

Abstract
The disintegrin and metalloprotease ADAM12 has important functions in normal physiology as well as in diseases, such as cancer. Little is known about how ADAM12 confers its pro-tumorigenic effect; however, its proteolytic capacity is probably a key component. Thus selective inhibition of ADAM12 activity may be of great value therapeutically and as an investigative tool to elucidate its mechanisms of action. We have previously reported the inhibitory profile of TIMPs (tissue inhibitor of metalloproteinases) against ADAM12, demonstrating in addition to TIMP-3, a unique ADAM-inhibitory activity of TIMP-2. These findings strongly suggest that it is feasible to design a TIMP mutant selectively inhibiting ADAM12. With this purpose, we characterized the molecular determinants of the ADAM12–TIMP complex formation as compared with known molecular requirements for TIMP-mediated inhibition of ADAM17/TACE (tumour necrosis factor α-converting enzyme). Kinetic analysis using a fluorescent peptide substrate demonstrated that the molecular interactions of N-TIMPs (N-terminal domains of TIMPs) with ADAM12 and TACE are for the most part comparable, yet revealed strikingly unique features of TIMP-mediated ADAM12 inhibition. Intriguingly, we found that removal of the AB-loop in N-TIMP-2, which is known to impair its interaction with TACE, resulted in increased affinity to ADAM12. Importantly, using a cell-based epidermal growth factor-shedding assay, we demonstrated for the first time an inhibitory activity of TIMPs against the transmembrane ADAM12-L (full-length ADAM12), verifying the distinctive inhibitory abilities of N-TIMP-2 and engineered N-TIMP-2 mutants in a cellular environment. Taken together, our findings support the idea that a distinctive ADAM12 inhibitor with future therapeutic potential can be designed.
Keywords: a disintegrin and metalloprotease 12 (ADAM12), ectodomain shedding, therapeutic target, tissue inhibitor of metalloproteinase (TIMP), tumour necrosis factor α-converting enzyme (TACE)
INTRODUCTION
ADAM12, a disintegrin and metalloprotease also known as meltrin α, is a multifunctional zinc-dependent enzyme and one of 13 catalytically active human ADAMs [1]. In humans, ADAM12 is expressed as a classical type 1 transmembrane form (ADAM12-L) and as a shorter secreted splice variant (ADAM12-S). Both forms consist of a pro domain, a catalytic domain, and cell-adhesive disintegrin, cysteine-rich and EGF (epidermal growth factor)-like domains, but ADAM12-S lacks the C-terminal transmembrane and cytoplasmic domains found in ADAM12-L [2]. A key feature of the ADAMs, including ADAM12, is their ability to ‘shed’ membrane-anchored proteins and thus regulate the availability of bioreactive molecules, such as cell-surface receptors, growth factors and cytokines [3,4]. Previous in vitro studies have implicated ADAM12-L in ectodomain shedding of several EGFR (EGF receptor) ligands [EGF, HB-EGF (heparin-binding EGF) and betacellulin] [5,6], the Notch ligand Delta-like 1 [7] and oxytocinase [8], whereas ADAM12-S can cleave insulin-like growth factor-binding protein-3 and -5 [9]. Whether cleavage of these substrates represents physiologically significant functions remains to be shown. However, ADAM12 appears to play an important pathogenic role in diseases such as osteoarthritis, cardiac hypertrophy and several human cancers [1], thus arguing in favour of selectively targeting its protease activity in efforts to design potential new therapies [10].
Known endogenous regulators of ADAM protease functions include certain members of the family of TIMPs (tissue inhibitor of metalloproteinases), which are mainly known for their inhibitory activity against the MMPs (matrix metalloproteinases). Four homologous human TIMPs (TIMP-1, TIMP-2, TIMP-3 and TIMP-4) have been characterized, all inhibiting metalloproteinases in a 1:1 stoichiometry by tightly binding to the active site of the enzymes. Structural analyses have revealed that the ~ 24 kDa TIMPs are composed of two very distinct N- and C-terminal domains, of which the N-terminal domain is responsible for most of the contact made with the active site of the enzyme [11,12]. The majority of MMPs are well inhibited by all TIMPs, except that TIMP-1 exhibits little activity towards MT-MMPs (membrane-type MMPs). Apart from TIMP-1, which is a good inhibitor of ADAM10 [13], TIMP-3 has been considered the sole endogenous ADAM modulator. Based on a series of mutagenesis studies, as well as the high-resolution co-crystal structure of N-TIMP-3 (N-terminal domain of TIMP-3) in complex with ADAM17/TACE (tumour necrosis factor α-converting enzyme), the TIMP reactive ridge has been localized to the N-terminal wedge, the AB-loop, and the CD- and EF-connecting loops of the molecule (Figure 1) [12,14].
Figure 1. Sequence alignments and co-crystal structure of TACE–N-TIMP-3.

(A) The co-crystal structure of the TACE–N-TIMP-3 complex (Protein Data Bank code 3CKI) was visualized using the PyMOL molecular viewer tool from LeNovo Scientific (blue ribbon, N-TIMP-3; red ribbon, TACE catalytic domain; purple sphere, catalytic zinc). Key secondary structural elements [strands (s) and helices (h)] and N-TIMP-3 residues are labelled. (B) ClustalW sequence alignment of ADAM12-Cat (AAC08703) and TACE-Cat (NP_003174). Identical residues are highlighted in grey and cysteine residues in yellow. The secondary structural elements, strands (s) and helices (h), are underlined with arrows and bars respectively, and the highly conserved Zn2+ co-ordination site is highlighted by a black box. (C) Sequence alignment of N-TIMP-1–3 (NP_003245, NP_003246 and NP_000353) using the same colour coding as in (B). The N-terminal wedge and the AB-, CD- and EF-loop regions are highlighted with black boxes.
Owing to the high structural similarities of their catalytic domains, metalloproteinases, including the ADAMs, are notoriously difficult to target selectively [15,16]. However, we recently discovered that TIMP-2 exhibits high inhibitory activity against ADAM12 [Ki (app) = 44 nM] [17]. This apparently unique TIMP inhibitory profile observed for ADAM12 (TIMP-3>TIMP-2≫TIMP-1) indicates that the engineering of an ADAM12-selective TIMP mutant should be possible. With this as our goal, in the present study we mapped and characterized the molecular determinants involved in ADAM12–TIMP complex formation. Using a quenched fluorescent peptide assay, we examined the inhibitory activity of wild-type and mutant forms of TIMPs and N-TIMPs against recombinant full-length ADAM12-S and truncated ADAM12-Cat, consisting of the pro and catalytic domains. By comparing our findings with the inhibitory profile of TIMPs against TACE, we demonstrated that, although the molecular interactions of TIMPs with ADAM12 and TACE are largely comparable, certain unique features of the ADAM12–TIMP interaction exist. Most remarkably, we found that removal of the AB-loop in N-TIMP-2, which significantly impairs its interaction with TACE, increased its affinity to ADAM12. Importantly, these in vitro data were confirmed in cell-based shedding assays using membrane-anchored EGF and HB-EGF as the cellular substrates and native ADAM proteases. Thus the reported results clearly encourage further efforts to develop a selective ADAM12 inhibitor that may have potential future experimental and/or therapeutic applications.
