

Abstract
Objective
Telomerase serves as a critical regulator of tissue renewal. Although telomerase activity is inducible in response to various environmental cues, it remains unknown whether telomerase is activated during the inflammatory remodeling underlying atherosclerosis formation. To address this question, we investigated in the present study the regulation of telomerase in macrophages and during atherosclerosis development in LDL-receptor-deficient mice.
Methods and Results
We demonstrate that inflammatory stimuli activate telomerase in macrophages by inducing the expression of the catalytic subunit telomerase reverse transcriptase (TERT). Reporter and chromatin immunoprecipitation assays identified a previously unrecognized NF-κB response element in the TERT promoter, to which NF-κB is recruited during inflammation. Inhibition of NF-κB signaling completely abolished the induction of TERT expression, characterizing TERT as a bona fide NF-κB target gene. Furthermore, functional experiments revealed that TERT-deficiency results in a senescent cell phenotype. Finally, we demonstrate high levels of TERT expression in macrophages of human atherosclerotic lesions and establish that telomerase is activated during atherosclerosis development in LDL-receptor-deficient mice.
Conclusion
These results characterize TERT as a previously unrecognized NF-κB target gene in macrophages and demonstrate that telomerase is activated during atherosclerosis. This induction of TERT expression prevents macrophage senescence and may have important implications for the development of atherosclerosis.
Keywords: Inflammation, Telomerase, Macrophages, Atherosclerosis, NF-κB
INTRODUCTION
Telomeres are repetitive DNA sequences that cap linear chromosomes to prevent degradation, end-to-end fusion, and rearrangement.1, 2 Owing to the biochemistry of incomplete DNA replication, each cycle of DNA replication leaves 50–200 bp at each 3′ end unreplicated. The solution to this ‘end-replication problem’ adopted by most organisms is the use of telomerase, a telomere-specific DNA polymerase that catalyzes the addition of TTAGGG repeats onto telomeric DNA through the activity of the telomerase reverse transcriptase (TERT).3 However, the majority of human adult somatic cells are thought to transcriptionally repress TERT resulting in telomere shortening and cellular senescence.4 In contrast, stem cells and most cancer cell lines constitutively overexpress TERT manifesting high levels of telomerase activity, which confers an apparently indefinite lifespan.5 Therefore, through its action in synthesizing telomeres at the end of the chromosome, constitutive telomerase activity is required to maintain the replicative capacity of progenitor cells during tissue renewal.6, 7
While most adult cells display low basal telomerase activity, increasing evidence indicates that the catalytic subunit TERT is tightly regulated in its expression and inducible in various cell types in response to changes in environmental cues.8 For example, in mature CD4+ and CD8+ T cells telomerase activity is highly induced following antigenic stimulation.9, 10 Similarly, it has recently been described that telomerase is reactivated upon maturation of dendritic cells.11 These observations indicate that telomerase activation may occur at critical stages of lymphoid and myeloid cell activation, and it has been postulated that this inducible telomerase activation may augment their function during immune responses.12–14
Intriguingly, telomerase activity has recently been demonstrated to be increased in neutrophils isolated from coronary atherosclerotic plaques of patients with unstable angina.15 At the cellular level, TERT is expressed in all cell types participating in the atherosclerotic disease process, including macrophages.16, 17, 18 However, whether telomerase is activated during atherosclerosis development and the putative molecular mechanisms underlying this activation remain unknown. In the current study, we sought to define the regulation and function of TERT in macrophages and investigate whether telomerase is activated during the inflammatory remodeling underlying atherosclerosis development.
MATERIALS AND METHODS
Reagents, plasmids and adenoviral constructs
Reagents were obtained from the following suppliers: LPS (Sigma-Aldrich); oxidized and non-oxidized LDL (oxLDL, Intracel); TNFα, IL-6 and IL-1β (Biolegend or R&D Systems). The CMV-DN-IκBα expression vector overexpressing a non-phosphorylatable N-terminal deletion mutant of IκBα was kindly provided by Dr. M. Chaisson.19 The adenovirus overexpressing a dominant-negative IκBαmutant (Ad-ΔN-IκBα) was obtained from Vector Biolabs. The adenovirus overexpressing GFP and the TERT reporter constructs were employed as described.20 The luciferase reporter construct driven by multiple copies of the NF-κB consensus sequence (8xNF-κB-Luc) was obtained from Clontech.
Cell culture
The human U937 monocyte and murine RAW 264.7 macrophage cell lines were obtained from American Type Culture Collection (ATCC) and cultured in DMEM or RPMI-1640 medium supplemented with 4 mM glutamine, 10 % FBS and antibiotics, respectively Differentiation of U937 monocytes into macrophages was induced by incubating monocytes for 16 h with 0.1 μM phorbol-12-myristate-13-acetate (PMA, Sigma-Aldrich) followed by 24 h incubation in complete media prior to treatment with inflammatory stimuli.
Mouse peritoneal macrophages (MPM) were elicited as described and incubated for 24 h in DMEM medium supplemented with 10 % lipoprotein protein-deficient serum (LPDS, Intracel).21 For adenoviral infections, 1.2×105 MPM were seeded in 6-well plates and viral stocks of Ad-GFP or Ad- N-IκBα (25 PFU) were immediately added to the cells. Four hours after infection, MPM were washed in PBS, reincubated in DMEM containing 1 % LPDS overnight, and stimulated as indicated. All experiments were repeated at least three times in duplicates or triplicates using different cell preparations.
