

Abstract
In our research on the role of apoptosis in the pathogenesis of the autoimmune disease systemic lupus erythematosus (SLE), we aim to evaluate the effects of early and late apoptotic cells and blebs on antigen presenting cells. This requires the in vitro generation of sufficiently large and homogeneous populations of early and late apoptotic cells. Here, we present a quick method encountered by serendipity that results in highly reproducible synchronized homogeneous apoptotic cell populations. In brief, granulocytic 32Dcl3 cells are incubated on ice for 2 h and subsequently rewarmed at 37°C. After 30–90 min at 37°C more than 80–90% of the cells become early apoptotic (Annexin V positive/propidium iodide negative). After 24 h of rewarming at 37°C 98% of the cells were late apoptotic (secondary necrotic; Annexin V positive/propidium iodide positive). Cells already formed apoptotic blebs at their cell surface after approximately 20 min at 37°C. Inter-nucleosomal chromatin cleavage and caspase activation were other characteristics of this cold-shock-induced process of apoptosis. Consequently, apoptosis could be inhibited by a caspase inhibitor. Finally, SLE-derived anti-chromatin autoantibodies showed a high affinity for apoptotic blebs generated by cold-shock. Overall, cold-shock induced apoptosis is achieved without the addition of toxic compounds or antibodies, and quickly leads to synchronized homogeneous apoptotic cell populations, which can be applied for various research questions addressing apoptosis.
Keywords: Apoptosis, Cold-shock, Autoantibodies, Apoptotic blebs, Lupus
Introduction
Apoptosis or programmed cell death is involved in tissue development and maintenance, which includes shaping of the immune system, and in cellular responses to stress and DNA damage. Several disorders are related to an aberrant apoptotic process. Roughly, too much (unwanted) apoptosis leads to cell loss disorders, like diabetes type 1, while too little apoptosis leads to cell proliferation disorders, like cancer [1–3]. So, many research fields address the role of apoptosis. In our research, we address the role of apoptosis in the development of the anti-chromatin autoimmune response in systemic lupus erythematosus (SLE) as will be briefly outlined below.
The hallmark of SLE is the presence of anti-nuclear antibodies to double stranded DNA, nucleosomes, histones and other DNA/RNA-binding proteins [4]. In patients with SLE, apoptotic cells and chromatin can be detected in the circulation and in several tissues, which may be explained by an aberrant apoptosis and/or reduced clearance [5–9]. During apoptosis autoantigens can be modified by several biochemical processes and they cluster in blebs formed at the apoptotic cell surface. Apoptotic blebs containing modified autoantigens can segregate and their contents can be released [10–12]. This released apoptotic material can be ingested by professional antigen presenting cells, the dendritic cells (DC), which then leads to DC maturation and presentation of modified autoantigens in an immunogenic way to autoreactive T cells [13, 14]. Activated T cells will stimulate (autoreactive) B cells producing anti-nuclear autoantibodies that form immune complexes with released apoptotic chromatin. In addition, autoreactive B cells can be rescued from deletion by accumulation and continuous presentation of apoptotic material on the surface of follicular dendritic cells in germinal centers, as well as by TLR-9 mediated activation by DNA and HMGB-1 [6, 15–17]. Deposition of the formed immune complexes in basement membranes leads to serious inflammation, like lupus nephritis. So, apoptosis can play two key roles in the pathogenesis of SLE. First, due to an aberrant apoptosis and/or insufficient removal of apoptotic cells, the immune system in SLE is activated by released (modified) apoptotic debris. Second, aberrant apoptosis may lead to the persistence of autoreactive T and B cells in SLE [6, 12, 18].
In our research we address the uptake by antigen presenting cells, i.e. dendritic cells, of early and late apoptotic blebs and cell bodies. Late and early apoptotic cell-derived blebs and cell bodies could differ in the amount and nature of apoptosis induced modifications [13, 14]. Early apoptotic cells are defined as annexin V (AnV) positive and propidium iodide (PI) negative, while late apoptotic or secondary necrotic cells are AnV and PI double positive. The generation of homogeneous preparations of early and late apoptotic cells is not straightforward. Apoptosis can be initiated in several ways, for instance by ligands binding to specific death receptors, like the TNF receptor family, by cellular stress through DNA damage, or by deprivation of growth factors or nutrients [19, 20]. The majority of experimental approaches to induce apoptosis lead to a mixture of cells in different stages of apoptosis. Triggering of the Fas–FasL pathway by incubation with anti-Fas antibodies is most likely one of the most well known methods to obtain a synchronized apoptotic cell population.
In our quest to prepare homogeneous preparations of early and late apoptotic cells, we found by serendipity that applying cold-shock at 0°C for 2 h followed by a re-warming at 37°C yielded highly homogeneous synchronized apoptotic cell suspensions, that served as a source for early and late apoptotic blebs and cell bodies. This method can easily be applied to address various research questions related to apoptosis.
