

Abstract
The yeast and fungal prions determine heritable and infectious traits, and are thus genes composed of protein. Most prions are inactive forms of a normal protein as it forms a self-propagating filamentous β – sheet - rich polymer structure called amyloid. Remarkably, a single prion protein sequence can form two or more faithfully inherited prion variants, in effect alleles of these genes. What protein structure explains this protein-based inheritance? Using solid-state NMR, we showed that the infectious amyloids of the prion domains of Ure2p, Sup35p and Rnq1p have an in-register parallel architecture. This structure explains how the amyloid filament ends can template the structure of a new protein as it joins the filament.
The yeast prions [PSI+] and [URE3] are not found in wild strains, indicating they are a disadvantage to the cell. Moreover, the prion domains of Ure2p and Sup35p have functions unrelated to prion formation, indicating that these domains are not present for the purpose of forming prions. Indeed, prion forming ability is not conserved, even within S. cerevisiae, suggesting that the rare formation of prions is a disease. The prion domain sequences generally vary more rapidly in evolution than does the remainder of the molecule, producing a barrier to prion transmission, perhaps selected in evolution by this protection.
Keywords: prion, amyloid, in-register parallel structure
Scrapie is a uniformly lethal neurodegenerative disease of sheep that has been known in Europe since at least the 18th century (Parry, 1983) and perhaps much longer in China (Wickner, 2005). Its transmissibility to sheep and goats (Cuille & Chelle, 1936, Cuille & Chelle, 1939) and the typical brain pathology gave it, and similar diseases of humans, cattle, deer and elk, the name transmissible spongiform encephalopathy (TSE). The recent epidemic of the bovine form of this disease (Wilesmith, 1988), and its fortunately rare transmission to humans (Will, et al., 1996) spotlighted these conditions, but their relation to non-infectious amyloid diseases such as Alzheimer's disease, Parkinson's disease, and type II diabetes may ultimately prove even more important. Early studies of the infectious agent were hampered by the year+ incubation periods and the expense of buying a flock of sheep for each experiment! The infection of mice with the scrapie agent improved matters (Chandler, 1961), but not until recent tissue culture infection methods (Klohn, et al., 2003) has the agent assay truly become simple.
Historically, the extreme UV-resistance of the scrapie agent first implied that any essential nucleic acid component must be much smaller than even the small RNA phages (Alper, et al., 1966, Alper, et al., 1967, Bellinger-Kawakara, et al., 1987), and led to the suggestion that the infectious agent had no essential nucleic acid (Alper, et al., 1967). Griffith then proposed what is essentially the modern 'protein-only' model (Griffith, 1967). The purification of the infectious agent showed a single major protein species, named PrP (Bolton, et al., 1982, Diringer, et al., 1983), and Prusiner coined the term "prion" to mean an "infectious protein", transmitting an infection without an essential nucleic acid component (Prusiner, 1982).
PrP is encoded by a chromosomal gene (Basler, et al., 1986, Locht, et al., 1986), the same (Carlson, et al., 1986, Hunter, et al., 1987) as the Sinc gene of mice, shown much earlier to control scrapie incubation period (Dickinson, et al., 1968). The finding that the gene affected in familial human TSE disease that produces infectious material for monkeys (Masters, et al., 1981) was the gene encoding PrP (Hsiao, et al., 1989), and that PrP determines the species barrier (Prusiner, et al., 1990) were important arguments for the prion model.
Amyloid formed from recombinant PrP is minimally infectious unless other lipid and nucleic acid components are included (Legname, et al., 2004, Deleault, et al., 2007, Makarava, et al., 2010, Wang, et al., 2010), leaving some residue of doubt about whether the TSE are truly prions. Clearly, PrP is the determinant of specificity and essential for infectivity, but it remains possible that minor RNA or lipid components contribute.
In contrast, the initial genetic evidence for yeast prions was convincing (Wickner, 1994, Masison, et al., 1997): unlike nucleic acid replicons, 1) curing of prions is reversible, meaning that they arise de novo in a cell presumed to be devoid of the putative replicon; 2) overproduction of the prion protein increases the frequency of prion generation; and 3) the propagation of the prion requires a chromosomal gene (encoding the prion protein) whose mutant phenotype is similar to that of the presence of the prion. These properties posited for a yeast prion were true of [URE3] and [PSI+] as prions of Ure2p and Sup35p, respectively (Wickner, 1994). None of the three genetic properties posited for yeast prions were known then (or even now) for TSEs. Similar evidence was obtained for the [Het-s] non-chromosomal gene of the filamentous fungus Podospora anserina being a prion of the HET-s protein (Coustou, et al., 1997). Amyloid formed in vitro from recombinant prion protein efficiently transmits the corresponding prion to yeast or fungal cells (Maddelein, et al., 2002, King & Diaz-Avalos, 2004, Tanaka, et al., 2004, Brachmann, et al., 2005, Patel & Liebman, 2007).
Studies of TSEs have revealed that many "strains" of the infectious agent could show quite distinct properties in the identical host (Bruce, 1993). One source of resistance by many to the proposals that the TSEs are infectious proteins has been the striking and consistent differences in the incubation periods, tissue distributions and other features of the disease produced by different strains of scrapie, and the lack of a plausible mechanism by which a protein could transmit its conformation from one molecule to another. Not only were there no known examples of heritable (or transmissible) traits being encoded in anything but nucleic acid sequence, but it was not clear how protein-only transmission of information could occur. For example, in reviewing the TSEs and proposing the term "prion" to describe them Prusiner (Prusiner, 1982) proposed that, if indeed the TSEs were cases of a "protein-only" infectious agent, the mechanism might be a) reverse translation, b) protein-dependent protein synthesis or c) a protein’s induction of transcription of its own gene. None of these proved to be the mechanism of the mammalian prion. While clear evidence of differences in protein conformation in different prion strains was obtained (Bessen & Marsh, 1994, Caughey, et al., 1998), no mechanism for the faithful propagation of such conformational differences was proposed (Fig. 1).
Fig. 1.
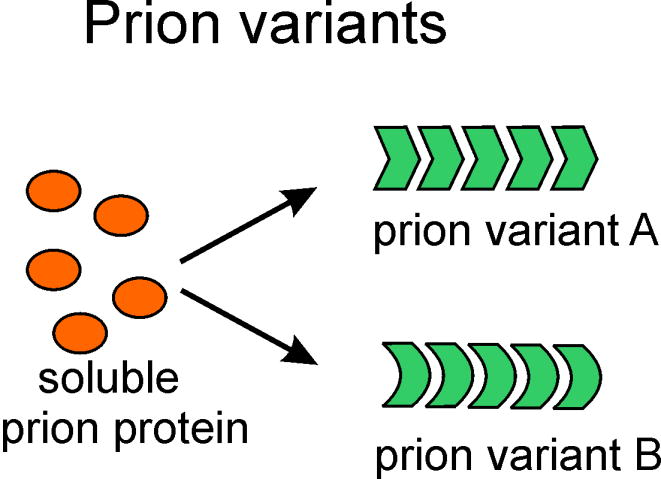
One protein sequence can produce several heritable prion variants. How can a protein structure be self-propagating? We have suggested that the in-register parallel β-sheet architecture, compatible with many different structures, is uniquely able to explain protein structure templating.
With the discovery of yeast prions (Wickner, 1994), and the existence of prion strains (or "variants") in yeast (Derkatch, et al., 1996), it was clear that the variant phenomenon was general, and did not reflect a nucleic acid component of the prions. Yeast prion variants are also based on different structures (see below) -amyloids- but what are these structures, and how are they faithfully propagated?
The spectrum of prions in yeast and fungi (Table 1)
Table 1.