Analysis of telomerase activity
Telomerase activity was analyzed using a quantitative Telomerase Detection Kit (Allied Biotech, Inc.) according to the manufacturer’s instructions. PCR reactions were performed using whole-cell extract containing 0.4 μg of protein and 36 cycles of denaturation/annealing/extension steps.
Reverse transcription and quantitative PCR
Transcript levels of target genes were quantified byreal-time RT-PCR as described.20 All primer sequences are provided in Supplemental Table I.
Western blotting
Western blotting was performed using antibodies for TERT (600–401–252, Rockland) or GAPDH (sc-25778, Santa Cruz Biotechnology) as described.20 The antibody detecting human TERT (600–401–252, Rockland) has previously been validated to specifically detect endogenous TERT expression.22
Transient transfection
RAW 264.7 cells were transfected using Lipofectamine2000 (Invitrogen) as described.21 Following transfection, cells were cultured in Opti-MEM supplemented with 10% LPDS overnight prior to stimulation with oxLDL or LPS. Luciferase activities were analyzed using the Dual-Luciferase Reporter Assay system (Promega).
Chromatin immunoprecipitation (ChIP) assays
ChIP assays were performed as described using the following antibodies: anti-p65 (sc-8008 or sc-372), anti-p50 (sc-8414), non-immune mouse or rabbit IgG (sc-2025 or sc-202, respectively), all from Santa Cruz Biotechnology.20, 21 Extracted DNA was PCR-amplified using primer pairs covering the indicated promoter elements (Supplemental Table I). Primer pairs surrounding the NF-κB element in the MCP-1 promoter or a distal region in the β-actin and TERT promoters served as positive and negative controls, respectively.
Immunochemistry
Segments of normal human coronary arteries or arteries containing atherosclerotic lesions were harvested from hearts during autopsy. Tissues were fixed in 4% phosphate-buffered formaldehyde and embedded in paraffin. Transverse sections (8 μm) were collected, and immunohistochemical analysis was performed using a TERT antibody(582005; Calbiochem) followed by counter-staining with hematoxylin as described.23 For confocal microscopy, sections were incubated with antibodies against TERT and CD68 (M0876, Dako Cytomation). Sections were subsequently incubated with Alexa 488-conjugated goat anti-rabbit IgG (A11008, Invitrogen) and Alexa 594-conjugated goat anti-mouse IgG (A11005, Invitrogen). All studies on human tissues were performed with the approval of the University of Kentucky Institutional Review Board.
Analysis of cellular senescence
TERT−/− mice were obtained from The Jackson Laboratory (stock number 005423). MPM from first generation TERT−/− and littermate wildtype mice were elicited as described.21 Cells from individual mice were pooled, plated on 6-well cell culture dishes, and maintained in DMEM media supplemented with 10 % FBS. Senescence-associated β-galactosidase activity was analyzed using the Senescent Cells Histochemical Staining Kit (CS0030, Sigma-Aldrich). Representative pictures were acquired, and the signal intensity was quantified using image analysis software. The signal intensity level of cyan staining was normalized to background level and expressed as the mean signal intensity ± SD. Experiments were repeated at least three times using different macrophage preparations. All studies on animals were performed with the approval of the University of Kentucky Institutional Animal Care and Use Committee.
Analysis of Telomerase Activity in vivo
Male LDL-receptor-deficient (LDLr−/−) mice were obtained from the Jackson Laboratory (stock number 002207). At 12 weeks of age, mice (n=8/group) were randomized to receive either standard laboratory chow diet or a diet supplemented with saturated fat and cholesterol (TD88137; Harlan Teklad). After 12 weeks of diet feeding, mice were sacrificed, and aortic tissues were analyzed for telomerase activity using a PCR-based telomeric repeat amplification protocol (TRAP, TeloTAGGG Telomerase PCR ELISA, Roche Applied Biosystems) as described.20
Statistics
Statistical differences between different groups were assessed by One-Way-Anova followed by Turkey test assays or Student’s t-test. P values <0.05 were considered to be statistically significant. Results are expressed as mean ± SD or SEM as indicated.
RESULTS
Proinflammatory stimuli induce TERT expression and telomerase activity in macrophages
Based on the evidence that TERT confers the catalytic activity of telomerase3 and our previous observation that other components of the telomerase complex are constitutively expressed but not regulated in vascular cells,24 we first investigated the regulation of TERT in macrophages. In these experiments, stimulation of differentiated human U937 macrophages with the proinflammatory mediators LPS, oxLDL, TNFα, or IL-1βsignificantly increased TERT mRNA expression (Fig. 1A) and telomerase activity (Fig. 1B). The induction of TERT transcript levels by LPS, oxLDL, and TNFα paralleled inducible telomerase activity. In contrast, IL-1β only modestly increased TERT mRNA, albeit a potent induction of telomerase activity, pointing to a potentially distinct posttranscriptional induction of telomerase activity by IL-1β. More detailed time-course experiments revealed that the induction of TERT expression and activity by oxLDL was transient (maximal levels after 3 h) (Supplemental Figure I) and dose-dependent (Fig. 1C-E). Furthermore, a comparable induction of TERT mRNA expression and telomerase activity by proinflammatory mediators was observed in primary MPM and human macrophages differentiated in vitro from monocytes of healthy donors (Supplemental Figure II-III). In concert, these studies establish a solid induction of TERT expression and telomerase activity in response to inflammatory stimulation of macrophages.
