


Abstract
FEN1, a key participant in DNA replication and repair, is the major human flap endonuclease that recognizes and cleaves flap DNA structures. Deficiencies in FEN1 function or deletion of the fen1 gene have profound biological effects, including the suppression of repair of DNA damage incurred from the action of various genotoxic agents. Given the importance of FEN1 in resolving abnormal DNA structures, inhibitors of the enzyme carry a potential as enhancers of DNA-interactive anticancer drugs. To facilitate the studies of FEN1 activity and the search for novel inhibitors, we developed a pair of complementary-readout homogeneous assays utilizing fluorogenic donor/quencher and AlphaScreen chemiluminescence strategies. A previously reported FEN1 inhibitor 3-hydroxy-5-methyl-1-phenylthieno[2,3-d]pyrimidine-2,4(1H,3H)-dione displayed equal potency in the new assays, in agreement with its published IC50. The assays were optimized to a low 4 µl volume and used to investigate a set of small molecules, leading to the identification of previously-unreported FEN1 inhibitors, among which aurintricarboxylic acid and NSC-13755 (an arylstibonic derivative) displayed submicromolar potency (average IC50 of 0.59 and 0.93 µM, respectively). The availability of these simple complementary assays obviates the need for undesirable radiotracer-based assays and should facilitate efforts to develop novel inhibitors for this key biological target.
INTRODUCTION
Human flap endonuclease 1 (FEN1) is an efficient structure-specific enzyme that recognizes and cleaves a 5′-unannealed DNA flap. It belongs to the RAD2 family of nucleases that metabolize DNA and is highly conserved in prokaryotes and eukaryotes (1–3). FEN1 is a key enzyme in DNA replication, repair and maintenance of genomic stability. 5′-flap removal by FEN1 is critical for Okazaki fragment processing during lagging strand DNA synthesis (4), long-patch base excision repair (LP BER) (5) and regulation of recombination [reviewed in (6)]. The importance of FEN1 in maintaining genomic stability is demonstrated by the phenotypes of cells deficient for the protein. Deletion of the yeast FEN1 gene, rad27, results in an increased frequency of short DNA (3–32 bp) repeats, micro- and mini-satellite formation, trinucleotide repeat expansion, spontaneous recombination events and a severe growth defect in association with cell cycle arrest in late S/G2 phase (6). Mice carrying a homozygous null genotype (fen1−/−) exhibit an early embryonic lethality (E4.5), indicating a requirement for normal development (7,8). Cells from fen1−/− blastocysts show increased apoptotic cell death after ionizing radiation treatment (8), and chicken cells lacking the fen1 gene are hypersensitive to DNA alkylating agents, e.g. methylmethane sulfonate (MMS) and N-methyl-N′-nitro-N-nitrosoguanidine (MNNG), as well as hydrogen peroxide (9). However, resistance to oxidative stress is only mildly affected in rad27-deletion yeast (10). These results indicate FEN1 deficiency leads to failure to repair DNA lesions generated by DNA alkylating agents, but the story is less clear with oxidizing compounds.
Several structure-specific 5′-flap substrates have been identified for FEN1. The nick-flap, a flap with its base adjacent to an upstream primer, is removed by FEN1, resulting in the production of a nick or a 1-nt gap (11,12). The double-flap with a 1-nt 3′-tail structure is also cleaved by FEN1 to generate only a single product, a nick that can be directly ligated. A substrate with this structure is optimal for FEN1 homologs of archeabacteria (13), yeast (14) and human (15) and is thought to be produced naturally in vivo as an intermediate of strand displacement DNA synthesis. The double-flap is bound with higher affinity, and cleaved with increased efficiency and specificity, compared to the nick-flap (6). A 5′-double-flap with a 10-nt 3′-tail is also efficiently cleaved by FEN1 in vitro and such double-flap structures may form during some homologous recombination (HR) events (16).
FEN1 is found to be increased in many human cancers, including lung cancer (17,18), gastric cancer (19), prostate cancer (20,21), pancreatic cancer (22), brain cancer (17,23) and breast cancer (24). These results suggest that FEN1 may, in part, be involved in tumor progression and development. In fact, FEN1 gene expression has been shown to be induced during cell proliferation and down-regulated during cell differentiation (25). In particular, elevation of FEN1 in hormone refractory human prostate cancer cells correlates with resistance to various anticancer agents, including ionizing radiation, doxorubicin, paclitaxel and vinblastine (26). Conversely, down-regulation of FEN1 has been shown to sensitize human glioblastoma cells to MMS, and the clinical drugs temozolomide (TMZ) and cisplatin (17). These studies suggest that the levels of FEN1 expression influence cancer cell function as it relates to proliferation potential, survival and apoptosis.
Most anticancer agents used in the clinic today introduce cytotoxic DNA lesions to destroy rapidly dividing cells (27). Cells have evolved a compilation of highly effective, conserved DNA repair systems to protect against both endogenous and exogenous DNA damage. However, these systems also process DNA lesions generated by anticancer drugs. Thus, BER, for example, has been shown to be an important factor in determining responsiveness to DNA-interactive drugs, such as alkylating agents (e.g. TMZ) and anti-metabolites (e.g. 5-fluorouracil and certain nucleoside analogs) (28). Moreover, tumor resistance to alkylating agents is common due to the increased levels of specific DNA repair enzymes (29). Human bladder carcinoma cells that lack flap endonuclease activity due to an amino acid point mutation in FEN1 (D181A) are highly susceptible to killing by the alkylating agent MMS (30), a compound that generates DNA substrates primarily processed by BER (31). Indeed, reduced FEN1 levels increase sensitivity of human cancer cells to the cytotoxicity of different alkylating agents (17). Moreover, it was recently shown that FEN1 can be a target in the selective killing of cancer cells via a mechanism involving ‘synthetic lethality’ (32). RAD54B-deficient human colorectal cancer cells, which are defective in HR, exhibit a proliferation defect and increased cellular cytotoxicity when FEN1 expression is reduced (33). The principle of synthetic lethality has been brought to the forefront and perhaps has been best exploited in the case of PARP-1 inhibitors, which promote the killing of BRCA-deficient (HR-defective) cancer cells (34,35). Therefore, FEN1 is a logical target for inactivation during both genetic-based and combinatorial anticancer treatment paradigms, and there is a need to develop functionally effective small molecule FEN1 inhibitors.
