

Abstract
Lignocellulosic biomass conversion inhibitors, furfural and HMF, inhibit microbial growth and interfere with subsequent fermentation of ethanol, posing significant challenges for a sustainable cellulosic ethanol conversion industry. Numerous yeast genes were found to be associated with the inhibitor tolerance. However, limited knowledge is available about mechanisms of the tolerance and the detoxification of the biomass conversion inhibitors. Using a robust standard for absolute mRNA quantification assay and a recently developed tolerant ethanologenic yeast Saccharomyces cerevisiae NRRL Y-50049, we investigate pathway-based transcription profiles relevant to the yeast tolerance and the inhibitor detoxification. Under the synergistic inhibitory challenges by furfural and HMF, Y-50049 was able to withstand the inhibitor stress, in situ detoxify furfural and HMF, and produce ethanol, while its parental control Y-12632 failed to function till 65 h after incubation. The tolerant strain Y-50049 displayed enriched genetic background with significantly higher abundant of transcripts for at least 16 genes than a non-tolerant parental strain Y-12632. The enhanced expression of ZWF1 appeared to drive glucose metabolism in favor of pentose phosphate pathway over glycolysis at earlier steps of glucose metabolisms. Cofactor NAD(P)H generation steps were likely accelerated by enzymes encoded by ZWF1, GND1, GND2, TDH1, and ALD4. NAD(P)H-dependent aldehyde reductions including conversion of furfural and HMF, in return, provided sufficient NAD(P)+ for NAD(P)H regeneration in the yeast detoxification pathways. Enriched genetic background and a well maintained redox balance through reprogrammed expression responses of Y-50049 were accountable for the acquired tolerance and detoxification of furfural to furan methanol and HMF to furan dimethanol. We present significant gene interactions and regulatory networks involved in NAD(P)H regenerations and functional aldehyde reductions under the inhibitor stress.
Keywords: Gene expression, Lignocellulosic ethanol, Pathway analysis, qRT-PCR standard, Stress tolerance
Introduction
Renewable biomass including lignocellulosic materials and agricultural residues contribute to low cost bioethanol production (Bothast and Saha 1997; Liu et al. 2008a). In order to utilize such materials by fermentative microorganisms, lingocellulosic biomass needs to be depolymerized into simple sugars. During lignocellulosic biomass hydrolysis pretreatment, numerous chemical by-products are generated which inhibit fermentative microorganisms (Klinke et al. 2004). Among many inhibitory compounds, 2-furaldehyde (furfural) and 5-hydroxymethyl-2-furaldehyde (5-hydroxymethylfurfural, HMF) are potent and representative inhibitors produced during biomass pretreatment, especially by economic dilute acid hydrolysis (Chung and Lee 1985; Taherzadeh et al. 1999; Larsson et al. 1999; Luo et al. 2002; Liu and Blaschek 2009). Furfural and HMF are formed by dehydration of pentoses and hexoses released from hemicelluloses and celluloses, respectively (Antal et al. 1991; Lewkowski 2001). These inhibitors damage cell walls and membranes, inhibit cell growth, reduce enzymatic activities, break down DNA, inhibit protein and RNA synthesis, and reduce ethanol production (Liu and Blaschek 2009; Sanchez and Bautista 1988; Khan and Hadi 1994; Modig et al. 2002). Few strains are able to withstand the inhibitory toxicity, and additional procedures are often required to remove or reduce the toxicity levels (Liu et al. 2008a, b; Liu and Blaschek 2009). Overcoming the inhibitor stress is one of the fundamental challenges in achieving a low-cost and sustainable lignocellulose-to-ethanol industry.
Saccharomyces cerevisiae is a traditional yeast used for industrial ethanol production but susceptible to aforementioned inhibitors and other stress conditions related to lignocellulosic biomass conversion. Yeast strains tolerant to single and combined inhibitors of furfural and HMF were recently developed (Liu et al. 2005, 2008a, b; Liu and Slininger 2005). A dose-dependent response of yeast to furfural and HMF has been characterized and a lag phase used to measure levels of strain tolerance (Taherzadeh et al. 2000; Liu et al. 2004). Furfural and HMF can be reduced to 2-furanmethanol (FM) and 2,5-bis-hydroxymethylfuran [furan-2,5-dimethanol (FDM)], respectively (Fig. 1) (Morimoto and Murakami 1967; Villa et al. 1992; Liu et al. 2004; Liu 2006). They can further break down to related organic acids (Taherzadeh et al. 2000; Nemirovskii et al. 1989; Palmqvist et al. 1999; Horvath et al. 2003). Under the inhibitor challenged conditions, once furfural and HMF fell to a certain lower level of concentrations, glucose consumption by yeast can be accelerated at a faster rate than would normally occur (Liu et al. 2004). Genomic adaptation is likely to happen at this stage (Liu 2006; Liu and Slininger 2006). Glycolysis and pentose phosphate pathway are major routes for glucose metabolisms that provide energy and important intermediate metabolites for biosynthesis and ethanol production. Unfortunately, important enzymes of glycolysis were inhibited by furfural (Banerjee et al. 1981). On the other hand, numerous genes and enzymes were reported to be associated with enhanced tolerance to furfural or HMF (Nilsson et al. 2005; Gorsich et al. 2006; Petersson et al. 2006; Almeida et al. 2008). It has been reported that multiple gene involved NAD(P)H-dependent aldehyde reductions is a mechanism of the detoxification of furfural and HMF (Liu et al. 2008b).
Fig. 1.

Conversion pathways of 2-furaldehyde (furfural) and 5-(hydroxymethyl)-2-furaldehyde (HMF) into 2-furanmethanol (FM) and furan-2,5-dimethanol (FDM) coupled with NADH and/or NADPH and catalyzed by multiple reductases and possible break down to varied organic acids
Genetic manipulation of one or a few genes is a common approach to improve a specific trait of yeast. However, when an integrated cell performance of QTLs (quantitative trait loci) or a group of balanced multiple functions is concerned, such methods often fall short in achieving satisfactory outcomes. For example, economic ethanol production and stress tolerance of yeast involving multiple genes is beyond the control of a small number of gene manipulations. By using an evolutionary adaptation procedure in laboratory settings mimicking natural selection under pressure, we developed tolerant ethanologenic yeast Saccharomyces cerevisiae NRRL Y-50049 that can in situ detoxify furfural and HMF while producing ethanol (Liu et al. 2008b). The tolerant yeast is able to grow on lignocellulosic hydrolysates pretreated by dilute acid hydrolysis containing high levels of inhibitors. Here, using a robust mRNA standard for qRT-PCR array assays, we investigate transcription dynamics and gene interactions during the lag phase in the two major pathways involving glucose metabolism under the furfural-HMF stress. Our results indicated that the tolerant yeast Y-50049 was able to withstand the inhibitor stress and in situ detoxify the inhibitors while producing ethanol through the reprogrammed transcription responses and altered metabolic pathways. A well maintained NAD(P)H redox balance appeared to be significant for numerous genes involved in the reprogrammed pathways.