Materials and methods
Cell culture and induction of apoptosis by cold-shock
Murine 32D clone 3 cells (32Dcl3), Jurkat cells and WEHI-3B cells were cultured in RPMI-1640 DM (Invitrogen Life Technologies, Breda, The Netherlands) with 10% fetal calf serum (FCS; Greiner Bio One, Alphen aan de Rijn, The Netherlands), 1% pyruvate, 1% glutamax, 1% penicillin/streptomycin (all from Invitrogen Life Technologies), and in the case of 32Dcl3 cells supplemented with 15% WEHI-3B-conditioned medium (DSMZ, Braunschweig, Germany) [21]. Cells were propagated every 2–3 days at 0.1×106 cells/ml. Apoptosis was induced by incubation of 1×106 cells/ml on ice for 2 h, unless indicated otherwise, followed by rewarming at 37°C in an incubator with 5% CO2 for periods of 30 min up to 24 h.
Flow cytometry
Apoptosis was determined by staining cells with Annexin V-FITC (AnV) and PI (Biovision, ITK Diagnostics, Uithoorn, The Netherlands) according to the manufacturer’s protocol and analyzed by flow cytometry (FACS-Calibur, BD, Alphen aan de Rijn, The Netherlands). Poly-caspase cleavage was examined by FLICA (Fluorochrome Inhibitor of Caspases) using the Vybrant FAM poly-caspases assay kit (Molecular Probes, Invitrogen), following the instructions of the manufacturer. To analyze whether cold-shock-induced apoptosis was mediated by caspases, cells were prior to cold-shock incubated with 100 μM Z-VAD-FMK (BD), which is a caspase inhibitor, and subsequently stained with labeled AnV and PI.
DNA isolation and agarose gel electrophoresis
DNA was isolated from apoptotic and control cells following standard procedures. Briefly, cells were pelleted and subsequently lyzed in 25 mM Tris/HCL (pH 8.0), 10 mM EDTA, 50 mM glucose, and incubated for 5 min at room temperature. DNA was extracted using Tris-saturated phenol:chloroform:isoamylalcohol (25:24:1) followed by chloroform:isoamylalcohol extraction, and subsequently precipitated with 100% ethanol and centrifuged for 5 min at 12,000×g. The DNA pellet was dissolved in DNase-free milliQ water. Samples were separated by electrophoresis on a 2% agarose TBE gel containing 0.5 μg/ml ethidium bromide. DNA was visualized using a UV transilluminator and photographed.
Autoantibody binding to cold-shock induced apoptotic cells
Binding of two SLE-derived monoclonal autoantibodies, KM-2 and #32, specific for acetylated histone H4 and nucleosomes, respectively, and SLE-derived plasma’s, to cold-shock induced apoptotic 32Dcl3 cells was determined [11, 22]. Briefly, cells were cytospinned for 5 min at 500×g and fixed with 2% paraformaldehyde, and permeabilized with 0.5% Triton X-100. Subsequently, cells were incubated with the indicated primary antibodies and an appropriate Alexa-488 conjugated secondary antibody (Molecular Probes, Invitrogen) followed by a DAPI staining to visualize DNA, according to the manufacturer’s instructions. Preparations were analyzed by fluorescent microscopy (Leica DM4000 B, Leica Lasertechnik GmbH, Heidelberg, Germany).
Results
Cold-shock induced apoptosis
As outlined, our research requires a method of apoptosis induction leading to synchronized populations of early and late apoptotic cells, preferably without the need for addition of antibodies or toxic compounds. We found that incubation of the granulocytic 32Dcl3 cells on ice followed by rewarming at 37°C, led to morphological changes, which started with shrinkage of cells, followed by loss of membrane integrity and the pronounced formation of characteristic apoptotic blebs that segregated from the remaining cell bodies at a later stage (Fig. 1a and 1b). Notably, these cellular changes occurred almost simultaneously in all cells. None of these morphological changes appeared when the cells were kept on ice, and they only developed when the cells were rewarmed at 37°C. Although 5 min on ice already led to apoptosis in some cells after rewarming at 37°C, a period between 1 and 2 h on ice induced apoptosis in virtual all cells. Notably, the formation of blebs at the cell surface started around 20 min after rewarming and lasted for about 60 min. The disintegrating cells and the segregating blebs stained positively with labeled AnV, which specifically binds to the re-oriented phospholipid phosphatidylserine (PS) that is a normal feature of early apoptosis (Fig. 1c).
Fig. 1.
