


Abstract
We propose a novel, efficient and intuitive approach of estimating mRNA abundances from the whole transcriptome shotgun sequencing (RNA-Seq) data. Our method, NEUMA (Normalization by Expected Uniquely Mappable Area), is based on effective length normalization using uniquely mappable areas of gene and mRNA isoform models. Using the known transcriptome sequence model such as RefSeq, NEUMA pre-computes the numbers of all possible gene-wise and isoform-wise informative reads: the former being sequences mapped to all mRNA isoforms of a single gene exclusively and the latter uniquely mapped to a single mRNA isoform. The results are used to estimate the effective length of genes and transcripts, taking experimental distributions of fragment size into consideration. Quantitative RT–PCR based on 27 randomly selected genes in two human cell lines and computer simulation experiments demonstrated superior accuracy of NEUMA over other recently developed methods. NEUMA covers a large proportion of genes and mRNA isoforms and offers a measure of consistency (‘consistency coefficient’) for each gene between an independently measured gene-wise level and the sum of the isoform levels. NEUMA is applicable to both paired-end and single-end RNA-Seq data. We propose that NEUMA could make a standard method in quantifying gene transcript levels from RNA-Seq data.
INTRODUCTION
The emerging RNA-Seq (whole transcriptome shotgun sequencing) technology has been replacing microarray-based expression profiling (1–6). Unlike microarrays, RNA-Seq is free of background hybridization and has less systematic bias (7). Its potential for discovery of novel mRNA isoforms is another major advantage. Moreover, RNA-Seq exhibits potentially unlimited dynamic range, more than five orders of magnitude, while microarrays have limited dynamic range due to background noise and saturation of signals (3,8).
Estimation of mRNA abundance from aggregated reads is not a trivial task. There is yet no standard protocol for measuring mRNA levels from RNA-Seq data. We show that a substantial improvement can be achieved in quantification accuracy by properly treating the gene length. Generally, the expected number of reads mapped on a gene is proportional to both its transcript abundance and length. Therefore, to obtain the mRNA expression level, the number of reads must be normalized by the effective length. Despite its importance, one of the major challenges in finding the right length is that the length of a gene is not well defined, since a gene may have two or more mRNA isoforms of different lengths. Another problem is that some genes have spuriously fewer unambiguously mapped reads, because they contain more repetitive sequences than others.
Previously reported approaches include ‘projective normalization,’ in which all reads mapped on a gene is divided by the total number of exonic base pairs to compute a gene’s total transcript level (3). This method has been proven to work only for single isoform genes, by Trapnell et al. (See its Supplementary Data) (6). Another approach, the ‘average length’ method that considers the average isoform length as the gene length, tends to underestimate the expression levels (6). Trapnell’s own approach (Cufflinks) is to treat the abundance-weighted average of isoform lengths as the gene length. Earlier, Sultan et al. had developed the concept of virtual length, the number of all uniquely mappable 27 nt from all the exons and splice junctions of each gene and used it for normalizing the number of reads uniquely mapped on the gene (1). Sultan’s method partly serves as a basis for developing our method, but our method solves the two major length problems of ambiguous length definition and repetitive sequences effectively.
For a precise definition of length, one needs to first clarify what exactly is to be quantified. To this end, we separated gene level quantification and mRNA isoform level quantification, by utilizing regions common to all the isoforms of a gene and specific to individual isoform, respectively. This concept is not new in the RNA quantification field. In fact, traditional methods such as northern blotting, RNase protection assay and quantitative RT–PCR rely on design of probes or primers common to or specific to a gene’s mRNA isoforms. However, such distinction has not been made in high-throughput quantification methods.
In this article, we propose a simple and intuitive algorithm that deals with the gene length effectively. According to the experimental and computer simulation tests, our method achieved accuracy far superior to other recently developed methods by implementing simple yet elegant concepts. In the following sections, we describe the overview of the algorithm and performance tests based on simulated and real data.
MATERIALS AND METHODS
Expected uniquely mappable area (EUMA)
The central idea of our method lies in precise estimation of effective length both at gene and isoform levels using informative reads only. To explain this concept more clearly, we first define an isoform-specific informative read as a read mapped only to a specific mRNA isoform. Likewise, a gene-wise informative read is defined as a read commonly mapped to all the mRNA isoforms of the gene but not to any other genes. The expected uniquely mappable area (EUMA) serves as the length for a gene’s common area (gEUMA) or for an isoform-specific area (iEUMA). The mapping is done on the transcriptome sequence (the whole set of known mRNA sequences) rather than the genome, to make the calculations straightforward.
