


Abstract
Alternative start AUG codons within a single transcript can contribute to diversity of the proteome; however, their functional significance remains controversial. Here, we provide comparative genomics evidence that alternative start codons are under negative selection in vertebrates, insects and yeast. In genes where the annotated start codon (sAUG) resides within the suboptimal nucleotide context, the downstream in-frame AUG codons (dAUG) among the first ∼30 codon sites are significantly more conserved between species than in genes where the sAUG resides within the optimal context. Proteomics data show that this difference is not an annotation artifact and that dAUGs are in fact under selection as alternative start sites. The key optimal, and sometimes suboptimal, context-determining nucleotides of both the sAUG and dAUGs are conserved. Selection for secondary start sites is stronger in genes with the weak primary start site. Genes with multiple conserved start sites are enriched for transcription factors, and tend to have longer 5′UTRs and higher degree of alternative splicing. Together, these results imply that the use of alternative start sites by means of leaky mRNA scanning is a functional mechanism under selection for increased efficiency of translation and/or for translation of different N-terminal protein variants.
INTRODUCTION
The multiplicity of functional protein isoforms emerges due to alternative splicing, alternative promoters or alternative translation start sites. The first two mechanisms are major sources of proteome diversity in higher eukaryotes (1–4), and may contribute to the development of complex organisms from genomes carrying only a few tens of thousand of protein-coding genes. In contrast, alternative use of translation start sites within a single transcript remains poorly investigated and commonly is not taken into account for evaluation of diversity of eukaryotic proteomes.
Nonetheless, the rapidly accumulating body of experimental data shows that dozens of messenger RNAs (mRNAs) could produce protein isoforms owing to the use of alternative translation initiation sites. The difference in the lengths of alternative N-termini of a protein can reach tens of codons, and sometimes can lead to a different targeting of the produced variants. For example, DNA ligase 1 (AtLIG1) is the only essential DNA ligase activity in Arabidopsis thaliana. The mitochondrial and nuclear forms of AtLIG1 are translated from a single mRNA species through translation initiation from either the first or second in-frame AUG codon, respectively. The nucleotide context around alternative start codons in the AtLIG1 transcripts shapes translation initiation to ensure balanced synthesis of both nuclear and mitochondrial AtLIG1 isoforms, probably via context-dependent leaky mRNA scanning (5). Similarly, alternative translation initiation signals (TISs) and leaky scanning are responsible for synthesis of mitochondrial and cytoplasmic isoforms of rat ornithine decarboxylase-antizyme (6) and human insulin-degrading enzyme (7), secretory and mitochondrial isoforms of human neuropeptide Y (8) and many other eukaryotic proteins (9). It is likely that the contribution of alternative starts of translation to eukaryotic proteomes is underestimated.
Initiation of translation of most eukaryotic mRNAs is likely to occur by linear scanning, although other mechanisms are also possible (10–12). According to the scanning model, 40S ribosomal subunits are recruited to the 5′-terminal cap structure, scan mRNA in the 5′-to-3′ direction and can initiate translation at the first AUG they encounter (10). The recognition of AUG triplet as a TIS depends on its nucleotide context. If the context is optimal, most 40S ribosomal subunits will recognize the AUG and initiate translation. In contrast, if the context is suboptimal, some 40S ribosomal subunits recognize the AUG as a TIS, but others may skip it, continue to scan in the 3′-direction and initiate translation at a downstream AUG (‘leaky scanning’ mechanism). The initiation/scanthrough ratio depends on the AUG context; in most eukaryotes, purine at position −3 has been found to increase the AUG recognition (10,13). Here, we study selection acting on primary and downstream AUG (dAUG) codons, attempting to elucidate their roles in generation of protein diversity.
METHODS
Multiple alignments of genome assemblies of 43 vertebrate species to Homo sapiens (hg18), 14 insect species to Drosophila melanogaster (dm3) and seven Saccharomyces species to Saccharomyces cerevisiae (sacCer2) were obtained from UCSC Genome Bioinformatics Site (http://genome.ucsc.edu). The canonical splicing variants of hg18 known gene genes in H. sapiens, FlyBase genes in D. melanogaster and SGD genes in S. cerevisiae (14) were used to map protein-coding genes of the corresponding species onto corresponding alignments. Multiple alignment of each coding region was then obtained by joining the aligned segments corresponding to exons of canonical genes. Three nucleotides upstream of the annotated start codon indicative of the start codon context were also appended to the alignment. Lengths of 5′ UTRs and numbers of alternative splicing variants for each gene were obtained from UCSC annotations (14). Evolutionary distances between species of vertebrates and insects were calculated from phylogenetic trees taken from UCSC Genome Bioinformatics Site. Evolutionary distances between species of yeast were taken from references (15,16).
In vertebrates, the context of the sAUG was assumed to be optimal if it followed either of the two consensus sequences: AnnAUGn or GnnAUGG, and suboptimal if it followed the consensus sequence YnnAUGH [Y = C or U; H = A, C or U; (9,10,17)]. Genes with sAUG not matching either of these two contexts were excluded from analysis. In insects and yeast, the context of sAUG was assumed to be optimal if it was preceded by purine at position −3, and suboptimal if it was preceded by pyrimidine at position −3 (18,19). Choice of slightly different context-determining positions from the literature, in addition to the key purine/pyrimidine at position −3, affected the results only marginally (data not shown). A total of 12 731 (1509), 11 535 (1638) and 5013 (1514) genes of H. sapiens, D. melanogaster and S. cerevisiae, respectively, had optimal (suboptimal) sAUG. The context of dAUG was always assumed to be optimal if it was preceded by purine at position −3, and suboptimal if it was preceded by pyrimidine at position −3.
For each codon position j of H. sapiens (D. melanogaster, S. cerevisiae) gene, conservation of AUG codons in other species of vertebrates (insects, yeast) was measured as
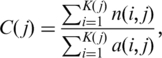 |
where K(j) is the number of H. sapiens (D. melanogaster, S. cerevisiae) genes with AUG codon at position j, n(i, j) is the number of species in which gene i had conserved sAUG and carried dAUG at position j, and a(i, j) is the number of species in which position j of gene i was covered by alignment. Nucleotide conservation was defined analogously. Conservation of presence of dAUG over the first l codons of a gene was measured as
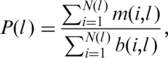 |
where N(l) is the number of H. sapiens (D. melanogaster, S. cerevisiae) genes with AUG codon in at least one of the positions 2, 3, … , l; m(i, l) is the number of species in which gene i had conserved sAUG and carried dAUG in at least one of the positions 2, 3, … , l; and b(I, l) is the number of species in which all codons at least up to codon l of gene i were covered by alignment.
All peptide sequences from proteomics experiments in H. sapiens, D. melanogaster and S. cerevisiae were downloaded from PRIDE (20) together with coordinates and accession numbers of the corresponding proteins. Because an AUG codon is necessary for translation initiation, peptides overlapping the region between sAUG and the nearest dAUG indicate that the sAUG served as translation start. Therefore, for the proteomics-supported data set, only those genes were chosen in which at least one peptide overlapped at least one amino acid between the sAUG and dAUG.
Enrichment of different annotation categories, including GO terms, sequence features and protein domains, was measured using DAVID (21,22).
RESULTS
Prevalence and conservation of dAUG codons
Initiation of translation at in-frame dAUG codons located downstream of the start AUG (sAUG) can give rise to N-truncated variants of the protein. As ribosomal scanning is more likely to leak in genes where the sAUG resides within the suboptimal context, dAUGs may be expected to function more often in such genes. Indeed, in each of the analyzed species—H. sapiens, D. melanogaster and S. cerevisiae—the dAUG codons are significantly overrepresented in 5′ regions of genes with suboptimal sAUGs, compared to the genes with optimal sAUGs (Figure 1), in agreement with previous results (19). The difference in frequency of dAUGs between these two classes of genes spans the first ∼30 codons of the coding sequence.
Figure 1.