To facilitate the discovery and development of FEN1 inhibitors, a robust and sensitive method for monitoring its catalytic activity is needed. Traditionally, in vitro studies of FEN1 have utilized radiolabeled substrate constructs in conjunction with gel electrophoretic separation, a method not suitable for large-scale testing of inhibitors. Recently, an assay based on fluorescently labeled nucleotides was used in a screen to indentify inhibitors of FEN1 (36). In this system, 5′-end TAMRA (6-carboxytetramethylrhodamine) and 3′-end VIC (proprietary fluorescent dye excited at 488 nm, peak emission at 552 nm) labeled oligodeoxynucleotides were annealed together with a 3′-end BHQ (Black Hole Quencher, a non-fluorescent dark quencher) labeled strand to create a FEN1 substrate. VIC and BHQ were in close proximity, and the signal remained quenched upon excitation. In the presence of the enzyme, the VIC dye-carrying strand was cleaved off, leading to an increase in the VIC fluorescence. However, the substrate was unnecessarily complicated by the use of three dye labels and its use of ‘green’ fluorescence detection made it susceptible to autofluorescence from small molecule library members (37). Here, we describe the development of a pair of complementary-readout miniaturized homogeneous assays for FEN1 activity utilizing red-shifted fluorogenic donor/quencher and AlphaScreen chemiluminescence strategies. Testing a set of small molecules in these assays led to the identification of new FEN1 inhibitors.
MATERIALS AND METHODS
Reagents
An amount of 1 M Tris–HCl, Tween-20, EDTA, NaCl, MgCl2 and dithiothreitol (DTT) were purchased from Sigma–Aldrich. Dimethyl sulfoxide (DMSO, certified ACS grade) was obtained from Fisher, Inc. Black and white solid-bottom 384-well and 1536-well plates were purchased from Greiner Bio One (Monroe, NC, USA). The AlphaScreen FITC/streptavidin detection kit was from PerkinElmer Life and Analytical Sciences (Waltham, MA, USA).
Small molecule inhibitors
Arylstibonic inhibitors (NSC 13744, NSC 13793, NSC 15596 and NSC 13755) were obtained from the National Cancer Institute Developmental Therapeutics Program Natural Products Repository, while aurintricarboxylic acid (ATA) was purchased from Sigma-Aldrich. 3-hydroxy-5-methyl-1-phenylthieno[2,3-d]pyrimidine-2,4(1H,3H)-dione (hereafter PTPD) was synthesized according to reported methods (36). The compounds were initially prepared as 10 mM DMSO stock solutions and were arrayed for testing as serial 2-fold dilutions at 7 µl per well in 1536-well Greiner polypropylene compound plates following previously described protocols (38).
FEN1 enzyme
Recombinant, untagged human FEN1 protein was purified from bacteria as previously described (1).
Oligodeoxynucleotide substrates
All oligodeoxynucleotides were purchased from Biosearch Technologies, Inc., (Novato, CA, USA). The double-stranded DNA substrate containing a double flap region used in the fluorogenic assay was prepared from three oligodeoxynucleotides: quencher (5′-CAC GTT GAC TAC CGC TCA ATC CTG ACG AAC ACA TC-BHQ-2), flap (5′-TAMRA-GA TGT CAA GCA GTC CTA ACT TTG AGG CAG AGT CCG C) and template (5′-GC GGA CTC TGC CTC AAG ACG GTA GTC AAC GTG-3′) strands by a standard annealing procedure (see below). For the AlphaScreen assay, a substrate containing a single flap strand was prepared from three oligodeoxynucleotides: adjacent (5′ biotin-TCA CCC TCG TAC GAC TCA), flap (5′-FITC-TTT TTT TTT TTT ATT CAT CAA CTG ACA TCT CCT AC) and template (5′-GT AGG AGA TGT CAG TTG ATG AAT TGA GTC GTA CGA GGG TGA-3′) strands by a standard annealing procedure (see below). Annealing of the oligodeoxynucleotide components was performed in 50 mM Tris pH 8.0, 100 mM KCl, 5 mM MgCl2 by first incubating the mixture at 95°C for 5 min, followed by gradual cooling to room temperature. The annealed double-stranded DNA substrates were then stored at −20°C as 50 µM stocks. High-resolution melting experiments that measure the increase in fluorescence intensity (corresponding to the relief in quenching) upon temperature-induced separation of the donor and quencher strands of the double-flap DNA substrate at 50 nM in the Roche LightCycler 480 instrument yielded a Tm of 53.5°C from a sextuplicate measurement (data not shown). While there is likely a fraction of the two flap strands which remain separated, the presence of a clear melting transition at 53.5°C confirms that the present substrate exists in a predominantly quenched (annealed) state at room temperature.
Fluorogenic assay in 384-well format
The FEN1 fluorogenic assay was carried out in a 40 µl reaction mixture in 50 mM Tris–HCl pH 8.0, 10 mM MgCl2, 1 mM DTT and 0.01% Tween-20. Specifically, 30 µl of either FEN1 at appropriate concentration or buffer (no-enzyme control) was pipetted into a 384-well plate; subsequently, 10 µl of substrate was added to start the reaction. Kinetic fluorescence data were collected on ViewLux high-throughput CCD imager (Perkin Elmer, Waltham, MA, USA) equipped with standard optics (excitation filter 525 nm and emission filter 598 nm).
Assay miniaturization in 1536-well format
Three microliters of reagent (buffer as negative control and FEN1 in the remainder of the plate) was dispensed by a BioRPTR nanoliter dispenser (Beckman Coulter Inc., Fullerton, CA) into a 1536-well solid-bottom black plate. Where applicable, inhibitor compounds were delivered as 23-nl aliquots of DMSO solutions via pintool transfer as described elsewhere (38,39); vehicle-only control consisted of 23 nl DMSO. The plate was incubated for 15 min at room temperature, and then 1 µl of substrate (50 nM final concentration) was added to initiate the reaction. The plate was transferred into ViewLux reader for kinetic fluorescence data collection; there was ∼50 s time lapse between the end of the substrate dispense and the commencement of the fluorescence kinetic read. IC50 values were calculated from the dose-response curve fits generated within GraphPad Prism using the fluorescence intensity change over the first 20 min of data collection and relating it to uninhibited and no-enzyme controls.