EXPERIMENTAL
Chemicals
Unless otherwise stated, all reagents and chemicals were from Sigma–Aldrich. Fluorescent-peptide substrates, Dabcyl-LAQAhomoPheRSK(FAM)-NH2 (hydrolysed by ADAMs) and Dabcyl-GPLGMRGK(FAM)-NH2 (hydrolysed by MMPs) were purchased from BioZyme Incorporated. The TACE inhibitor TAPI-2 and PMA were from Calbiochem.
Plasmids
The mammalian expression vector pCEP-4 and the Escherichia coli expression vector pRSET-C were from Invitrogen. Wild-type and catalytically inactive (E351Q point mutation) full-length human ADAM12-L constructs in the pcDNA3.1 vector have been described previously [18,19]. The cDNA constructs encoding pro-EGF or pro-HB-EGF fused to AP (alkaline phosphatase) in the pRC/CMV expression vector (AP–EGF and AP–HB-EGF) were provided by Dr Shigeki Higashiyama (Ehime University Graduate School of Medicine, Ehime, Japan) and Dr Michael Freeman (Children’s Hospital Boston, MA, U.S.A.) respectively.
Production of recombinant proteins
Human ADAM12 cDNAs (NM_021641) encoding either full-length ADAM12-S (amino acids 1–707) or the ADAM12-Cat (amino acids 1–419) in the pCEP4 plasmid were expressed in HEK (human embryonic kidney)-293 EBNA cells and recombinant proteins were purified as previously described [17,20]. Human MMP-1, MMP-2, MMP-3 and MT1-MMP were expressed in E. coli, purified, and the active site titrated as described previously [21,22]. Recombinant ADAM17 ectodomain (TACE651; TACE-long), was expressed from baculovirus (a gift of Dr David Becherer, GlaxoSmithKline, Research Triangle Park, NC, U.S.A.) and purified by Dr Peter Stanley according to the method of Milla et al. [23], with minor modifications. The mature catalytic domain of ADAM17 (TACE477GHHis6; TACE-Cat) was also prepared using the baculovirus system and was a gift of Dr Becherer. Expression and purification of human full-length TIMP-1 and TIMP-2, as well as N-TIMP-3, were all as previously described [24–26]. N-TIMP-1 and N-TIMP-2 in the pRSET-C E. coli expression vector and all variants of these (N-TIMP-1TACE, N-TIMP-1[V4S], N-TIMP-1[V4A], N-TIMP-1[T98L], N-TIMP-1[TIMP-2-ABloop], N-TIMP-1[TIMP-3-ABloop], N-TIMP-2TACE, N-TIMP-2TACE[L100E], N-TIMP-2-ΔABloop and N-TIMP-2TACE[F34G]) were generated as previously reported [14,27,28] and transformed into E. coli BL21(DE3). Cells were grown in LB (Luria–Bertani) medium containing 50 μg/ml carbenicillin until D600 = 0.8, then protein expression was induced by adding 1 mM isopropyl β-D-thiogalactoside followed by cultivation for 4 h at 37 °C. Cells were harvested by centrifugation, resuspended in 50 mM Tris/HCl, pH 8.0, and lysed by sonication (MSE Soniprep 150; five pulses of 30 s on ice). Inclusion bodies were collected by centrifugation for 15 min and washed once in 50 mM Tris/HCl, pH 8.0, containing 150 mM NaCl, twice in water, once in propan-2-ol, and once in water, then resuspended in inclusion body buffer containing 5 M guanidine hydrochloride, 50 mM Tris/HCl, pH 8.0, 0.2 mM GSH (reduced), 0.8 mM GSSG (oxidized) and 500 mM NaCl. Resuspended inclusion bodies were in vitro refolded essentially as previously described [27–29]. All proteins were purified by Ni-NTA (Ni2+-nitrilotriacetate) and cation-exchange chromatography and finally dialysed into 25 mM Hepes, pH 7.5, containing 100 mM NaCl, 0.05 % Brij35 and 0.02 %sodium azide. To determine the concentration of active N-TIMPs in each preparation, the inhibitor was titrated against a known amount of either MMP-1 or MMP-2. In addition to the active-site titrations, proteins were analysed by reducing and non-reducing SDS/PAGE on 15 %polyacrylamide gels followed by staining with Coomassie Brilliant Blue R-250. Single bands shifting in mobility under reduced and non-reduced conditions indicated that correct folding had occurred.
Kinetic analysis
Apparent inhibition constants [Ki (app)] were measured using a fluorescent peptide assay [30]. In brief, 5 μl of recombinant human ADAM12-Cat (0.1 nM final concentration) or full-length ADAM12-S (0.25 nM final concentration) was mixed with 5 μl of TIMPs at various concentrations and 90 μl of 20 mM Tris/HCl, pH 8.0, containing 20 mM NaCl, 1 mM CaCl2 and 0.05 % Brij35 and pre-incubated for 2 h at 37 °C. Reactions were initiated by adding 5 μl of the quenched fluorescent-peptide substrate (10 μM final concentration) and steady-state velocities (Vs) were monitored on a BMG Fluorostar Optima fluorimeter (excitation at 485 nM and emission at 530 nM). To determine Ki (app) values, Vs was plotted against TIMP concentration and data were fitted to the Morrison equation describing tight-binding inhibition using the computer software GraphPad Prism 5.0 (GraphPad Software) [17,30,31].
Cell culture
Human 293-VnR cells were kindly provided by Dr Archana Sanjay (Temple University School of Medicine, Philadelphia, PA, U.S.A.) and cultured in Dulbecco’s modified Eagle’s medium supplemented with 10 %fetal bovine serum, 1 % L-glutamine and 1 % penicillin/streptomycin (Invitrogen) as previously described [18]. The cells were transiently transfected with AP–EGF together with wild-type ADAM12-L or ADAM12-L (E351Q) using FuGENE® 6 Transfection Reagent (Roche Diagnostics) according to the manufacturer’s instructions. Human HT1080 cells (from American Type Culture Collection) were cultured and transfected with the AP–HB-EGF construct as described above. At 48 h after transfection, cells were trypsinized then stained for cell-surface AP expression using the mouse monoclonal anti-AP (clone 8B6) ascites fluid (Sigma–Aldrich) and Alexa Fluor® 488 donkey anti-mouse IgG (Invitrogen). The 10 % highest-expressing cells were isolated by FACS and maintained in culture by selection with 500 μg/ml geneticin (G418, Invitrogen).