Materials and methods
Yeast strains, medium, and culture conditions
Ethanologenic yeast strain S. cerevisiae NRRL Y-12632 and its tolerant derivative NRRL Y-50049 by environmental engineering (patent culture deposit) were used in this study (Agricultural Research Service Culture Collection, Peoria, IL, USA). Yeast strains were recovered from a lyophilized stock, maintained on yeast extract medium, incubated on a fleaker system, cultured, and treated by furfural and HMF at a final concentration of 20 mM each using procedures as previously described (Liu et al. 2004, 2005). Two replicated experiments were carried out for each strain and condition.
Sample collection and analysis
Cell growth was monitored by absorbance at OD600 during the fermentation. The time point at the addition of the inhibitors after 6 h growth was designated as 0 time point. Samples were taken at 0, 1, 16, 24, 42, 48, and 65 h after the inhibitor treatment. Metabolic conversion profiles including glucose, furfural, HMF, ethanol, FM, FDM, and acetic acid of each sample were analyzed using a Waters high-performance liquid chromatography (HPLC) equipped with an Aminex Fast Acid column (Bio-Rad Laboratories, Hercules, CA) and a refractive index detector as previously described (Liu et al. 2004, 2005, 2008b). Yeast cells were harvested and the frozen cell samples were stored at −80°C until use.
Standard of mRNA controls
Five external mRNA species, beta-2-microglobulin (B2M), major latex protein (MSG), chlorophyll A-B binding protein of LHCI type III precursor (CAB), ribulose bisphosphate carboxylase small chain 1 precursor (RBS1), and beta-actin (ACTB), were synthesized in vitro as described previously (Liu and Slininger 2007). A control mix was prepared consisting of accurately calibrated mRNA transcripts of MSG, CAB, RBS1, and ACTB at 0.1, 1, 10, and 1,000 pg per μl, respectively. This control mix served as a calibration standard. A standard curve was constructed for each set of qRT-PCR run. A reaction with corresponding primers but without a template of B2M was used to serve as a non-template negative control. The control gene CAB from soybean at an input mRNA of 1 pg was designated as a sole exogenous RNA reference to set up a manual threshold for data acquisition and analysis of each qRT-PCR run.
Gene selection and primer design
Based on transcriptome profiling of ethanologenic yeast under the inhibitor stress conditions using microarray (Liu and Slininger 2005; Liu 2006), glycolysis and pentose phosphate pathways were chosen for qRT-PCR array assays. Genes involved in these two pathways were selected according to the KEGG database (Kanehisa et al. 2006). PCR primers were designed based on sequence source of Saccharomyces genome database (Fisk et al. 2006) with an aid of primer screening using Primer 3 software (Rozen and Skaletsky 2000). The length of designed amplicons of all test genes ranged from 100 to 150 bp (Supplementary Table 1). Only those significantly affected genes were reported in this study.
Reverse transcription incorporated with mRNA control
The total RNA was isolated from each of two biological and two technical replications using a protocol as previously described (Liu and Slininger 2007). RNA integrity was verified by gel electrophoresis and NanoDrop Spectrophotometer ND-100 (NanoDrop Technologies, Inc., Wilmington, DE). Reverse transcription reaction was prepared by adding 1 μl of a control mix consisting of a set of the above-mentioned accurately calibrated mRNA transcripts into 2 μg of a host total RNA, 0.5 μg of oligo (dT)18, and 1 μ of 10 mM of dNTP mix. The volume was adjusted by water to 13 μl, then mixed well and incubated at 65°C for 5 min. The reaction tubes were chilled on ice for at least 1 min and the following was added: 4 μl 5X first strand buffer, 1 μl of 0.1 M DTT, 1 μl SuperScript III (200 U/μl) (Invitrogen, CA), and 1 μl RNaseOUT (40 U/μl) (Invitrogen, CA). The final volume of the reaction was 20 μl. The volume of this reverse transcription reaction can be proportionally enlarged to 80 μl for consistent performance in our laboratory. The reaction was incubated at 50°C for 1 h, 70°C for 15 min, and 4°C to end the reaction using a PCR cycler.
Real time qRT-PCR
SYBR Green iTaq PCR master mix (BioRad Laboratories) was applied for each qRT-PCR reaction. For each reaction, a total of 25 μl was used consisting of 12.5 μl 2X SYBR Green MasterMix, 0.5 μl each of forward and reverse primer (10 μM each), 0.25 μl cDNA template and 11.25 μl H2O. On each 96-well plate, reactions of qRT-PCR were carried out with two replications for each control gene except for the control CAB of three replications. All reactions of tested target gene were run duplicated. Ctrl_B2M served as a non template negative control for each plate. PCR was run on an ABI 7500 real time PCR system using a defined profile.
Data analysis
The mean value of three CAB amplifications on a plate was designated and used as a constant reference to set up a manual threshold at 26 Ct (cycle number) for data analysis. This sole reference served as a constant standard for data acquisition and analysis for each and every qRT-PCR run. The data then were exported to an Excel file and treated using a custom-programmed macro Visual Basic function. Statistical analyses were performed using statistical function tools of Microsoft Excel. A standard curve was constructed for each plate run and a master equation was generated using the sum of data. Comparison of variation between individual standard curve and the master equation was done to validate a standard application. PCR amplification efficiency for each reaction run was calculated as previously described (Livak and Schmittgen 2001; Liu and Saint 2002; Applied Biosystem, 2006).
Gene interactions and regulatory networks
All selected genes were grouped into 32 clusters based on expression time course for the yeast using a hierarchical variable clustering algorithm in R (R Development Core Team 2008), with the absolute Pearson correlation coefficient greater than 0.95. This step eliminated the competition of all highly linearly correlated genes during modeling. Each cluster was represented by a gene most similar to other genes in the cluster. Only the representatives were used in the network modeling. The expression levels of representatives were quantified into three levels of low, normal, and high concentrations. A recently developed generalized logical network model (Song et al. 2009)—a discrete-time dynamical system model—was applied. This model represents nonlinear temporal interactions among genes and is reconstructed by searching for statistically significant temporal associations among genes over time courses. The associations as directed edges were detected by applying chi-square tests for each node on the transitions occurring at each time point. The network reconstruction controls the false positive rate, making it unique from other modeling approaches, such as Bayesian networks (Friedman 2004). The Markovian orders of both 0 and −1 were searched. A Markovian order of −k signifies that the expression of one gene at time t is examined for association with gene expression of another gene at up to k time points back into history. The maximum number of parents per node was 2. The program was written in C++ with parallel computing capability and tested on both Windows XP and SuSE Linux operating systems.