AlphaScreen-based assay development and optimization
Beads’ binding capacity to the FITC/biotin flap substrate, also known as hook point because at analyte concentrations greater than the peak value the luminescence signal decays due to saturation of the beads’ binding capacity, was determined by substrate titration against the anti-FITC-acceptor and streptavidin-donor AlphaScreen beads (final concentration 16 µg/ml). An amount of 10 µl of 80 µg/ml stock solution of anti-FITC-acceptor and streptavidin-donor AlphaScreen beads was added to 40 µl of FITC/biotin flap substrate in 384-well solid white plate and incubated for 20 min at room temperature. Plates were read on EnVision plate reader (PerkinElmer) equipped with AlphaScreen optical detection module. After determination of the hook point, the biotin/FITC double-labeled substrate (1 nM) was blended with its unlabeled counterpart to produce a 50 nM final concentration, hereinafter referred to as AlphaScreen substrate. All further assay optimization in the 384-well format was carried out in a total reaction volume of 40 µl. Briefly, 30 µl of FEN1 at the appropriate concentration or buffer (no-enzyme control) was added into a 384-well plate; subsequently, 10 µl of substrate was added to initiate the enzymatic reaction which was then allowed to proceed for the necessary time. AlphaScreen signal was measured 20 min after addition of 10 µl bead homogenate containing 80 µg/ml each of streptavidin-coated donor beads and anti-FITC antibody-coated acceptor beads.
The AlphaScreen assay was further miniaturized to a final volume of 5 µl in the 1536-well format. Three µl of FEN1 was dispensed into a 1536-well plate and test compounds were delivered as 23-nl aliquots of DMSO solutions via pintool transfer. The plate was incubated for 15 min at room temperature, and after dispensing of 1 µl of substrate, the enzyme reaction was allowed to proceed for 5 min at room temperature. AlphaScreen beads were then added and the signal was measured on EnVision plate reader (PerkinElmer) after a 20-min incubation at room temperature.
Determination of the FEN1 cleavage site
To determine the site of cleavage of the fluorogenic substrate, FEN1 (100 nM) was incubated with the double-flap oligonucleotide substrate (50 nM) for 15 min at room temperature (∼23°C) under the conditions used for the HTS assay. Reactions were stopped, and DNAs were separated on a 15% polyacrylamide denaturing gel. Visualization was executed using a Typhoon 9410 Variable Mode Imager and the direct green-excited fluorescence setting. In turn, the AlphaScreen assay substrate (500 nM) was incubated with FEN1 (30 nM) for 15 min at room temperature (∼23°C) under the conditions used for the HTS assay. Visualization was executed using a standard UV light box. Control TAMRA- and FITC-labeled oligodeoxynucleotides corresponding to a range of possible cleavage fragments of the two substrates (at the flap position or one or more positions upstream or downstream) were synthesized and included on the gels as migration markers.
RESULTS
Fluorogenic FEN1 assay principle
We exploited the ability of FEN1 to process DNA substrates containing double-flap structures in order to develop a kinetic fluorogenic donor/quencher assay for identification of FEN1 inhibitors operating in the red-shifted light detection spectral region. A double-stranded DNA substrate containing a pair of flap strands was prepared from three oligodeoxynucleotide strands as depicted in Figure 1. The 5′-end of the downstream flap strand was labeled with 6-TAMRA as the fluorophore donor, whereas the 3′-end of the quencher flap strand was labeled with a BHQ-2 (Figure 1B). In order to maintain proximity between the TAMRA label on the 5′-flap strand and the BHQ-2 matching quencher on the adjacent flap strand, a five-base nucleotide stretch was designed to provide complementarity between the 5′-fluorophore region and 3′-quencher region (bases directly adjacent to the fluorophore and quencher labels indicated by dotted lines in Figure 1A). Upon flap strand cleavage by FEN1 of the annealed three stranded substrate, a short single stranded product labeled with 6-TAMRA is enzymatically released, causing the fluorophore emission to increase (Figure 1A).
Figure 1.

Fluorogenic FEN1 assay. (A) FEN1 cleaves the 5′-end of flap strand of the substrate construct (broken arrow) to liberate a short fluorophore-labeled single-stranded fragment causing increased fluorescence signal (denoted by gold flash). Dotted line indicates region of complementarity between the 5′-fluorophore region and 3′-quencher region; flap length is indicated by the number in italics. F can be any fluorophore, and Q represents any compatible quench molecule. (B) Structures of the 6-TAMRA fluorophore and BHQ-2 quencher used here. (C) Sequences of template, quencher and flap strands that make up the fluorogenic assay DNA substrate. Colors correspond to substrate image depicted in (A).
Fluorogenic assay optimization and miniaturization
In an initial test, 50 nM substrate was incubated in the absence or presence of 20 nM FEN1 in a 384-well plate at a 40 µl total reaction volume, with fluorescence being monitored as a function of reaction time (Figure 2A). The assay signal increased with time only in the presence of FEN1 and reached a 7-fold higher level relative to the no-enzyme control. The DNA substrate was further evaluated by recording the reaction progress at different substrate concentrations (Figure 2B). The increase in substrate supplied led to a direct increase in initial rate and in final signal amplitude, supporting multiple turnover for the enzyme. From these data, a Km of 59 ± 7 nM was estimated (Figure 2C), in general agreement with values previously determined for this enzyme using a single flap substrate in a radiolabel-based assay (12,40), validating the use of the present substrate construct. A substrate concentration of 50 nM was chosen for the subsequent experiments. We selected a relatively high enzyme concentration during these investigations as we wished to observe the completion of the reaction within a relatively short time frame, to avoid complications from reagent evaporation in the microtiter plate. Lower enzyme concentrations do produce signal, as well, but the corresponding data collection (to attain the same change in fluorescence) requires longer reaction times and the dilute enzyme solutions generally have lower overnight stability, making large-scale high throughput screening difficult (data not shown). The new tripartite double-flap substrate was stable upon storage at −20°C and exhibited consistent low fluorescence upon repeated freeze-thaw cycles (data not shown).
Figure 2.

Demonstration of the fluorogenic assay in 384-well plate format. (A) The assay was run at room temperature by following the fluorescence signal in kinetic mode in the presence (20 nM) or absence of FEN1 protein over a 30-min time period (N = 3, individual plots from the triplicate experiments are shown and indicated by the numbers in parentheses). Substrate concentration = 50 nM. (B) Fluorescence signal as a function of time at different fluorogenic substrate concentrations (averages and standard deviations from triplicate measurements shown). (C) Kinetic data from Panel B plotted to estimate Km. Initial rate data were fitted to the Michaelis–Menten equation using GraphPad Prism ver. 5.
The fluorogenic assay was miniaturized to a 4 µl final volume. The assay protocol consisted of dispensing 3 µl of enzyme solution into the plate, followed by the addition of 1 µl substrate to start the reaction: a robust fluorescence intensity rise and low well-to-well variation were observed (Figure 3A). Figure 3B demonstrates that the assay reagents, as formulated as working stocks, were stable over a 16 h storage period with a high average assay Z′ factor of 0.75. The Z′ factor is a statistical score calculated based on assay signal window and standard deviation: values >0.5 (maximum Z′ factor attainable is equal to 1.0) are indicative of a robust assay (41).