Shedding assays
ADAM12-mediated AP–EGF shedding was determined by co-transfection of 293-VnR cells with either wild-type or catalytically inactive (E351Q) full-length ADAM12-L together with AP–EGF as previously described [18]. At 24 h after transfection, cells were seeded into 24-well plates and the following day washed twice with SFM (serum-free medium), treated with recombinant TIMP inhibitors or vehicle control as indicated for an initial 15 min, then incubated for 1 h with 250 μl of fresh SFM with or without inhibitor. For photometric quantification of AP–EGF shedding, cell-conditioned medium was harvested and the cell layer lysed in 250 μl of 1 % Triton X-100 in PBS. Conditioned SFM or cell lysate (50 μl) was mixed with 50 μl of a 2 mg/ml solution of the AP substrate 4-nitrophenyl and each well assayed in duplicate in a 96-well plate. After incubation at 37 °C for 1–2 h, the amount of product was quantified by measuring the absorbance at 405 nm. All treatments were performed in triplicate. AP–EGF shedding in cells transfected with wild-type or catalytically inactive ADAM12 was calculated as AP activity in the conditioned medium divided by AP activity in the medium and corresponding cell lysate after subtracting the background signal from non-transfected cells. The value obtained for wild-type ADAM12-L was then divided by the value for catalytically inactive ADAM12-L (E351Q), and the ADAM12-mediated cleavage calculated as the percentage of that observed in control-treated cells. To ensure equal expression levels of wild-type and catalytically inactive ADAM12-L, cell lysates from triplicate wells were pooled and examined by SDS/PAGE and Western blot as previously described [18,19].
As a measurement of TACE-mediated shedding, HT1080 cells stably expressing AP–HB-EGF were seeded in 96-well plates, then the confluent cell layer was treated 24 h later with 400 nM PMA or vehicle control for 30 min, and AP activity was measured in quadruplicate as described above. All data were analysed by one-way ANOVA with Dunnett’s or Bonferroni’s post tests for multiple comparisons, with P<0.05 considered statistically significant.
RESULTS AND DISCUSSION
Non-catalytic domains of ADAM12 regulate the binding affinity to TIMP-2 and -3
Together, the four TIMP inhibitors regulate the activity of all catalytically active ADAMs, ADAMTS (ADAM with thrombospondin repeats) and MMPs, which share high structural similarity in their catalytic domains [32]. Apart from a few exceptions, soluble MMPs are inhibited by TIMP-1–4, transmembrane MT-MMPs by TIMP-2–4, and ADAMs and ADAMTS by TIMP-3. However, we recently demonstrated that in contrast with other ADAMs, ADAM12 is also inhibited by TIMP-2 [17]. In that earlier study, the inhibitory activities of TIMP-1–3 were evaluated using the general substrate Cm-Tf (S-carboxymethylated transferrin), which at the time of the study was the only feasible substrate for kinetic analysis of ADAM12. Cm-Tf is in some ways a suboptimal substrate, as it generates degradation products that undergo further processing. Thus in the present study we utilized a novel quenched fluorescent-peptide assay to examine the apparent binding constants [Ki (app)] of wild-type and mutant forms of TIMPs and N-TIMPs against ADAM12-S (Table 1). Because our previous data revealed that the non-catalytic C-terminal domains of ADAM12-S regulate the catalytic activity, we tested various inhibitors against both full-length wild-type ADAM12-S and the truncated ADAM12-Cat that consists of the pro and catalytic domains. The Ki (app) values produced corresponded very well with our previous results using Cm-Tf as a substrate [17], supporting the use of Cm-Tf as a reliable substrate for kinetic studies.
Table 1. Apparent inhibition constants [Ki (app), nM] of wild-type TIMPs, N-TIMPs and N-TIMP mutants against ADAM12.
Wild-type full-length and N-terminal forms of N-TIMP-1–3, as well as N-TIMP mutants were tested against ADAM12-S and ADAM12-Cat and compared with inhibition constants for TACE-Cat or the full-length extracellular part of TACE (TACE-long) taken from the literature. The mutants of N-TIMP-1TACE (V4S/V69L/T98L/TIMP-3-ABloop) and N-TIMP-2TACE (S2T/A70S/V71L/TIMP-3-ABloop) were originally designed to increase the binding affinity against TACE. AB-loop mutations represent substitutions or deletion of the AB-loop region or a point mutation at amino acid 34 (F34G) in the AB-loop. Data are presented as the mean (n ≥ 3) ± S.D. N/A, not available.
| ADAM12-Cat | ADAM12-S | TACE-Cat | TACE-long | |
|---|---|---|---|---|
| N-TIMP-1 | 778 ± 90 | >500 | 356 ± 87 [23] | |
| TIMP-1 | 741 ± 62 | 223 ± 30 | ||
| N-TIMP-2 | 5.5 ± 0.7 | 69.7 ± 8.8 | 893 ± 126 [24] | |
| TIMP-2 | 2.5 ± 0.3 | 84.0 ± 13.6 | ||
| N-TIMP-3 | 0.93 ± 0.09 | 4.4 ± 0.4 | 0.22 ± 0.07 [25] | 1.75 ± 0.20 [25] |
| N-TIMP-1TACE | 1.9 ± 0.18 | 16.4 ± 2.2 | 0.14 ± 0.06 [30] | |
| N-TIMP-2TACE | 0.65 ± 0.02 | 6.0 ± 0.6 | 1.49 ± 0.31 [24] | |
| N-TIMP-1[V4S] | N/A | 122 ± 12.6 | 216 ± 44 [23] | |
| N-TIMP-1[V4A] | N/A | 190 ± 30.7 | 497 ± 180 [30] | |
| N-TIMP-1[T98L] | 11.2 ± 2.1 | 21.7 ± 2.3 | 21.1 ± 4.1 [30] | |
| N-TIMP-2TACE[L100E] | 4.1 ± 0.37 | 22.0 ± 2.8 | 35.9 ± 3.7 [24] | |
| N-TIMP-1[TIMP-2-ABloop] | 247 ± 65.6 | >500 | 57.9 ± 33 [30] | |
| N-TIMP-1[TIMP-3-ABloop] | N/A | 66.0 ± 4.0 | 67 ± 17 [23] | 452 ± 86 [23] |
| N-TIMP-2TACE[F34G] | N/A | 18.0 ± 2.2 | 21.6 ± 2.9 [24] | |
| N-TIMP-2-ΔABloop | 2.5 ± 0.2 | 28.1 ± 2.4 | >9000 |
N-TIMP-3 was the most active inhibitor of both ADAM12-S [Ki (app) = 4.4 nM] and ADAM12-Cat [Ki (app) = 0.93 nM] (Table 1). The kinetic analysis further revealed that the inhibitory activity of N-TIMP-2 [Ki (app) = 5.5 nM] and TIMP-2 [Ki (app) = 2.5 nM] against ADAM12-Cat greatly surpassed that of N-TIMP-2 [Ki (app) = 69.7 nM] and TIMP-2 [Ki (app) = 84.0 nM] against full-length ADAM12-S. As expected, N-TIMP-1 and TIMP-1 were the poorest inhibitors of ADAM12 activities, with Ki (app) values in the 0.2–0.8 μM range. On the basis of these results, the N-terminal domains of TIMP-2 and TIMP-3 appear to be sufficient for the inhibition of ADAM12-S. In addition, as previously demonstrated and in agreement with what has been previously shown for TACE [30], deletion of the C-terminal disintegrin and cysteine-rich domains in ADAM12 strengthened the inhibitory activity, which is most dramatically seen for TIMP-2 (34-fold, Table 1).