Yeast and fungal prions.
| Prion | Protein | Normal protein function | Prion manifestation | Refs. |
|---|---|---|---|---|
| [URE3] | Ure2p | Nitrogen catabolism: In the presence of a rich N source, Ure2p binds the positive transcription factor Gln3p, keeping it in the cytoplasm. | Inappropriate derepression of enzymes and transporters for the utilization of poor nitrogen sources. | (Lacroute, 1971) (Wickner, 1994)(Turoscy & Cooper, 1987) |
| [PSI+] | Sup35p | Translation termination, mRNA turnover | Increased readthrough of translation termination codons | (Cox, 1965)(Wickner, 1994) |
| [PIN+] | Rnq1p | none known | Increased frequency of generation of [PSI+] and [URE3] prions. | (Derkatch, et al., 1997)(Sondheimer & Lindquist, 2000) (Derkatch, et al., 2001) |
| [β] | Prb1p | Vacuolar protease PrB; prion form is active PrB, not amyloid | Poor sporulation, poor survival in stationary phase | (Zubenko, et al., 1982)(Roberts & Wickner, 2003) |
| [SWI+] | Swi1p | subunit of SWI-SNF chromatin remodeling complex | Partially defective Swi1- phenotype such as poor growth on raffinose, galactose or glycerol | (Du, et al., 2008) |
| [MCA] | Mca1p | Metacaspase homolog. ?apoptosis? | (Nemecek, et al., 2009) | |
| [OCT+] | Cyc8p | Transcription co-repressor | Derepressed invertase, Cyc2p, other proteins | (Patel, et al., 2009) |
| [MOT3] | Mot3p | Transcription repressor of genes derepressed under anaerobiasis | Derepression of "anaerobic genes". | (Alberti, et al., 2009) |
| [GAR] | Pma1p, Std1p | plasma membrane proton pump; glucose signalling | Resistance to glucose-repression | (Brown & Lindquist, 2009) |
| [Het-s] | HET-s | No known non-prion function | Prion form necessary for heterokaryon incompatibility | (Coustou, et al., 1997) |
The yeast amyloid-based prions include [URE3], a prion of Ure2p, a regulator of nitrogen catabolism (Fig. 2)(Wickner, 1994); [PSI+], a prion of Sup35p, a subunit of the translation termination factor (Wickner, 1994); [PIN+], a prion of Rnq1p of unknown normal function (Derkatch, et al., 2001); [SWI+], a prion of Swi1p, a component of the SWI-SNF chromatin remodeling complex (Du, et al., 2008); [MCA], a prion of MCA1p, the yeast metacaspase homolog (Nemecek, et al., 2009); [OCT+], a prion of Cyc8, a subunit of the Tup1-Cyc8 transcription repressor (Patel, et al., 2009); and [MOT3] a prion of Mot3p, a transcription factor (Alberti, et al., 2009). The yeast [β] prion is a self-propagating active vacuolar protease B, which can be essential for the activation of its inactive precursor protein (Zubenko, et al., 1982, Roberts & Wickner, 2003). [Het-s], a prion of the HET-s protein of the filamentous fungus Podospora anserina (Coustou, et al., 1997), is a mediator of heterokaryon incompatibility, and is unusual in that it functions in its prion form, but has no known function in its non-prion form.
Fig. 2.

Yeast and fungal prions. The most widely studied prions of S. cerevisiae and Podospora anserina are diagramed. For the yeast prion proteins Ure2p and Sup35p, prion amyloid formation prevents normal function, thus producing a phenotype. The normal function of Rnq1p is unknown. The normal function of the HET-s protein is its prion function.
Biology of yeast and fungal prions
Species barriers
Mammalian prions of one species may be unable to infect another species, or only do so with dramatically increased incubation period (reviewed in (Collinge & Clarke, 2007)). This 'species barrier' is a result of sequence differences between the PrP proteins of the respective species (Prusiner, et al., 1990). A similar species barrier between yeast species has likewise been observed (Chernoff, et al., 2000, Kushnirov, et al., 2000, Santoso, et al., 2000, Chen, et al., 2007, Edskes, et al., 2009).
Prion variants
Prion variants of [PSI+], [URE3] and [PIN+] were first observed as differences in intensity of the prion phenotype (strong or weak) and in the stability in propagation of the prion during growth (Derkatch, et al., 1996, Schlumpberger, et al., 2001, Bradley, et al., 2002, Brachmann, et al., 2005). However, [PSI+] variants also differ in whether they can propagate in cells expressing a given mutant of the prion domain (King, 2001), a result that corresponds logically to the species barrier in mammalians. Variants of [URE3] show dramatic differences in the actual species barrier among different species of Saccharomyces (Edskes, et al., 2009). Chaperones are important in prion propagation (reviewed by (Sharma & Masison, 2009)) and these effects also vary depending on the prion variant (Kushnirov, et al., 2000). Different [PSI+] variant amyloids show different distribution along the prion domain sequence of rates of hydrogen-deuterium exchange, indicating that as in mammals, yeast prion variants are based on self-propagating structural variants (Toyama, et al., 2007).
Are yeast and fungal prions a help or a hindrance?
Heterokaryon incompatibility of fungi is like transplantation incompatibility in mammals, preventing vegetative fusion of genetically distinct fungal strains, apparently for the purpose of blocking the spread of fungal viruses (Saupe, et al., 2000). Since the [Het-s] prion mediates heterokaryon incompatibility in Podospora (Coustou, et al., 1997), it was proposed that it may actually be a beneficial prion (Wickner, 1997). This theme was applied to prions of yeast when it was noted that certain [PSI+] strains were more stress-resistant than the corresponding [psi-] strain (Eaglestone, et al., 1999). However, further observations did not support a general stress-resistance of [PSI+] cells (True & Lindquist, 2000). Indeed, in a screen of many phenotypes, there was, except for hypersensitivity to 5 mM Zn2+, no phenotype consistently imparted by being [PSI+], and in most cases, being [PSI+] was detrimental (True & Lindquist, 2000). Finally, in a re-examination using the same strains it was found that less than half of the differences were reproducible (Namy, et al., 2008). Even if these experiments had shown that a prion conferred a consistent benefit for yeast under some specific culture condition, this would not prove that the prion was of benefit to yeast unless a) this condition could be shown to be a significant part of the niche of this species in the wild, b) yeast carrying this prion could be isolated from that niche, and c) this benefit outweighed any detriment that would be encountered under other conditions (Partridge & Barton, 2000). It seems clear that [PSI+] should produce inappropriate readthrough of translation termination codons of many genes, and thus cause the production of many non-functional or mis-functional proteins, so it is not surprising that no consistent advantage of being [PSI+] has been detected.
Infectious elements, such as viruses, bacteria or prions, are easily found in the wild even if they are lethal, simply because their rate of spread out-runs the damage they do to their hosts. For example, chronic wasting disease, a prion infection of deer and elk, is found in several percent of animals in large areas of the US, and scrapie of sheep has been a problem for at least centuries (Parry, 1983), if not millenia (Wickner, 2005). Certainly if an infectious element is a benefit to its host, and is stable (like the [PSI+] and [URE3] variants claimed to help their hosts) it will quickly spread in the population as infectivity and benefit work together. The mitochondria, which originated as a bacterium infecting another cell, is an example. Thus, if an infectious entity is NOT found in wild isolates, one can then safely conclude that it is a detriment to its host. We surveyed seventy wild yeast strains isolated from several continents and many different environments, and none had [URE3] or [PSI+], indicating that both are diseases (Nakayashiki, et al., 2005).
It is suggested that [PSI+] formation may be induced under certain stress conditions to relieve the cells of the stress by altering translation (Tyedmers, et al., 2008). However, it was found that under four of the six stress conditions inducing [PSI+] formation, [PSI+] was a detriment to the cells, rather than helping them survive. This indicates that the [PSI+] induction was not an adaptive response, but may have been due to chaperones being occupied with dealing with the stress, and were not available to prevent prion formation. In the two other [PSI+] “induction” conditions where the presence of [PSI+] was reported to favor cell survival, the authors failed to rule out the possibility that this condition was not inducing [PSI+] appearance but was simply selecting [PSI+] cells already in the population before the stress, thus explaining the modestly increased fraction of surviving cells that were [PSI+].