Figure 3.

Fluorogenic assay miniaturization to 1536-well format. (A) Real-time reaction monitoring (N = 264), with and without FEN1. (B) Reagent stability test: bottles containing enzyme (or not), buffer and substrate working solutions were stored in a 4°C refrigerator and the reagents were assayed periodically using the miniaturized assay protocol (N = 256). Key assay statistical parameters, signal window and Z′ factor, are shown.
Chemiluminescence assay for FEN1 activity
A single-flap DNA structure served as a template for the design of the chemiluminescent substrate. In order to install binding sites for the donor and acceptor beads, the 5′-end of the flap strand was labeled with a fluorescein tag (fluorescein isothiocyanate, FITC) and the adjacent upstream strand was labeled with biotin at its 5′-end (Figure 4A). The 5′-biotin moiety served as a recognition point for the streptavidin-coated donor bead, while the FITC label on the 5′-end of the flap strand was recognized by an anti-fluorescein antibody-coated acceptor bead. Thus, intact substrate, when mixed with chemiluminescent donor and acceptor beads, was expected to yield a high signal, while FEN1-catalyzed flap strand cleavage would result in a decreased signal due to the separation of biotin and fluorescein tags. Inhibition of the FEN1 reaction would result in a higher signal than the uninhibited control.
Figure 4.

Chemiluminescence-based Assay. (A) Schematic representation of the flap DNA substrate; flap length is indicated by the number in italics. The three deoxyoligonucleotide sequences are shown. The biotin and fluorescein (FITC) labels introduced for bead recognition are indicated on the 5′-ends of the flap and upstream strands, respectively. The FEN1 cleavage site is indicated by the broken arrow. Upon red-shifted light excitation (λ680) of the donor bead, singlet oxygen is generated. If singlet oxygen encounters an acceptor bead within its traveling range, it triggers the emission of blue-shifted light from the acceptor bead (λ520–620). (B) Substrate titration against beads (N = 2). A strong dose-dependent signal associated with a ‘hook-shaped’ curve was observed as expected (see text for details), in contrast to the unlabeled control (empty triangles), which elicited only a background signal. (C) Concentration dependence of the cleavage reaction on FEN1 protein (N = 2).
We first analyzed the beads’ binding capacity to the FITC/biotin flap substrate by titration against a mix of anti-FITC-acceptor and streptavidin-donor AlphaScreen beads in a 40 µl total reaction volume. The concentration-response curve (Figure 4B) exhibited a maximum response ∼2 nM followed by a signal decrease to background levels as the substrate concentration was further increased. This biphasic behavior (also referred to as hook point) is characteristic of the AlphaScreen assay format, as well as similar-format ELISA type immunoassays, and is due to the saturation of the beads’ binding capacity. Only a background signal was detected when the unlabeled version of the substrate (i.e. devoid of biotin and FITC tags) was used (Figure 4B, empty triangles), indicating that the detection signal was strictly dependent upon the presence of the corresponding recognition tags and that no detectable nonspecific bead-substrate interaction was present. To attain a total substrate concentration close to the Km value for FEN1 and to more closely match the reaction conditions of the fluorogenic assay, the biotin/FITC double-labeled reagent (1 nM) was blended with its unlabeled counterpart to produce a 50 nM final substrate concentration, akin to the presence of only a small fraction of radiolabeled (‘hot’) molecule in the background of excess unlabeled (‘cold’) counterpart when conducting radiotracer-based assays.
To demonstrate the assay reaction and its dependency on FEN1 enzyme, we performed a titration of FEN1 against 50 nM substrate in a 384-well plate format in a total volume of 40 µl. The assay signal decreased in a dose-dependent manner with increasing concentration of FEN1 (Figure 4C), with flap cleavage reaching completion at ≥10 nM protein (reaction signal became undistinguishable from that obtained with buffer background). The optimal concentration for AlphaScreen FEN1 assay was selected as 3 nM, balancing the needs for ample signal window and minimal substrate conversion.
The assay was further miniaturized to a final volume of 4 µl in 1536-well format. The enzyme reaction (3 µl enzyme dispensed into an empty well, 23 nl inhibitor solution added via pin-transfer as necessary, and 1 µl substrate addition initiating the reaction) was performed as described in ‘Materials and Methods’ section. To observe the AlphaScreen signal at the end of the enzymatic reaction period, 1 µl of bead mix was added and the signal was measured after a 20-min bead incubation. A strong signal window of over 8.0 (defined as the signal ratio between catalyzed and uncatalyzed reactions) and a high Z′ factor of 0.89 were obtained (Supplementary Figure S1).
Enzymatic processing of the fluorogenic and chemiluminescence substrates
To determine the site of cleavage of the present substrates, we performed the FEN1 reactions and analyzed the products by gel electrophoresis followed by gel imaging. The reaction utilizing the fluorogenic double-flap substrate was analyzed by fluorescence gel imaging of the TAMRA label and processing of the AlphaScreen single-flap substrate was monitored by detection of the FITC-labeled flap strand. Control TAMRA- and FITC-labeled oligodeoxynucleotides corresponding to a range of possible cleavage fragments (at the flap position or one or more positions upstream or downstream) were synthesized and included on the gel as migration markers (Figure 5). We found that both substrates are cleaved in the presence of FEN1 in their corresponding flap regions. The cleavage position of double-flap substrate is approximately two bases downstream of the flap (Figure 5A), while the incision point of the single-flap chemiluminescence substrate is either one or two bases downstream (Figure 5B).
Figure 5.

FEN1 cleavage site. (A) HTS assay substrate. FEN1 was incubated with the HTS oligonucleotide substrate as described in ‘Materials and Methods’ section. Reactions were stopped, and DNAs were separated and visualized as described in the text; shown is a representative gel image. Arrow denotes the site of FEN1 incision, which appears to be around ‘+2’. NE = no enzyme control reaction with HTS substrate. FLS = 5′-TAMRA-GA TGT CAA GCA GTC CTA ACT TTG AGG CAG AGT CCG C-3′; FLP = 5′-TAMRA-GA TGT CAA GCA GTC CTA ACT-3′; FLP-1 = 5′-TAMRA-GA TGT CAA GCA GTC CTA AC-3′; FLP + 1 = 5′-TAMRA-GA TGT CAA GCA GTC CTA ACT T-3′. (B) AlphaScreen assay substrate. FEN1 was incubated with the AlphaScreen substrate as described in ‘Materials and Methods’ section. Visualization was executed using a standard UV light box, and shown is a representative gel image. Arrows denote the two sites of incision, which appear to be around ‘+1’ and ‘+2’. See above for further details. α-S = 5′-FITC-TTT TTT TTT TTT ATT CAT CAA CTG ACA TCT CCT AC-3′; α-P = 5′-FITC-TTT TTT TTT TTT-3′; α-P-1 = 5′-FITC-TTT TTT TTT TT-3′; α-P + 1 = 5′-FITC-TTT TTT TTT TTT A-3′.