Figure 1. Inflammatory stimuli induce TERT expression and telomerase activity in human macrophages.



Differentiated human U937 macrophages were treated for 3 h with the indicated proinflammatory stimuli. (A–B) TERT mRNA expression levels (A) and telomerase activity (B) were analyzed in response to treatment with LPS (100 ng/ml), oxidized LDL (oxLDL, 50 μg/ml), TNFα (50 ng/ml), or IL-1β (20 ng/ml). RNase-treatment or heat-inactivation of the LPS-induced samples served as negative controls for the telomerase activity assay. (C-E) Dose-dependent induction of TERT transcript levels (C), protein expression (D), and telomerase activity (E) in response to treatment of macrophages with oxidized LDL. TERT mRNA expression was analyzed by real-time RT-PCR and normalized to transcript levels of the house-keeping gene TBP. Whole cell proteins were analyzed for TERT protein expression by Western blotting. Molecular weight markers are indicated on the left while the arrow on the right indicates TERT expression. Cohybridization for GAPDH was performed to control for equal protein loading. The autoradiographs shown are representative of three independently performed experiments. All quantitative data are presented as mean ± SEM percent increase compared to untreated cells (n=3, * P<0.05).
NF-κB signaling mediates the induction of TERT expression in response to oxidized LDL and LPS
To investigate the transcriptional mechanisms involved in the induction of TERT expression in macrophages, we analyzed 5′-deletion series of a luciferase reporter driven by a 2 kb TERT promoter fragment. As depicted in Fig. 2A, treatment of macrophages with oxLDL significantly induced the activity of the −2.0 kb, but not the +18 bp, TERT promoter. OxLDL-dependent TERT promoter activation was maintained upon 5′-deletion to −776 bp, indicating that a 202 bp promoter region between −776 and −574 conferred the induction of the TERT promoter activity by oxLDL.
Figure 2. Inflammatory stimuli transactivate the TERT promoter through a NF-κB-dependent signaling pathway.



(A) RAW 264.7 macrophages were transfected with reporter constructs driven by the indicated TERT promoter fragments. Transfection of a reporter driven by multiple NF-κB sites (8xNF-κBLuc) served as control. Following transfection, macrophages were incubated in 10 % LPDS and treated with oxidized LDL (oxLDL, 10 μg/ml). Cells were harvested after 24 h and analyzed for luciferase activities. (B and C) RAW 264.7 macrophages were cotransfected with the −776TERTLuc, +18 TERTLuc, or 8xNF-κBLuc reporter construct and either an empty CMV vector or a CMV-DN-IκBβ expression vector. Following transfection, macrophages were cultured in 10 % LPDS and treated with (B) oxidized LDL (oxLDL, 10μg/ml) or (C) LPS (100 ng/ml). Following stimulation, cells were harvested and analyzed for luciferase activities as in (A). Data are expressed as normalized luciferase activities and presented as mean ± SD percent increase over basal TERT promoter activity (* P < 0.05 vs. untreated cells (A) or CMV empty vector (B–C)). (D) Peritoneal macrophages were infected with 25 PFU Ad-GFP or Ad-ΔN-IκBβ and treated with LPS (100 ng/ml) for 24 h. TERT mRNA expression was analyzed and normalized to TFIIB transcript levels. Data are presented as mean ± SD percent increase compared to untreated Ad-GFP-infected cells (n=3, * P < 0.05 vs. vehicle).
Sequence analysis of this TERT promoter region identified a highly conserved NF-κB response element at −592/−580. Moreover, transient transfection of a NF-κB-driven reporter construct (8xNF-κB-Luc) confirmed a potent increase in NF-κB transcriptional activity in response to oxLDL (Fig. 2A lower panel). To more definitively address the role of NF-κB signaling for the activation of the TERT promoter in macrophages, we next performed transactivation assays using a TERT promoter construct (−776TERTLuc) cotransfected with an expression vector overexpressing an IκBα super-repressor (CMV-DN- IκBα). Macrophages overexpressing this mutant completely repressed NF-κB activity (Fig. 2B and C, right panel). As shown in Fig. 2B and C, transfection of this IκBα mutant inhibited the transactivation of the TERT promoter induced by oxLDL or LPS, pointing to a central role of the NF-κB pathway in the activation of the TERT promoter in macrophages.
To confirm a key role of NF-κB signaling for inducible TERT expression, we next overexpressed a phosphorylation-resistant mutant of IκBα using adenoviral-mediated transduction (Ad-ΔN- IκBα). Using this approach, we noted that the induction of TERT mRNA expression by LPS in MPM was completely inhibited in cells overexpressing the IκBα mutant (Fig. 2D). Collectively, these results demonstrate that inflammatory stimuli induce NF-B activation leading to the subsequent transcription of the TERT gene.
NF-κB is recruited to the TERT promoter in macrophages
We next performed ChIP assays using primer pairs that cover the NF-κB site at −592/−580 in the human TERT promoter to determine whether NF- κB subunits are recruited to the endogenous TERT promoter. These assays confirmed that LPS, oxLDL, and TNFα induce the recruitment of both NF- κB subunits to the consensus site in the human TERT promoter (Fig. 3). Similarly, ChIP analysis in primary MPM revealed that oxLDL induces a strong recruitment of p65 and p50 to a region encompassing a NF-κB site at −250/−238 in the murine TERT promoter (Supplemental Figure IV). Taken together, these experiments demonstrate that inflammatory stimuli induce NF- κB recruitment to the endogenous TERT promoter.