Results
Robust standard and master equation for absolute quantification of mRNA abundance
To ensure reproducibility and comparability of qRT-PCR data, we used an improved robust external mRNA control CAB, a unique soybean gene showing no homologous DNA sequence with microbial genomes including yeast, as a fixed threshold for data acquisition (see Materials and methods). Using CAB as a sole reference to set a manual threshold at 26 Ct for data acquisition, raw data were normalized and analyzed for the entire PCR reactions in 38 individual 96-well plate runs. A standard curve was constructed for each of 38 individual plates applying the universal quality control genes MSG, CAB, RBS1, and ACTB. Since variations of the slopes and intercepts of the regression lines were statistically ignorable (Table 1, Supplementary Table 2), a master equation was further established using the pooled data for all reference control reactions (Fig. 2) and validated by the accurate estimation for known input mRNA (Table 1). Based on robust performance of the universal controls, we obtained a master equation of standard curve using the total sum data as follows:
| (1) |
where X represents mRNA (log pg) and Y equals qRT-PCR cycle number (Ct) estimated for all reactions performed on the ABI 7500 real time PCR System. Average amplification efficiency of the PCR was 90% for all reactions using the master equation, which fell into valid efficiency ranges of qRT-PCR assay as described previously (Applied Biosystem, 2006). Thus, mRNA mass of all reactions regardless experimental conditions was estimated using the validated master equation. The external RNA control has been recognized having numerous advantages over the variable housekeeping gene controls for expression analysis (Baker et al. 2005; External RNA Control Consortium 2005). The robust mRNA control system and application of the master equation by this study provided consistent reference for the qRT-PCR assay and allowed comparisons of data derived from various experimental sets. Such a robust reference has become a necessary tool for pathway-based qRT-PCR array analysis.
Table 1.
Robust performance of standard control genes using CAB as sole reference to set a manual threshold at 26 Ct and a master equation derived from 38 replicated plate reactions on Applied Biosystems 7,500 real time PCR System
| Control gene | Reference Ct | Mean Ct | Stdev | Estimated mRNA (pg) | Input mRNA (pg) | Amplification efficiency |
|---|---|---|---|---|---|---|
| MSG | 29.349 | 0.189 | 0.105 | 0.1 | 0.95 | |
| CAB | 26.0 | 25.942 | 0.18 | 0.939 | 1 | 0.94 |
| RBS1 | 22.267 | 0.111 | 9.97 | 10 | 1.00 | |
| ACTB | 15.072 | 0.027 | 1017.448 | 1000 | 0.98 |
Fig. 2.
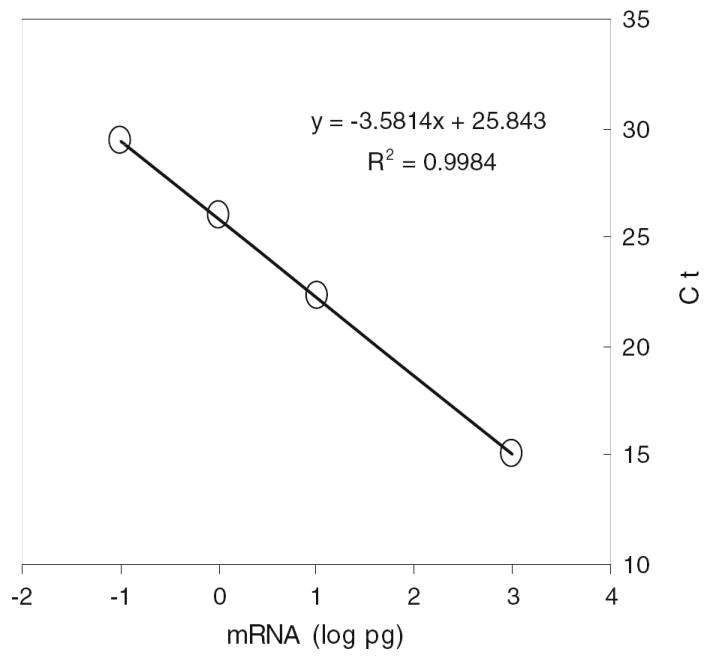
Functional performance of universal RNA controls for real time qRT-PCR assays. Robust calibration control genes of MSG, CAB, RBS1, and ACTB at 0.1, 1, 10, and 1,000 pg over 38 individual 96-well reaction plates for Saccharomyces cerevisiae NRRL Y-50049 and NRRL Y-12632 treated with and without combined inhibitors of furfural and HMF at a final concentration of 20 mM each demonstrated highly fitted linear relationship between the mRNA input (log pg) and cycle numbers (Ct) by a master equation for assays on ABI 7500 real time PCR System. Standard deviation of the slope and the intercept of the master equation based on 38 individual standard curves under varied experimental conditions was 0.0549 and 0.1896, respectively
Cell growth and metabolic conversion profiles
Cell growth was inhibited and an extended lag phase observed for both strains treated by combined inhibitors of furfural and HMF, each at a final concentration of 20 mM (Fig. 3a). After 42 h, tolerant strain Y-50049 showed a dramatic recovery of cell growth and quickly entered into a log phase. In contrast, strain Y-12632 failed to recover and was unable to establish a viable culture till 65 h after incubation. Similarly, metabolic profiles of these strains demonstrated delayed metabolic conversion process as measured by HPLC analysis. Y-50049 showed accelerated glucose consumption at 42 h after a slow initiation response and completed ethanol production no more than 65 h. Y-12632, on the other hand, showed no glucose consumption activity at all (Fig. 3b). As expected, for Y-50049 cultures, concentrations of furfural and HMF were reduced with increased corresponding conversion products of FM and FDM, respectively (Fig. 3c, d). In contrast, the control parental strain Y-12632 failed to recover and was unable to establish a viable culture until 65 h after incubation. The cell growth and metabolic profiles are highly correlated with the mRNA expression profiles related to glycolysis and pentose phosphate pathways. In addition, a small amount of acetic acid was observed to be produced earlier for Y-50049 than that of Y-12632 (data not shown).
Fig. 3.