Our algorithm can be described in two steps—the sample-independent pre-processing step and the sample-dependent data analysis step as shown in Figure 1. We take the case of paired-end reads as an illustration example since it is more complicated due to the presence of variable fragment size.
Figure 1.

Algorithm overview, for paired-end RNA-Seq. (a) Calculation of gU and iU tables. First, all possible APEs are computationally made from the transcriptome sequence. The length d of an APE is fixed at each round. APEs are mapped back to the transcriptome sequence and classified into groups representing gene-wise (orange) and isoform-wise (green and violet) informative reads. APEs mapped on multiple genes (grey) are not used. For each mRNA isoform, APEs specific to the isoform are counted (iUd,i). For each gene, gene-wise informative APEs are counted (gUd,g). This procedure (from extraction of APEs to calculation of iUd,i and gUd,g) is repeated for every d, ranging from 37 to 250 bp in case of the 36-bp data. As a result, we obtain matrices gUd,g and iUd,i. (b) Calculation of EUMA and expression levels. Real RNA-Seq reads are mapped to the transcriptome sequence. For each gene gEUMA is computed by averaging gUd,i over all d, with weight P(d). iEUMA is computed likewise. P(d) is the probability distribution obtained from all mapped reads from the experiment. Then, for each gene, reads that are mapped to all of the gene's mRNA isoforms and not mapped to any other mRNA isoforms are counted (gNIR). Likewise, for each mRNA isoform, reads that are specifically mapped to the mRNA isoform are counted (iNIR). Finally, gNIR and iNIR are divided by gEUMA and iEUMA, to produce the mRNA abundance at the gene and isoform levels, respectively.
Sample-independent pre-processing step
To cover the entire space of sequence reads, we generate all possible distinct artificial paired-end reads (APEs) from the given transcriptome sequence (Figure 1a). The fragment size is systematically varied up to a certain limit to cover the experimental condition. After mapping APEs on the transcriptome sequence with perfect match, APEs are classified into three groups—gene-wise informative reads, isoform-specific informative reads, and all others including multi-reads that are mapped on multiple genes. The number of distinct informative reads are stored into matrices gUd,g and iUd,i for gene and isoform level estimation, respectively. This step takes a significant computing time to deal with all possible APEs for whole transcriptome sequences with a variable fragment size. But this needs to be done only once for a given transcriptome model and read length.
Sample-dependent data analysis step
The experimental paired-end reads are mapped on the transcriptome sequence using the same criteria. We use the experimental probability distribution of fragment size Ps(d), as the weighting function to calculate EUMA as follows (Figure 1b):
| (1) |
| (2) |
where gUd,g and iUd,i represent the number of distinct informative APEs of fragment size d for gene g and isoform i, respectively. Note that EUMA is estimated independently for each gene and isoform. The sample-dependency is taken into account via the experimental distribution of cDNA fragment size
Modification for single end reads is straight-forward. Since we do not have the variable fragment size, gU and iU become the number of total distinct informative reads for genes and isoforms, respectively. Then EUMA is independent of samples and can be pre-computed.
Calculation of mRNA abundance FVKM and LVKM by NEUMA
Surprisingly, use of the right reads has not gained as much attention as finding the right length to normalize them. Our NEUMA (Normalization by EUMA) algorithm matches these two in a coherent and effective way. The key is that only informative RNA-Seq reads are used to estimate the mRNA abundance. We apply the same procedure and criteria as APEs to obtain the number of informative reads (NIRs) among experimental data (gNIR and iNIR for gene and isoform levels, respectively, in Figure 1b). Note that NIRs may include reads of an identical sequence multiple times unlike APEs. These NIRs are divided by the corresponding normalization factor EUMA to produce mRNA abundance.
The mRNA expression level is often given by RPKM (reads per kilobase per million sequenced reads) (3) and FPKM (fragments per kilobase per million sequenced reads) (6). We introduce a comparable measure named FVKM (fragments per virtual kilobase per million sequenced reads) defined as FVKM = NIR/{EUMA(in kb) · N}, where N represents the total number of mRNA-mapped reads in the sample (in million). Note that the unit of EUMA is base pairs since it serves as the effective length. The gene and isoform levels are reported as gFVKM and iFVKM, respectively, as shown in Figure 1b.