Prevalence of dAUG. Fraction of dAUG codons among all codons, in each codon position relative to the start of the coding region (0 corresponds to sAUG) in H. sapiens (A), D. melanogaster (B) and S. cerevisiae (C), in genes with optimal (gray squares, dashed line) and suboptimal (black diamonds, solid line) contexts of sAUG.
If dAUGs are under selection to enable translation from a downstream alternative start site, their presence in the beginning of the coding sequence will be conserved in interspecies divergence. For each gene with a dAUG in the 5′-end in H. sapiens, D. melanogaster or S. cerevisiae, we asked whether a dAUG was also present somewhere in the 5′-end of the orthologous gene among each of the 43 other vertebrate species, 14 other insect species or six other yeast species, correspondingly. As expected, the presence of a dAUG codon in the beginning of the coding sequence was significantly better conserved in genes with suboptimal sAUG than in genes with optimal sAUG (Table 1). This phylogenetic footprint was pronounced for the first ∼20–30 codons of the genes, and was evident at all the evolutionary distances considered (Figure 2).
Table 1.
Conservation of presence of dAUG in 5′-ends of genes with sAUG in optimal versus suboptimal context
| Codons | Conservation of presence of dAUG among these codons |
|||||
|---|---|---|---|---|---|---|
| Vertebrates |
Insects |
Yeast |
||||
| Optimal (%) | Suboptimal (%) | Optimal (%) | Suboptimal (%) | Optimal (%) | Suboptimal (%) | |
| 2–5 | 76.3 | 91.7*** | 75.5 | 87.7*** | 67.2 | 77.5*** |
| 2–10 | 76.1 | 91.0*** | 76.0 | 85.5*** | 74.2 | 78.7* |
| 2–20 | 79.5 | 91.7*** | 78.0 | 87.1*** | 79.1 | 81.0 |
| 2–100 | 93.6 | 95.2*** | 93.7 | 95.2*** | 92.4 | 91.1* |
Asterisks denote significance of difference between optimal and suboptimal sAUG (chi-square test): ***P < 0.001; **P < 0.01; *P < 0.05.
Figure 2.