If the prion domains of prion proteins had no function other than prion formation, and were conserved through a long span of evolution, then one could argue that prion formation may be conserved for some purpose that we do not know about. However, prion domains of Ure2p and Sup35p each have non-prion functions and the ability to form prions is not generally conserved. The prion domain of Ure2p is important for the nitrogen regulation function of the molecule in that Ure2p is rapidly degraded if it lacks this region and nitrogen catabolite repression is leaky (Shewmaker, et al., 2007). Likewise, the prion domain of Sup35p interacts with the polyA binding protein and is necessary for the normal shortening of the polyA structure of mRNAs and thus for normal mRNA turnover (Hoshino, et al., 1999, Hosoda, et al., 2003). Moreover, the prion-forming ability of Sup35p is not well conserved even within Saccharomyces cerevisiae isolates and fully 25% of strains examined have a large deletion that prevents them from becoming [PSI+] (Resende, et al., 2003). The Ure2p prion domain is apparently conserved within S. cerevisiae (Edskes & Wickner, 2002), but S. paradoxus is unable to form the [URE3] prion (Talarek, et al., 2005), and the Ure2p of S. castelii is unable to convert to [URE3], at least in S. cerevisiae (Edskes, et al., 2009). Thus prion formation is sporadic, and not well conserved, and the prion domains have non-prion functions, indicating that prions are molecular malfunctions.
Human populations are polymorphic for residue 129 of PrP with about half of alleles having Val and half a Met residue. Heterozygous individuals rarely get either spontaneous or infectious CJD, leading Mead et al. to propose that this polymorphism arose to protect humans from this disease in an era when cannibalism was not rare (Mead, et al., 2003). The Ure2p and Sup35p prion domains also vary more rapidly than does the remainder of the molecule (although both prion domains have non-prion functions), and this results in a species barrier between the intermating species of the Saccharomyces genus (Chen, et al., 2007, Edskes, et al., 2009). We suggested that the Q/N - rich prion domains are preserved in evolution because they have functions (protein stabilization, mRNA turnover), but that, as in the human case, they vary rapidly (within limits imposed by these functions) to prevent prion infection (Edskes, et al., 2009).
The [Het-s] prion of Podospora anserina constitutes an interesting comparison with the yeast prions. [Het-s] is found in 80% of wild het-s strains (Dalstra, et al., 2003), suggesting that it is beneficial to its host, as mentioned above. However, in addition to heterokaryon incompatibility, [Het-s] also determines a 'meiotic drive' system that promotes the inheritance of the prion-forming allele, het-s, over the het-S allele (Dalstra, et al., 2003). Meiotic drive is a phenomenon in which a chromosomal gene promotes its own inheritance by inactivating germ cells carrying the opposite allele. The meiotic drive allele may be quite detrimental to the organism, but it can often spread in the wild by cheating on meiosis. Thus, it is not clear whether the [Het-s] prion is widespread because heterokaryon incompatibility is important to Podospora, or because of the meiotic drive effect. In either case, the HET-s protein has evolved to be a prion with a specific effect on the cell, and thus a specific structure.
The [β] prion is not an amyloid but is just the active form of vacuolar protease B, normally synthesized as an inactive precursor which is activated by cleavage by protease A (Jones, 1991). In the absence of protease A, active protease B can activate its own precursor (Zubenko, et al., 1982)(Roberts & Wickner, 2003). A cell which lacks active protease B remains so, but can be 'infected' by transfer of cytoplasm from a cell that has it. Thus, in the absence of protease A, the active mature form of protease B is a prion, called [β] (Roberts & Wickner, 2003). [β] is important for cell survival in stationary phase and for sporulation and meiosis, both processes requiring protein turnover (Roberts & Wickner, 2003). This is clearly a case of a beneficial prion.
Shuffled prion domains can still form prions
The N-terminal prion domain of Ure2p is unstructured in the native form and changes to β-sheet in the formation of the infectious amyloid (Taylor, et al., 1999, Baxa, et al., 2003, Baxa, et al., 2005, Pierce, et al., 2005); we assume that the prion domain of Sup35p is likewise unstructure in the native form, and it certainly is largely β-sheet in the prion form (e.g. (King, et al., 1997)). The C-terminal nitrogen regulation domain of Ure2p (residues ~94-354) does not change substantially (Baxa, et al., 2002, Bai, et al., 2004, Loquet, et al., 2009), and the C-terminal part of Sup35p is likewise unaltered by amyloid formation (Krzewska, et al., 2007).
In order to determine whether there were specific sequences in the Ure2p or Sup35p prion domains which were essential for prion formation, we shuffled each domain, without altering their amino acid content, inserted the shuffled domain in place of the normal prion domain, and selected prion-containing cells, either those arising spontaneously, or those induced by overproduction of the shuffled prion domain. We were surprised to find that each of the 5 shuffled Ure2ps and each of the 5 shuffled Sup35p proteins could become a prion in yeast (Ross, et al., 2004, Ross, et al., 2005). This remarkable result would appear, on the surface, to contradict the fact that even a single amino acid change can, in some cases, constitute a 'species barrier' and block propagation of a prion from one protein to another. For example, a single amino acid change in the Sup35p prion domain can block propagation of the usual [PSI+] from the wild type Sup35 (Doel, et al., 1994), but the mutant Sup35p can form its own [PSI+] (Kochneva-Pervukhova, et al., 1998).
Amyloid is β sheet - rich, with the β strands perpendicular to the long axis of the filaments, but amyloid can have any of four types of architecture: 1) antiparallel, 2) parallel in-register, 3) parallel out-of-register or 4) β-helix (Fig 3A). Except for the parallel in register structure (Fig. 3B), different amino acid residues are apposed and interact with each other. For such a structure to require near identical sequence for propagation, there would have to be some relation between the apposed residues, perhaps complementarity (large with small, positive with negative charge) or identity (both hydrophobic or both hydrophilic). This relation would almost certainly be lost on shuffling the amino acid sequence. In contrast, the parallel in-register architecture pairs identical residues of different molecules. In this case, shuffling the sequence would not prevent identical residues from having the same interactions; the only difference is that they would be ordered in a different sequence (Fig. 3B). Such a structure would be favored by a peptide composed of residues that have favorable interactions with themselves, such as hydrophobic or hydrophilic residues, but would not be expected if charged residues were abundant. In fact, charged residues are scarce in the known prion domains. Postulating a parallel in-register architecture for prion amyloids is not without precedent, as this structure has been demonstrated for amyloid of the Aβ peptide and Tau involved in Alzheimer's disease, the amyloid of amylin involved in type II diabetes mellitus, alpha-synuclein involved in several neurodegenerative diseases (Balbach, et al., 2002, Der-Sarkissian, et al., 2003, Luca, et al., 2007, Margittai & Langen, 2008).
Fig. 3.

A. The four types of β-sheet architecture. Only the in-register parallel form results in a labeled atom in a residue of one molecule being about 4.7 angstroms from the same atom of the same residue of an adjacent molecule. The solid-state NMR experiments measure this distance. The large dot represents a single amino acid residue labeled with 13C at its carbonyl carbon. B. If a prion domain can be shuffled and still be a prion, it is likely to have an in-register parallel architecture. The specificity of prion propagation requires that interacting residues have some relation to each other. If the architecture is antiparallel, β helix or out of register parallel, that relation would have to be one of non-identity in most cases. Shuffling the sequence would disrupt this relation. However, in the case of a parallel in-register β sheet, it is identical residues whose side-chains interact to determine the sequence-specific prion propagation. Shuffling the sequence does not prevent this same interaction, although the residues will be in a different sequence, identical residues can still interact to produce an amyloid filament.
Verification of parallel in-register β-sheet architecture of yeast prion amyloids
Because of their non-crystalline nature, large size and insolubility, solid-state nuclear magnetic resonance (ssNMR) is the best method to address the structure of amyloid filaments (Tycko, 2006). Labelling the carbonyl carbon of specific groups of residues with 13C provides a probe of both secondary structure of the labeled residues and of their distance from the next closest labeled residue. Compared to random coil, residues in β-sheet structure show a shift in carbonyl 13C resonant frequencies to lower values, while those in α-helix shift to higher values (Wishart, et al., 1991). This confirmed measurements showing that the Ure2p, Sup35p and Rnq1p prion domain amyloids were largely β-sheet structures (Shewmaker, et al., 2006, Baxa, et al., 2007, Wickner, et al., 2008).