During the above experiments, we observed that the fluorogenic double-flap substrate was less preferred by FEN1 than the chemiluminescence substrate, as the reaction using the former required higher concentration of enzyme compared with the latter (Figure 5). This is consistent with the mechanism of FEN1 engagement of its target DNA. Specifically, recent studies by Gloor et al. (42) and Stewart et al. (43) have shown that the enzyme first recognizes the double-stranded DNA portion of the substrate at the base of the flap and then threads the 5′-flap for cleavage, with the binding interactions of FEN1 with the substrate's first base of the 3′-flap being a critical step in reaction initiation (44). Furthermore, X-ray crystallographic evidence indicates that FEN1 recognizes single nucleotide 3′-flaps and makes several contacts with the sugar moiety of the 3′-flap, especially the free 3′-hydroxyl terminus (45). The present fluorogenic substrate is less preferred because it contains a multinucleotide 3′-flap and thus is unable to facilitate key contacts with the 3′-hydroxyl. Overall, the fluorophore/quencher-labeled double-flap structure presents more impediments to FEN1 binding (a long 3′-flap and both a donor and a quencher tag present at the loading site) than the singly labeled single-flap chemiluminescence substrate (no second/3′-flap and only one label at the loading site).
Small molecule inhibitor investigations
There are only a few FEN1 inhibitors reported in the literature and most of these compounds are not commercially available. In order to assess the recently-reported hydroxyurea-based inhibitors, we resynthesized 3-hydroxy-5-methyl-1-phenylthieno[2,3-d]pyrimidine-2,4(1H,3H)-dione (PTPD) described in Tumey et al. (36) (details on the preparation and characterization of PTPD are available in Supplementary Data). When PTPD was tested in each of the above assays, a strong concentration-dependent inhibition was observed (Figure 6), with the present study revealing IC50 values close to those reported originally (36). DMSO, a small molecule frequently used as a solvent in inhibition studies, was tested and found to not inhibit FEN1 at concentrations up to 0.67% (data not shown).
Figure 6.

Small molecule inhibitor investigations. Concentration-response plots for the molecules investigated using (A) the kinetic fluorogenic assay and (B) the chemiluminescence AlphaScreen assay (N = 2). Compound structures and IC50 values derived from these plots are presented in Table 1.
We next tested aurintricarboxylic acid (ATA), a compound previously reported to be a broad-specificity inhibitor of multiple classes of DNA-processing enzymes (46,47), and a small set of compounds sharing the arylstibonic core, which had recently been reported by Seiple et al. (48) as inhibitors of the human abasic endonuclease APE1. ATA inhibited FEN1 at submicromolar potency and the IC50 values obtained in the two independent assays were nearly identical, indicating that the fluorogenic and AlphaScreen formats afforded similar sensitivity to inhibitors (Figure 6 and Table 1). The arylstibonic series yielded robust concentration-dependent inhibition and a range of IC50 potencies (Figure 6). Again, the IC50 values derived from the two assays showed very little assay-to-assay variation (Table 1).
Table 1.
Small molecule inhibitors
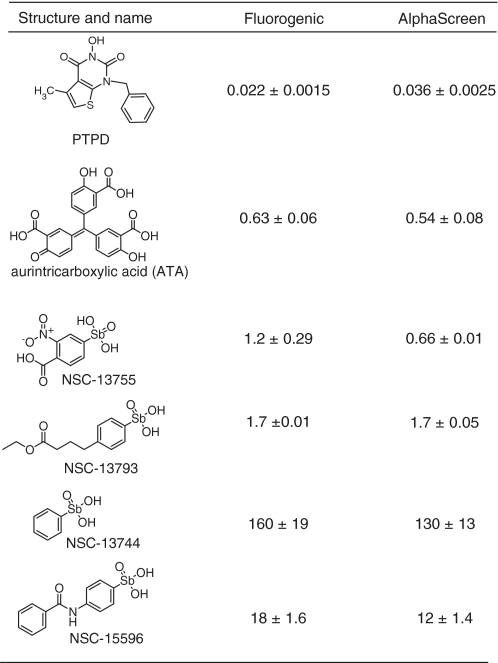 |
IC50 values (µM) are shown for both assay formats.
Lastly, to provide additional validation of the new assays developed here, we tested four out of the six compounds from above in a standard FEN1 assay with a 32P-labeled single-flap substrate prepared as described previously (49). Robust concentration-dependent inhibition was observed for the four compounds in the 32P-based assay (Supplementary Figure S2); moreover, the rank order of inhibitory potency was maintained across all three assay platforms.
DISCUSSION
Recent studies have indicated a role for FEN1 in cancer progression and therapeutic agent resistance, suggesting that the protein might be a sound target in certain anticancer treatment paradigms. However, simple methods for monitoring FEN1 activity have been lacking. Moreover, reliance on a single assay to discover and develop inhibitors can lead to retention of undesired false positives. Ideally, a second assay, operating at the same level of biochemical complexity, but utilizing a different signal output, would be used to validate previously-uncharacterized inhibitors. The present work provides two simple homogeneous assays for monitoring FEN1 activity and inhibition that operate under orthogonal principles. The fluorogenic donor-quencher assay follows the generation of reaction product in real time, while the chemiluminescence-based assay monitors the consumption of substrate using a simple flap structure. The selection of TAMRA as the assay fluorophore donor was driven primarily by the desire to place the detection wavelength within a red-shifted region of the light spectrum in order to avoid compound library autofluorescence which is dominant in the shorter wavelength regions. BHQ-2 is a highly efficient non-emitting quencher whose absorption spectrum overlaps well with the emission of TAMRA. Furthermore, both fluorophore and quencher moieties have been incorporated in a number of assays for DNA repair proteins and other targets and have been used successfully in at least four large-scale HTS campaigns [PubChem Assay IDs 2517, 1490, 2528 and 2549 and (50–52)].