Conservation of presence of dAUG. Conservation of presence of H. sapiens (A), D. melanogaster (B) and S. cerevisiae (C) dAUG was assessed in each species of vertebrates (A), insects (B) and yeast (C), in codon positions 2–5, depending on log phylogenetic distance of the species to H. sapiens (A), D. melanogaster (B) and S. cerevisiae (C), in genes with optimal (gray squares) and suboptimal (black diamonds) contexts of sAUG. Phylogenetic distances are in units Ds.
To test whether our findings are affected by annotation errors, we analyzed conservation of dAUGs only among those genes in which the annotated sAUG was supported by proteomics data (see ‘Methods’ section). Proteomic support for sAUG was available, respectively, for 30.8% (17.1%), 47.6% (44.7%) and 17.2% (8.3%) of H. sapiens, D. melanogaster and S. cerevisiae genes with optimal (suboptimal) context of sAUG. The results on conservation of dAUGs obtained only for the proteomics-supported subset of genes were similar to those obtained for all genes (Table 2).
Table 2.
Conservation of presence of dAUG in 5′-ends of genes with sAUG in optimal versus suboptimal context, in genes with sAUG supported by a peptide sequence
| Codons | Conservation of presence of dAUG among these codons |
|||||
|---|---|---|---|---|---|---|
| Vertebrates |
Insects |
Yeast |
||||
| Optimal (%) | Suboptimal (%) | Optimal (%) | Suboptimal (%) | Optimal (%) | Suboptimal (%) | |
| 2–5 | 79.1 | 92.4*** | 78.3 | 85.3** | 92.3 | 100.0 |
| 2–10 | 79.0 | 91.7*** | 77.3 | 83.8*** | 88.3 | 100.0 |
| 2–20 | 83.6 | 93.6*** | 79.1 | 86.9*** | 88.2 | 94.3 |
| 2–100 | 95.1 | 95.3 | 94.0 | 95.7*** | 93.3 | 93.1 |
Asterisks denote significance of difference between optimal and suboptimal sAUG (chi-square test): ***P < 0.001; **P < 0.01; *P < 0.05.
Besides the conservation of the presence of a dAUG, enabling translation of an N-truncated variant, the conservation of each individual dAUG between species was also increased in genes with a suboptimal sAUG, compared to genes with an optimal sAUG. Again, this difference in conservation decreased rapidly with distance between sAUG and dAUG (Figure 3).
Figure 3.

Conservation of individual dAUGs. Fraction of dAUG codons in each position (0 corresponds to sAUG) conserved between H. sapiens and other vertebrate species, in genes with optimal (gray squares) and suboptimal (black diamonds) contexts of sAUG. Power curve fit is provided for visual reference. (A) First 45 codons; (B) first 600 codons.
Prevalence and conservation of contexts of sAUG
In line with the previous findings (9), 89.4% of all classified H. sapiens genes, 87.6% of D. melanogaster genes and 76.8% of S. cerevisiae genes contained the sAUG in the optimal context. In each analyzed group of species, purine at position −3 relative to sAUG, which is the primary determinant of the optimal context, was significantly more conserved than the surrounding purines (Figure 4, left column). The conservation of pyrimidines in this position—the primary determinant of the suboptimal context of sAUG—was always lower than the conservation of purines. Still, conservation of pyrimidines in this position was somewhat elevated, compared to that of pyrimidines in the surrounding positions, in vertebrates, although not in insects or yeast (Figure 4, right column). The suboptimal context of the sAUG was slightly more conserved in genes in which a dAUG was present within 30 codons (Figure 5, right column).
Figure 4.

Conservation of context-determining nucleotides of sAUGs. Conservation of purines in genes with optimal context of sAUG (left column), and pyrimidines in genes with suboptimal context of sAUG (right column), in the vicinity of sAUG. Origin at the x-axis corresponds to the adenine of the sAUG. Top line, Homo sapiens to other vertebrates; middle line, D. melanogaster to other insects; bottom line, S. cerevisiae to other yeast. Cross denotes the conservation of the nucleotide in –3 which determines the context of the sAUG. Dashes denote the conservation of the nucleotides of sAUG, which always equals 1.
Figure 5.