Distance measurements were used to distinguish the in-register parallel architecture fromβ-helix, antiparallel, or parallel out-of-register structures. The former predicts a distance of ~0.5 nm from one labeled carbonyl carbon atom to the same atom on the adjacent molecule in the filament (Fig. 3A). This is just the distance between strands in a β-sheet and if residues of adjacent molecules are aligned (in-register) then this will be the distance observed between labeled atoms. Each of the other possible structures predict a much greater distance (Fig. 3A). The distance measurement is done using a dipolar recoupling experiment (Tycko, 2007). "Magic angle spinning" - spinning the sample at an angle of 54.74 degrees relative to the direction of the magnetic field - is used to eliminate dipole-dipole interactions (the nuclei directly interacting as two tiny magnets). Selected dipole-dipole interactions are re-established by a series of radiowave pulses. These interactions result over time in the loss of alignment of the 13C nuclear spins, and thus the decay of the NMR signal. The rate of this signal decay is proportional to the inverse of the cube of the distance to the next labeled nucleus (Fig 4B).
Fig. 4.

Solid-state NMR data demonstrates the in-register parallel architecture for the yeast prion amyloids - the prion domain of Rnq1 in this case (Wickner, et al., 2008). The rapid decay of the NMR signal for 1-13C-Tyr or 1-13C-Leu labeled molecules indicates that these atoms are ~5 angstroms from their nears labeled neighbor (B). To show that that nearest neighbor is in a different molecule, labeled molecules are diluted with unlabeled molecules and the observed results (B, inverted blue triangles) are in accord with the expected results (inverted empty green triangles) as diagrammed in (A) for the nearest neighbor being in a different molecule. To determine if the structure is really in-register, methyl-13C-Ala labeled molecules were examined. Even a single residue out of register would result in a slow signal decay (C). The rapid decay confirms the in-register parallel architecture (D). Similar results have been obtained for the prion domains of Sup35p (Shewmaker, et al., 2006) and Ure2p (Baxa, et al., 2007).
The material for these experiments was amyloid formed in vitro from recombinant prion domains of Sup35p, Ure2p and Rnq1p, labeled with one amino acid carrying 13C specifically in the carbonyl position. Because of their relatively large size, these peptides cannot be synthesized, and must be made in E. coli. Due to the vicisitudes of bacterial metabolism, it is only practical to label certain amino acids without concern about leakage of label into other residues or dilution of labeled amino acid with endogenously synthesized material. For this purpose, Leu, Ile, Val, Phe, Tyr, and Met have been most useful. For Ala-3-13C, a large amount of labeled amino acid and a short labeling period produces only ~60% labeling, but not cross-labeling of other amino acids. The key point is that each labeled amyloid preparation is highly infectious for yeast, transmitting the respective prion (King & Diaz-Avalos, 2004, Tanaka, et al., 2004, Brachmann, et al., 2005, Patel & Liebman, 2007).
In each case, we found that the rate of signal decay reflected a distance of about 0.5 nm, consistent with a parallel in-register β-sheet architecture (Fig. 4B) (Shewmaker, et al., 2006, Baxa, et al., 2007, Wickner, et al., 2008). However, it was critical to establish that this distance was an intermolecular distance, because we are unable to label single residues. For this purpose, we diluted the fully amino acid-labeled sample with an unlabeled sample. If the measured distance were an intramolecular distance, then we would expect no effect on the rate of signal decay (Fig. 4A). However, if we had an in-register parallel β-sheet structure, the rate of signal decay should be substantially deminished because in most cases, the neighboring molecules of the labeled molecule would be unlabeled, and decay would be promoted only by more distant 13C nuclei. Indeed, as shown for example by Fig 4B, this was found in each case, showing that the nearest neighbor was in a different molecule, again indicative of the in-register parallel structure.
To finally confirm the in-register aspect, it was necessary to use Ala-3-13C labeled amyloid. The side chains in a β-strand point alternately in opposite directions (Fig. 4C). Thus, while an in-register structure structure would give a distance in the dipolar recoupling experiment of about 0.5 nm, this distance is >0.8 nm if the strands are even a single residue out of register. The result was as expected for an in-register structure in each case (Fig. 4D) (Shewmaker, et al., 2006, Baxa, et al., 2007, Wickner, et al., 2008).
That shuffled prion domains could still be prions originally suggested the in-register parallel structure (Ross, et al., 2005), so we examined whether in fact the shuffled prion domains have this structure (Shewmaker, et al., 2008). Indeed, by the same NMR methods used to study the normal sequences, we showed that two of the shuffled Ure2p prion domains and one shuffled Sup35p prion domain each have an in-register parallel β-sheet structure (Shewmaker, et al., 2008).
Although the amyloid formed in vitro from recombinant prion domains is highly infectious for yeast, it generally produces an array of prion variants on infection (e.g. (Brachmann, et al., 2005)). Corresponding to this demonstrated genetic heterogeneity, the 2-dimensional 13C-13C ssNMR spectra show rather broad peaks indicative of structural heterogeneity (Shewmaker, et al., 2006, Baxa, et al., 2007, Wickner, et al., 2008). It has been found that Sup35NM filaments formed at 37C produce, on transfection into yeast, mainly a weak [PSI+] variant, while fibers formed at 4C produce mainly a strong variant (Tanaka, et al., 2004). Differences in the distribution of slow-exchanging amide hydrogens between the 37C and 4C filaments indicate structural differences between these preparations (Toyama, et al., 2007). We used the same solid-state NMR methods to examine such preparations and found that both had an in-register parallel structure (Shewmaker, et al., 2009). This study also confirmed that drying of samples did not alter their infectivity or their properties as judged by ssNMR.
Again, the [Het-s] prion of Podospora anserina provides an illuminating contrast to the yeast prions (Saupe, 2007). Only a single variant of [Het-s] has been described, and, correspondingly, the ssNMR peaks of a 2D 13C-13C homonuclear dipolar coupling experiment of amyloid of HET-s217–289 (the prion domain) are remarkably sharp, probably sharper than for any other amyloid described, suggesting that a single well defined structure is formed (Ritter, et al., 2005). This structure is a 2-turn β-helix, formed by direct partial repeats in the peptide sequence (Wasmer, et al., 2008).
In-register parallel structure explains inheritance of variant information
The in-register parallel architecture was hypothesized to explain the shuffleability of prion domains of Ure2p and Sup35p (Ross, et al., 2005). We then verified, using solid-state NMR, that this is in fact the architecture of the various infectious amyloids of Ure2p, Sup35p and Rnq1p (Shewmaker, et al., 2006, Baxa, et al., 2007, Wickner, et al., 2008). We still do not know the detailed structure of any prion amyloid. But it is clear that the in-register parallel architecture can explain the inheritance of prion variant information (Wickner, et al., 2007, Wickner, et al., 2008, Wickner, et al., 2008), as discussed below.
What holds the structure in-register?
The in-register parallel β-sheet structure (Fig. 5) is held in-register by several types of bonding between the side chains of identical residues of adjacent molecules:
Fig. 5.
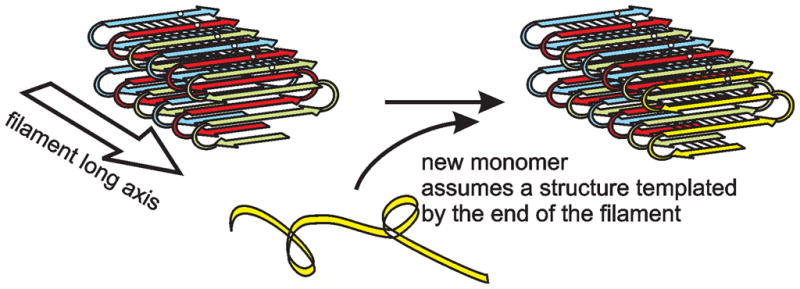
In-register parallel structure explains the ability of each yeast prion amyloid to faithfully template any of several "variant" structures. We suggest that variants differ in the locations of the turns (the folds of the sheet). Side chain–side chain interactions along the filament axis enforce the same locations for turns in the molecule newly joining the end of the filament as those in the previous molecule. The black dots represent a particular residue, say Gln46, in a prion domain sequence.
the β-zipper structure of lines of glutamine or asparagine residues consisting of hydrogen bonds between their sidechain amide hydrogen and sidechain carbonyl oxygen (Perutz, et al., 1994, Chan, et al., 2005, Nelson, et al., 2005). This forms a strip of hydrogen bonds running the length of the amyloid filament.
hydrogen bonds between serine or threonine hydroxyl hydrogen and hydroxyl oxygen of the same residue of the next molecule up or down the filament.
hydrophobic interactions between identical residues.