For convenience in analysis and comparison with other mRNA abundance estimates, we adopt a new term, LVKM (log FVKM), representing the log2(x + 1) transformation on the FVKM values. The gLVKM and iLVKM values are computed from gFVKM and iFVKM, respectively. In the following comparison studies in the Results section, we applied the same log2(x + 1) transformation to the equivalent units such as RPKM and FPKM.
Methods
RNA-Seq data generation
cDNA library was prepared using random primed mRNA fragments using commercial kits and following manufacturer’s protocols. Sequencing of the library was carried out with the Illumina genome analyzer II under the standard protocols. Two human gastric cancer cell lines, MKN-28 and MKN-45 were used to generate 36-bp paired-end RNA-Seq data set. Detailed protocol is available upon request.
Quantitative RT–PCR
Total RNA was isolated from MKN-28 and MKN-45 cell lines using RNASpin® (iNtRON Biotechnology, Seongnam, Korea) and each cDNA was synthesized using oligo-dT primer and Improm-II reverse mRNA isoformaseTM (Promega, Madison, WI). Real-time quantitative PCR was performed with Bio-Rad iCycler iQTM5 instrument and iQTM SYBR® Green supermix (Bio-Rad Laboratories, Hercules, CA, USA) according to the manufacturer’s instructions. The relative expression level of each gene was normalized by that of GAPDH.
Mapping of paired-end RNA-Seq data
NEUMA
Mapping of the paired end sequences was done using Bowtie (9) (version 0.12.5) on the hg19 Refseq RNA sequences downloaded from the UCSC Genome Browser (http://genome.ucsc.edu) on 11 March 2010. For 36-bp paired-end data, Bowtie was run with the –v 0 –a −maxins 250 option, i.e. retrieving all aligned positions, allowing only perfect matches and cDNA fragment size from 37 up to 250 bp. For 50-bp data, the same option was used except for maxins = 400 (cDNA fragment size ranging from 51 to 400 bp). The same range of cDNA fragment sizes was used for generation of APEs. We mapped reads to all 32 774 RNA isoforms corresponding to 21 660 genes, then we used the abundance estimates of only 18 909 coding genes (29 754 mRNA isoforms), considering the poly-A selection step in the RNA-Seq procedure.
ERANGE
ERANGE 3.2 was run on top of Cistematic 3.0 and Bowtie 0.12.5, on the human genome sequence (hg19). Annotation files (gene_info.db and knownGene.txt) were matched to the versions of the software and the genome. Default options were used in mapping (-v 2 -k 11 -m 10 -t –best) and other steps for analyzing RNA-Seq data. Repeat mask option was used with the rmsk.txt file (hg19) obtained from the UCSC Genome Browser.
TOPHAT and cufflinks
TOPHAT version 1.0.14 and Cufflinks version 0.8.3 were used on hg19 refGenes data. TOPHAT was run with the option of -m 2 -g 10. For paired-end runs, the inner mate distances (–r) were set to 60 for the MKN-28 and MKN-45 data (36-bp), and to 160 for the 50-bp simulated data. The -closure-search option was used for paired-end runs. The mapping results from TOPHAT were fed to Cufflinks. For TOPHAT estimation of expression levels, we used TOPHAT version 1.0.11, the most recent version that produces abundance estimates. Bowtie 0.12.5 was used for all TOPHAT runs.
Gene selection for quantitative RT–PCR
The focus of the selection scheme was (i) to randomly select genes that can representatively cover all range of transcript levels, (ii) to include both a set of genes whose estimates were different among methods and a set whose abundance estimates were consistent among the methods and (iii) to have the same set of genes for MKN-28 and MKN-45.
For each of the two cell lines, reciprocal pair-wise linear regressions were performed to compute residual z-scores between NEUMA, TOPHAT and ERANGE-based expression values. Then, all genes were ranked by residual z-scores from the NEUMA-TOPHAT regressions and by z-scores from the NEUMA-ERANGE regressions. The union was taken for the top 600 genes from the two rankings to report a ‘variable’ set in each cell line. The variable set represents genes whose transcript level estimation was widely inconsistent among the three methods. Likewise, the union of bottom 1500 genes was taken to make an ‘invariable’ set for each cell line. The invariable set represents genes whose transcript level estimation was most consistent among the three methods.