Figure 3. Inflammatory stimuli induce NF-κB recruitment to the TERT promoter in macrophages.
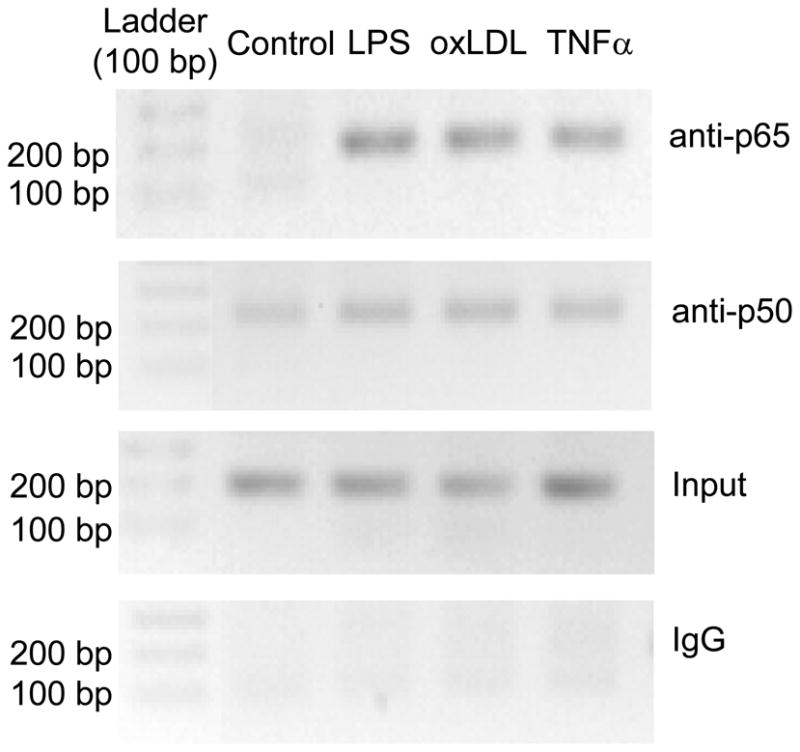
Differentiated U937 macrophages were treated for 2.5 h with LPS (100 ng/ml), oxLDL (50 μg/ml), or TNFα (50 ng/ml). Chromatin immunoprecipitations were performed with antibodies raised against p65 or p50, followed by PCR amplification using primer pairs that cover the proximal NF-κB site in the human TERT promoter. Controls included PCR performed on non-precipitated genomic DNA (Input) or on chromatin immunoprecipitated with non-immune IgG. The agarose gels shown are representative of three independently performed experiments.
Genetic TERT deficiency induces macrophage senescence
To investigate the functional role of inducible TERT expression in the macrophage, we next analyzed phenotypes in cells isolated from mice genetically deficient for the TERT locus. As depicted in Fig. 4A, MPM isolated from first generation TERT-deficient mice significantly upregulated senescence-associated β-galactosidase activity, an established biomarker of replicative senescence.25 Furthermore, TERT-deficient macrophages revealed increased transcript levels of several genes causally involved in replicative senescence,26 including p16, p21, and the retinoblastoma protein (Fig. 4C). Collectively, these experiments indicate that TERT deletion in macrophages induces cellular senescence.
Figure 4. Genetic deletion of TERT results in macrophage senescence.


(A–B) MPM from TERT−/− and littermate TERT+/+ mice were isolated and stained for senescence-associated β-galactosidase activity as described “Materials and Methods”. (A) Representative images showing β-galactosidase activity in TERT+/+ and TERT−/− macrophages (objective magnification ×20). (B) Quantitative analysis of β-galactosidase activity in TERT+/+ and TERT−/− macrophages. Data are presented as mean ± SEM percent increase compared to TERT+/+ macrophages (n=10, * P<0.05). (C) p16, p21, and retinoblastoma protein (RB) transcript levels in wildtype and TERT−/− macrophages. Target gene mRNA expression was analyzed by real-time RT-PCR and normalized to transcript levels of the house-keeping gene TFIIB. Data are presented as mean ± SEM percent increase compared to wildtype macrophages (n=3, * P<0.05).
TERT is expressed in macrophages of human coronary artery atherosclerotic lesions
To corroborate that TERT is expressed in complex atherosclerotic lesions in vivo, we performed immunohistochemical analysis on human coronary arteries. Consistent with earlier reports,18 TERT expression was negligible in normal human coronary arteries (Fig. 5A) but highly expressed in atherosclerotic lesions (Fig. 5B). Particularly high levels of TERT expression were observed in the macrophage-rich shoulder region of advanced atherosclerotic lesions (Fig. 5B). Using immunofluorescent analysis, Fig. 5C depicts representative colocalization of TERT with macrophage CD68 immunoreactivity in the atherosclerotic lesion. These observations confirm that TERT is expressed in macrophages of human atherosclerotic lesions.
Figure 5. TERT is expressed in macrophages of human coronary artery atherosclerotic lesions.