The only interactions between identical residues that are unfavorable are between identical charged residues, and such residues are few in the known prion domains, while Gln, Asn, Ser and Thr residues are particularly abundant.
What are the differences among prion variants?
We have proposed that variants differ in the location of the turns connecting the β-strands, the extent of the β-sheet structure or, potentially, the way in which protofilaments associate. Variants of Aβ structure have either two or three protofilaments associating to form a filament (Paravastu, et al., 2008), but the mass per unit length of prion domains of Ure2p, Sup35p and Rnq1p are each one monomer per 4.7 angstroms (Baxa, et al., 2003, Diaz-Avalos, et al., 2005, Chen, et al., 2009), making this unlikely in the yeast prion case.
How does prion amyloid assemble?
Assembly of Sup35 prion domain amyloid occurs by addition of monomers (Collins, et al., 2004). Amyloid of the [URE3] prion (at least) is assembled by conversion of the unstructured prion domain in the native molecules (Pierce, et al., 2005) into highly structured polymers, with the structure assumed by new monomers templated by the structure of the last monomer on the end of the filament (Fig. 5). Thus, in the same way that DNA sequence of the parental strand templates the sequence of the new strand, the conformation of a protein molecule at the end of the filament templates the conformation of a new molecule as it joins the end of the filament. Turns in the template become turns in the new molecule. Intramolecular side chain-side chain interactions perpendicular to the filament axis will also be reproduced in the new molecule. We now see a mechanism by which proteins can act as genes, but it remains to be determined exactly what are the structures of different prion variants.
Conclusions
The rapidly increasing number of yeast prions, and the in-depth studies of their mechanisms of generation and propagation, is revealing an array of pathobiological phenomena that is aiding efforts to understand mammalian prion and non-prion amyloid diseases. Our finding an in-register parallel architecture for the infectious amyloids of the prion domains of Ure2p, Sup35p and Rnq1p has clear implications for the mechanism that underly the otherwise mysterious fact that proteins are capable of templating their own self-propagating conformation.
Acknowledgments
We were fortunate to be able to collaborate with Rob Tycko (NIDDK, NIH) in our solid-state NMR studies. This work was supported by the Intramural Program of the National Institute of Diabetes Digestive and Kidney Diseases of the National Institutes of Health.
References
- 1.Alberti S, Halfmann R, King O, Kapila A, Lindquist S. A systematic survey identifies prions and illuminates sequence features of prionogenic proteins. Cell. 2009;137:146 – 158. doi: 10.1016/j.cell.2009.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alper T, Haig DA, Clarke MC. The exceptionally small size of the scrapie agent. Biochem Biophys Res Commun. 1966;22:278 – 284. doi: 10.1016/0006-291x(66)90478-5. [DOI] [PubMed] [Google Scholar]
- 3.Alper T, Cramp WA, Haig DA, Clarke MC. Does the agent of scrapie replicate without nucleic acid? Nature. 1967;214:764 – 766. doi: 10.1038/214764a0. [DOI] [PubMed] [Google Scholar]
- 4.Bai M, Zhou JM, Perrett S. The yeast prion protein Ure2 shows glutathione peroxidase activity in both native and fibrillar forms. J Biol Chem. 2004;279:50025–50030. doi: 10.1074/jbc.M406612200. [DOI] [PubMed] [Google Scholar]
- 5.Balbach JJ, Petkova AT, Oyler NA, Antzutkin ON, Gordon DJ, Meredith SC, Tycko R. Supramolecular structure in full-length Alzheimer's beta-amyloid fibrils: Evidence for a parallel beta-sheet organization from solid-state nuclear magnetic resonance. Biophys J. 2002;83:1205–1216. doi: 10.1016/S0006-3495(02)75244-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Basler K, Oesch B, Scott M, et al. Scrapie and cellular PrP isoforms are encoded by the same chromosomal locus. Cell. 1986;46:417 – 428. doi: 10.1016/0092-8674(86)90662-8. [DOI] [PubMed] [Google Scholar]
- 7.Baxa U, Speransky V, Steven AC, Wickner RB. Mechanism of inactivation on prion conversion of the Saccharomyces cerevisiae Ure2 protein. Proc Natl Acad Sci U S A. 2002;99:5253 – 5260. doi: 10.1073/pnas.082097899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baxa U, Taylor KL, Wall JS, Simon MN, Cheng N, Wickner RB, Steven A. Architecture of Ure2p prion filaments: the N-terminal domain forms a central core fiber. J Biol Chem. 2003;278:43717 –43727. doi: 10.1074/jbc.M306004200. [DOI] [PubMed] [Google Scholar]
- 9.Baxa U, Wickner RB, Steven AC, Anderson D, Marekov L, Yau W-M, Tycko R. Characterization of β-sheet structure in Ure2p1-89 yeast prion fibrils by solid state nuclear magnetic resonance. Biochemistry. 2007;46:13149 – 13162. doi: 10.1021/bi700826b. [DOI] [PubMed] [Google Scholar]
- 10.Baxa U, Cheng N, Winkler DC, et al. Filaments of the Ure2p prion protein have a cross-beta core structure. J Struct Biol. 2005;150:170–179. doi: 10.1016/j.jsb.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 11.Bellinger-Kawakara C, Cleaver JE, Diener TO, Prusiner SB. Purified scrapie prions resist inactivation by UV irradiation. J Virol. 1987;61:159 – 166. doi: 10.1128/jvi.61.1.159-166.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bessen RA, Marsh RF. Distinct PrP properties suggest the molecular basis of strain variation in transmissible mink encephalopathy. J Virol. 1994;68:7859 – 7868. doi: 10.1128/jvi.68.12.7859-7868.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bolton DC, McKinley MP, Prusiner SB. Identification of a protein that purifies with the scrapie prion. Science. 1982;218:1309 – 1311. doi: 10.1126/science.6815801. [DOI] [PubMed] [Google Scholar]
- 14.Brachmann A, Baxa U, Wickner RB. Prion generation in vitro: amyloid of Ure2p is infectious. Embo J. 2005;24:3082 – 3092. doi: 10.1038/sj.emboj.7600772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bradley ME, Edskes HK, Hong JY, Wickner RB, Liebman SW. Interactions among prions and prion "strains" in yeast. Proc Natl Acad Sci U S A. 2002;99 (Suppl 4):16392 – 16399. doi: 10.1073/pnas.152330699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brown JC, Lindquist S. A heritable switch in carbon source utilization driven by an unusual yeast prion. Genes Dev. 2009;23:2320 – 2332. doi: 10.1101/gad.1839109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bruce ME. Scrapie strain variation and mutation. Br Med Bull. 1993;49:822 – 838. doi: 10.1093/oxfordjournals.bmb.a072649. [DOI] [PubMed] [Google Scholar]
- 18.Carlson GA, Kingsbury DT, Goodman PA, et al. Linkagae of prion protein and scrapie incubation time genes. Cell. 1986;46:503 – 511. doi: 10.1016/0092-8674(86)90875-5. [DOI] [PubMed] [Google Scholar]
- 19.Caughey B, Raymond GJ, Bessen RA. Strain-dependent differences in beta-sheet conformations of abnormal prion protein. J Biol Chem. 1998;273:32230 – 32235. doi: 10.1074/jbc.273.48.32230. [DOI] [PubMed] [Google Scholar]
- 20.Chan JCC, Oyler NA, Yau W-M, Tycko R. Parallel β-sheets and polar zippers in amyloid fibrils formed by residues 10--39 of the yeast prion protein Ure2p. Biochemistry. 2005;44:10669 – 10680. doi: 10.1021/bi050724t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chandler RL. Encephalopathy in mice produced by inoculation with scrapie brain material. Lancet. 1961;1:107 – 108. doi: 10.1016/s0140-6736(61)92008-6. [DOI] [PubMed] [Google Scholar]