Next, all genes were divided into eight groups, according to the MKN-28 and MKN-45 transcript levels estimated by NEUMA. In each group, a final variable gene set was computed by intersecting MKN-28- and MKN-45 variable sets, and a final invariable set was obtained by intersecting MKN-28- and MKN-45- invariable sets. Two or three variable genes and one invariable gene were randomly chosen for each group.
Simulation of RNA-Seq
To assess the accuracy of the NEUMA estimates, we simulated RNA-Seq experiments using the Flux Simulator (http://flux.sammeth.net/simulator.html) (build 20100702). Flux Simulator provides an in silico production of the experimental pipelines for RNA-Seq, adopting a set of parameters. We set NB_MOLECULE (total number of RNA molecules in the sample) to be 50 millions, reverse-transcribed cDNA molecule range from 30 to 1000 bp and read length of 36 and 50 bp. Other parameters were set to mimic true experimental conditions. We simulated sequencing several times to produce technical replicates from the same ‘sample’, i.e. random assignment of mRNA abundances, and obtained about 114 million reads in total for four samples. Flux Simulator produced RNA-Seq reads from the UCSC hg19 whole genome sequence and mRNA isoform annotation (UCSC, refGene). Prediction accuracy was computed on genes and mRNA isoforms that exist in both genomic (refGene) and transcriptomic (RefSeq) references.
RESULTS
Analysis of human RNA-Seq data with the RefSeq mRNA models
To assess the performance of our NEUMA method, we produced 36-bp paired-end RNA-Seq data for two human gastric cancer cell lines, MKN-28 and MKN-45, whose total numbers of reads were 7.56 and 1.54 millions, respectively. These two data sets represent cases with relatively high and low mapping percentage (61.3 and 12.3%, respectively on total RefSeq RNAs).
For MKN-28, out of the 1.86 million reads mapped on RefSeq mRNAs with perfect match, 1.68 and 1.17 million reads were gene-wise and isoform-wise informative (i.e. total gNIR and iNIR in Figure 1b), respectively. For MKN-45, we had 0.19 million mapped reads with the total gNIR and iNIR to be 0.17 and 0.12 millions, respectively. Note that these numbers are not mutually exclusive since genes with a single isoform are counted in both cases.
Not all genes or isoforms are measurable because the number of distinct informative reads is not sufficient for reliable estimation of mRNA abundance. We define ‘measurable’ genes or isoforms as those whose EUMA value is above a certain cutoff. This implies that these mRNA sequences have significant portion of informative regions to estimate the expression level. It is important to distinguish unmeasurable genes and isoforms from unexpressed ones. ‘Unmeasurable’ means that our method cannot estimate the expression reliably, whereas ‘unexpressed’ means that no informative reads are observed experimentally even though the gene has enough mappable area. In the final output, measurable but unexpressed genes are reported as zero whereas unmeasurable genes are removed. Total numbers of measurable genes and mRNA isoforms were 18 680 and 26515 in the MKN-28 data, respectively. The corresponding numbers for the MKN-45 cell line data were 18 666 and 26 454, almost independent of sample size.
Comparison of various methods
Performance of the NEUMA method was evaluated in two ways by analyzing real paired-end RNA-Seq data for two gastric cancer cell lines and by using computationally generated data sets. We compared the prediction accuracy of NEUMA with three other publicly available, recently developed, and widely used programs—ERANGE (3), TOPHAT (10) and Cufflinks (6). These programs have been used commonly to obtain mRNA expression levels (RPKM and FPKM), in combination with their other major functionalities such as detection of novel genes or isoforms. We have restricted the comparison to quantification accuracy only.
Quantitative RT–PCR
In an effort to gain a comparative perspective on accuracy among different programs, the gene’s total transcript level estimated from the RNA-Seq data was compared with quantitative RT–PCR results. We selected 27 genes for quantitative RT–PCR experiments in such a way that a wide range of mRNA expression levels could be covered (see the Method section for more details). The results of quantitative RT–PCR measurements were reliable with the mean standard error of 0.078. Details of quantitative RT–PCR experiment are given in Supplementary Table 1.