The excellent stability of the fluorogenic assay coupled with a robust statistical performance in the 1536-well plate format indicates that the assay can be deployed reliably. The ability to collect kinetic data, allowing an assessment of the rate of fluorescence change, as opposed to collecting endpoint data only, makes the assay signal less susceptible to variations in liquid dispenser performance. Furthermore, kinetic reads make it possible to flag false positive compounds which interfere with the light detection by autofluorescence or quenching, as such compounds will shift the starting well fluorescence up or down, but will have no effect on the rate of fluorescence change.
In turn, the AlphaScreen (Amplified Luminescence Proximity Homogeneous Assay) assay uses a bead-based chemistry combined with chemiluminescence detection in order to detect low concentrations of analyte. Due to their colloidal size and properties, AlphaScreen beads are conveniently processed by liquid dispensers in the same manner as regular homogeneous reagents. Upon excitation with 680-nm light, reagents embedded within the donor bead generate singlet oxygen, which diffuses in solution within its short lifetime. If an acceptor bead is within close proximity (200 nm), the singlet oxygen triggers light emission in the 520- to 620-nm range from rubrene contained within that bead. In the FEN1 assay described herein, AlphaScreen is used in a ‘reverse’ mode to monitor the decrease of the substrate analyte: inhibition of the enzymatic reaction is being detected as a signal gain relative to uninhibited reaction control, making this assay configuration particularly resistant to interference from signal quenchers, such as intensely colored substances and singlet oxygen scavengers.
To validate our pair of newly developed FEN1 assays, we tested a dose response of PTPD, a previously-reported inhibitor (36), which was resynthesized for the present study. The compound displayed similar potency in the present assays as reported earlier, thus validating the set-up developed here. To extend the present methodology to additional small molecules, we tested a dose response of ATA, a compound previously reported to be a broad-specificity inhibitor of multiple classes of DNA-processing enzymes such as polymerases and nucleases. For example, in our previous studies, ATA displayed potent inhibition against Tdp1 (53), the DNA polymerases beta, eta and iota (50), and against the major abasic endonuclease APE1 (51). Thus, we wished to ascertain whether such a generic inhibitor will suppress FEN1 activity, and if it did, whether its inhibitory potency will be similar across the two assay methods under consideration. As anticipated, ATA inhibited FEN1 at submicromolar potency; furthermore, the IC50 values obtained in the two independent assays were nearly identical, indicating that the fluorogenic and AlphaScreen formats afforded similar sensitivities to inhibitors.
Lastly, we tested a small set of structurally-related compounds (based on the arylstibonic core), which had recently been reported as inhibitors of APE1, the main enzyme for repairing abasic sites within the genome (48). These APE1 inhibitors were found to inhibit FEN1, as well: notably, the IC50 values derived herein against FEN1 showed very little assay-to-assay variation, despite the differences in the assay format (detection modality, as well as different sequence and structure of the flap substrate). Moreover, the inter-assay concordance of IC50 values remained good within a broad range of inhibitor potencies (including the region above the maximum compound concentration tested where lack of high-concentration data made curve fitting less robust and where only extrapolated IC50 values could be obtained), indicating that the present two assays are not only capable of detecting a wide range of compound potencies, but are also equally sensitive to inhibition across that broad potency range. Furthermore, the comparative testing of the four inhibitors using the radiogel method served as additional validation of the new assays presented here.
In summary, we describe two homogeneous assays to enable investigations of FEN1 enzymatic activity that should be easily adaptable to other enzymes capable of processing similar DNA structures. Unique features of each format include the ability to collect kinetic data with the fluorogenic assay and the high signal and insensitivity to autofluorescence of the chemiluminescence method. We anticipate the fluorogenic method to be used for most enzymological and inhibitor investigations, with the AlphaScreen counterpart being deployed when newly-discovered inhibitors need to be validated by an orthogonal readout approach.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Molecular Libraries Initiative of the National Institutes of Health Roadmap for Medical Research [grants 1 R03 MH092154-01 (D.M.W.III) and U54MH084681 (to A.S.)]; Intramural Research Programs of National Institute on Aging and National Human Genome Research Institute, National Institutes of Health. Funding for open access charge: National Institutes of Health Intramural funding.
Conflict of interest statement. None declared.
REFERENCES
- 1.Lee BI, Wilson DM., 3rd The RAD2 domain of human exonuclease 1 exhibits 5′ to 3′ exonuclease and flap structure-specific endonuclease activities. J. Biol. Chem. 1999;274:37763–37769. doi: 10.1074/jbc.274.53.37763. [DOI] [PubMed] [Google Scholar]
- 2.Shen B, Singh P, Liu R, Qiu J, Zheng L, Finger LD, Alas S. Multiple but dissectible functions of FEN-1 nucleases in nucleic acid processing, genome stability and diseases. Bioessays. 2005;27:717–729. doi: 10.1002/bies.20255. [DOI] [PubMed] [Google Scholar]
- 3.Lieber MR. The FEN-1 family of structure-specific nucleases in eukaryotic DNA replication, recombination and repair. Bioessays. 1997;19:233–240. doi: 10.1002/bies.950190309. [DOI] [PubMed] [Google Scholar]