- 22.Chen B, Newnam GP, Chernoff YO. Prion species barrier between the closely related yeast proteins is detected despite coaggregation. Proc Natl Acad Sci U S A. 2007;104:2791–2796. doi: 10.1073/pnas.0611158104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen B, Thurber KR, Shewmaker F, Wickner RB, Tycko R. Measurement of amyloid fibril mass-per-length by tilted-beam transmission electron microscopy. Proc Natl Acad Sci USA. 2009 doi: 10.1073/pnas.0907821106. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chernoff YO, Galkin AP, Lewitin E, Chernova TA, Newnam GP, Belenkiy SM. Evolutionary conservation of prion-forming abilities of the yeast Sup35 protein. Molec Microbiol. 2000;35:865 – 876. doi: 10.1046/j.1365-2958.2000.01761.x. [DOI] [PubMed] [Google Scholar]
- 25.Collinge J, Clarke AR. A general model of prion strains and their pathogenicity. Science. 2007;318:930 – 936. doi: 10.1126/science.1138718. [DOI] [PubMed] [Google Scholar]
- 26.Collins SR, Douglass A, Vale RD, Weissman JS. Mechanism of prion propagation: amyloid growth occurs by monomer addition. Plos Biol. 2004;2:1582 – 1590. doi: 10.1371/journal.pbio.0020321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Coustou V, Deleu C, Saupe S, Begueret J. The protein product of the het-s heterokaryon incompatibility gene of the fungus Podospora anserina behaves as a prion analog. Proc Natl Acad Sci USA. 1997;94:9773 – 9778. doi: 10.1073/pnas.94.18.9773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cox BS. PSI, a cytoplasmic suppressor of super-suppressor in yeast. Heredity. 1965;20:505 – 521. [Google Scholar]
- 29.Cuille J, Chelle PL. Pathologie animale. La maladie dite tremblant du mouton est-elle inoculable? Compt Rend Acad Sci (Paris) 1936;203:1552 – 1554. [Google Scholar]
- 30.Cuille J, Chelle PL. Experimental transmission of trembling to the goat. C R Seances Acad Sci. 1939;208:1058 – 1060. [Google Scholar]
- 31.Dalstra HJP, Swart K, Debets AJM, Saupe SJ, Hoekstra RF. Sexual transmission of the [Het-s] prion leads to meiotic drive in Podospora anserina. Proc Natl Acad Sci U S A. 2003;100:6616 – 6621. doi: 10.1073/pnas.1030058100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Deleault NR, Harris BT, Rees JR, Supattapone S. Formation of native prions from minimal components in vitro. Proc Natl Acad Sci U S A. 2007;104:9741 – 9746. doi: 10.1073/pnas.0702662104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Der-Sarkissian A, Jao CC, Chen J, Langen R. Structural organization of α-synuclein fibrils studied by site-directed spin labeling. J Biol Chem. 2003;278:37530 – 37535. doi: 10.1074/jbc.M305266200. [DOI] [PubMed] [Google Scholar]
- 34.Derkatch IL, Bradley ME, Hong JY, Liebman SW. Prions affect the appearance of other prions: the story of [PIN] Cell. 2001;106:171 – 182. doi: 10.1016/s0092-8674(01)00427-5. [DOI] [PubMed] [Google Scholar]
- 35.Derkatch IL, Chernoff YO, Kushnirov VV, Inge-Vechtomov SG, Liebman SW. Genesis and variability of [PSI] prion factors in Saccharomyces cerevisiae. Genetics. 1996;144:1375 – 1386. doi: 10.1093/genetics/144.4.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Derkatch IL, Bradley ME, Zhou P, Chernoff YO, Liebman SW. Genetic and environmental factors affecting the de novo appearance of the [PSI+] prion in Saccharomyces cerevisiae. Genetics. 1997;147:507 – 519. doi: 10.1093/genetics/147.2.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Diaz-Avalos R, King CY, Wall JS, Simon M, Caspar DLD. Strain–specific morphologies of yeast prion amyloids. Proc Natl Acad Sci U S A. 2005;102:10165 – 10170. doi: 10.1073/pnas.0504599102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dickinson AG, Meikle VMH, Fraser H. Identification of a gene which controls the incubation period of some strains of scrapie in mice. J Comp Path. 1968;78:293 – 299. doi: 10.1016/0021-9975(68)90005-4. [DOI] [PubMed] [Google Scholar]
- 39.Diringer H, Gelderblom H, Hilmert H, Ozel M, Edelbluth C, Kimberlin RH. Scrapie infectivity, fibrils and low molecular weight protein. Nature. 1983;306:476 – 478. doi: 10.1038/306476a0. [DOI] [PubMed] [Google Scholar]
- 40.Doel SM, McCready SJ, Nierras CR, Cox BS. The dominant PNM2− mutation which eliminates the [PSI] factor of Saccharomyces cerevisiae is the result of a missense mutation in the SUP35 gene. Genetics. 1994;137:659 – 670. doi: 10.1093/genetics/137.3.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Du Z, Park K-W, Yu H, Fan Q, Li L. Newly identified prion linked to the chromatin-remodeling factor Swi1 in Saccharomyces cerevisiae. Nat Genet. 2008;40:460 – 465. doi: 10.1038/ng.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Eaglestone SS, Cox BS, Tuite MF. Translation termination efficiency can be regulated in Saccharomyces cerevisiae by environmental stress through a prion-mediated mechanism. EMBO J. 1999;18:1974 – 1981. doi: 10.1093/emboj/18.7.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Edskes HK, Wickner RB. Conservation of a portion of the S. cerevisiae Ure2p prion domain that interacts with the full - length protein. Proc Natl Acad Sci USA. 2002;99 (Suppl 4):16384–16391. doi: 10.1073/pnas.162349599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Edskes HK, McCann LM, Hebert AM, Wickner RB. Prion variants and species barriers among Saccharomyces Ure2 proteins. Genetics. 2009;181:1159 – 1167. doi: 10.1534/genetics.108.099929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Griffith JS. Self-replication and scrapie. Nature. 1967;215:1043 – 1044. doi: 10.1038/2151043a0. [DOI] [PubMed] [Google Scholar]
- 46.Hoshino S, Imai M, Kobayashi T, Uchida N, Katada T. The eukaryotic polypeptide chain releasing factor (eRF3/GSPT) carrying the translation termination signal to the 3'-poly(A) tail of mRNA. J Biol Chem. 1999;274:16677 – 16680. doi: 10.1074/jbc.274.24.16677. [DOI] [PubMed] [Google Scholar]
- 47.Hosoda N, Kobayashii T, Uchida N, Funakoshi Y, Kikuchi Y, Hoshino S, Katada T. Translation termination factor eRF3 mediates mRNA decay through the regulation of deadenylation. J Biol Chem. 2003;278:38287 – 38291. doi: 10.1074/jbc.C300300200. [DOI] [PubMed] [Google Scholar]
- 48.Hsiao K, Baker HF, Crow TJ, et al. Linkage of a prion protein missense variant to Gerstmann-Straussler syndrome. Nature. 1989;338:342 – 345. doi: 10.1038/338342a0. [DOI] [PubMed] [Google Scholar]
- 49.Hunter N, Hope J, McConnell I, Dickinson AG. Linkage of the scrapie-associated fibril protein (PrP) gene and Sinc using congenic mice and restriction fragment length polymorphism analysis. J Gen Virol. 1987;68:2711 – 2716. doi: 10.1099/0022-1317-68-10-2711. [DOI] [PubMed] [Google Scholar]
- 50.Jones EW. Three proteolytic systems in the yeast Saccharomyces cerevisiae. J Biol Chem. 1991;266:7963 – 7966. [PubMed] [Google Scholar]
- 51.King C-Y, Tittmann P, Gross H, Gebert R, Aebi M, Wuthrich K. Prion-inducing domain 2-114 of yeast Sup35 protein transforms in vitro into amyloid-like filaments. Proc Natl Acad Sci USA. 1997;94:6618 – 6622. doi: 10.1073/pnas.94.13.6618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.King CY. Supporting the structural basis of prion strains: induction and identification of [PSI] variants. J Mol Biol. 2001;307:1247–1260. doi: 10.1006/jmbi.2001.4542. [DOI] [PubMed] [Google Scholar]