Figures 2 and 3 show the comparison between quantitative RT–PCR and prediction results from RNA-Seq data for two cell lines. The squared correlation coefficient r2 values of the NEUMA method were over 0.90 for both cell lines, whereas those of other methods ranged from 0.62 to 0.79. The scatter plot shows that the agreement is uniformly superior for NEUMA method regardless of the expression level. Comparison between the two cell lines shows that NEUMA’s performance is sound even at low sequencing coverage, being almost independent of the number of sequence reads.
Figure 2.

Scatter plots of gene’s total transcript level measured by RT–qPCR (log2-transformed) versus estimation from RNA-Seq [log2(x + 1)-transformed RPKM, FPKM and FVKM] for human gastric cancer cell line MKN-28. Four different RNA-Seq processing methods were compared: (a) NEUMA (FVKM), (b) Cufflinks (FPKM), (c) TOPHAT (RPKM) and (d) ERANGE (RPKM).
Figure 3.

Scatter plots of gene’s total transcript level measured by RT–qPCR (log2-transformed) versus estimation from RNA-Seq [log2(x + 1)-transformed RPKM, FPKM and FVKM] for human gastric cancer cell line MKN-45. Four different RNA-Seq processing methods were compared: (a) NEUMA (FVKM), (b) Cufflinks (FPKM), (c) TOPHAT (RPKM) and (d) ERANGE (RPKM).
Abundant genes
As an indirect measure of prediction accuracy, we selected top 30 highly expressed genes from MKN-28 data and examined how many of those were well-known abundant genes such as GAPDH, ACTB and ribosomal genes. NEUMA’s top 30 genes included 18 such cases, whereas other methods predicted significantly fewer abundant genes (10, 7, 4 for Cufflinks, TOPHAT and ERANGE, respectively). The results are shown in Supplementary Table 2.
Simulation
Computer simulation is frequently used to estimate prediction accuracy. Using the Flux Simulator program, we generated 50-bp paired-end RNA-Seq data (see the ‘Methods’ section for more details). The data set consists of five technical replicates of different sizes. The numbers of generated mate pairs in the sample ranged from 1 to 8 millions. Each replicate data set was analyzed by using the NEUMA, ERANGE, TOPHAT, and Cufflinks pipelines.
The resulting mRNA abundances were compared with the true values used in simulation. We used the log-transformed version, i.e. log2(x + 1), to calculate the prediction accuracy (Squared Pearson correlation coefficient between the true and estimated abundances, r2), where x indicates the abundance equivalent for each method (FVKM for NEUMA, RPKM for ERANGE and TOPHAT, and FPKM for Cufflinks). The results at gene and isoform levels are shown in Figure 4a and b, respectively. Our NEUMA method demonstrated a significantly better performance than the other methods in all range of sample size. The gene-level estimation achieved prediction accuracy 0.82 with NEUMA, compared to 0.79 for the second (Cufflinks) for sample size 7 millions. NEUMA performs pretty well in the isoform-level estimation as well with r2 = 0.77, whereas other methods showed substantial decrease (0.66 for Cufflinks and 0.31 for TOPHAT for the same sample size). Note that the agreement generally increases with sample size although saturation was observed.
Figure 4.

Comparison of four methods in prediction accuracy as a function of total number of reads. Prediction accuracy was defined as the Pearson correlation coefficient between true and estimated mRNA abundances. The x-axis denotes the total number of reads generated in each simulation for technical replicates. (a) Gene-level estimation for 50-bp paired-end RNA-Seq data. (b) Isoform-level estimation for 50-bp paired-end RNA-Seq data. (c and d) Gene and isoform-level estimation for 36-bp single-end RNA-Seq data. ERANGE does not report mRNA isoform abundances and was excluded from isoform analyses.
We also tested the single-end case with a set of 36-bp RNA-Seq data. Again, NEUMA outperformed other methods significantly (Figure 4c and d), achieving the levels of accuracy similar to that in the paired-end case.
Quantitative RT–PCR and computer simulation tests provide complementary assessments, since the former provides a real sample validation for a limited number of genes, whereas the latter offers comparison with true theoretical mRNA abundance for as many genes and mRNA isoforms as in the known transcriptome model. Our NEUMA method showed a dramatic improvement over the other methods in both aspects.