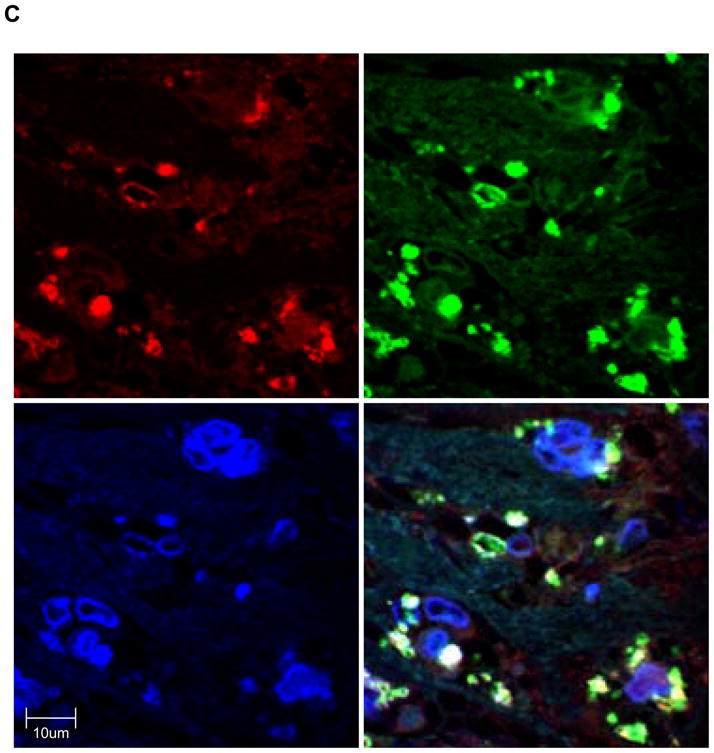
Segments of normal (A) or atherosclerotic human coronary arteries (B) were harvested during autopsy. Paraffin-embedded sections were stained for TERT followed by hematoxylin counterstaining. Black scale bars indicate the respective distance; objective magnifications are ×20 (upper panels) and ×100 (lower panels). Note the lack of TERT staining in normal coronary arteries compared to the abundant immunoreactivity against TERT in the shoulder region of atherosclerotic lesions. (C) Representative immunofluorescence images (objective magnification ×100) of an atherosclerotic lesion from a human coronary artery showing macrophages stained for CD68 (red, left upper panel) and TERT (green, upper right panel). Sections were counterstained with DAPI to visualize nuclei (blue, lower left panel) and merged to show the composite staining (lower right panel). Note the colocalization of CD68 and TERT immunoreactivity (lower right panel, yellow staining).
Telomerase activity is increased during atherosclerosis formation
The increased expression of TERT in macrophages of atherosclerotic lesions raised the question whether TERT expression in atherosclerosis is functional. To address this question, we analyzed TERT activity in aortic tissues isolated from LDLr−/− mice fed a control diet or an atherogenic Western Diet for 12 weeks. Quantification of vascular telomerase activity (Fig. 6, left panel) revealed that the development of atherosclerosis was associated with a more than 13-fold increase in telomerase activity. A typical Southern blot from four representative samples is shown in Fig. 6, right panel, and confirms increased vascular telomerase activity in LDLr−/− mice fed an atherogenic diet. In concert, these observations demonstrate that telomerase is activated during atherosclerosis development, a process driven primarily by macrophages.
Figure 6. Telomerase is activated in LDL-receptor-deficient mice fed an atherogenic diet.

Male LDL-receptor−/− mice were fed a standard laboratory diet or an atherogenic Western diet (n=8/group). After 12 weeks, telomerase activity was analyzed in aortic tissues using the telomeric repeat amplification protocol assay and quantified by ELISA (left panel). Results are presented as mean ± SEM (* P < 0.05 vs. control diet). Characteristic 6-bp telomerase-specific laddering determined by Southern blotting from two representative mice per group is depicted on the right panel.
DISCUSSION
Although it is well established that somatic cells repress TERT resulting in telomere attrition,4 accumulating evidence suggests that many cell types are capable to re-activate telomerase in response to environmental cues9–11. While telomerase activation has been described during adaptive immune responses and enhances immune function of lymphoid cells,12–14 little is known about the regulation and function of telomerase during macrophage inflammation. In the current study, we provide initial evidence that inflammatory mediators induce TERT expression and telomerase activation during macrophage inflammation. TERT is expressed in macrophages of human atherosclerotic lesions, and we further establish that telomerase is activated during experimental atherosclerosis formation. Moreover, functional experiments demonstrate that genetic TERT-deficiency in macrophages induces a phenotype of cellular senescence, pointing to a potentially important role of inducible TERT expression during inflammation.
TERT confers the catalytic activity of telomerase and is primarily regulated through transcriptional mechanisms.3, 8 Consistent with this evidence was our observation that the induction of TERT transcript levels by oxidized LDL, LPS, or TNFα in macrophages paralleled a concomitant telomerase activation. Considering these findings, we sought to define the transcriptional mechanisms underlying inducible TERT expression in the macrophage. Using 5′-deletion analysis, we identified a proximal promoter region that conferred TERT transcription. Sequence analysis of this region revealed a highly conserved NF-κB response element, and inhibition of NF-κB signaling completely abolished TERT expression during macrophage inflammation. Moreover, NF-κB was rapidly recruited to this site in response to inflammatory stimuli, characterizing TERT as a previously unrecognized bona fide NF-κB target gene in macrophages. Although these findings establish that NF-κB signaling induces TERT transcription in macrophages, we cannot exclude the possibility that stimulus-dependent posttranscriptional/posttranslational mechanisms might contribute to telomerase activation in this cell type. Interestingly, IL-1β only modestly increased TERT transcript levels but potently induced telomerase activity, pointing to possible distinct mechanisms governing IL-1β-induced telomerase activation in macrophages. Therefore, potential posttranscriptional/posttranslational mechanisms of telomerase activation by IL-1β in macrophages remain to be further investigated, but may involve Akt-dependent phosphorylation and nuclear import of TERT, which have both been implicated in telomerase activation in certain cell types.27, 28
An intriguing question that arises from the observation that TERT is expressed during macrophage inflammation relates to the regulation of telomerase activity during inflammatory remodeling underlying atherosclerosis formation. Our observations presented herein indicate that macrophages in the shoulder region of advanced human atherosclerotic plaques express high levels of TERT. These findings are consistent with recent data published by Matthews et al.,18 who noted that TERT expression is readily detectable in lesional macrophages. In contrast, other cell types present in the human advanced atherosclerotic lesion, including endothelial cells17 and SMC of the fibrous cap18, are thought to express low levels of TERT. Using LDLr-deficient mice our data extends these observations and identifies a more than 13-fold increase in telomerase activity during lesion formation. Similarly, Narducci et al. recently documented high telomerase activity in neutrophils isolated from coronary artery plaques of patients with unstable angina.15 In concert with our observations in LDLr-deficient mice, these studies provide compelling evidence for the activation of telomerase during atherosclerosis development and support the notion that this telomerase activation originates primarily from macrophages.