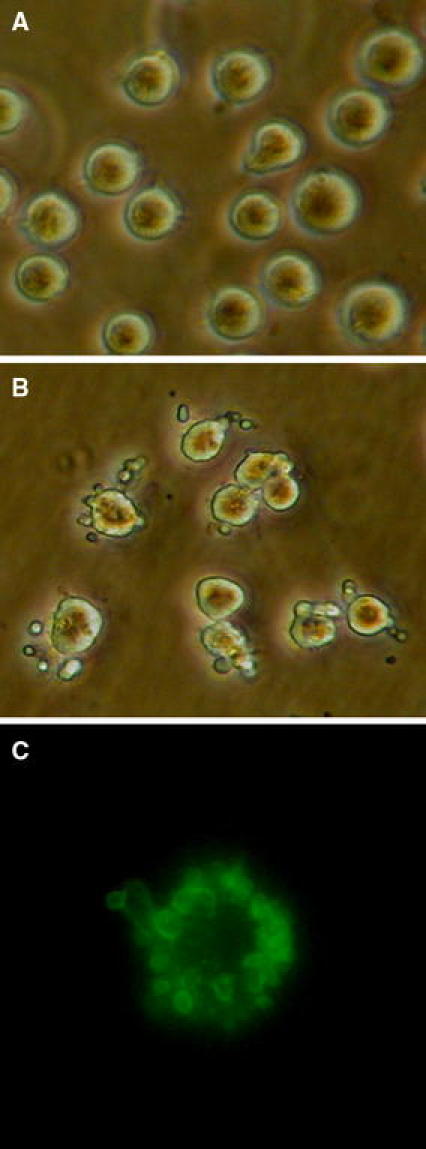
Morphological changes in 32Dcl3 cells after cold-shock induced apoptosis. a Representative picture of control 32Dcl3 cells. b Representative picture of 32Dcl3 cells exposed to cold-shock by incubation for 2 h at 0°C followed by rewarming at 37°C for 2 h. Note the formation of typical apoptotic blebs at the cell surface. c Staining of apoptotic blebs with FITC-labeled Annexin V (green) indicating the reorientation of the phospholipid phosphatidylserine in apoptotic membranes. Magnification ×40 (Color figure online)
We examined the course of apoptosis during the rewarming period following the cold-shock on ice by determining the staining with labeled AnV and propidium iodide (PI). As mentioned, AnV staining already occurs in an early stage of apoptosis, while the DNA intercalating compound PI only can enter late apoptotic cells. As depicted in Fig. 2a, at the start of the rewarming period nearly all cells were AnV−/PI−. After 30 min of rewarming at 37°C, already 82% of the cells were AnV+/PI− (Fig. 2b), which increased to 92% after 90 min (Fig. 2c). Since the cells at this stage remained PI-negative, they can be considered as early apoptotic cells, indicating a highly synchronized induction and rate of apoptosis. After 5 h the number of late apoptotic cells (AnV+/PI+) increased to 37% (Fig. 2d), and after 24 h, nearly all cells were AnV+/PI+ indicating they completely proceeded to the late apoptotic stage (Fig. 2e). These results were highly reproducible as revealed by the small standard deviations for four separate experiments (Fig. 2f). It appeared that cold-shock-induced apoptosis also occurred in other cell lines like Jurkat and WEHI3B cells (Fig. 3). However, compared to 32Dcl3 cells (Fig. 2f), cold-shock induced apoptosis only in part of the cells, in both Jurkat and WEHI-3B cells, resulting in less homogenous apoptotic cell populations.
Fig. 2.
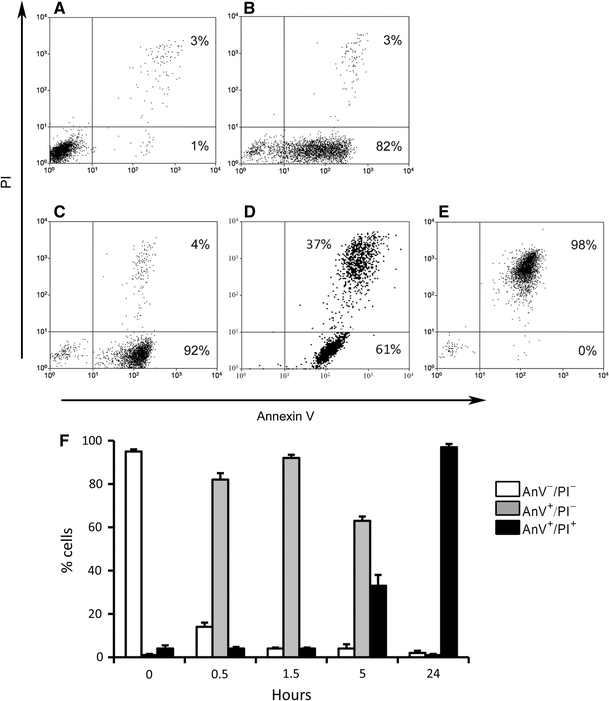
Kinetics of cold-shock induced apoptosis. Apoptosis induced by cold-shock in 32Dcl3 cells examined in flow cytometry by simultaneous staining for labeled annexin V (AnV) and propidium iodide (PI). a Prior to cold-shock most cells are not apoptotic. b 30 min after rewarming a high percentage of cells is early apoptotic. c After 90 min an even higher percentage of cells is early apoptotic. d After 5 h the percentage of late apoptotic cells increases e 24 h after cold-shock and rewarming most cells are late apoptotic. The average (±SD) of four experiments is depicted in panel f X-axis; hours of rewarming
Fig. 3.