Consistency coefficient between gene-wise and isoform-wise estimates
For genes with multiple isoforms, mRNA abundance can be calculated for the gene and all of its isoforms if their corresponding EUMA values are above the cutoff value. We call these genes completely measurable. For these genes, we can compare the mRNA abundance estimated at the gene level with the sum of its entire isoforms’ expression estimates (i.e. gFVKM versus ∑iFVKM). These two numbers are expected to be the same if the catalog of mRNA isoform is complete.
We propose the ‘consistency coefficient’ that could measure the agreement between mRNA abundance estimated at the gene level and the sum of its isoform abundances. The index is defined as the Pearson correlation coefficient within the completely measurable genes as follows:
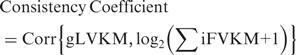 |
(3) |
NEUMA uses disjoint areas and reads to compute gene’s total transcript level and isoform-specific expression levels. In an ideal situation, i.e. with sufficiently large number of uniformly distributed sequenced reads and comprehensive knowledge of isoform catalog, the consistency coefficient would be close to 1. Large deviation could indicate that any of these conditions were not satisfied or that the experiment might be flawed. Thus, we expect that the consistency coefficient could serve as a measure of data quality.
To test the hypothesis, we used computer simulation to examine whether the consistency coefficient and prediction accuracy are proportional. Based on in silico 36-bp and 50-bp paired-end RNA-Seq data sets with technical replicates of different sizes, the consistency coefficient showed strong correlation (r2 = 0.895) with the prediction accuracy, regardless of the read length (Figure 5). It is also noteworthy that NEUMA’s accuracy improved considerably with longer reads. This confirms that the consistency coefficient well reflects the data quality and prediction accuracy.
Figure 5.

Plot of prediction accuracy versus consistency coefficient for technical replicates of four simulated 36- and 50-bp paired-end RNA-Seq samples. Each data point represents different number of sequence reads generated (labeled in million).
In the MKN-28 and MKN-45 samples, we obtained 4316 and 4271 completely measurable genes, respectively. The consistency indices were 0.90 and 0.64 for MKN-28 and MKN-45, respectively. As described earlier, the MKN-45 data had a lower percentage of mapped reads than MKN-28. These two facts suggest a relatively poor quality of the MKN-45 data. However, it should be pointed out that NEUMA’s performance was not significantly affected according to the quantitative RT–PCR results.
Sensitivity of EUMA cutoff
Throughout this study, we have used the cutoff value of 50 bp for both gEUMA and iEUMA. The numbers of measurable genes and isoforms at 50-bp cutoff were 18 680 (98.8%) and 26 515 (89.1%), respectively in MKN-28. These numbers are close to the maximum (at cutoff 0bp), which are 18763 (99.2%) and 27 952 (93.9%), respectively, as shown in Figure 6. The coverage of isoforms decreases rapidly in the range of 50- to 200-bp cutoff in both MKN-28 and MKN-45.
Figure 6.

The percent of measurable genes and isoforms as a function of EUMA cutoff in two MKN cell lines.
To demonstrate the sensitivity of NEUMA at the default cutoff, we offer the case of RPS24 gene in MKN-28 as an example. The RPS24 gene has six isoforms with subtle differences as shown in Figure 7. Five of its six isoforms, including the top three most highly expressed ones, had marginal iEUMA values at the default cutoff (Table 1). Nevertheless, the sum of iFVKM values (1018.78) was extremely close to the independently measured gFVKM value (1010.172) based on gEUMA = 426.68. Since EUMA is an expected number of positions per mate distance, the total number of possible mapping pairs for all distances is much larger than 50 in the paired-end case.
Figure 7.

Isoform structure of RPS24 gene. All six mRNA isoforms have unique regions.
Table 1.
NEUMA result for the six isoforms of the RPS24 gene
| RPS24 mRNA isoform | iFVKM | iEUMA (bp) |
|---|---|---|
| NM_001142283 | 0 | 63.56 |
| NM_001026 | 815.545 | 64.50 |
| NM_001142285 | 0.235 | 2282.69 |
| NM_001142284 | 0 | 59.57 |
| NM_001142282 | 152.249 | 63.46 |
| NM_033022 | 50.749 | 63.46 |
The comprehensive scanning of the mRNA models for generation of APEs allows high gene and isoform coverage. The total numbers of gene-wise and isoform-wise informative reads were over 10 and 8.7 billions, respectively using the RefSeq transcriptome sequence. For both MKN-28 and MKN-45 data sets, genes and mRNA isoforms contained on average 87 and 51% of the mapped reads as informative reads, respectively.