- 53.King CY, Diaz-Avalos R. Protein-only transmission of three yeast prion strains. Nature. 2004;428:319 – 323. doi: 10.1038/nature02391. [DOI] [PubMed] [Google Scholar]
- 54.Klohn PC, Stoltze L, Flechsig E, Enari M, Weissmann C. A quantitative, highly sensitive cell-based infectivity assay for mouse scrapie prions. Proc Natl Acad Sci USA. 2003;100:11666 – 11671. doi: 10.1073/pnas.1834432100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kochneva-Pervukhova NV, Paushkin SV, Kushnirov VV, Cox BS, Tuite MF, Ter-Avanesyan MD. Mechanism of inhibition of Ψ+ prion determinant propagation by a mutation of the N-terminus of the yeast Sup35 protein. Embo J. 1998;17:5805–5810. doi: 10.1093/emboj/17.19.5805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Krzewska J, Tanaka M, Burston SG, Melki R. Biochemical and functional analysis of the assembly of full-length Sup35p and its prion-forming domain. J Biol Chem. 2007;282:1679 – 1686. doi: 10.1074/jbc.M608110200. [DOI] [PubMed] [Google Scholar]
- 57.Kushnirov VV, Kochneva-Pervukhova NV, Cechenova MB, Frolova NS, Ter-Avanesyan MD. Prion properties of the Sup35 protein of yeast Pichia methanolica. EMBO J. 2000;19:324 – 331. doi: 10.1093/emboj/19.3.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kushnirov VV, Kryndushkin D, Boguta M, Smirnov VN, Ter-Avanesyan MD. Chaperones that cure yeast artificial [PSI+] and their prion-specific effects. Curr Biol. 2000;10:1443 – 1446. doi: 10.1016/s0960-9822(00)00802-2. [DOI] [PubMed] [Google Scholar]
- 59.Lacroute F. Non-Mendelian mutation allowing ureidosuccinic acid uptake in yeast. J Bacteriol. 1971;106:519 – 522. doi: 10.1128/jb.106.2.519-522.1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Legname G, Baskakov IV, Nguyen H-OB, Reisner D, Cohen FE, DeArmond SJ, Prusiner SB. Synthetic mammalian prions. Science. 2004;305:673–676. doi: 10.1126/science.1100195. [DOI] [PubMed] [Google Scholar]
- 61.Locht C, Chesebro B, Race R, Keith JM. Molecular cloning and complete sequence of prion protein cDNA from mouse brain infected with the scrapie agent. Proc Natl Acad Sci USA. 1986;83:6372 –6376. doi: 10.1073/pnas.83.17.6372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Loquet A, Bousset L, Gardiennet C, et al. Prion fibrils of Ure2p assembled under physiological conditions contain highly ordered, natively folded molecules. J Mol Biol. 2009;394:108 – 118. doi: 10.1016/j.jmb.2009.09.016. [DOI] [PubMed] [Google Scholar]
- 63.Luca S, Yau W-M, Leapman R, Tycko R. Peptide conformation and supramolecular organization in amylin fibrils: constraints from solid-state NMR. Biochemistry. 2007;46:13505 – 13522. doi: 10.1021/bi701427q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Maddelein ML, Dos Reis S, Duvezin-Caubet S, Coulary-Salin B, Saupe SJ. Amyloid aggregates of the HET-s prion protein are infectious. Proc Natl Acad Sci U S A. 2002;99:7402–7407. doi: 10.1073/pnas.072199199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Makarava N, Kovacs GG, Bocharova O, et al. Recombinant prion protein induces a new transmissible prion disease in wild-type animals. Acta Neuropathol. 2010;119:177 – 187. doi: 10.1007/s00401-009-0633-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Margittai M, Langen R. Fibrils with parallel in-register structure constitute a major class of amyloid fibrils: molecular insights from electron paramagnetic resonance spectroscopy. Q Rev Biophys. 2008;41:265 – 297. doi: 10.1017/S0033583508004733. [DOI] [PubMed] [Google Scholar]
- 67.Masison DC, Maddelein M-L, Wickner RB. The prion model for [URE3] of yeast: spontaneous generation and requirements for propagation. Proc Natl Acad Sci USA. 1997;94:12503–12508. doi: 10.1073/pnas.94.23.12503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Masters CL, Gajdusek DC, Gibbs CJ. Creutzfeldt-Jakob disease virus isolations from the Gerstmann-Straussler syndrome with an analysis of the various forms of amyloid plaque deposition in the virus-induced spongiform encephalopathies. Brain. 1981;104:559 – 588. doi: 10.1093/brain/104.3.559. [DOI] [PubMed] [Google Scholar]
- 69.Mead S, Stumpf MP, Whitfield J, et al. Balancing selection at the prion protein gene consistent with prehistoric kurulike epidemics. Science. 2003;300:640–643. doi: 10.1126/science.1083320. [DOI] [PubMed] [Google Scholar]
- 70.Nakayashiki T, Kurtzman CP, Edskes HK, Wickner RB. Yeast prions [URE3] and [PSI+] are diseases. Proc Natl Acad Sci U S A. 2005;102:10575–10580. doi: 10.1073/pnas.0504882102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Namy O, Galopier A, Martini C, Matsufuji S, Fabret C, Rousset C. Epigenetic control of polyamines by the prion [PSI+] Nat Cell Biol. 2008;10:1069 – 1075. doi: 10.1038/ncb1766. [DOI] [PubMed] [Google Scholar]
- 72.Nelson R, Sawaya MR, Balbirnie M, Madsen AO, Riekel C, Grothe R, Eisenberg D. Structure of the cross-β spine of amyloid-like fibrils. Nature. 2005;435:773–778. doi: 10.1038/nature03680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nemecek J, Nakayashiki T, Wickner RB. A prion of yeast metacaspase homolog (Mca1p) detected by a genetic screen. Proc Natl Acad Sci USA. 2009;106:1892 – 1896. doi: 10.1073/pnas.0812470106. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 74.Paravastu AK, Leapman RD, Yau WM, Tycko R. Molecular structural basis for polymorphism in Alzheimer's β-amyloid fibrils. Proc Natl Acad Sci USA. 2008;105:18349 – 18354. doi: 10.1073/pnas.0806270105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Parry HB. Scrapie disease in sheep - historical, clinical, epidemiological, pathological and practical aspects of the natural disease. Academic Press; London: 1983. [Google Scholar]
- 76.Partridge L, Barton NH. Evolving evolvability. Nature. 2000;407:457–458. doi: 10.1038/35035173. [DOI] [PubMed] [Google Scholar]
- 77.Patel BK, Liebman SW. "Prion proof" for [PIN+]: infection with in vitro-made amyloid aggregates of Rnq1p-(132–405) induces [PIN+] J Mol Biol. 2007;365:773 – 782. doi: 10.1016/j.jmb.2006.10.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Patel BK, Gavin-Smyth J, Liebman SW. The yeast global transcriptional co-repressor protein Cyc8 can propagate as a prion. Nat Cell Biol. 2009;11:344 – 349. doi: 10.1038/ncb1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Perutz MF, Johnson T, Suzuki M, Finch JT. Glutamine repeats as polar zippers: their possible role in inherited neurodegenerative diseases. Proc Natl Acad Sci USA. 1994;91:5355 – 5358. doi: 10.1073/pnas.91.12.5355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pierce MM, Baxa U, Steven AC, Bax A, Wickner RB. Is the prion domain of soluble Ure2p unstructured? Biochemistry. 2005;44:321–328. doi: 10.1021/bi047964d. [DOI] [PubMed] [Google Scholar]
- 81.Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216:136 – 144. doi: 10.1126/science.6801762. [DOI] [PubMed] [Google Scholar]
- 82.Prusiner SB, Scott M, Foster D, et al. Transgenic studies implicate interactions between homologous PrP isoforms in scrapie prion replication. Cell. 1990;63:673 – 686. doi: 10.1016/0092-8674(90)90134-z. [DOI] [PubMed] [Google Scholar]
- 83.Resende CG, Outeiro TF, Sands L, Lindquist S, Tuite MF. Prion protein gene polymorphisms in Saccharomyces cerevisiae. Mol Microbiol. 2003;49:1005 – 1017. doi: 10.1046/j.1365-2958.2003.03608.x. [DOI] [PubMed] [Google Scholar]