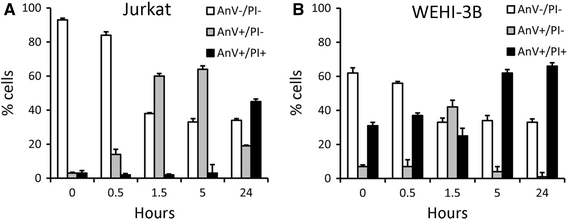
Cold-shock induced apoptosis in Jurkat and WEHI-3B cells. Apoptosis induced by cold-shock in Jurkat cells (a) and WEHI-3B cells (b) cells examined in flow cytometry by simultaneous staining for labeled annexin V (AnV) and propidium iodide (PI). Compared to 32Dcl3 cells (Fig. 2f), cold-shock induces apoptosis only in subset of the cells, in both Jurkat and WEHI-3B cells, resulting in less homogenous and synchronized apoptotic cell populations. The average (±SD) of three experiments is depicted. X-axis; hours of rewarming
Cold-shock induced apoptosis is mediated by caspase and associated with chromatin fragmentation
Activation of caspases (cystein-aspartic-acid-proteases) and subsequent signaling cascades is frequently involved in the induction and execution phases of the apoptotic process. Therefore, we determined the activation of caspases in cold-shock-induced apoptosis using the FLICA (Fluorochrome Inhibitor of Caspases) method, in which the fluorochrome binds covalently to the cleaved intracellular caspases. 1 h after start of the rewarming period caspase cleavage could be detected in about 75% of the cells (Fig. 4a), which shows that cold-shock induced apoptosis is caspase dependent. Indeed, the addition of the caspase inhibitor Z-VAD-FMK completely inhibited the cold-shock induced apoptosis as analyzed by AnV staining 2 h after start of the rewarming (Fig. 4b). Also after 4 h and 24 h of rewarming cold-shock induced apoptosis was still completely inhibited (Fig. 4c).
Fig. 4.
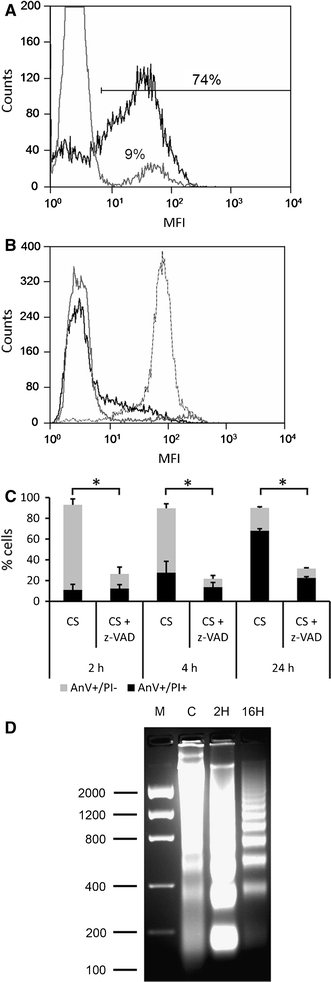
Cold-shock activates caspase and induces chromatin fragmentation. a FLICA staining: activation of caspases was analyzed by flow cytometry using the FLICA method, in which the fluorochrome binds covalently to the cleaved intracellular caspases. Cells were either kept on ice (gray line) or rewarmed to 37°C (bold black line) for 2 h. Y-axis cell count. b Annexin V staining: the caspase inhibitor Z-VAD-FMK was added 30 min prior to cold-shock (solid black line). DMSO as Z-VAD-FMK solvent control was added also 30 min prior to cold-shock yielding apoptotic cells (dotted gray line). Untreated cells kept on ice were included as a control (solid gray line). Y-axis cell count. c Inhibition of cold-shock induced apoptosis by Z-VAD-FMK (Z-VAD). Staining for annexin V (AnV) and propidium iodide was measured 2 h (2 h), 4 h (4 h) and 24 h (24 h) after start of the rewarming period following cold-shock. Gray bars, AnV+/PI−; Black bars AnV+/PI+. The average (±SD) of three experiments is depicted. *P < 0.05 (Student’s t-test). d DNA was isolated from control culture (C) and apoptotic cells 2 h (2H) and 16 h (16H) after start of the rewarming period following cold-shock. DNA laddering was observed after 2 and 16 h, indicating inter-nucleosomal chromatin cleavage. Marker (M) expressed as base pairs
In addition to apoptotic blebbing, re-orientation of PS, and caspase activity, one of the hallmarks of apoptosis is the inter-nucleosomal cleavage of chromatin by endonucleases, such as caspase-activated DNase (CAD) or endonuclease G (Endo G) [23]. We analyzed the DNA extracted from several 32Dcl3 cultures before and after cold-shock induced apoptosis, which revealed the characteristic banding pattern (“DNA laddering”) that represents multitudes of the mononucleosomal DNA size of approximately 180 base pairs (Fig. 4d). Cold-shock induced apoptosis resulted already in a marked chromatin fragmentation within 2 h after the start of the rewarming period. Notably, chromatin fragmentation seemed to increase further in time, whereas later on the mononucleosomal band disappeared from the late apoptotic cells suggesting leakage into the culture supernatant at this stage.