DISCUSSION
More details of NEUMA algorithm
Adjustment of mRNA abundance at the gene level
Some unmeasurable genes (gEUMA below cutoff) may still be quantified by adopting the sum of all of its measurable isoforms as the gene-level abundance. Other genes may be underestimated due to unknown isoforms, in which case the gene-level estimate can be corrected for the sum of all known isoform estimates. Accordingly, NEUMA takes the representative gene-level mRNA abundance as the larger one between the original gene-level estimate (gFVKM) and the sum of all of its measurable iFVKM values. The NEUMA program reports final gLVKM values based on the adjusted estimates. In the MKN-28 and MKN-45 samples, 1560 and 848 genes are affected by this adjustment, respectively.
All the gLVKM values used in the study are after the final adjustment of gene-level estimates. The consistency coefficient and the RPS24 gene analysis were based on the original estimates.
Distribution of read mate distance, P(d)
As shown in Supplementary Figure 1, the cDNA fragment length distribution is usually sharp and rarely goes beyond a certain point. Thus, one can identify a safe range of mate distance d for computation of EUMA. We used cDNA fragment length 250 and 400 bp as distance upper-bounds for 36 and 50 bp cases, respectively.
Computation time
Computation of the gU and iU tables is the time limiting step, but this needs to be done only once for a given transcriptome model and read length. Given these pre-computed tables, calculating the mRNA abundances (gLVKM and iLVKM) is a fast process. This step took about 10 min on a Linux platform with 2.66 GHz CPU and 16 Gb memory for a sample data of ∼2 million reads. The preprocessing step for 21 660 hg19 RefSeq genes takes about an hour on a cluster of 559 2.66 GHz cores, involving extensive memory swaps.
Availability
The program source code (written in Perl scripts) is available at http://neuma.kobic.re.kr. Pre-computed tables iU and gU for read lengths of 36 and 50 bp for human are also available at the website, along with the raw sequence data for the two human cell lines and simulated RNA-Seq reads.
Transcriptome mapping versus genome mapping
The use of the transcriptome sequence makes calculation fast and straightforward, though the concepts of NEUMA can be applied on the genome sequence as well. Artificial read generation and mapping is done on the same reference for computation of EUMA. Reads spanning exons and splice junctions can be handled equally with no complications. Mate pair distances are obtained without concerning introns. Despite its disadvantage of lacking the ability to identify and incorporate unknown transcripts, we suggest that using the transcriptome sequence is practically useful for quantification purpose.
Use of multi-reads
We did not use reads mapped on multiple genes (multi-reads) for quantification. A commonly used strategy of dealing with multi-reads is to distribute them in proportion to the abundance of unique reads, either directly or iteratively. Likewise, we could distribute multi-reads in proportion to the gFVKM values. However, we propose that this is unnecessary and even rather harmful. In theory, the multi-reads distributed in proportion to gFVKM should be scattered around the gFVKM value. In other words, the accuracy of a normalized multi-read count is limited by the accuracy of the unique-read based estimate. Any difference between the gFVKM and a normalized multi-read count comes from noise and therefore incorporation of this difference does not improve the estimation but only worsens it.
Flux simulator
The Flux Simulator program uses a genome sequence and a gene annotation file (UCSC refGene) identical to the reference used by the other programs. Thus, the prediction accuracy computed by Cufflinks and TOPHAT are based on the assumption that they were run given the perfect knowledge of all isoforms. However, NEUMA uses the transcript sequence library, which is slightly different from the transcriptome source that Flux Simulator used, in isoform annotation and mRNA nucleotide sequence. Note that this setting is disadvantageous to NEUMA. Nevertheless, NEUMA shows a superior performance to the other methods.
For comparison between prediction accuracy and consistency coefficient, we ran NEUMA on filtered data that contains only reads sourced from the mRNA accessions present in the RefSeq library, i.e. assuming that we have a perfect knowledge of all isoforms.