- 4.Turchi JJ, Huang L, Murante RS, Kim Y, Bambara RA. Enzymatic completion of mammalian lagging-strand DNA replication. Proc. Natl Acad. Sci. USA. 1994;91:9803–9807. doi: 10.1073/pnas.91.21.9803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Klungland A, Lindahl T. Second pathway for completion of human DNA base excision-repair: reconstitution with purified proteins and requirement for DNase IV (FEN1) EMBO J. 1997;16:3341–3348. doi: 10.1093/emboj/16.11.3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu Y, Kao HI, Bambara RA. Flap endonuclease 1: a central component of DNA metabolism. Annu. Rev. Biochem. 2004;73:589–615. doi: 10.1146/annurev.biochem.73.012803.092453. [DOI] [PubMed] [Google Scholar]
- 7.Kucherlapati M, Yang K, Kuraguchi M, Zhao J, Lia M, Heyer J, Kane MF, Fan K, Russell R, Brown AM, et al. Haploinsufficiency of Flap endonuclease (Fen1) leads to rapid tumor progression. Proc. Natl Acad. Sci. USA. 2002;99:9924–9929. doi: 10.1073/pnas.152321699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Larsen E, Gran C, Saether BE, Seeberg E, Klungland A. Proliferation failure and gamma radiation sensitivity of Fen1 null mutant mice at the blastocyst stage. Mol. Cell Biol. 2003;23:5346–5353. doi: 10.1128/MCB.23.15.5346-5353.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Matsuzaki Y, Adachi N, Koyama H. Vertebrate cells lacking FEN-1 endonuclease are viable but hypersensitive to methylating agents and H2O2. Nucleic Acids Res. 2002;30:3273–3277. doi: 10.1093/nar/gkf440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hansen RJ, Friedberg EC, Reagan MS. Sensitivity of a S. cerevisiae RAD27 deletion mutant to DNA-damaging agents and in vivo complementation by the human FEN-1 gene. Mut. Res. 2000;461:243–248. doi: 10.1016/s0921-8777(00)00056-2. [DOI] [PubMed] [Google Scholar]
- 11.Harrington JJ, Lieber MR. The characterization of a mammalian DNA structure-specific endonuclease. EMBO J. 1994;13:1235–1246. doi: 10.1002/j.1460-2075.1994.tb06373.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Murante RS, Huang L, Turchi JJ, Bambara RA. The calf 5′- to 3′-exonuclease is also an endonuclease with both activities dependent on primers annealed upstream of the point of cleavage. J. Biol. Chem. 1994;269:1191–1196. [PubMed] [Google Scholar]
- 13.Kaiser MW, Lyamicheva N, Ma W, Miller C, Neri B, Fors L, Lyamichev VI. A comparison of eubacterial and archaeal structure-specific 5′-exonucleases. J. Biol. Chem. 1999;274:21387–21394. doi: 10.1074/jbc.274.30.21387. [DOI] [PubMed] [Google Scholar]
- 14.Kao HI, Henricksen LA, Liu Y, Bambara RA. Cleavage specificity of Saccharomyces cerevisiae flap endonuclease 1 suggests a double-flap structure as the cellular substrate. J. Biol. Chem. 2002;277:14379–14389. doi: 10.1074/jbc.M110662200. [DOI] [PubMed] [Google Scholar]
- 15.Friedrich-Heineken E, Henneke G, Ferrari E, Hubscher U. The acetylatable lysines of human Fen1 are important for endo- and exonuclease activities. J. Mol. Biol. 2003;328:73–84. doi: 10.1016/s0022-2836(03)00270-5. [DOI] [PubMed] [Google Scholar]
- 16.Harrington JJ, Lieber MR. DNA structural elements required for FEN-1 binding. J. Biol. Chem. 1995;270:4503–4508. doi: 10.1074/jbc.270.9.4503. [DOI] [PubMed] [Google Scholar]
- 17.Nikolova T, Christmann M, Kaina B. FEN1 is overexpressed in testis, lung and brain tumors. Anticancer Res. 2009;29:2453–2459. [PubMed] [Google Scholar]
- 18.Sato M, Girard L, Sekine I, Sunaga N, Ramirez RD, Kamibayashi C, Minna JD. Increased expression and no mutation of the Flap endonuclease (FEN1) gene in human lung cancer. Oncogene. 2003;22:7243–7246. doi: 10.1038/sj.onc.1206977. [DOI] [PubMed] [Google Scholar]
- 19.Kim JM, Sohn HY, Yoon SY, Oh JH, Yang JO, Kim JH, Song KS, Rho SM, Yoo HS, Kim YS, et al. Identification of gastric cancer-related genes using a cDNA microarray containing novel expressed sequence tags expressed in gastric cancer cells. Clin. Cancer Res. 2005;11:473–482. [PubMed] [Google Scholar]
- 20.LaTulippe E, Satagopan J, Smith A, Scher H, Scardino P, Reuter V, Gerald WL. Comprehensive gene expression analysis of prostate cancer reveals distinct transcriptional programs associated with metastatic disease. Cancer Res. 2002;62:4499–4506. [PubMed] [Google Scholar]
- 21.Lam JS, Seligson DB, Yu H, Li A, Eeva M, Pantuck AJ, Zeng G, Horvath S, Belldegrun AS. Flap endonuclease 1 is overexpressed in prostate cancer and is associated with a high Gleason score. BJU Int. 2006;98:445–451. doi: 10.1111/j.1464-410X.2006.06224.x. [DOI] [PubMed] [Google Scholar]
- 22.Iacobuzio-Donahue CA, Maitra A, Olsen M, Lowe AW, van Heek NT, Rosty C, Walter K, Sato N, Parker A, Ashfaq R, et al. Exploration of global gene expression patterns in pancreatic adenocarcinoma using cDNA microarrays. Am. J. Pathol. 2003;162:1151–1162. doi: 10.1016/S0002-9440(10)63911-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Krause A, Combaret V, Iacono I, Lacroix B, Compagnon C, Bergeron C, Valsesia-Wittmann S, Leissner P, Mougin B, Puisieux A. Genome-wide analysis of gene expression in neuroblastomas detected by mass screening. Cancer Lett. 2005;225:111–120. doi: 10.1016/j.canlet.2004.10.035. [DOI] [PubMed] [Google Scholar]
- 24.Singh P, Yang M, Dai H, Yu D, Huang Q, Tan W, Kernstine KH, Lin D, Shen B. Overexpression and hypomethylation of flap endonuclease 1 gene in breast and other cancers. Mol. Cancer Res. 2008;6:1710–1717. doi: 10.1158/1541-7786.MCR-08-0269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim IS, Lee MY, Lee IH, Shin SL, Lee SY. Gene expression of flap endonuclease-1 during cell proliferation and differentiation. Biochim. Biophys. Acta. 2000;1496:333–340. doi: 10.1016/s0167-4889(00)00029-x. [DOI] [PubMed] [Google Scholar]
- 26.Freedland SJ, Pantuck AJ, Paik SH, Zisman A, Graeber TG, Eisenberg D, McBride WH, Nguyen D, Tso CL, Belldegrun AS. Heterogeneity of molecular targets on clonal cancer lines derived from a novel hormone-refractory prostate cancer tumor system. Prostate. 2003;55:299–307. doi: 10.1002/pros.10226. [DOI] [PubMed] [Google Scholar]
- 27.Helleday T, Petermann E, Lundin C, Hodgson B, Sharma RA. DNA repair pathways as targets for cancer therapy. Nat. Rev. Cancer. 2008;8:193–204. doi: 10.1038/nrc2342. [DOI] [PubMed] [Google Scholar]