Comparison of cell growth and metabolic conversion profiles of Saccharomyces cerevisiae NRRL Y-50049 and NRRL Y-12632 over time in response to a combined inhibitor treatment of furfural and HMF at a final concentration of 20 mM each after 6 h incubation on YM medium. a Cell growth as measured by OD600 for Y-50049 (filled circle) and Y-12632 (open circle). b Glucose consumption (filled circle) and ethanol conversion (open circle) for Y-50049 versus glucose (filled square) and ethanol (open square) for Y-12632. c HMF (filled diamond) and its conversion product furandimethanol (FDM) (open diamond) for Y-50049 versus HMF (filled triangle) and FDM (open triangle) for Y-12632. d Furfural (filled diamond) and its conversion product furan methanol (FM) (open diamond) for Y-50049 versus furfural (filled triangle) and FM (Δ) for Y-12632
Enriched genetic background of tolerant strain Y-50049
The tolerant strain NRRL Y-50049 is derived from wild type Y-12632 and a stable culture that carries different genetic background from its parental strain. Y-50049 displayed enhanced gene expression profiles distinct from the control Y-12632. At least 16 genes, including HXK1, HXK2, GLK1, TDH1, TDH3, LAT1, PDC6, ADH4, ALD2, ALD4, ZWF1, SOL3, RBK1, TAL1, NQL1, and PRS2 demonstrated significantly higher levels of mRNA transcripts prior to the inhibitor treatment compared with that of the parental strain Y-12632 (Figs. 4, 5). Such differences of the enriched genetic background were obvious as measured 6 h after cultivation and before the inhibitors were added. Many of these genes showed 4–6-fold enhanced expressions. Examples included hexoskinase encoding genes HXK1 and HXK2, glyceraldehyde-3-phosphate dehydrogenase gene TDH1, dihydrolipoamide acetyltransferase component (E2) of pyruvate dehydrogenase complex gene LAT, major mitochondrial aldehyde dehydrogenase gene ALD4 and TAL1 that encodes transaldolase.
Fig. 4.
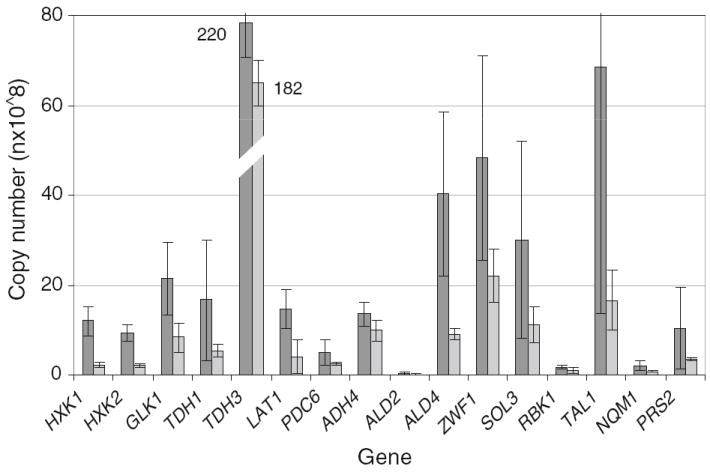
Comparison of expression levels in total gene copy numbers per 0.25 μl probe reaction for selected genes of Saccharomyces cerevisiae NRRL Y-50049 and NRRL Y-12632 under normal conditions 6 h after cultivation before furfural and HMF were added on a YM medium. Mean values are presented with error bars representing variations of two standard deviations
Fig. 5.
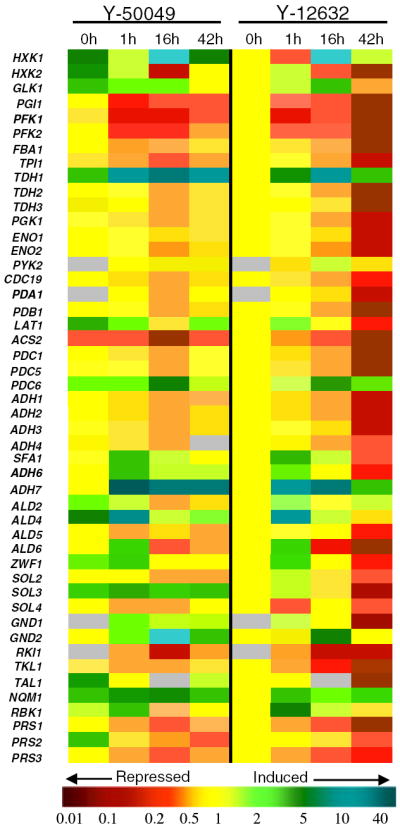
Comparison of mRNA expression for selected genes relevant in glucose metabolic pathways of Saccharomyces cerevisiae NRRL Y-50049 and NRRL Y-12632 under challenges of furfural and HMF at 20 mM each from 0 to 42 h after the treatment. Quantitative expression for each gene at each time point was expressed in fold changes against that of Y-12632 at 0 h. Yellow indicates no change; green indicates enhanced expression and red for repressed expression in quantitative scales as indicated by a color bar at the bottom. Expression data at a time point marked with gray indicate missing data or data need to be confirmed
Tolerant response to the furfural-HMF stress by strain Y-50049
Distinct dynamic transcriptome response can be detected immediately after the inhibitor challenges. At 1 h after the inhibitor treatment, PGI1, PFK1, PFK2, FBA1, and TPI1, significant genes involved in catalyzing glucose-6-phosphate to glyceraldedyde-3-phosphate at the earlier steps of glycolysis, were repressed for both strains (Fig. 5). Y-12632 showed continued inhibition in cell growth and expression of increased number of genes over time and failed to function eventually. However, Y-50049 was able to withstand and recover from the inhibitor challenge. HXK1, GLK1, TDH1, LAT1, PDC6, ALD4, SFA1, ADH6, ADH7, and ALD4 showed significantly enhanced transcription levels for Y-50049 compared with that of Y-12632 (Fig. 5, Supplementary Table 3). Among these, glyceraldehyde-3-phosphate dehydrogenase gene TDH1 and alcohol dehydrogenases gene ADH7 displayed significantly high transcript copy numbers of 18- and 35-fold, respectively, compared with the Y-12632 background. Gene transcription levels of PGK1, ENO1, ENO2, PYK2, CDC19, PDA1, and PDB1 encoding varied enzymes for phosphate transferring and extended pyruvate metabolisms did not show significant changes at 1 h after the inhibitor challenge. Transcription levels were low at 16 h for numerous genes in glycolysis and pentose phosphate pathways for both strains. However, many genes were recovered to normal or near normal function levels at 42 h for Y-50049 (Fig. 5, Supplementary Table 3). In contrast, most genes of Y-12632 were continued repressed by the furfural and HMF treatment that led to eventually no viable cell functions after 42 h (Figs. 3, 5).