- 84.Ritter C, Maddelein ML, Siemer AB, et al. Correlation of structural elements and infectivity of the HET-s prion. Nature. 2005;435:844–848. doi: 10.1038/nature03793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Roberts BT, Wickner RB. A class of prions that propagate via covalent auto-activation. Genes Dev. 2003;17:2083 – 2087. doi: 10.1101/gad.1115803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ross ED, Baxa U, Wickner RB. Scrambled prion domains form prions and amyloid. Mol Cell Biol. 2004;24:7206–7213. doi: 10.1128/MCB.24.16.7206-7213.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ross ED, Minton AP, Wickner RB. Prion domains: sequences, structures and interactions. Nat Cell Biol. 2005;7:1039–1044. doi: 10.1038/ncb1105-1039. [DOI] [PubMed] [Google Scholar]
- 88.Ross ED, Edskes HK, Terry MJ, Wickner RB. Primary sequence independence for prion formation. Proc Natl Acad Sci U S A. 2005;102:12825 – 12830. doi: 10.1073/pnas.0506136102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Santoso A, Chien P, Osherovich LZ, Weissman JS. Molecular basis of a yeast prion species barrier. Cell. 2000;100:277 – 288. doi: 10.1016/s0092-8674(00)81565-2. [DOI] [PubMed] [Google Scholar]
- 90.Saupe SJ. A short history of small s: a prion of the fungus Podospora anserina. Prion. 2007;1:110 –115. doi: 10.4161/pri.1.2.4666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Saupe SJ, Clave C, Begueret J. Vegetative incompatibility in filamentous fungi: Podospora and Neurospora provide some clues. Curr Opin Microbiol. 2000;3:608–612. doi: 10.1016/s1369-5274(00)00148-x. [DOI] [PubMed] [Google Scholar]
- 92.Schlumpberger M, Prusiner SB, Herskowitz I. Induction of distinct [URE3] yeast prion strains. Mol Cell Biol. 2001;21:7035–7046. doi: 10.1128/MCB.21.20.7035-7046.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sharma D, Masison DC. Hsp70 structure, function, regulation and influence on yeast prions. Prot & Peptide Lett. 2009;16:571 – 581. doi: 10.2174/092986609788490230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Shewmaker F, Wickner RB, Tycko R. Amyloid of the prion domain of Sup35p has an in-register parallel β-sheet structure. Proc Natl Acad Sci USA. 2006;103:19754 – 19759. doi: 10.1073/pnas.0609638103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Shewmaker F, Ross ED, Tycko R, Wickner RB. Amyloids of shuffled prion domains that form prions have a parallel in-register β-sheet structure. Biochemistry. 2008;47:4000–4007. doi: 10.1021/bi7024589. [DOI] [PubMed] [Google Scholar]
- 96.Shewmaker F, Mull L, Nakayashiki T, Masison DC, Wickner RB. Ure2p function is enhanced by its prion domain in Saccharomyces cerevisiae. Genetics. 2007;176:1557 – 1565. doi: 10.1534/genetics.107.074153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Shewmaker F, Kryndushkin D, Chen B, Tycko R, Wickner RB. Two prion variants of Sup35p have in-register β-sheet structures, independent of hydration. Biochemistry. 2009;48:5074–5082. doi: 10.1021/bi900345q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sondheimer N, Lindquist S. Rnq1: an epigenetic modifier of protein function in yeast. Molec Cell. 2000;5:163 – 172. doi: 10.1016/s1097-2765(00)80412-8. [DOI] [PubMed] [Google Scholar]
- 99.Talarek N, Maillet L, Cullin C, Aigle M. The [URE3] prion is not conserved among Saccharomyces species. Genetics. 2005;171:23–54. doi: 10.1534/genetics.105.043489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Tanaka M, Chien P, Naber N, Cooke R, Weissman JS. Conformational variations in an infectious protein determine prion strain differences. Nature. 2004;428:323 – 328. doi: 10.1038/nature02392. [DOI] [PubMed] [Google Scholar]
- 101.Taylor KL, Cheng N, Williams RW, Steven AC, Wickner RB. Prion domain initiation of amyloid formation in vitro from native Ure2p. Science. 1999;283:1339 – 1343. doi: 10.1126/science.283.5406.1339. [DOI] [PubMed] [Google Scholar]
- 102.Toyama BH, Kelly MJ, Gross JD, Weissman JS. The structural basis of yeast prion strain variants. Nature. 2007;449:233 – 237. doi: 10.1038/nature06108. [DOI] [PubMed] [Google Scholar]
- 103.True HL, Lindquist SL. A yeast prion provides a mechanism for genetic variation and phenotypic diversity. Nature. 2000;407:477–483. doi: 10.1038/35035005. [DOI] [PubMed] [Google Scholar]
- 104.Turoscy V, Cooper TG. Ureidosuccinate is transported by the allantoate transport system in Saccharomyces cerevisiae. J Bacteriol. 1987;169:2598 – 2600. doi: 10.1128/jb.169.6.2598-2600.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Tycko R. Molecular structure of amyloid fibrils: insights from solid-state NMR. Quart Revs Biophys. 2006;1:1–55. doi: 10.1017/S0033583506004173. [DOI] [PubMed] [Google Scholar]
- 106.Tycko R. Symmetry-based constant-time homonuclear dipolar recoupling in solid-state NMR. J Chem Phys. 2007;126:064506. doi: 10.1063/1.2437194. [DOI] [PubMed] [Google Scholar]
- 107.Tyedmers J, Madariaga ML, Lindquist S. Prion switching in response to environmental stress. PLoS Biol. 2008;6:e294. doi: 10.1371/journal.pbio.0060294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wang F, Wang X, Yuan C-G, Ma J. Generating a prion with bacterially expressed recombinant prion protein. Science. 2010;327:1132 – 1135. doi: 10.1126/science.1183748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wasmer C, Lange A, Van Melckebeke H, Siemer AB, Riek R, Meier BH. Amyloid fibrils of the HET-s(218–279) prion form a beta solenoid with a triangular hydrophobic core. Science. 2008;319:1523 –1526. doi: 10.1126/science.1151839. [DOI] [PubMed] [Google Scholar]
- 110.Wickner RB. [URE3] as an altered URE2 protein: evidence for a prion analog in S. cerevisiae. Science. 1994;264:566 – 569. doi: 10.1126/science.7909170. [DOI] [PubMed] [Google Scholar]
- 111.Wickner RB. A new prion controls fungal cell fusion incompatibility. Proc Natl Acad Sci USA. 1997;94:10012 – 10014. doi: 10.1073/pnas.94.19.10012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wickner RB. Scrapie in ancient China? Science. 2005;309:874. doi: 10.1126/science.309.5736.874b. [DOI] [PubMed] [Google Scholar]
- 113.Wickner RB, Dyda F, Tycko R. Amyloid of Rnq1p, the basis of the [PIN+] prion, has a parallel in-register β-sheet structure. Proc Natl Acad Sci U S A. 2008;105:2403 – 2408. doi: 10.1073/pnas.0712032105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wickner RB, Edskes HK, Shewmaker F, Nakayashiki T. Prions of fungi: inherited structures and biological roles. Nat Microbiol Rev. 2007;5:611–618. doi: 10.1038/nrmicro1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wickner RB, Shewmaker F, Kryndushkin D, Edskes HK. Protein inheritance (prions) based on parallel in-register β-sheet amyloid structures. Bioessays. 2008;30:955 – 964. doi: 10.1002/bies.20821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Wilesmith JW. Bovine spongiform encephalopathy. Vet Rec. 1988;122:614. doi: 10.1136/vr.122.25.614-a. [DOI] [PubMed] [Google Scholar]
- 117.Will RG, Ironside JW, Zeidler M, et al. A new variant of Creutzfeldt-Jakob disease in the UK. Lancet. 1996;347:921 – 925. doi: 10.1016/s0140-6736(96)91412-9. [DOI] [PubMed] [Google Scholar]
- 118.Wishart DS, Sykes BD, Richards FM. Relationship between nuclear magnetic resonance chemical shift and protein secondary structure. J Mol Biol. 1991;222:311 – 333. doi: 10.1016/0022-2836(91)90214-q. [DOI] [PubMed] [Google Scholar]
- 119.Zubenko GS, Park FJ, Jones EW. Genetic properties of mutations at the PEP4 locus in Saccharomyces cerevisiae. Genetics. 1982;102:679 – 690. doi: 10.1093/genetics/102.4.679. [DOI] [PMC free article] [PubMed] [Google Scholar]