Binding of autoantibodies to cold-shock induced apoptotic cells and blebs
As outlined, nuclear autoantigens may be modified during apoptosis and lead to maturation of dendritic cells in SLE [6, 11, 12]. To evaluate whether apoptotic cell bodies and blebs generated by cold-shock-induced turbo apoptosis contain autoantigens targeted in SLE, we used SLE-derived autoantibodies as probes. 1 h after the start of the rewarming period the lupus mouse-derived autoantibody #32, which is directed against nucleosomes, specifically stained cold-shock-induced apoptotic cells and blebs (Fig. 5a). The staining by antibody #32 co-localized with the DNA staining by DAPI, in particular in the regions that stained less intense for DAPI, indicating a more open chromatin structure. Similar results were obtained with the lupus-derived monoclonal autoantibody KM-2 recognizing a specific acetylation pattern on histone H4 at lysine residues 8, 12 and 16 (Fig. 5b). We have shown recently that this specific acetylation pattern on histone H4 was associated with apoptosis using other methods of apoptosis induction [11]. Finally, we showed that plasma’s of 20-weeks-old diseased lupus (MRL/lpr) mice also stained cold-shock induced apoptotic blebs (Fig. 5c). Taken together, these data show that nuclear material is translocated to apoptotic blebs and modified during cold-shock induced apoptosis.
Fig. 5.
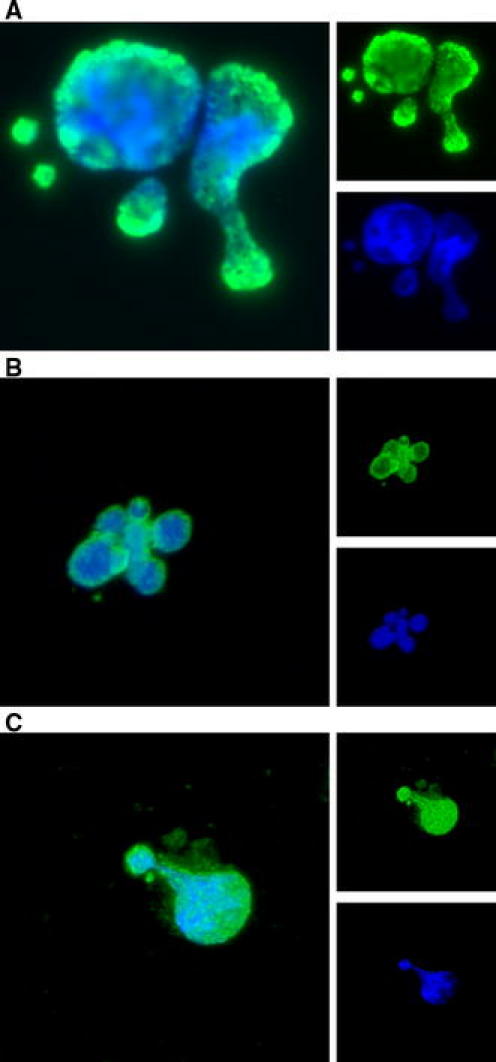
SLE-derived autoantibodies bind to cold-shock induced apoptotic cells and blebs. Cold-shock apoptotic cells and blebs were probed with SLE-derived autoantibodies (small upper-right panels; green) and DNA was stained with DAPI (small lower-right panels; blue). a Binding of the SLE-derived nucleosome-specific monoclonal autoantibody #32. b Binding of the SLE-derived monoclonal autoantibody KM-2 (recognizing the apoptosis-associated acetylation of histone H4 at K8, 12, 16). c Binding of plasma of diseased, 20-weeks-old, SLE (MRL/lpr) mice. Merge is indicated in the large left panels. Magnification ×40 (Color figure online)
Discussion
We describe an experimental system that was discovered by serendipity, which is characterized by a very fast, synchronized and complete induction of apoptosis. After storing the granulocyte 32Dcl3 cells on ice for a brief period and returning them at 37°C, we observed excessive cellular blebbing, which hinted to apoptosis. Further characterization indeed confirmed the occurrence of apoptosis. Reorientation of the phospholipid PS was demonstrated by binding of AnV. Notably, about 90 min after the start of the rewarming period about 90% of the cells stained positive for Annexin V, whereas the cells were still impermeable for propidium iodide. Other hallmarks characteristic for apoptosis included chromatin fragmentation and activation of caspases. Notably, cold-shock-induced apoptosis could be completely inhibited by a caspase inhibitor. Finally, we could demonstrate binding of lupus-derived autoantibodies to apoptotic blebs of cold-shock-induced apoptotic cells. Notably, one of the tested autoantibodies, KM-2, recognizes an apoptosis-associated acetylation motif on histone H4, as we previously showed for cells made apoptotic with anti-Fas or camptothecin [11]. This suggests that apoptosis induced autoantigen modification by cold-shock resembles those obtained by other apoptosis-inducing methods.