Limiting TERT expression results in cellular senescence, a state that is accompanied by changes in cell morphology, function, and gene expression programs.29 In consonance with this notion, we observed that TERT-deficiency in macrophages induces senescence-associated β-galactosidase activity and gene expression of p16, p21, and the retinoblastoma protein, both established biomarkers of cellular senescence.25, 26 This finding indicates that transient TERT expression during inflammation may prevent senescence of the macrophage, potentially to maintain cellular responsiveness during immunity. Indeed, senescent macrophages display altered immune functions,30 and overexpression of TERT in fibroblasts has recently been demonstrated to increase IL-1β, IL-6, and IL-8 expression,31 indicating that TERT may not only prevent senescence but also participate in a feed-forward loop for inflammatory gene expression. Considering that TERT extends lifespan without increasing telomere length while TERT disruption induces senescence without telomere shortening,6, 32, 33 it has been questioned whether the activity of TERT to prevent senescence is dependent on its function to elongate telomeres.34 Alternatively, there is an increasing appreciation of telomere-independent activities of TERT,34 and recent experiments demonstrated that overexpression of TERT activates gene expression programs and induces chromatin remodeling.35, 36 Although the molecular mechanisms underlying these non-canonical functions remain unclear, the broader changes in gene expression affected by TERT point to a regulation of signaling pathways and/or transcriptional mechanisms. With respect to inflammatory signaling, this concept is further supported by the finding that TERT physically interacts with NF-κB,37 which may provide a reasonable mechanism underlying the activation of inflammatory genes by TERT.31
The expression and activation of TERT in macrophages presented in this study provide important justification for further investigating the physiological function of TERT in inflammation. Moreover, the activation of telomerase in atherosclerosis described here, the demonstration that telomere attrition prevents atherosclerosis in apoE-deficient mice,38 combined with the reported telomere shortening in circulating leukocytes of patients with coronary artery disease39 indicate that the association between TERT expression, telomerase activation, and telomere length are more complex than previously anticipated. Continued investigation of the role of TERT in vascular biology will likely provide new insights into how TERT functions during atherosclerosis development.
Supplementary Material
Acknowledgments
a) Acknowledgments – none.
b) Sources of Funding – This work was supported by the American Recovery and Reinvestment Act of 2009 (NIH/NHLBI 1R01HL091924-01A109 to D.B.). F. Gizard and Y. Zhao were supported by Fellowship Grants from the American Heart Association (0725313B and 0815514D, respectively).
Footnotes
Disclosure – The authors have no relationships to disclose that could be perceived as real or apparent conflict(s) of interest.
References
- 1.Blackburn EH. Switching and Signaling at the Telomere. Cell. 2001;106:661–673. doi: 10.1016/s0092-8674(01)00492-5. [DOI] [PubMed] [Google Scholar]
- 2.Fuster JJ, Andres V. Telomere Biology and Cardiovascular Disease. Circ Res. 2006;99:1167–1180. doi: 10.1161/01.RES.0000251281.00845.18. [DOI] [PubMed] [Google Scholar]
- 3.Blackburn EH, Greider CW, Henderson E, Lee MS, Shampay J, Shippen-Lentz D. Recognition and elongation of telomeres by telomerase. Genome. 1989;31:553–560. doi: 10.1139/g89-104. [DOI] [PubMed] [Google Scholar]
- 4.Harley CB, Futcher AB, Greider CW. Telomeres shorten during ageing of human fibroblasts. Nature. 1990;345:458–460. doi: 10.1038/345458a0. [DOI] [PubMed] [Google Scholar]
- 5.Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu CP, Morin GB, Harley CB, Shay JW, Lichtsteiner S, Wright WE. Extension of life-span by introduction of telomerase into normal human cells. Science. 1998;279:349–352. doi: 10.1126/science.279.5349.349. [DOI] [PubMed] [Google Scholar]
- 6.Flores I, Cayuela ML, Blasco MA. Effects of telomerase and telomere length on epidermal stem cell behavior. Science. 2005;309:1253–1256. doi: 10.1126/science.1115025. [DOI] [PubMed] [Google Scholar]
- 7.Lee HW, Blasco MA, Gottlieb GJ, Horner JW, 2nd, Greider CW, DePinho RA. Essential role of mouse telomerase in highly proliferative organs. Nature. 1998;392:569–574. doi: 10.1038/33345. [DOI] [PubMed] [Google Scholar]