Some significant genes involved in the pentose phosphate pathway such as ZWF1, SOL3, GND1, GND2, NQM1, and RBK1 were induced expressed for both strains by furfural and HMF. ZWF1 encoding enzyme to catalyze the first oxidative reduction of NADP+ to NADPH was induced significantly higher for Y-50049 at 1 h. Among three members of SOL gene family, SOL3 showed the most significant and consistent induced expression over time. GND1 and GND2 encoding enzyme catalyzing the second oxidative phase in releasing of NADPH were consistently enhanced expressed, especially for GND2 which reached significantly high levels of more than 10-fold increase at 16 h for Y-50049.
Pathways of in situ detoxification of furfural and HMF by strain Y-50049
Since the metabolic conversion dynamics of the inhibitors were highly corresponding to the induced abundant transcripts of all these reductase genes under the furfural-HMF stress we constructed the in situ detoxification pathways by the tolerant Y-50049 in relationship to the altered glycolysis and pentose phosphate pathways (Fig. 6). The enhanced expression of ZWF1 appeared to drive glucose metabolism in favor of pentose phosphate pathway over glycolysis at an earlier step of the glucose metabolism. Consequently, all other cofactor NAD(P)H regenerating steps involving ZWF1, GND1, GND2, and TDH1 were up-regulated for Y-50049. Aldehyde reduction enzyme encoding genes ALD4, ALD6, ADH6, ADH7, and SFA1 displayed significantly high abundant transcripts induced at the earlier time points in the presence of furfural and HMF (Fig. 5). These accelerated NAD(P)H-dependent reductions of acetaldehyde, furfural, and HMF would generate sufficient NAD+ and NADP+. In return, the supplementary NAD(P)+ supplies provided the necessary cofactors needed for oxidative reactions or NAD(P)H regenerations by Zwf1p, Gnd1p, Gnd2p, Tdh1p, and Ald4p. Thus, an NAD(P)+/NAD(P)H-dependent redox balance is likely well maintained in the altered pathways for in situ detoxification of furfural and HMF.
Fig. 6.

A schematic illustration of glucose metabolic pathways and conversion of furfural and HMF by tolerant Saccharomyces cerevisiae NRRL Y-50049 inferred by metabolic profiling analysis and quantitative mRNA expression analysis compared with a wild type strain NRRL Y-12632. Black arrowed lines and letters indicate normal or near normal levels of reactions, expressions or pathways, green indicates enhanced, and red for repressed expressions, reactions, or pathways. Bolded lines and letters indicate the levels of expression and pathways are statistically significant. Key steps of enhanced NAD(P)H regenerations are circled in blue and significant aldehyde reductions circled in orange
Interactions and regulatory networks of selected genes for tolerant strain Y-50049
Although our in situ detoxification pathways, based on known pathways, are derived from qualitatively observing gene expression patterns, we show further here that our pathway model is also quantitatively supported by those statistically significant interactions reconstructed from the observed data using independent data-driven gene regulatory network modeling without prior knowledge. The generalized logical network modeling approach (Song et al. 2009) assesses the statistical significance of potential gene interactions using a statistical chi-square test, and it only selects the most significant interaction for each gene. It explicitly controls the false positive rate, which is an improvement from previous network modeling approaches (Song and Liu 2007). Generalized logical network models constructed based on expression data were distinctly different between the two strains of Y-50049 and Y-12632 (data not shown). High levels of transcription and enhanced earlier expressions of HXK1, HXK2, and GLK1 appeared to be centers of statistically significant interactions (p value ≤ 0.05) with other genes. Among these, consistent interactions relevant to the observed yeast tolerance and detoxification included interactions of HXK1 with ZWF1, ALD4, ALD6, ADH6, and RBK1; HXK2 with RBK1, SFA1, TDH3, and PDC6; and GLK1 with TDH1 and ADH7. Following the first step of glycolysis pumped by HXK1, HXK2, and GLK1, interactions between the groups of cofactor regenerations and the functional reductases were highly significant (Fig. 7). These results were consistent and provide statistical significance support to our proposed detoxification pathways. The significant relationships among GLK1, TDH1, and ADH7 (Figs. 6, 7) represent a typical model of the redox balance and the detoxification of furfural and HMF by the tolerant yeast.
Fig. 7.
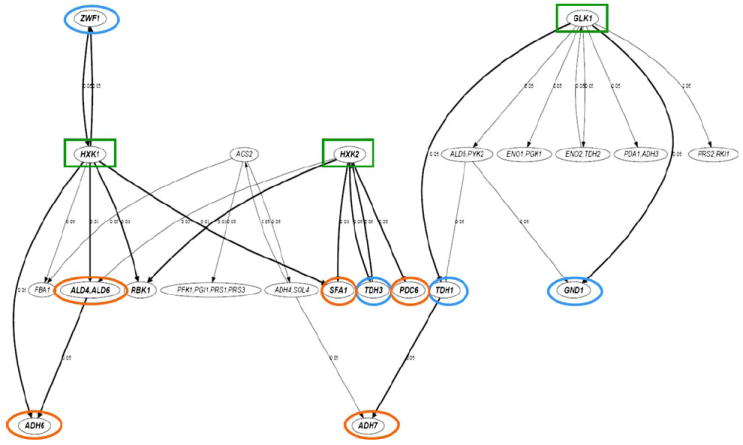
A generalized logical network model for selected genes of Saccharomyces cerevisiae NRRL Y50049 based on quantitative dynamic expression data using pathway-base qRT-PCR array analysis. The nodes in the network represent the selected genes. A node with two or more gene labels suggests that the time courses of the represented genes are highly linearly correlated and thus treated equally. The directed edges in the network indicate statistically significant (p value ≤ 0.5) nonlinear temporal associations among the genes. The p value of each interaction is shown next to the corresponding edge. Arrowed lines indicate direction of influence in an interaction. The thicker edges denote those interactions that are consistent with the proposed pathways of glucose metabolisms and detoxification of furfural and HMF. Highlighted nodes with gene names in bold font are the end points of those thicker lines that are biologically relevant. Key genes involving NAD(P)H regenerations are circled in blue and significant genes involving aldehyde reductions consistent with the reprogrammed pathways circled in orange. Three important enhanced expressed genes involving the initial step of glucose phosphorylation and energy release are boxed in green
Discussion
Applying an enhanced gene expression tool, a pathway-based qRT-PCR array validated by the robust external mRNA controls, we presented unique pathways to demonstrate mechanisms of the acquired tolerance and the in situ detoxification of furfural and HMF by the tolerant ethanologenic yeast S. cerevisiae NRRL Y-50049 based on metabolic profiling analyses and dynamics of absolute quantification of mRNA transcripts. The recent development of the tolerant yeast (Liu et al. 2008b) and the identification and synthesis of HMF metabolic conversion product FDM (Liu et al. 2004) are necessary to make this study possible.