To our knowledge this fast and synchronized generation of such a homogeneous population of early apoptotic cells, without the addition of antibodies or any other agents, has not been described before. Moreover, our approach allows the generation and separation of early and late apoptotic cells and blebs. Recently, we have used this method to compare effects of early and late apoptotic blebs and cell bodies on immune cells [13]. Previously, cold-shock and apoptosis has been studied in other cell types [24–29]. There are a number of important differences compared to our current work: (i) cold-shock-induced apoptosis was not as fast, massive (complete), and synchronized; (ii) the apoptotic process was not always formally proven by morphological and biochemical analysis; (iii) the effect of rewarming was evaluated only incidentally. Our procedure, including the 32Dcl3 cell line we use, may explain why our experimental system to induce apoptosis is so fast and synchronized. Notably, other cell types, such as Jurkat and WEHI-3B, which we subjected to cold-shock and rewarming, also went into apoptosis, but yielded less homogenous apoptotic cell populations. In addition to cold-shock at 0–4°C, hypothermic conditions, that is a modest lowering of the temperature to 25–35°C, also may lead to cellular stress and apoptosis [30]. To our knowledge there seems no clear physiological relevance of apoptosis induction by cold-shock or hypothermia. Cold-shock-induced apoptosis may be relevant for tissues and organs that are stored on ice prior to transplantation, in particular when the initial protection by cold is counteracted by apoptosis-mediated tissue damage through rewarming after transplantation [31]. Indeed, apoptosis is present after ischemia followed by reperfusion in clinical organ transplantation. However, the contribution of cold storage and rewarming of the organ to the apoptotic process is unclear, since apoptosis has also been demonstrated after normothermic ischemia [32].
The molecular mechanisms underlying and regulating cold-shock-induced apoptosis are poorly understood. Conditions of cold induce the expression of certain cold-shock proteins [30, 33–35]. Cold-shock proteins may stabilize other proteins or specific RNA transcripts, or may act as specific transcription factor for other cold-shock responsive genes. Therefore, cold-shock induced apoptosis may be the result of an insufficient protective capacity mediated by cold-shock proteins. Several studies have revealed that reactive oxygen species (ROS) play a role in apoptosis induced by conditions of cold and that apoptosis could be prevented by antioxidants [28, 30, 36, 37]. With the involvement of ROS, cold-shock-induced apoptosis enters the more established pathways recognized in apoptosis [20, 37].
In conclusion, we have described an efficient and fast method to induce synchronized apoptosis that allows the generation of homogeneous preparations of early and late apoptotic cells. This method can be applied as a powerful tool to evaluate the specific effects of early versus late apoptotic cells on other cells, such as immune cells. Research questions addressing cellular and molecular processes related to apoptosis also may benefit from the method we describe here.
Acknowledgments
The author thank Drs. J. Greenberger (University of Pittsburgh School of Medicine, Pittsburgh, USA) and S. Baker (Children’s Hospital of Philadelphia, Philadelphia, USA) for providing the 32Dcl3 cells. This work was supported by the Ph.D. program of the Radboud University Nijmegen Medical Center, the Dutch Kidney Foundation (grant C05.2119) and the Dutch Arthritis Association (grant 09-1-308).
Conflict of interest
The authors declare that they have no conflict of interest.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Footnotes