As revealed by the co-crystal structure of the TACE–N-TIMP-3 complex, as well as earlier mutagenesis studies, the TIMP reactive ridge is located exclusively in the N-terminal domain (Figure 1 and [12,14]). However, the C-terminal domains of both the inhibitor and the enzyme have been shown to regulate the affinity of the complex formation between ADAMs and TIMPs [33,34]. Model docking experiments of snake venom metalloproteinases VAP-1 and -2 with N-TIMP-3 suggest that the cysteine-rich domain approaches the active-site cleft, potentially interfering with the bound TIMP inhibitor [12]. Thus our findings may be explained by steric clashes between TIMPs and the cysteine-rich domains. In contrast with our findings, the N-terminal domains of TIMP-1 and TIMP-3 have been reported to be insufficient for the inhibition of ADAM10 [34], indicating the presence of yet unmapped interaction sites in TIMP-1 and TIMP-3 outside the catalytic cleft.
N-TIMP mutagenesis studies
As mentioned above, previous detailed mutagenesis studies have addressed the inactivity of TIMP-1, -2 and -4 against TACE [27,28,35]. Thus we used these findings, together with sequence alignments supported by the co-crystal structure of TACE–N-TIMP-3 (Figure 1 and [12]) to predict a number of ADAM12-interacting sites located in the TIMP reactive ridge. Interaction sites that were found to be identical in TIMP-2 and TIMP-3, but substituted in the weaker inhibitor TIMP-1, were selected and examined. Specifically, we hypothesized that a polar residue in position 4, as well as a leucine residue in the EF-loop (position 94 in N-TIMP-3), would be critical for binding to ADAM12. In addition, we chose to investigate the contribution of the highly variable AB-loops to binding, because the AB-loop region of TIMP-3 has proved to be critical for the interaction with TACE [28,29]. The previously reported N-TIMP mutants N-TIMP-1TACE (V4S/V69L/T98L/TIMP-3-ABloop) [27] and N-TIMP-2TACE (S2T/A70S/V71L/TIMP-3-ABloop) [28], both of which have high activity towards TACE, encompass affinity-optimizing mutations in all of the regions we expected to be essential for potent inhibition of ADAM12. We therefore initiated our mutagenesis study by addressing whether these TACE-directed mutants would also be improved in their reactivity towards ADAM12.
Indeed, both N-TIMP-1TACE and N-TIMP-2TACE proved to be excellent ADAM12 inhibitors, far exceeding the inhibitory potency of their wild-type counterparts (Table 1). Compared with the wild-type forms, N-TIMP-1TACE was improved more than 30-fold [Ki (app) = 16.4 nM compared with >500 nM] and N-TIMP-2TACE showed a ~ 10-fold enhancement [Ki (app) = 6.0 nM compared with 69.7 nM for ADAM12-S]. The positive influence on binding of the TACE-directed mutants was even more pronounced for the interaction between ADAM12-Cat and N-TIMP-1TACE, which displayed a >400-fold increase when compared with wild-type N-TIMP-1. It is also noteworthy that N-TIMP-2TACE, with a Ki (app) value of 0.65 nM for ADAM12-Cat, is the best ADAM12 inhibitor reported so far. The improved binding to ADAM12-S strongly suggests that our initial structural predictions were correct, and we next proceeded to dissect the contribution of the individual mutations to the binding affinity observed.
Adverse effects of N-TIMP-1 Val4 on the ADAM12 interaction
The first six amino acids of TIMPs constitute the N-terminal reactive ridge that inserts into the primed site of the enzyme’s catalytic cleft. The co-crystal structure of TACE–N-TIMP-3 demonstrated that the side chain of position 4 projects into the S3′ pocket, and mutational analysis has indicated a preference for polar residues in this position [14]. When we compared the position 4 residues of TIMP-1, -2 and -3 (Figure 1C), we noted that the serine residue found in TIMP-2 and TIMP-3 was substituted with a valine residue in the TIMP-1 sequence. Because this difference could indicate that ADAM12 prefers a polar residue in position 4, we tested the valine-to-serine mutant (N-TIMP-1[V4S]) to mimic the naturally occurring polar amino acid found in TIMP-2 and TIMP-3. This substitution resulted in a weakly improved binding affinity for ADAM12-S [Ki (app) of 122 nM compared with >500 nM for wild-type N-TIMP-1] (Table 1). Intriguingly, our kinetic analysis indicated that mutation to the less hydrophobic aliphatic alanine residue (V4A) was sufficient to overcome the unfavourable effects of valine [Ki (app) = 190 nM].
The fact that mutation to the less hydrophobic alanine residue had almost the same improved binding capacity as the serine substitution contradicts previous TACE data, which showed that alanine decreased the affinity of N-TIMP-1 against TACE [14,27]. The S3′ pocket of TACE is a wide and relatively deep depression that is not filled by any of the naturally occurring residues (valine and serine) [36]. However, since the same scenario is true for ADAM12, where our modelled structure of ADAM12 predicts an even deeper hydrophobic pocket with an 8.6 Å (1 Å = 0.1 nm) depression resembling that of TACE (results not shown), it does not explain the observed finding.
A leucine residue in the EF-loop is important for the ADAM12 interaction
TIMP-2 and -3 contain a leucine residue in the EF-loop (Figure 1C), and replacement of the corresponding Thr98 on N-TIMP-1 with a leucine residue (T98L) successfully transforms N-TIMP-1 into a potent tight binding inhibitor of both TACE and MT1-MMP [29]. To elucidate the impact of this particular leucine residue on the TIMP–ADAM12 interaction and to examine whether the T98L mutation in N-TIMP-1TACE could be the key residue explaining the superior fold improvement of N-TIMP-1TACE over N-TIMP-2TACE, where a leucine residue in this position is naturally occurring, we examined the N-TIMP-1[T98L] mutant. As observed for other metalloproteinases otherwise insensitive to N-TIMP-1, the mutation of Thr98 to leucine (T98L) transformed N-TIMP-1 into an active inhibitor of ADAM12-S and ADAM12-Cat [Ki (app) = 21.7 nM and 11.2 nM respectively] (Table 1).