- 8.Ducrest AL, Szutorisz H, Lingner J, Nabholz M. Regulation of the human telomerase reverse transcriptase gene. Oncogene. 2002;21:541–552. doi: 10.1038/sj.onc.1205081. [DOI] [PubMed] [Google Scholar]
- 9.Weng NP, Levine BL, June CH, Hodes RJ. Regulated expression of telomerase activity in human T lymphocyte development and activation. J Exp Med. 1996;183:2471–2479. doi: 10.1084/jem.183.6.2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hathcock KS, Weng N-p, Merica R, Jenkins MK, Hodes R. Cutting Edge: Antigen-Dependent Regulation of Telomerase Activity in Murine T Cells. J Immunol. 1998;160:5702–5706. [PubMed] [Google Scholar]
- 11.Ping L, Asai A, Okada A, Isobe K, Nakajima H. Dramatic increase of telomerase activity during dendritic cell differentiation and maturation. J Leukoc Biol. 2003;74:270–276. doi: 10.1189/jlb.0103014. [DOI] [PubMed] [Google Scholar]
- 12.Herrera E, Samper E, Martin-Caballero J, Flores JM, Lee HW, Blasco MA. Disease states associated with telomerase deficiency appear earlier in mice with short telomeres. EMBO J. 1999;18:2950–2960. doi: 10.1093/emboj/18.11.2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ujike-Asai A, Okada A, Du Y, Maruyama M, Yuan X, Ishikawa F, Motoo Y, Isobe K, Nakajima H. Large defects of type I allergic response in telomerase reverse transcriptase knockout mice. J Leukoc Biol. 2007;82:429–35. doi: 10.1189/jlb.1006638. [DOI] [PubMed] [Google Scholar]
- 14.Herrera E, Martinez AC, Blasco MA. Impaired germinal center reaction in mice with short telomeres. Embo J. 2000;19:472–481. doi: 10.1093/emboj/19.3.472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Narducci ML, Grasselli A, Biasucci LM, Farsetti A, Mule A, Liuzzo G, La Torre G, Niccoli G, Mongiardo R, Pontecorvi A, Crea F. High telomerase activity in neutrophils from unstable coronary plaques. J Am Coll Cardiol. 2007;50:2369–2374. doi: 10.1016/j.jacc.2007.08.048. [DOI] [PubMed] [Google Scholar]
- 16.Minamino T, Kourembanas S. Mechanisms of telomerase induction during vascular smooth muscle cell proliferation. Circ Res. 2001;89:237–243. doi: 10.1161/hh1501.094267. [DOI] [PubMed] [Google Scholar]
- 17.Minamino T, Miyauchi H, Yoshida T, Ishida Y, Yoshida H, Komuro I. Endothelial Cell Senescence in Human Atherosclerosis: Role of Telomere in Endothelial Dysfunction. Circulation. 2002;105:1541–1544. doi: 10.1161/01.cir.0000013836.85741.17. [DOI] [PubMed] [Google Scholar]
- 18.Matthews C, Gorenne I, Scott S, Figg N, Kirkpatrick P, Ritchie A, Goddard M, Bennett M. Vascular Smooth Muscle Cells Undergo Telomere-Based Senescence in Human Atherosclerosis: Effects of Telomerase and Oxidative Stress. Circ Res. 2006;99:156–164. doi: 10.1161/01.RES.0000233315.38086.bc. [DOI] [PubMed] [Google Scholar]
- 19.Scatena M, Almeida M, Chaisson ML, Fausto N, Nicosia RF, Giachelli CM. NF-kappa B Mediates alpha vbeta 3 Integrin-induced Endothelial Cell Survival. J Cell Biol. 1998;141:1083–1093. doi: 10.1083/jcb.141.4.1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gizard F, Nomiyama T, Zhao Y, Findeisen HM, Heywood EB, Jones KL, Staels B, Bruemmer D. The PPARalpha/p16INK4a pathway inhibits vascular smooth muscle cell proliferation by repressing cell cycle-dependent telomerase activation. Circ Res. 2008;103:1155–1163. doi: 10.1161/CIRCRESAHA.108.186205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nakamachi T, Nomiyama T, Gizard F, Heywood EB, Jones KL, Zhao Y, Fuentes L, Takebayashi K, Aso Y, Staels B, Inukai T, Bruemmer D. PPARalpha agonists suppress osteopontin expression in macrophages and decrease plasma levels in patients with type 2 diabetes. Diabetes. 2007;56:1662–1670. doi: 10.2337/db06-1177. [DOI] [PubMed] [Google Scholar]
- 22.Wu YL, Dudognon C, Nguyen E, Hillion J, Pendino F, Tarkanyi I, Aradi J, Lanotte M, Tong JH, Chen GQ, Segal-Bendirdjian E. Immunodetection of human telomerase reverse-transcriptase (hTERT) re-appraised: nucleolin and telomerase cross paths. J Cell Sci. 2006;119:2797–2806. doi: 10.1242/jcs.03001. [DOI] [PubMed] [Google Scholar]
- 23.Nomiyama T, Nakamachi T, Gizard F, Heywood EB, Jones KL, Ohkura N, Kawamori R, Conneely OM, Bruemmer D. The NR4A Orphan Nuclear Receptor NOR1 Is Induced by Platelet-derived Growth Factor and Mediates Vascular Smooth Muscle Cell Proliferation. J Biol Chem. 2006;281:33467–33476. doi: 10.1074/jbc.M603436200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ogawa D, Nomiyama T, Nakamachi T, Heywood EB, Stone JF, Berger JP, Law RE, Bruemmer D. Activation of peroxisome proliferator-activated receptor gamma suppresses telomerase activity in vascular smooth muscle cells. Circ Res. 2006;98:e50–59. doi: 10.1161/01.RES.0000218271.93076.c3. [DOI] [PubMed] [Google Scholar]
