [DOI] [PubMed] [Google Scholar]
- Liu J, Marchase RB, Chatham JC. Increased O-GlcNAc levels during reperfusion lead to improved functional recovery and reduced calpain proteolysis. Am J Physiol Heart Circ Physiol. 2007;293:H1391–H1399. doi: 10.1152/ajpheart.00285.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love DC, Hanover JA. The hexosamine signaling pathway: deciphering the “O-GlcNAc code”. Sci STKE. 2005;312:re13. doi: 10.1126/stke.3122005re13. [DOI] [PubMed] [Google Scholar]
- Marsh SA, Dell'italia LJ, Chatham JC. Interaction of diet and diabetes on cardiovascular function in rats. Am J Physiol Heart Circ Physiol. 2009;296:282–H292. doi: 10.1152/ajpheart.00421.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy T, Champattanachai V, Marchase RB, Chatham JC. Glucosamine inhibits angiotensin II induced cytoplasmic Ca2+ elevation in neonatal cardiomyocytes via protein-associated O-GlcNAc. Am J Physiol Cell Physiol. 2006;290:C57–C65. doi: 10.1152/ajpcell.00263.2005. [DOI] [PubMed] [Google Scholar]
- Nissen SE, Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N Engl J Med. 2007;356:2457–2471. doi: 10.1056/NEJMoa072761. [DOI] [PubMed] [Google Scholar]
- O'Connell TD, Ni YG, Lin K-M, Han H, Yan X. Isolation and culture of adult mouse cardiac myocytes for signaling studies. AfCS Research Reports 1. 2003 online. [Google Scholar]
- Ohba T, Watanabe H, Murakami M, Takahashi Y, Iino K, Kuromitsu S, Mori Y, Ono K, Iijima T, Ito H. Upregulation of TRPC1 in the development of cardiac hypertrophy. J Mol Cell Cardiol. 2007;42:498–507. doi: 10.1016/j.yjmcc.2006.10.020. [DOI] [PubMed] [Google Scholar]
- Pang Y, Hunton DL, Bounelis P, Marchase RB. Hyperglycemia inhibits capacitative calcium entry and hypertrophy in neonatal cardiomyocytes. Diabetes. 2002;51:3461–3467. doi: 10.2337/diabetes.51.12.3461. [DOI] [PubMed] [Google Scholar]
- Pang Y, Bounelis P, Chatham JC, Marchase RB. The hexosamine pathway is responsible for the inhibition by diabetes of phenylephrine-induced inotropy. Diabetes. 2004;53:1074–1081. doi: 10.2337/diabetes.53.4.1074. [DOI] [PubMed] [Google Scholar]
- Petrova R, Yamamoto Y, Muraki K, Yonekura H, Sakurai S, Watanabe T, Li H, Takeuchi M, Makita Z, Kato I, Takasawa S, Okamoto H, Imaizumi Y, Yamamoto H. Advanced glycation endproduct-induced calcium handling impairment in mouse cardiac myocytes. J Mol Cell Cardiol. 2002;34:1425–1431. doi: 10.1006/jmcc.2002.2084. [DOI] [PubMed] [Google Scholar]
- Poornima IG, Parikh P, Shannon RP. Diabetic cardiomyopathy: the search for a unifying hypothesis. Circ Res. 2006;98:596–605. doi: 10.1161/01.RES.0000207406.94146.c2. [DOI] [PubMed] [Google Scholar]
- Preis SR, Hwang SJ, Coady S, Pencina MJ, D'Agostino RB, Sr, Savage PJ, Levy D, Fox CS. Trends in all-cause and cardiovascular disease mortality among women and men with and without diabetes mellitus in the Framingham Heart Study, 1950 to 2005. Circulation. 2009;119:1728–1735. doi: 10.1161/CIRCULATIONAHA.108.829176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren J, Gintant GA, Miller RE, Davidoff AJ. High extracellular glucose impairs cardiac E-C coupling in a glycosylation-dependent manner. Am J Physiol Heart Circ Physiol. 1997;273:H2876–H2883. doi: 10.1152/ajpheart.1997.273.6.H2876. [DOI] [PubMed] [Google Scholar]
- Ren J, Dong F, Cai GJ, Zhao P, Nunn JM, Wold LE, Pei J. Interaction between age and obesity on cardiomyocyte contractile function: role of leptin and stress signaling. PLoS One. 2010;5:e10085. doi: 10.1371/journal.pone.0010085. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Rodrigues B, McNeill JH. The diabetic heart: metabolic causes for the development of a cardiomyopathy. Cardiovasc Res. 1992;26:913–922. doi: 10.1093/cvr/26.10.913. [DOI] [PubMed] [Google Scholar]
- Rosenkranz AC, Hood SG, Woods RL, Dusting GJ, Ritchie RH. B-type natriuretic peptide prevents acute hypertrophic responses in the diabetic rat heart: importance of cyclic GMP. Diabetes. 2003;52:2389–2395. doi: 10.2337/diabetes.52.9.2389. [DOI] [PubMed] [Google Scholar]
- Shan D, Marchase RB, Chatham JC. Overexpression of TRPC3 increases apoptosis but not necrosis in response to ischemia–reperfusion in adult mouse cardiomyocytes. Am J Physiol Cell Physiol. 2008;294:C833–C841. doi: 10.1152/ajpcell.00313.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiojima I, Walsh K. Regulation of cardiac growth and coronary angiogenesis by the Akt/PKB signaling pathway. Genes Dev. 2006;20:3347–3365. doi: 10.1101/gad.1492806. [DOI] [PubMed] [Google Scholar]
- Slawson C, Housley MP, Hart GW. O-GlcNAc cycling: how a single sugar post-translational modification is changing the way we think about signaling networks. J Cell Biochem. 2006;97:71–83. doi: 10.1002/jcb.20676. [DOI] [PubMed] [Google Scholar]
- Tanaka Y, Kashiwagi A, Saeki Y, Shigeta Y. Abnormalities in cardiac alpha 1-adrenoceptor and its signal transduction in streptozocin-induced diabetic rats. Am J Physiol. 1992;263:E425–E429. doi: 10.1152/ajpendo.1992.263.3.E425. [DOI] [PubMed] [Google Scholar]
- Taylor RP, Parker GJ, Hazel MW, Soesanto Y, Fuller W, Yazzie MJ, McClain DA. Glucose deprivation stimulates O-GlcNAc modification of proteins through up-regulation of O-linked N-acetylglucosaminyltransferase. J Biol Chem. 2008;283:6050–6057. doi: 10.1074/jbc.M707328200. [DOI] [PubMed] [Google Scholar]
- Teo CF, Ingale S, Wolfert MA, Elsayed GA, Not LG, Chatham JC, Wells L, Boons GJ. Glycopeptide-specific monoclonal antibodies suggest new roles for O-GlcNAc. Nat Chem Biol. 2010;6:338–343. doi: 10.1038/nchembio.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Chatham JC. The onset of diabetes in Zucker diabetic fatty (ZDF) rats leads to improved recovery of function following ischemia in the isolated perfused heart. Am J Physiol Endocrin Met. 2004;286:E725–E736. doi: 10.1152/ajpendo.00295.2003. [DOI] [PubMed] [Google Scholar]