The robust performance using the control gene CAB as sole standard of a fixed manual threshold setting and thereafter the use of the master equation provided reliable and consistent absolute quantification reference for real time qRT-PCR assays. As demonstrated by this study, such a standard measurement is independent from toxic treatment, culture conditions, varied strains, and different set of reactions that allow comparison of data obtained from different conditions and sources. It has significant advantages over variable housekeeping genes used in qRT-PCR assays (Baker et al. 2005; The External RNA Control Consortium 2005). Such a development significantly improved reliability, reproducibility, and throughput of the qRT-PCR and simplified the conventional practice of the assay.
Redox metabolism, in the form of interconversion of the pyridine-nucleotide co-factors NADH/NAD+ and NADPH/NADP+, plays a key role in the metabolism of yeast. NADH is required in respiration and fermentative pathway in conversion of pyruvate to CO2 and ethanol; and NADPH is mainly required for the synthesis of amino acids and nucleotides. Furfural conversion to furan methanol and HMF to furan dimethanol are NAD(P)H-dependent reduction activities by multiple enzymes (Petersson et al. 2006; Song and Liu 2007; Liu et al. 2008b; Song et al. 2009). These reduction reactions consume NADH and NADPH. Therefore, a well maintained redox balance is critical for efficient conversion and in situ detoxification of furfural and HMF. A major source of NADPH production in yeast is through the oxidative phase of pentose phosphate pathway. Since expressions of ZWF1, SOL3, GND1, and GND2 were up-regulated at earlier time points, pentose phosphate pathway became a dominant reprogrammed path for glucose metabolisms and inhibitor conversions for Y-50049. These genes along with enhanced expressed TDH1 are significant for NADPH regenerations to supply necessary cofactors needed for acetaldehyde conversion and reduction of furfural and HMF. We previously observed that deletion mutants of ZWF1 and GND1 were highly sensitive to furfural and HMF (Gorsich et al. 2006). Significant effect of phosphogluconate dehydrogenase in redox metabolism has been demonstrated (Bro et al. 2004). Apparently, enriched genetic background by aforementioned genes and a well maintained redox balance through the reprogrammed expression responses of Y-50049 were likely accountable for the acquired tolerance and detoxification of the inhibitors.
Glycolysis and pentose phosphate pathway are closely related pathways for glucose metabolism. This close relationship is of such significant importance that the two pathways cannot be viewed separately in the view of yeast tolerance and detoxification of furfural and HMF. For Y-50049, after initiation stage of phosphorylation of glucose by enzyme encoding genes HXK1, HXK2, and GLK1, glucose metabolism apparently shifted toward pentose phosphate pathways than glycolysis due to significantly induced expression of ZWF1, SOL3, GND1, and GND2 and repression of glycolytic enzyme phosphoglucose isomerase pathway. Such a pathway shift appeared to be significant for Y-50049 to carry continued glucose consumption and detoxification metabolism under the inhibitor stress.
It is known that furfural and HMF inhibit yeast growth and cause delayed glucose consumption at a tolerable dose (Taherzadeh et al. 2000; Liu et al. 2004, 2005). The reprogrammed transcription responses and the altered metabolic pathways presented in this study can also explain such observations that yeast has an accelerated glucose conversion rate once cells recovered from the inhibitor challenges than would normally occur without furfural and HMF. As demonstrated in this study, inhibition of glucose phosphorylation, together with repression of PFK1, PFK2, PYK2 and CDC19 expressions was likely to be the mechanism of the delay of glycolysis by furfural and HMF. This delayed biological process in yeast is also attributed to a lack of ATP, NAD(P)H, and carbon intermediate metabolites necessary to support cell growth and reproduction (Fisk et al. 2006; Liu 2006). Comparing the susceptible parental strain Y12632, gene transcription levels of PGK1, ENO1, ENO2, PYK2, CDC19, PDA1, and PDB1 encoding varied enzymes for extended pyruvate metabolisms did not show significant effect by the inhibitor challenge at earlier time points for the tolerant Y-50049. Transcription levels were low for numerous other genes in glycolysis and pentose phosphate pathways for both strains. However, most of these genes were able to recover to normal function levels at 42 h for Y-50049, whereas those for Y-12632 continued to be repressed by the furfural and HMF treatment that eventually led to no viable cell functions after 42 h.
Over 300 genes were found to be statistically significant in response to the inhibitor challenges for the ethanologenic yeast genome (Liu 2006). Numerous members of PDR gene family and key transcription factors played significant roles in regulating global transcription response to the bioethanol conversion inhibitors (Song and Liu 2007; Song et al. 2009). Puzzles of many complex pathways at the genome level remain unresolved. Nonetheless, regarding the in situ detoxification of furfural and HMF metabolic pathways involving glycolysis and pentose phosphate pathway, at least three significant groups of events of the reprogrammed evolutionary engineering are accountable for the acquired tolerance of Y-50049. First, initial high abundant transcript levels of at least 16 genes were critical enablers for Y-50049 to withstand the inhibitor stress. This represents the enriched genetic characteristics of Y-50049. Second, more than a dozen genes immediately responded with induced expression, specifically, the genes with continued enhanced expression over time enabled Y-50049 to function and detoxify furfural and HMF into corresponding furan methanol with a well maintained redox balance. Third, the integrated transcription response and reprogrammed regulatory networks, including those significantly enhanced expressed and as well as many initially repressed but able to recover to normal function levels at a later time, which allowed Y-50049 to maintain a balanced biological process to complete ethanol fermentation. In the absence of such reprogrammed transcription responses at the genome level as shown by the control yeast Y-12632, continued inhibition and repression by furfural and HMF led the yeast to lost function and eventually death. Our results guide continued efforts in developing stress tolerant ethanologenic yeast for a sustainable lignocellulosic biomass-to-ethanol industry.
Supplementary Material
Acknowledgments
We thank Scott Weber and Stephanie Thompson for technical assistance and Pat Slininger for discussions. This work was supported by the National Research Initiative of the USDA Cooperative State Research, Education and Extension Service, grant number 2006-35504-17359.
The mention of trade names or commercial products in this article is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the US Department of Agriculture.
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s00438-009-0461-7) contains supplementary material, which is available to authorized users.
Contributor Information
Z. Lewis Liu, U.S. Department of Agriculture, Agricultural Research Service, National Center for Agricultural Utilization Research, 1815 N University, Peoria, IL 61604, USA, ZLewis.Liu@ars.usda.gov.