- 25.Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O, Peacocke M, Campisi J. A Biomarker that Identifies Senescent Human Cells in Culture and in Aging Skin in vivo. Proc Natl Acad Sci U S A. 1995;92:9363–9367. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alcorta DA, Xiong Y, Phelps D, Hannon G, Beach D, Barrett JC. Involvement of the cyclin-dependent kinase inhibitor p16 (INK4a) in replicative senescence of normal human fibroblasts. Proc Natl Acad Sci U S A. 1996;93:13742–13747. doi: 10.1073/pnas.93.24.13742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kang SS, Kwon T, Kwon DY, Do SI. Akt Protein Kinase Enhances Human Telomerase Activity through Phosphorylation of Telomerase Reverse Transcriptase Subunit. J Biol Chem. 1999;274:13085–13090. doi: 10.1074/jbc.274.19.13085. [DOI] [PubMed] [Google Scholar]
- 28.Liu K, Hodes RJ, Weng N-p. Cutting Edge: Telomerase Activation in Human T Lymphocytes Does Not Require Increase in Telomerase Reverse Transcriptase (hTERT) Protein But Is Associated with hTERT Phosphorylation and Nuclear Translocation. J Immunol. 2001;166:4826–4830. doi: 10.4049/jimmunol.166.8.4826. [DOI] [PubMed] [Google Scholar]
- 29.Campisi J. Senescent Cells, Tumor Suppression, and Organismal Aging: Good Citizens, Bad Neighbors. Cell. 2005;120:513–522. doi: 10.1016/j.cell.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 30.Lloberas J, Celada A. Effect of aging on macrophage function. Exp Gerontol. 2002;37:1325–1331. doi: 10.1016/s0531-5565(02)00125-0. [DOI] [PubMed] [Google Scholar]
- 31.Kanzaki Y, Onoue F, Sakurai H, Ide T. Telomerase upregulates expression levels of interleukin (IL)-1alpha, IL-1beta, IL-6, IL-8, and granulocyte-macrophage colony-stimulating factor in normal human fibroblasts. Biochem Biophys Res Commun. 2003;305:150–154. doi: 10.1016/s0006-291x(03)00717-4. [DOI] [PubMed] [Google Scholar]
- 32.Sarin KY, Cheung P, Gilison D, Lee E, Tennen RI, Wang E, Artandi MK, Oro AE, Artandi SE. Conditional telomerase induction causes proliferation of hair follicle stem cells. Nature. 2005;436:1048–1052. doi: 10.1038/nature03836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Masutomi K, Yu EY, Khurts S, Ben-Porath I, Currier JL, Metz GB, Brooks MW, Kaneko S, Murakami S, DeCaprio JA, Weinberg RA, Stewart SA, Hahn WC. Telomerase maintains telomere structure in normal human cells. Cell. 2003;114:241–253. doi: 10.1016/s0092-8674(03)00550-6. [DOI] [PubMed] [Google Scholar]
- 34.Chang S, DePinho RA. Telomerase extracurricular activities. Proc Natl Acad Sci U S A. 2002;99:12520–12522. doi: 10.1073/pnas.212514699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Choi J, Southworth LK, Sarin KY, Venteicher AS, Ma W, Chang W, Cheung P, Jun S, Artandi MK, Shah N, Kim SK, Artandi SE. TERT promotes epithelial proliferation through transcriptional control of a Myc- and Wnt-related developmental program. PLoS Genet. 2008;4:e10. doi: 10.1371/journal.pgen.0040010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Masutomi K, Possemato R, Wong JMY, Currier JL, Tothova Z, Manola JB, Ganesan S, Lansdorp PM, Collins K, Hahn WC. The telomerase reverse transcriptase regulates chromatin state and DNA damage responses. Proc Natl Acad Sci U S A. 2005;102:8222–8227. doi: 10.1073/pnas.0503095102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Akiyama M, Hideshima T, Hayashi T, Tai Y-T, Mitsiades CS, Mitsiades N, Chauhan D, Richardson P, Munshi NC, Anderson KC. Nuclear Factor-{kappa}B p65 Mediates Tumor Necrosis Factor {alpha}-induced Nuclear Translocation of Telomerase Reverse Transcriptase Protein. Cancer Res. 2003;63:18–21. [PubMed] [Google Scholar]
- 38.Poch E, Carbonell P, Franco S, Diez-Juan A, Blasco MA, Andres V. Short telomeres protect from diet-induced atherosclerosis in apolipoprotein E-null mice. FASEB J. 2003:03-0710fje. doi: 10.1096/fj.03-0710fje. [DOI] [PubMed] [Google Scholar]
- 39.Brouilette SW, Moore JS, McMahon AD, Thompson JR, Ford I, Shepherd J, Packard CJ, Samani NJ. Telomere length, risk of coronary heart disease, and statin treatment in the West of Scotland Primary Prevention Study: a nested case-control study. The Lancet. 2007;369:107–114. doi: 10.1016/S0140-6736(07)60071-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.