- 28.McNeill DR, Lam W, DeWeese TL, Cheng YC, Wilson DM., 3rd Impairment of APE1 function enhances cellular sensitivity to clinically relevant alkylators and antimetabolites. Mol. Cancer Res. 2009;7:897–906. doi: 10.1158/1541-7786.MCR-08-0519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reed AM, Fishel ML, Kelley MR. Small-molecule inhibitors of proteins involved in base excision repair potentiate the anti-tumorigenic effect of existing chemotherapeutics and irradiation. Future Oncol. 2009;5:713–726. doi: 10.2217/FON.09.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shibata Y, Nakamura T. Defective flap endonuclease 1 activity in mammalian cells is associated with impaired DNA repair and prolonged S phase delay. J. Biol. Chem. 2002;277:746–754. doi: 10.1074/jbc.M109461200. [DOI] [PubMed] [Google Scholar]
- 31.Wyatt MD, Pittman DL. Methylating agents and DNA repair responses: Methylated bases and sources of strand breaks. Chem. Res. Toxicol. 2006;19:1580–1594. doi: 10.1021/tx060164e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Iglehart JD, Silver DP. Synthetic lethality–a new direction in cancer-drug development. N. Engl. J. Med. 2009;361:189–191. doi: 10.1056/NEJMe0903044. [DOI] [PubMed] [Google Scholar]
- 33.McManus KJ, Barrett IJ, Nouhi Y, Hieter P. Specific synthetic lethal killing of RAD54B-deficient human colorectal cancer cells by FEN1 silencing. Proc. Natl Acad. Sci. USA. 2009;106:3276–3281. doi: 10.1073/pnas.0813414106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434:917–921. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 35.Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434:913–917. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 36.Tumey LN, Bom D, Huck B, Gleason E, Wang J, Silver D, Brunden K, Boozer S, Rundlett S, Sherf B, et al. The identification and optimization of a N-hydroxy urea series of flap endonuclease 1 inhibitors. Bioorg. Med. Chem. Lett. 2005;15:277–281. doi: 10.1016/j.bmcl.2004.10.086. [DOI] [PubMed] [Google Scholar]
- 37.Simeonov A, Jadhav A, Thomas CJ, Wang Y, Huang R, Southall NT, Shinn P, Smith J, Austin CP, Auld DS, et al. Fluorescence spectroscopic profiling of compound libraries. J. Med. Chem. 2008;51:2363–2371. doi: 10.1021/jm701301m. [DOI] [PubMed] [Google Scholar]
- 38.Michael S, Auld D, Klumpp C, Jadhav A, Zheng W, Thorne N, Austin C, Inglese J, Simeonov A. A robotic platform for quantitative high-throughput screening. Assay Drug Dev. Technol. 2008;6:637–657. doi: 10.1089/adt.2008.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Inglese J, Auld DS, Jadhav A, Johnson RL, Simeonov A, Yasgar A, Zheng W, Austin CP. Quantitative high-throughput screening: a titration-based approach that efficiently identifies biological activities in large chemical libraries. Proc. Natl Acad. Sci. USA. 2006;103:11473–11478. doi: 10.1073/pnas.0604348103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Negritto MC, Qiu J, Ratay DO, Shen B, Bailis AM. Novel function of Rad27 (FEN-1) in restricting short-sequence recombination. Mol. Cell Biol. 2001;21:2349–2358. doi: 10.1128/MCB.21.7.2349-2358.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang JH, Chung TD, Oldenburg KR. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 42.Gloor JW, Balakrishnan L, Bambara RA. Flap endonuclease 1 mechanism analysis indicates flap base binding prior to threading. J. Biol. Chem. 2010 doi: 10.1074/jbc.M110.165902. doi:10.1074/jbc.M110.165902; [25 August 2010, Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stewart JA, Campbell JL, Bambara RA. Dna2 is a structure-specific nuclease, with affinity for 5′-flap intermediates. Nucleic Acids Res. 2010;38:920–930. doi: 10.1093/nar/gkp1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Finger LD, Blanchard MS, Theimer CA, Sengerova B, Singh P, Chavez V, Liu F, Grasby JA, Shen B. The 3′-flap pocket of human flap endonuclease 1 is critical for substrate binding and catalysis. J. Biol. Chem. 2009;284:22184–22194. doi: 10.1074/jbc.M109.015065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chapados BR, Hosfield DJ, Han S, Qiu J, Yelent B, Shen B, Tainer JA. Structural basis for FEN-1 substrate specificity and PCNA-mediated activation in DNA replication and repair. Cell. 2004;116:39–50. doi: 10.1016/s0092-8674(03)01036-5. [DOI] [PubMed] [Google Scholar]
- 46.Bina-Stein M, Tritton TR. Aurintricarboxylic acid is a nonspecific enzyme inhibitor. Mol. Pharmacol. 1976;12:191–193. [PubMed] [Google Scholar]
- 47.Blumenthal T, Landers TA. The inhibition of nucleic acid-binding proteins by aurintricarboxylic acid. Biochem. Biophys. Res. Comm. 1973;55:680–688. doi: 10.1016/0006-291x(73)91198-4. [DOI] [PubMed] [Google Scholar]
- 48.Seiple LA, Cardellina JH, 2nd, Akee R, Stivers JT. Potent inhibition of human apurinic/apyrimidinic endonuclease 1 by arylstibonic acids. Mol. Pharmacol. 2008;73:669–677. doi: 10.1124/mol.107.042622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McNeill DR, Narayana A, Wong HK, Wilson DM., 3rd Inhibition of Ape1 nuclease activity by lead, iron, and cadmium. Environ Health Perspect. 2004;112:799–804. doi: 10.1289/ehp.7038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dorjsuren D, Wilson DM, III, Beard WA, McDonald JP, Austin CP, Woodgate R, Wilson SH, Simeonov A. A real-time fluorescence method for enzymatic characterization of specialized human DNA polymerases. Nucleic Acids Res. 2009;37:e128. doi: 10.1093/nar/gkp641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Simeonov A, Kulkarni A, Dorjsuren D, Jadhav A, Shen M, McNeill DR, Austin CP, Wilson DM., III Identification and characterization of inhibitors of human apurinic/apyrimidinic endonuclease APE1. PLoS One. 2009;4:e5740. doi: 10.1371/journal.pone.0005740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yasgar A, Foley T, Jadhav A, Inglese J, Burkart M, Simeonov A. A strategy to discover inhibitors of Bacillus subtilis surfactin-type phosphopantetheinyl transferase. Mol. BioSystems. 2010;6:365–375. doi: 10.1039/b913291k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Marchand C, Lea W, Jadhav A, Dexheimer T, Austin C, Inglese J, Pommier Y, Simeonov A. Identification of phosphotyrosine mimetic inhibitors of human tyrosyl-DNA phosphodiesterase I by a novel AlphaScreen high-throughput assay. Mol. Cancer Ther. 2009;8:240–248. doi: 10.1158/1535-7163.MCT-08-0878. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.