Menggen Ma, U.S. Department of Agriculture, Agricultural Research Service, National Center for Agricultural Utilization Research, 1815 N University, Peoria, IL 61604, USA; Department of Computer Science, New Mexico State University, P.O. Box 30001, MSC CS, Las Cruces, NM 88003, USA.
Mingzhou Song, Department of Computer Science, New Mexico State University, P.O. Box 30001, MSC CS, Las Cruces, NM 88003, USA.
References
- Almeida JRM, Roder A, Modig T, Laadan B, Liden G, Gorwa-Grauslund M. NADH- versus NADPH-coupled reduction of 5-hydroxymethylfurfural HMF and its implications on product distribution in Saccharomyces cerevisiae. Appl Microbiol Biotechnol. 2008;78:939–945. doi: 10.1007/s00253-008-1364-y. [DOI] [PubMed] [Google Scholar]
- Antal MJ, Leesomboon T, Mok WS, Richards GN. Mechanism of formation of 2-furaldehyde from d-xylose. Carbohydr Res. 1991;217:71–85. doi: 10.1016/0008-6215(90)84096-d. [DOI] [PubMed] [Google Scholar]
- Applied Biosystem. Amplification efficiency of TagMan gene expression assays. 2006:5. Application Note Publication 127AP05-03. [Google Scholar]
- Baker SC, Bauer SR, Beyer RP, Brenton JD, Bromley B, Burrill J, Causton H, Conley MP, Elespuru R, Fero M, Foy C, Fuscoe J, Gao X, Gerhold DL, Gilles P, Goodsaid F, Guo X, Hackett J, Hockett RD, Ikonomi P, Irizarry RA, Kawasaki ES, Kaysser-Kranich T, Kerr K, Kiser G, Koch WH, Lee KY, Liu C, Liu ZL, Lucas A, Manohar CF, Miyada G, Modrusan Z, Parkes H, Puri RK, Reid L, Ryder TB, Salit M, Samaha RR, Scherf U, Sendera TJ, Setterquist RA, Shi L, Shippy R, Soriano JV, Wagar EA, Warrington JA, Williams M, Wilmer F, Wilson M, Wolber PK, Wu X, Zadro R. The external RNA controls consortium: a progress report. Nat Methods. 2005;2:731–734. doi: 10.1038/nmeth1005-731. [DOI] [PubMed] [Google Scholar]
- Banerjee N, Bhatnagar R, Viswanathan L. Inhibition of glycolysis by furfural in Saccharomyces cerevisiae. Eur J Appl Microbiol Biotechnol. 1981;11:226–228. [Google Scholar]
- Bothast RJ, Saha BC. Ethanol production from agricultural biomass substrate. Adv Appl Microbiol. 1997;44:261–286. [Google Scholar]
- Bro C, Regenberg B, Nielsen J. Genome-wide transcriptional response of a Saccharomyces cerevisiae strain with an altered redox metabolism. Biotechnol Bioeng. 2004;85:269–276. doi: 10.1002/bit.10899. [DOI] [PubMed] [Google Scholar]
- Chung IS, Lee YY. Ethanol fermentation of crude acid hydrolyzate of cellulose using high-level yeast inocula. Biotechnol Bioeng. 1985;27:308–315. doi: 10.1002/bit.260270315. [DOI] [PubMed] [Google Scholar]
- Fisk DG, Ball CA, Dolinski K, Engel SR, Hong EL, Issel-Tarver L, Schwartz K, Sethuraman A, Botstein D, Cherry JM. Saccharomyces cerevisiae S288C genome annotation: a working hypothesis. Yeast. 2006;23:857–865. doi: 10.1002/yea.1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman N. Inferring cellular networks using probabilistic graphical models. Science. 2004;303:799–805. doi: 10.1126/science.1094068. [DOI] [PubMed] [Google Scholar]
- Gorsich SW, Dien BS, Nichols NN, Slininger PJ, Liu ZL, Skory C. Tolerance to furfural-induced stress is associated with pentose phosphate pathway genes ZWF1, GND1, RPE1, and TKL1 in Saccharomyces cerevisiae. ApplMicrobiol Biotechnol. 2006;71:339–349. doi: 10.1007/s00253-005-0142-3. [DOI] [PubMed] [Google Scholar]
- Horvath IS, Franzen CJ, Taherzadeh MJ, Niklasson C, Liden G. Effects of furfural on the respiratory metabolism of Saccharomyces cerevisiae in glucose-limited chemostats. Appl Environ Microbiol. 2003;69:4076–4086. doi: 10.1128/AEM.69.7.4076-4086.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Goto S, Hattori M, Aoki-Kinoshita KF, Itoh M, Kawashima S, Katayama T, Araki M, Hirakawa M. From genomics to chemical genomics: new developments in KEGG. Nucleic Acids Res. 2006;34:D354–D357. doi: 10.1093/nar/gkj102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan QA, Hadi SA. Inactivation and repair of bacteriophage lambda by furfural. Biochem Mol Biol Int. 1994;32:379–385. [PubMed] [Google Scholar]
- Klinke HB, Thomsen AB, Ahring BK. Inhibition of ethanol-producing yeast and bacteria by degradation products produced during pre-treatment of biomass. Appl Microbiol Biotechnol. 2004;66:10–26. doi: 10.1007/s00253-004-1642-2. [DOI] [PubMed] [Google Scholar]
- Larsson S, Palmqvist E, Hahn-Hägerdal B, Tengborg C, Stenberg K, Zacchi G, Nilvebrant N. The generation of fermentation inhibitors during dilute acid hydrolysis of softwood. Enzyme Microb Technol. 1999;24:151–159. [Google Scholar]
- Lewkowski J. Synthesis, chemistry and applications of 5-hydroxymethylfurfural and its derivatives. Arkivoc. 2001;1:17–54. [Google Scholar]
- Liu ZL. Genomic adaptation of ethanologenic yeast to biomass conversion inhibitors. Appl Microbiol Biotechnol. 2006;73:27–36. doi: 10.1007/s00253-006-0567-3. [DOI] [PubMed] [Google Scholar]
- Liu ZL, Blaschek HP. Lignocellulosic biomass conversion to ethanol by Saccharomyces. In: Vertes A, Qureshi N, Yukawa H, Blaschek H, editors. Biomass to biofuels. Wiley; West Sussex: 2009. pp. 17–36. [Google Scholar]
- Liu W, Saint DA. A new quantitative method of real time reverse transcription polymerase chain reaction assay based on simulation of polymerase chain reaction kinetics. Analytical Biochem. 2002;302:52–59. doi: 10.1006/abio.2001.5530. [DOI] [PubMed] [Google Scholar]
- Liu ZL, Slininger PJ. CAB International. Development of genetically engineered stress tolerant ethanologenic yeasts using integrated functional genomics for effective biomass conversion to ethanol. In: Outlaw J, Collins K, Duffield J, editors. Agriculture as a producer and consumer of energy. Wallingford, UK: 2005. pp. 283–294. [Google Scholar]