Related methods
In consistent with the rising demand for a reliable quantification system based on RNA-Seq data, many research groups have been working on this problem. For example, Jiang and Wong (11) modeled reads falling on multiple isoforms as a Poisson variable and used estimated expression parameters using a Bayesian approach. Howard and Heber (12) and Bohnert et al. (13) explore the position bias of reads on a transcript. Recently, Srivastava and Chen (14) estimated position-wise parameters in a two-parameter generalized Poisson model. Our system is free of any probabilistic model and assumes uniform sampling of reads in an RNA-Seq experiment. Combination of these approaches with the concepts of NEUMA may lead to a more reliable abundance estimation.
More challenges
As pointed out in Trapnell et al. (6), measuring mRNA isoform abundance can benefit from knowledge of correct isoform structures. This is true as well for traditional methods such as Northern blotting and RT–qPCR. Like any other methods, the NEUMA method is not free of errors coming from ignorance of some isoforms. Therefore, it is crucial to identify all existing isoforms in the long run, for accurate quantification of a transcriptome. There have been active efforts towards this direction recently (6,15). Advancement in quantification scheme and detection of novel isoforms are two important and complementary aspects of high-throughput global expression profiling.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
This work was supported by Korea Research Institute of Bioscience and Biotechnology Research Initiative Program [to S.L.]; ‘Systems Biology Infrastructure Establishment Grant’ provided by Gwangju Institute of Science & Technology in 2010 through Ewha Research Center for Systems Biology [to S.L.]; and Korea Healthcare Technology R&D Project [A084417 to C.K.]. Funding for open access charges: Korea Research Institute of Bioscience and Biotechnology Research Initiative Program.
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
The authors thank Dr Sung-Min Ahn and Dr Seong-Jin Kim at Gachon University of Medicine and Science, Korea, for providing the RNA-Seq data in the MKN-28 and MKN-45 cell lines. The authors thank Eujin Kwak at KOBIC for her help with preparation of figures and website. The authors also thank Dr Namshin Kim and Dr In-Sun Chu at Korean Bioinformation Center (KOBIC), Korea, Dr Gaurav Sablok at JNV University, India, members of Dr Changwon Kang’s lab and two anonymous reviewers for their insightful comments.
REFERENCES
- 1.Sultan M, Schulz MH, Richard H, Magen A, Klingenhoff A, Scherf M, Seifert M, Borodina T, Soldatov A, Parkhomchuk D, et al. A global view of gene activity and alternative splicing by deep sequencing of the human transcriptome. Science. 2008;321:956–960. doi: 10.1126/science.1160342. [DOI] [PubMed] [Google Scholar]
- 2.Nagalakshmi U, Wang Z, Waern K, Shou C, Raha D, Gerstein M, Snyder M. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science. 2008;320:1344–1349. doi: 10.1126/science.1158441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 4.Ramskold D, Wang ET, Burge CB, Sandberg R. An abundance of ubiquitously expressed genes revealed by tissue transcriptome sequence data. PLoS Comput. Biol. 2009;5:e1000598. doi: 10.1371/journal.pcbi.1000598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456:470–476. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marioni JC, Mason CE, Mane SM, Stephens M, Gilad Y. RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome Res. 2008;18:1509–1517. doi: 10.1101/gr.079558.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jiang H, Wong WH. Statistical inferences for isoform expression in RNA-Seq. Bioinformatics. 2009;25:1026–1032. doi: 10.1093/bioinformatics/btp113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Howard BE, Heber S. Towards reliable isoform quantification using RNA-SEQ data. BMC Bioinformatics. 2010;11(Suppl. 3):S6. doi: 10.1186/1471-2105-11-S3-S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bohnert R, Behr J, Rätsch G. Transcript quantification with RNA-Seq data. BMC Bioinformatics. 2009;10:P5. [Google Scholar]
- 14.Srivastava S, Chen L. A two-parameter generalized Poisson model to improve the analysis of RNA-seq data. Nucleic Acids Res. 2010;38:e170. doi: 10.1093/nar/gkq670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Richard H, Schulz MH, Sultan M, Nurnberger A, Schrinner S, Balzereit D, Dagand E, Rasche A, Lehrach H, Vingron M, et al. Prediction of alternative isoforms from exon expression levels in RNA-Seq experiments. Nucleic Acids Res. 2010;38:e112. doi: 10.1093/nar/gkq041. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.