Previous studies have shown that the EF-loop leucine residue is a prerequisite for high-affinity inhibition of TACE to occur, because substitution of Leu100 in N-TIMP-2TACE with any existing amino acid led to poorer Ki (app) values [27]. To confirm this effect for ADAM12, we examined the inhibitory potency of the N-TIMP-2TACE[L100E] mutant. Even though the L100E mutant showed reduced reactivity with ADAM12-S and ADAM12-Cat [Ki (app) = 22.0 nM and 4.1 nM respectively], the impairment was less pronounced than against TACE-Cat, where the mutation caused a ~ 24-fold decrease in the Ki (app) value (Table 1 and [29]). In the TACE–N-TIMP-3 co-crystal structure, the leucine residue protrudes from the EF-loop into a hydrophobic groove on TACE [12]; thus the relatively mild impact of the L100E mutation could suggest compensatory electrostatic interactions of Glu100 with sites in ADAM12.
N-TIMP-2 AB-loop deletion enhances the interaction with ADAM12
The AB-loop of TIMP-3 is of major importance for the inhibition of TACE [12]. It protrudes from the surface of the inhibitor and binds in a unique hydrophobic groove on TACE, with most direct intermolecular contacts being made via the phenyl group of the phenylalanine residue at position 34 (Figure 1A). Only TIMP-3 harbours a phenylalanine residue in this position, and kinetic studies have shown that TACE clearly favours the AB-loop of TIMP-3 over that of TIMP-2 [28]. To address the contribution of the AB-loop to ADAM12 binding, we first tested a N-TIMP-1 mutant whose short AB-loop (Thr32Thr33Leu34Tyr35) was replaced with either that of TIMP-2 (Asn33Asp34Ile35Tyr36Gly37Asn38Pro39) or TIMP-3 (Phe34Gly35Thr36). Whereas N-TIMP-1[TIMP-2-ABloop] remained a poor inhibitor of ADAM12-S, the binding capacity was improved 3-fold over that of N-TIMP-1 against ADAM12-Cat (Table 1). Transfer of the AB-loop of TIMP-3 to N-TIMP-1 in the N-TIMP-1[TIMP-3-ABloop] mutant transformed the inhibitor into a high-affinity inhibitor of ADAM12-S with a Ki (app) of 66.0 nM (Table 1). To explain the preference for the AB-loop of TIMP-3, we examined the importance of the phenylalanine residue at position 34. To this end, we used N-TIMP-2TACE as a scaffold and mutated phenylalanine to glycine (F34G). With a Ki (app) value of 18.0 nM against ADAM12-S, the F34G mutant is weakly but substantially impaired in its binding to ADAM12-S compared with N-TIMP-2TACE [Ki (app) = 6.0 nM] (Table 1). As a final experiment designed to investigate the role of the AB-loop in the interaction between ADAM12 and TIMPs, we evaluated an N-TIMP-2-ΔABloop mutant. In this mutant, the AB-loop region of TIMP-2 (Asn33Asp34Ile35Tyr36Gly37Asn38Phe39) was completely deleted. Inexplicably, the ΔABloop mutant showed a 2–3-fold increase in affinity against ADAM12-S [Ki (app) = 28.1 nM] and ADAM12-Cat [Ki (app) = 2.5 nM] (Table 1), which is in direct contrast with the interaction with TACE and MT1-MMP, where removal of the AB-loop of N-TIMP-2 was shown to have radical negative consequences [14,22].
These findings, together with the fact that ADAM12, unlike TACE, is well inhibited by wild-type TIMP-2, suggest major differences in their interactions with the AB-loop. In the TACE–N-TIMP-3 co-crystal structure, the phenyl group of the phenylalanine residue at position 34 in the AB-loop makes a number of direct contacts with a unique hydrophobic groove on the surface of TACE formed by the side chains of Tyr352, Val353, Tyr369 and Leu380. In the region between sIV and sV (Figure 1B), the sequence divergence between ADAM12 and ADAM17 is very large, indicating that the hydrophobic pocket accommodating Phe34 is a unique structural feature of TACE. Therefore the structural delineation of ADAM12–N-TIMP-3 is required to explain exactly why the F34G mutation decreases the Ki (app) value of N-TIMP-2TACE approx. 3-fold.
To validate these unexpected findings that N-TIMP-2-ΔABloop showed a ~ 2.5-fold increase in its affinity towards ADAM12 when compared with N-TIMP-2 wild-type, we reassessed previously determined Ki (app) values for this mutant against a number of soluble and membrane-type MMPs (MMP1, MMP2, MMP3 and MT1-MMP). These studies confirmed the values already provided in the literature, demonstrating that deletion of the AB-loop of TIMP-2 has little impact on the interaction with soluble MMPs, but is detrimental to the interaction with cell-surface-bound metalloproteinases (results not shown). Hence, the exception to this rule appears to be ADAM12, and consequently N-TIMP-2-ΔABloop appears to be a selective inhibitor of ADAM12.
TIMPs are functional inhibitors of ADAM12-mediated EGF shedding
The in vitro-established unique inhibitory profile for ADAM12 may offer a possibility to exploit TIMPs to discover hitherto unknown ADAM12-mediated cell-surface shedding events. We therefore used a cell-based shedding assay to examine whether the findings from our kinetic studies of soluble ADAM12-S reflected TIMP inhibition of the transmembrane ADAM12-L. For this purpose, a pro-EGF–AP fusion protein (AP–EGF) was used as a substrate, allowing the quantitative assessment of EGF shedding as AP activity in the conditioned cell culture medium. Wild-type ADAM12-L or the corresponding catalytic site E351Q mutant was co-expressed with AP–EGF in 293-VnR cells, and ADAM12 protease activity was determined as the ratio of EGF-shedding in cells transfected with wild-type ADAM12-L compared with catalytically inactive ADAM12-L. Both N-TIMP-2 and N-TIMP-3 inhibited EGF-shedding in a dose-dependent manner (Figure 2A). In agreement with our in vitro data, an almost complete inhibition of shedding activity was observed at 100 nM of N-TIMP-3 treatment, whereas N-TIMP-2 at an equimolar concentration inhibited activity ~ 40 %.
Figure 2. Effect of TIMPs on cell-based ADAM-mediated ectodomain shedding activity.