- Liu ZL, Slininger PJ. Transcriptome dynamics of ethanologenic yeast in response to 5-hydroxymethylfurfural stress related to biomass conversion to ethanol. In: Mendez-Vilas A, editor. Modern multidisciplinary applied microbiology: exploiting microbes and their interactions. Wiley-VCH; Weinheim: 2006. pp. 679–685. [Google Scholar]
- Liu ZL, Slininger PJ. Universal external RNA controls for microbial gene expression analysis using microarray and qRT-PCR. J Microbiol Methods. 2007;68:486–496. doi: 10.1016/j.mimet.2006.10.014. [DOI] [PubMed] [Google Scholar]
- Liu ZL, Slininger PJ, Dien BS, Berhow MA, Kurtzman CP, Gorsich SW. Adaptive response of yeasts to furfural and 5-hydroxymethylfurfural and new chemical evidence for HMF conversion to 2,5-bis-hydroxymethylfuran. J Ind Microbiol Biotechnol. 2004;31:345–352. doi: 10.1007/s10295-004-0148-3. [DOI] [PubMed] [Google Scholar]
- Liu ZL, Slininger PJ, Gorsich SW. Enhanced biotransformation of furfural and 5-hydroxymethylfurfural by newly developed ethanologenic yeast strains. Appl Biochem Biotechnol. 2005:121–124. 451–460. [PubMed] [Google Scholar]
- Liu ZL, Saha BC, Slininger PJ. Lignocellulosic biomass conversion to ethanol by Saccharomyces. In: Wall J, Harwood C, Demain A, editors. Bioenergy. ASM Press; Washington DC: 2008a. pp. 17–36. [Google Scholar]
- Liu ZL, Moon J, Andersh AJ, Slininger PJ, Weber S. Multiple gene mediated NAD(P)H-dependent aldehyde reduction is a mechanism of in situ detoxification of furfural and HMF by ethanologenic yeast Saccharomyces cerevisiae. Appl Microbiol Biotechnol. 2008b;81:743–753. doi: 10.1007/s00253-008-1702-0. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Luo C, Brink D, Blanch HW. Identification of potential fermentation inhibitors in conversion of hybrid poplar hydrolyzate to ethanol. Biomass Bioenergy. 2002;22:125–138. [Google Scholar]
- Modig T, Liden G, Taherzadeh MJ. Inhibition effects of furfural on alcohol dehydrogenase, aldehyde dehydrogenase and pyruvate dehydrogenase. Biochem J. 2002;363:769–776. doi: 10.1042/0264-6021:3630769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimoto S, Murakami M. Studies on fermentation products from aldehyde by microorganisms: the fermentative production of furfural alcohol from furfural by yeasts (part I) J Ferm Technol. 1967;45:442–446. [Google Scholar]
- Nemirovskii V, Gusarova L, Rakhmilevich Y, Sizov A, Kostenko V. Pathways of furfurol and oxymethyl furfurol conversion in the process of fodder yeast cultivation. Biotekhnologiia. 1989;5:285–289. [Google Scholar]
- Nilsson A, Gorwa-Grauslund MF, Hahn-Hagerdal B, Liden G. Cofactor dependence in furan reduction by Saccharomyces cerevisiae in fermentation of acid-hydrolyzed lignocellulose. Appl Environ Microbiol. 2005;71:7866–7871. doi: 10.1128/AEM.71.12.7866-7871.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmqvist E, Almeida JS, Hahn-Hagerdal B. Influence of furfural on anaerobic glycolytic kinetics of Saccharomyces cerevisiae in batch culture. Biotechnol Bioeng. 1999;62:447–454. doi: 10.1002/(sici)1097-0290(19990220)62:4<447::aid-bit7>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- Petersson A, Almeida JR, Modig T, Karhumma K, Hahn-Hägerdal B, Gorwa-Grauslund MF. A 5-hydroxymethylfurfural reducing enzyme encoded by the Saccharomyces cerevisiae ADH6 gene conveys HMF tolerance. Yeast. 2006;23:455–464. doi: 10.1002/yea.1370. [DOI] [PubMed] [Google Scholar]
- R Development Core Team. R foundation for statistical computing; Vienna, Austria: 2008. R: a language and environment for statistical computing. http://www.R-project.org. [Google Scholar]
- Rozen S, Skaletsky H. Bioinformatics methods and protocols. In: Krawetz S, Misener S, editors. Methods in molecular biology. Humana Press; Totowa: 2000. pp. 365–386. [DOI] [PubMed] [Google Scholar]
- Sanchez B, Bautista J. Effects of furfural and 5-Hydroxymethylfurfrual on the fermentation of Saccharomyces cerevisiae and biomass production from Candida guilliermondii. Enzyme Microb Technol. 1988;10:315–318. [Google Scholar]
- Song M, Liu ZL. A linear discrete dynamic system model for temporal gene interaction and regulatory network influence in response to bioethanol conversion inhibitor HMF for ethanologenic yeast. Lect Notes Bioinfomatics. 2007;4532:77–95. [Google Scholar]
- Song M, Ouyang Z, Liu ZL. Discrete dynamic system modeling for gene regulatory networks of HMF tolerance for ethanologenic yeast. IET Sys Biology. 2009;3:203–218. doi: 10.1049/iet-syb.2008.0089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taherzadeh MJ, Gustafsson L, Niklasson C, Liden G. Conversion of furfural in aerobic and anaerobic batch fermentation of glucose by Saccharomyces cerevisiae. J Biosci Bioeng. 1999;87:169–174. doi: 10.1016/s1389-1723(99)89007-0. [DOI] [PubMed] [Google Scholar]
- Taherzadeh MJ, Gustafsson L, Niklasson C, Liden G. Physiological effects of 5-hydroxymethylfurfural on Saccharomyces cerevisiae. Appl Microbiol Biotechnol. 2000;53:701–708. doi: 10.1007/s002530000328. [DOI] [PubMed] [Google Scholar]
- The External RNA Control Consortium. Proposed methods for testing and selecting ERCC external RNA controls. BMC Genomics. 2005;6:150. doi: 10.1186/1471-2164-6-150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villa GP, Bartroli R, Lopez R, Guerra M, Enrique M, Penas M, Rodriquez E, Redondo D, Iglesias I, Diaz I. Microbial transformation of furfural to furfuryl alcohol by Saccharomyces cerevisiae. Acta Biotechnol. 1992;12:509–512. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.