J. H. Fransen and J. W. Dieker contributed equally to this work.
References
- 1.Edinger AL, Thompson CB. Death by design: apoptosis, necrosis and autophagy. Curr Opin Cell Biol. 2004;16:663–669. doi: 10.1016/j.ceb.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 2.Hengartner MO. The biochemistry of apoptosis. Nature. 2000;407:770–776. doi: 10.1038/35037710. [DOI] [PubMed] [Google Scholar]
- 3.Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26:239–257. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cervera R, Vinas O, Ramos-Casals M, Garcia-Carrasco M, Siso A, Machuca Y, Ramirez F, Font J, Vives J, Ingelmo M, Burlingame RW. Antichromatin antibodies in systemic lupus erythematosus: a useful marker for lupus nephropathy. Arthritis Rheum. 2001;44:S247. doi: 10.1002/1529-0131(200102)44:2<247::AID-ANR40>3.0.CO;2-U. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dieker JWC, van der Vlag J, Berden JHM. Deranged removal of apoptotic cells: its role in the genesis of lupus. Nephrol Dial Transplant. 2004;19:282–285. doi: 10.1093/ndt/gfg485. [DOI] [PubMed] [Google Scholar]
- 6.Munoz LE, van Bavel C, Franz S, Berden J, Herrmann M, van der Vlag J. Apoptosis in the pathogenesis of systemic lupus erythematosus. Lupus. 2008;17:371–375. doi: 10.1177/0961203308089990. [DOI] [PubMed] [Google Scholar]
- 7.van Bavel CC, Fenton KA, Rekvig OP, van der Vlag J, Berden JH. Glomerular targets of nephritogenic autoantibodies in systemic lupus erythematosus. Arthritis Rheum. 2008;58:1892–1899. doi: 10.1002/art.23626. [DOI] [PubMed] [Google Scholar]
- 8.Herrmann M, Voll RE, Zoller OM, Hagenhofer M, Ponner BB, Kalden JR. Impaired phagocytosis of apoptotic cell material by monocyte-derived macrophages from patients with systemic lupus erythematosus. Arthritis Rheum. 1998;41:1241–1250. doi: 10.1002/1529-0131(199807)41:7<1241::AID-ART15>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 9.Licht R, Dieker JWC, Jacobs CWM, Tax WJM, Berden JHM. Decreased phagocytosis of apoptotic cells in diseased SLE mice. J Autoimmun. 2004;22:139–145. doi: 10.1016/j.jaut.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 10.Dieker J, Cisterna B, Monneaux F, Decossas M, van der Vlag J, Biggiogera M, Muller S. Apoptosis-linked changes in the phosphorylation status and subcellular localization of the spliceosomal autoantigen U1–70 K. Cell Death Differ. 2008;15:793–804. doi: 10.1038/sj.cdd.4402312. [DOI] [PubMed] [Google Scholar]
- 11.Dieker JW, Fransen JH, van Bavel CC, Briand JP, Jacobs CW, Muller S, Berden JH, van der Vlag J. Apoptosis induced acetylation of histones is pathogenic in systemic lupus erythematosus. Arthritis Rheum. 2007;56:1921–1933. doi: 10.1002/art.22646. [DOI] [PubMed] [Google Scholar]
- 12.Rosen A, Casciola-Rosen L. Autoantigens in systemic autoimmunity: critical partner in pathogenesis. J Intern Med. 2009;265:625–631. doi: 10.1111/j.1365-2796.2009.02102.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fransen JH, Hilbrands LB, Jacobs CW, Adema GJ, Berden JH, van der Vlag J. Both early and late apoptotic blebs are taken up by DC and induce IL-6 production. Autoimmun. 2009;42:325–327. doi: 10.1080/08916930902828049. [DOI] [PubMed] [Google Scholar]
- 14.Fransen JH, Hilbrands LB, Ruben J, Stoffels M, Adema GJ, van der Vlag J, Berden JH. Mouse dendritic cells matured by ingestion of apoptotic blebs induce T cells to produce interleukin-17. Arthritis Rheum. 2009;60:2304–2313. doi: 10.1002/art.24719. [DOI] [PubMed] [Google Scholar]
- 15.Baumann I, Kolowos W, Voll RE, Manger B, Gaipl U, Neuhuber WL, Kirchner T, Kalden JR, Herrmann M. Impaired uptake of apoptotic cells into tingible body macrophages in germinal centers of patients with systemic lupus erythematosus. Arthritis Rheum. 2002;46:191–201. doi: 10.1002/1529-0131(200201)46:1<191::AID-ART10027>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 16.Tian J, Avalos AM, Mao SY, Chen B, Senthil K, Wu H, Parroche P, Drabic S, Golenbock D, Sirois C, Hua J, An LL, Audoly L, La Rosa G, Bierhaus A, Naworth P, Marshak-Rothstein A, Crow MK, Fitzgerald KA, Latz E, Kiener PA, Coyle AJ. Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nature Immunol. 2007;8:487–496. doi: 10.1038/ni1457. [DOI] [PubMed] [Google Scholar]
- 17.Viglianti GA, Lau CM, Hanley TM, Miko BA, Shlomchik MJ, Rothstein AM. Activation of autoreactive B cells by CpG dsDNA. Immun. 2003;19:837–847. doi: 10.1016/S1074-7613(03)00323-6. [DOI] [PubMed] [Google Scholar]
- 18.Fransen JH, Hilbrands LB, Koeter CM, Berden JH, van der Vlag J. The role of apoptosis and removal of apoptotic cells in the genesis of systemic lupus erythematosus. Arch of Med Sci. 2009;5(3A):S466–S477. [Google Scholar]