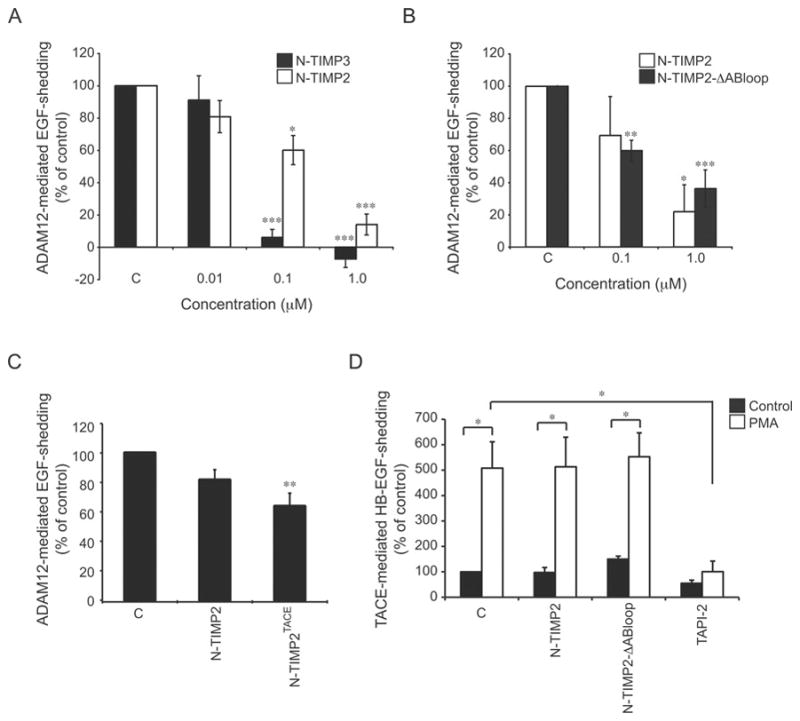
(A–C) At 48 h after transfection with either wild-type ADAM12-L or the corresponding catalytic site E351Q mutant and AP–EGF, 293-VnR cells were treated with vehicle control (C), wild-type N-TIMP-3 (N-TIMP3), N-TIMP-2, N-TIMP-2 with the AB-loop deleted (N-TIMP2-ΔABloop), or the N-TIMP-2TACE mutant (N-TIMP2TACE) recombinant protein for 1 h at the indicated concentration (in panel C, a concentration of 0.1 μM was used). ADAM12-mediated EGF shedding was calculated as AP activity in the cell media divided by AP activity in the media plus corresponding cell lysate from cells transfected with AP–EGF and wild-type ADAM12-L relative to cells transfected with AP–EGF and catalytically inactive ADAM12-L (E351Q). (D) HT1080 cells stably expressing AP–HB-EGF were treated with 400 nM PMA or vehicle control in combination with 0.1 μM recombinant wild-type N-TIMP-2 (N-TIMP2) or N-TIMP-2 with the AB-loop deleted (N-TIMP2-ΔABloop) mutant for 30 min. The TACE inhibitor TAPI-2 (10 μM) was used as a positive control. All data are shown as the mean of 3–5 independent experiments, each performed in triplicate, and error bars represent the S.E.M. *P< 0.05, **P< 0.01 and ***P< 0.005 relative to control treated cells.
We next tested whether the intriguing ability of the N-TIMP-2-ΔABloop mutant to selectively inhibit ADAM12 over TACE and MT1-MMP in vitro was also observed in the cell-based assay. Because some variation between assays exists, we repeated the N-TIMP-2 titration in parallel with the N-TIMP-2-ΔABloop experiment to allow for a direct comparison. The N-TIMP-2-ΔABloop inhibited shedding of AP-EGF to a similar extent to what we observed for wild-type N-TIMP-2 (~ 65 % inhibition at 1 μM concentration) (Figure 2B). We also tested N-TIMP-2TACE, which had also exhibited improved inhibitory capacity as compared with wild-type N-TIMP-2 in vitro. Although it showed a tendency towards increased inhibitory activity, N-TIMP-2TACE was not statistically significantly different from wild-type N-TIMP-2 (Figure 2C). The finding that neither N-TIMP-2TACE nor N-TIMP-2-ΔABloop showed improved inhibitory activity against ADAM12-mediated AP-EGF shedding as compared with wild-type N-TIMP-2 may indicate that the TIMP mutant is titrated by other metalloproteinases expressed by the cells, or alternatively, that the interaction of ADAM12 with the cell-bound AP–EGF substrate modifies the TIMP interaction. Importantly, however, the N-TIMP-2-ΔABloop mutant had no effect on PMA-induced AP–HB-EGF shedding, which is mediated by TACE and as expected could be blocked with the TACE inhibitor TAPI-2 (Figure 2D). Thus taken together, these findings confirm the specificity of N-TIMP-2 and N-TIMP-2-ΔABloop against ADAM12 that we observed in vitro.
In conclusion, we have identified an ADAM12-selective inhibitory profile of wild-type and mutant TIMPs, which could possibly be used to discover novel ADAM12 substrates and thereby reveal new biological implications of this protease. For example, the prominent cell-surface protein MT1-MMP is shed from the cell-surface by an unidentified ADAM enzyme [37]. Shedding of MT1-MMP can be inhibited by TIMP-3, to a lesser extent by TIMP-2, and weakly by TIMP-1, thus resembling the inhibitory profile of ADAM12. Moreover, the difference in the inhibitory behaviour of full-length TIMPs, N-TIMPs and mutant N-TIMPs towards ADAM12, ADAM10 and TACE is encouraging with respect to the design of selective inhibitors. In particular, the results obtained for the TIMP-2-ΔABloop mutant is of great value, and we hope that this mutation, in combination with other ADAM12-selective mutations, could aid the design of a highly selective ADAM12 inhibitor with future therapeutic applications.
Acknowledgments
We thank Dr Archana Sanjay, Dr Shigeki Higashiyama (Ehime University School of Medicine, Ehime, Japan) and Dr Michael Freeman (Children’s Hospital Boston, Boston, MA, U.S.A.) for providing the 293-VnR cells, AP-tagged EGF constructs, and HB-EGF constructs respectively. We thank Dr Peter Stanley (Cambridge Research Institute, Cambridge, U.K.) for the recombinant ADAM17 ectodomain. We also thank Dr Reidar Albrechtsen (Department of Biomedical Sciences and BRIC, University of Copenhagen) for critical reading of the manuscript and Linda Raab for editorial assistance prior to submission of the manuscript.
FUNDING
The work was supported by the Danish Medical Research Council, the Danish Cancer Society, and the Friis, Munksholm, Lundbeck, and Novo Nordisk Foundations to M.K. and U.W.; National Institutes of Health [grant number AR40449 (to H.N.); and Biotechnology and Biological Sciences Research Council (BBSRC) UK and Cancer Research UK to G.M.
Abbreviations used
- ADAM
a disintegrin and metalloprotease
- ADAM12-Cat
truncated ADAM12 protein consisting of only the pro and catalytic domains
- ADAM12-L
full-length ADAM12
- ADAM12-S
shorter secreted form of ADAM12
- ADAMTS
ADAM with thrombospondin repeats
- AP
alkaline phosphatase
- Cm-Tf
S-carboxymethylated transferrin
- EGF
epidermal growth factor
- HB-EGF
heparin-binding EGF
- MMP
matrix metalloproteinase
- MT-MMP
membrane-type MMP
- N-TIMP
N-terminal domain of TIMP
- SFM
serum-free medium
- TACE
tumour necrosis factor α-converting enzyme
- TIMP
tissue inhibitor of metalloproteinase
Footnotes
AUTHOR CONTRIBUTION
Jonas Jacobsen generated most of the recombinant proteins and performed all the kinetic peptide substrate assays. Marie Kveiborg performed all the cell-based experiments and Meng-Huee Lee created most of the TIMP expression constructs. Gillian Murphy and Hideaki Nagase designed and co-ordinated the study and Ulla Wewer contributed with discussion of the study. Marie Kveiborg and Jonas Jacobsen prepared the manuscript in consultation with Gillian Murphy, Hideaki Nagase and Ulla Wewer.
References
- 1.Kveiborg M, Albrechtsen R, Couchman JR, Wewer UM. Cellular roles of ADAM12 in health and disease. Int J Biochem Cell Biol. 2008;40:1685–1702. doi: 10.1016/j.biocel.2008.01.025. [DOI] [PubMed] [Google Scholar]
- 2.Gilpin BJ, Loechel F, Mattei MG, Engvall E, Albrechtsen R, Wewer UM. A novel, secreted form of human ADAM 12 (meltrin alpha) provokes myogenesis in vivo. J Biol Chem. 1998;273:157–166. doi: 10.1074/jbc.273.1.157. [DOI] [PubMed] [Google Scholar]
- 3.Blobel CP. Remarkable roles of proteolysis on and beyond the cell surface. Curr Opin Cell Biol. 2000;12:606–612. doi: 10.1016/s0955-0674(00)00139-3. [DOI] [PubMed] [Google Scholar]
- 4.Murphy G. The ADAMs: signalling scissors in the tumour microenvironment. Nat Rev Cancer. 2008;8:929–941. doi: 10.1038/nrc2459. [DOI] [PubMed] [Google Scholar]
- 5.Kurisaki T, Masuda A, Sudo K, Sakagami J, Higashiyama S, Matsuda Y, Nagabukuro A, Tsuji A, Nabeshima Y, Asano M, et al. Phenotypic analysis of meltrin alpha (ADAM12)-deficient mice: involvement of meltrin alpha in adipogenesis and myogenesis. Mol Cell Biol. 2003;23:55–61. doi: 10.1128/MCB.23.1.55-61.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Horiuchi K, Le Gall S, Schulte M, Yamaguchi T, Reiss K, Murphy G, Toyama Y, Hartmann D, Saftig P, Blobel CP. Substrate selectivity of epidermal growth factor-receptor ligand sheddases and their regulation by phorbol esters and calcium influx. Mol Biol Cell. 2007;18:176–188. doi: 10.1091/mbc.E06-01-0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dyczynska E, Sun D, Yi H, Sehara-Fujisawa A, Blobel CP, Zolkiewska A. Proteolytic processing of delta-like 1 by ADAM proteases. J Biol Chem. 2007;282:436–444. doi: 10.1074/jbc.M605451200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ito N, Nomura S, Iwase A, Ito T, Kikkawa F, Tsujimoto M, Ishiura S, Mizutani S. ADAMs, a disintegrin and metalloproteinases, mediate shedding of oxytocinase. Biochem Biophys Res Commun. 2004;314:1008–1013. doi: 10.1016/j.bbrc.2003.12.183. [DOI] [PubMed] [Google Scholar]
- 9.Loechel F, Fox JW, Murphy G, Albrechtsen R, Wewer UM. ADAM 12-S cleaves IGFBP-3 and IGFBP-5 and is inhibited by TIMP-3. Biochem Biophys Res Commun. 2000;278:511–515. doi: 10.1006/bbrc.2000.3835. [DOI] [PubMed] [Google Scholar]
- 10.Jacobsen J, Wewer UM. Targeting ADAM12 in human disease: head, body or tail? Curr Pharm Des. 2009;15:2300–2310. doi: 10.2174/138161209788682389. [DOI] [PubMed] [Google Scholar]
- 11.Fernandez-Catalan C, Bode W, Huber R, Turk D, Calvete JJ, Lichte A, Tschesche H, Maskos K. Crystal structure of the complex formed by the membrane type 1-matrix metalloproteinase with the tissue inhibitor of metalloproteinases-2, the soluble progelatinase A receptor. EMBO J. 1998;17:5238–5248. doi: 10.1093/emboj/17.17.5238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wisniewska M, Goettig P, Maskos K, Belouski E, Winters D, Hecht R, Black R, Bode W. Structural determinants of the ADAM inhibition by TIMP-3: crystal structure of the TACE-N-TIMP-3 complex. J Mol Biol. 2008;381:1307–1319. doi: 10.1016/j.jmb.2008.06.088. [DOI] [PubMed] [Google Scholar]
- 13.Amour A, Knight CG, Webster A, Slocombe PM, Stephens PE, Knauper V, Docherty AJ, Murphy G. The in vitro activity of ADAM-10 is inhibited by TIMP-1 and TIMP-3. FEBS Lett. 2000;473:275–279. doi: 10.1016/s0014-5793(00)01528-3. [DOI] [PubMed] [Google Scholar]
- 14.Lee MH, Maskos K, Knauper V, Dodds P, Murphy G. Mapping and characterization of the functional epitopes of tissue inhibitor of metalloproteinases (TIMP)-3 using TIMP-1 as the scaffold: a new frontier in TIMP engineering. Protein Sci. 2002;11:2493–2503. doi: 10.1110/ps.0216202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hu J, Van den Steen PE, Sang QX, Opdenakker G. Matrix metalloproteinase inhibitors as therapy for inflammatory and vascular diseases. Nat Rev Drug Discov. 2007;6:480–498. doi: 10.1038/nrd2308. [DOI] [PubMed] [Google Scholar]
- 16.Overall CM, Kleifeld O. Towards third generation matrix metalloproteinase inhibitors for cancer therapy. Br J Cancer. 2006;94:941–946. doi: 10.1038/sj.bjc.6603043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jacobsen J, Visse R, Sorensen HP, Enghild JJ, Brew K, Wewer UM, Nagase H. Catalytic properties of ADAM12 and its domain deletion mutants. Biochemistry. 2008;47:537–547. doi: 10.1021/bi701629c. [DOI] [PubMed] [Google Scholar]