- 19.Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Science. 1998;281:1305–1308. doi: 10.1126/science.281.5381.1305. [DOI] [PubMed] [Google Scholar]
- 20.Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281:1309–1312. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- 21.Greenberger JS, Sakakeeny MA, Humphries RK, Eaves CJ, Eckner RJ. Demonstration of permanent factor-dependent multipotential (erythroid-neutrophil-basophil) hematopoietic progenitor-cell lines. Proc Natl Acad Sci USA. 1983;80:2931–2935. doi: 10.1073/pnas.80.10.2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dieker JW, Sun YJ, Jacobs CW, Putterman C, Monestier M, Muller S, van der Vlag J, Berden JH. Mimotopes for lupus-derived anti-DNA and nucleosome-specific autoantibodies selected from random peptide phage display libraries: facts and follies. J Immunol Methods. 2005;296:83–93. doi: 10.1016/j.jim.2004.10.010. [DOI] [PubMed] [Google Scholar]
- 23.Widlak P, Garrard WT. Discovery, regulation, and action of the major apoptotic nucleases DFF40/CAD and endonuclease G. J Cell Biochem. 2005;94:1078–1087. doi: 10.1002/jcb.20409. [DOI] [PubMed] [Google Scholar]
- 24.Gregory CD, Milner AE. Regulation of cell-survival in burkitt-lymphoma—implications from studies of apoptosis following cold-shock treatment. Int J Cancer. 1994;57:419–426. doi: 10.1002/ijc.2910570321. [DOI] [PubMed] [Google Scholar]
- 25.Kruman II, Gukovskaya AS, Petrunyaka VV, Beletsky IP, Trepakova ES. Apoptosis of murine Bw 5147 thymoma cells induced by cold shock. J Cell Physiol. 1992;153:112–117. doi: 10.1002/jcp.1041530115. [DOI] [PubMed] [Google Scholar]
- 26.Nagle WA, Soloff BL, Moss AJ, Henle KJ. Cultured chinese hamster-cells undergo apoptosis after exposure to cold but nonfreezing temperatures. Cryobiology. 1990;27:439–451. doi: 10.1016/0011-2240(90)90021-U. [DOI] [PubMed] [Google Scholar]
- 27.Perotti M, Toddei F, Mirabelli F, Vairetti M, Bellomo G, Mcconkey DJ, Orrenius S. Calcium-dependent dna fragmentation in human synovial-cells exposed to cold shock. FEBS Letters. 1990;259:331–334. doi: 10.1016/0014-5793(90)80040-P. [DOI] [PubMed] [Google Scholar]
- 28.Rauen U, Polzar B, Stephan H, Mannherz HG, de Groot H. Cold-induced apoptosis in cultured hepatocytes and liver endothelial cells: mediation by reactive oxygen species. FASEB J. 1999;13:155–168. doi: 10.1096/fasebj.13.1.155. [DOI] [PubMed] [Google Scholar]
- 29.Soloff BL, Nagle WA, Moss AJ, Henle KJ, Crawford JT. Apoptosis induced by cold shock invitro is dependent on cell-growth phase. Biochem Biophys Res Commun. 1987;145:876–883. doi: 10.1016/0006-291X(87)91046-1. [DOI] [PubMed] [Google Scholar]
- 30.Al-Fageeh MB, Smales CM. Control and regulation of the cellular responses to cold shock: the responses in yeast and mammalian systems. Biochem J. 2006;397:247–259. doi: 10.1042/BJ20060166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Burns AT, Davies DR, McLaren AJ, Cerundolo L, Morris PJ, Fuggle SV. Apoptosis in ischemia/reperfusion injury of human renal allografts. Transplantation. 1998;66:872–876. doi: 10.1097/00007890-199810150-00010. [DOI] [PubMed] [Google Scholar]
- 32.Wolfs TGAM, de Vries B, Walter SJ, Peutz-Kootstra CJ, van Heurn LWE, Oosterhof GON, Buurman WA. Apoptotic cell death is initiated during normothermic ischemia in human kidneys. Am J Transplant. 2005;5:68–75. doi: 10.1111/j.1600-6143.2004.00657.x. [DOI] [PubMed] [Google Scholar]
- 33.Cullen KE, Sarge KD. Characterization of hypothermia-induced cellular stress response in mouse tissues. J Biol Chem. 1997;272:1742–1746. doi: 10.1074/jbc.272.43.27266. [DOI] [PubMed] [Google Scholar]
- 34.Al-Fageeh MB, Marchant RJ, Carden MJ, Smales CM. The cold-shock response in cultured mammalian cells: Harnessing the response for the improvement of recombinant protein production. Biotechnol Bioeng. 2006;93:829–835. doi: 10.1002/bit.20789. [DOI] [PubMed] [Google Scholar]
- 35.Storey KB. Mammalian hibernation. Transcriptional and translational controls. Adv Exp Med Biol. 2003;543:21–38. [PubMed] [Google Scholar]
- 36.Plin C, Tillement JP, Berdeaux A, Morin D. Resveratrol protects against cold ischemia-warm reoxygenation-induced damages to mitochondria and cells in rat liver. Eur J Pharmacol. 2005;528:162–168. doi: 10.1016/j.ejphar.2005.10.044. [DOI] [PubMed] [Google Scholar]
- 37.Wagener FADT, Dekker D, Berden JH, Scharstuhl A, van der Vlag J. The role of reactive oxygen species in apoptosis of the diabetic kidney. Apoptosis. 2009;14:1451–1458. doi: 10.1007/s10495-009-0359-1. [DOI] [PMC free article] [PubMed] [Google Scholar]