- 18.Stautz D, Sanjay A, Hansen MT, Albrechtsen R, Wewer UM, Kveiborg M. ADAM12 localizes with c-Src to actin-rich structures at the cell periphery and regulates Src kinase activity. Exp Cell Res. 2010;316:55–67. doi: 10.1016/j.yexcr.2009.09.017. [DOI] [PubMed] [Google Scholar]
- 19.Sundberg C, Thodeti CK, Kveiborg M, Larsson C, Parker P, Albrechtsen R, Wewer UM. Regulation of ADAM12 cell-surface expression by protein kinase C epsilon. J Biol Chem. 2004;279:51601–51611. doi: 10.1074/jbc.M403753200. [DOI] [PubMed] [Google Scholar]
- 20.Wewer UM, Morgelin M, Holck P, Jacobsen J, Lydolph MC, Johnsen AH, Kveiborg M, Albrechtsen R. ADAM12 is a four-leafed clover: the excised prodomain remains bound to the mature enzyme. J Biol Chem. 2006;281:9418–9422. doi: 10.1074/jbc.M513580200. [DOI] [PubMed] [Google Scholar]
- 21.Chung L, Shimokawa K, Dinakarpandian D, Grams F, Fields GB, Nagase H. Identification of the (183)RWTNNFREY(191) region as a critical segment of matrix metalloproteinase 1 for the expression of collagenolytic activity. J Biol Chem. 2000;275:29610–29617. doi: 10.1074/jbc.M004039200. [DOI] [PubMed] [Google Scholar]
- 22.Lee MH, Rapti M, Murphy G. Unveiling the surface epitopes that render tissue inhibitor of metalloproteinase-1 inactive against membrane type 1-matrix metalloproteinase. J Biol Chem. 2003;278:40224–40230. doi: 10.1074/jbc.M305678200. [DOI] [PubMed] [Google Scholar]
- 23.Milla ME, Leesnitzer MA, Moss ML, Clay WC, Carter HL, Miller AB, Su JL, Lambert MH, Willard DH, Sheeley DM, et al. Specific sequence elements are required for the expression of functional tumor necrosis factor-alpha-converting enzyme (TACE) J Biol Chem. 1999;274:30563–30570. doi: 10.1074/jbc.274.43.30563. [DOI] [PubMed] [Google Scholar]
- 24.Huang W, Suzuki K, Nagase H, Arumugam S, Van Doren SR, Brew K. Folding and characterization of the amino-terminal domain of human tissue inhibitor of metalloproteinases-1 (TIMP-1) expressed at high yield in E. coli. FEBS Lett. 1996;384:155–161. doi: 10.1016/0014-5793(96)00304-3. [DOI] [PubMed] [Google Scholar]
- 25.Kashiwagi M, Tortorella M, Nagase H, Brew K. TIMP-3 is a potent inhibitor of aggrecanase 1 (ADAM-TS4) and aggrecanase 2 (ADAM-TS5) J Biol Chem. 2001;276:12501–12504. doi: 10.1074/jbc.C000848200. [DOI] [PubMed] [Google Scholar]
- 26.Troeberg L, Tanaka M, Wait R, Shi YE, Brew K, Nagase H. E. coli. expression of TIMP-4 and comparative kinetic studies with TIMP-1 and TIMP-2: insights into the interactions of TIMPs and matrix metalloproteinase 2 (gelatinase A) Biochemistry. 2002;41:15025–15035. doi: 10.1021/bi026454l. [DOI] [PubMed] [Google Scholar]
- 27.Lee MH, Rapti M, Knauper V, Murphy G. Threonine 98, the pivotal residue of tissue inhibitor of metalloproteinases (TIMP)-1 in metalloproteinase recognition. J Biol Chem. 2004;279:17562–17569. doi: 10.1074/jbc.M312589200. [DOI] [PubMed] [Google Scholar]
- 28.Lee MH, Rapti M, Murphy G. Delineating the molecular basis of the inactivity of tissue inhibitor of metalloproteinase-2 against tumor necrosis factor-alpha-converting enzyme. J Biol Chem. 2004;279:45121–45129. doi: 10.1074/jbc.M406611200. [DOI] [PubMed] [Google Scholar]
- 29.Lee MH, Rapti M, Murphy G. Total conversion of tissue inhibitor of metalloproteinase (TIMP) for specific metalloproteinase targeting: fine-tuning TIMP-4 for optimal inhibition of tumor necrosis factor-α-converting enzyme. J Biol Chem. 2005;280:15967–15975. doi: 10.1074/jbc.M500897200. [DOI] [PubMed] [Google Scholar]
- 30.Moss ML, Rasmussen FH. Fluorescent substrates for the proteinases ADAM17, ADAM10, ADAM8, and ADAM12 useful for high-throughput inhibitor screening. Anal Biochem. 2007;366:144–148. doi: 10.1016/j.ab.2007.04.043. [DOI] [PubMed] [Google Scholar]
- 31.Williams JW, Morrison JF. The kinetics of reversible tight-binding inhibition. Methods Enzymol. 1979;63:437–467. doi: 10.1016/0076-6879(79)63019-7. [DOI] [PubMed] [Google Scholar]
- 32.Nagase H, Visse R, Murphy G. Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc Res. 2006;69:562–573. doi: 10.1016/j.cardiores.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 33.Lee MH, Verma V, Maskos K, Becherer JD, Knauper V, Dodds P, Amour A, Murphy G. The C-terminal domains of TACE weaken the inhibitory action of N-TIMP-3. FEBS Lett. 2002;520:102–106. doi: 10.1016/s0014-5793(02)02776-x. [DOI] [PubMed] [Google Scholar]
- 34.Rapti M, Atkinson SJ, Lee MH, Trim A, Moss M, Murphy G. The isolated N-terminal domains of TIMP-1 and TIMP-3 are insufficient for ADAM10 inhibition. Biochem J. 2008;411:433–439. doi: 10.1042/BJ20071430. [DOI] [PubMed] [Google Scholar]
- 35.Lee MH, Verma V, Maskos K, Nath D, Knauper V, Dodds P, Amour A, Murphy G. Engineering N-terminal domain of tissue inhibitor of metalloproteinase (TIMP)-3 to be a better inhibitor against tumour necrosis factor-alpha-converting enzyme. Biochem J. 2002;364:227–234. doi: 10.1042/bj3640227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maskos K, Fernandez-Catalan C, Huber R, Bourenkov GP, Bartunik H, Ellestad GA, Reddy P, Wolfson MF, Rauch CT, Castner BJ, et al. Crystal structure of the catalytic domain of human tumor necrosis factor-alpha-converting enzyme. Proc Natl Acad Sci USA. 1998;95:3408–3412. doi: 10.1073/pnas.95.7.3408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Toth M, Sohail A, Mobashery S, Fridman R. MT1-MMP shedding involves an ADAM and is independent of its localization in lipid rafts. Biochem Biophys Res Commun. 2006;350:377–384. doi: 10.1016/j.bbrc.2006.09.052. [DOI] [PubMed] [Google Scholar]