Conservation of context-determining nucleotides of sAUGs in genes with optimal dAUG. Conservation of purines in genes with optimal context of sAUG (left column), and pyrimidines in genes with suboptimal context of sAUG (right column), in the vicinity of sAUG, in genes with an optimal dAUG among the first 30 codons after sAUG. Origin at the x-axis corresponds to the adenine of the sAUG. Top line, Homo sapiens to other vertebrates; middle line, D. melanogaster to other insects; bottom line, S. cerevisiae to other yeast. Cross denotes the conservation of the nucleotide in –3 which determines the context of the sAUG. Dashes denote the conservation of the nucleotides of sAUG, which always equals 1.
Prevalence and conservation of contexts of dAUGs
Alternative initiation of translation can serve to produce two alternative protein isoforms. If this phenomenon is frequent, we expect to see an interplay between the contexts of the two AUG codons within a gene. Specifically, a conceivable way to balance the output of two isoforms is to have a suboptimal sAUG followed by an optimal dAUG. Indeed, a dAUG that follows closely a suboptimal sAUG is slightly more likely to be optimal than a dAUG that follows an optimal sAUG (chi-square, vertebrates: P = 0.0055; insects: P < 10−4; yeast: P = 0.010; Figure 6).
Figure 6.

Prevalence of optimal context of dAUG. Fraction of dAUGs in optimal context (with purine at position –3, relative to dAUG) among all dAUGs, in genes with optimal (gray squares) and suboptimal (black diamonds) contexts of sAUG, depending on the position of dAUG relative to sAUG. Power curve fit is provided for visual reference. (A) Homo sapiens; (B) D. melanogaster; (C), S cerevisiae.
Finally, we analyzed the pattern of interspecies conservation of the context of the dAUG, depending on whether the sAUG was in optimal or in suboptimal context. Because translation is more likely to be initiated at a dAUG located after a suboptimal than after an optimal sAUG, the selection on the context of a dAUG after a suboptimal sAUG is expected to be higher. Indeed, the optimal context of dAUG was more conserved in genes with the suboptimal context of sAUG than in genes with the optimal context of sAUG (Table 3). Conversely, there was no difference in conservation of the suboptimal context of dAUG between genes with different contexts of sAUG (Table 3).
Table 3.
Conservation of the nucleotide determining the optimal or the suboptimal context of dAUG (purine or pyrimidine at position –3 relative to dAUG, correspondingly), in genes with different contexts of sAUG
| Nucleotide at position –3 relative to dAUG | Purine |
Pyrimidine |
||
|---|---|---|---|---|
| Optimal sAUG (%) | Suboptimal sAUG (%) | Optimal sAUG (%) | Suboptimal sAUG (%) | |
| Vertebrates | 97.7 | 98.6*** | 97.0 | 96.6 |
| Insects | 95.7 | 97.4*** | 95.2 | 95.5 |
| Yeast | 97.0 | 96.9 | 96.0 | 95.1 |
Asterisks denote significance of difference between optimal and suboptimal sAUG (chi-square test): ***P < 0.001; **P < 0.01; *P < 0.05.
Table shows the percentage of conserved context-determining purines and pyrimidines for dAUGs in codon positions 2–30 (0 corresponds to sAUG; codon 1 was not analyzed as it always carries a purine—the adenine of the sAUG—at position –3).
Characteristics of genes with alternative initiation of translation
We analyzed the enrichment of different gene sets among the genes with multiple potential TISs in vertebrates. Genes with suboptimal sAUG were enriched for olfactory receptors and for transcription regulation (Table 4). When the genes with suboptimal sAUG were considered as a background set, genes with dAUGs among them still showed significant enrichment for transcription regulation (Table 5). Finally, genes involved in transcription regulation were also overrepresented among the genes with conserved dAUGs, compared to all genes with dAUGs (Table 6).
Table 4.
Gene set enrichment in genes with suboptimal context of sAUG in human
| Category | Term | Count | % | P-value |
|---|---|---|---|---|
| PIR_SUPERFAMILY | PIRSF003152:G protein-coupled olfactory receptor, class II | 41 | 3.17 | 8.43E-08 |
| KEGG_PATHWAY | hsa04740:Olfactory transduction | 46 | 3.55 | 1.78E-07 |
| UP_SEQ_FEATURE | splice variant | 555 | 42.86 | 3.16E-07 |
| SP_PIR_KEYWORDS | alternative splicing | 555 | 42.86 | 9.02E-07 |
| INTERPRO | IPR000725:Olfactory receptor | 47 | 3.63 | 1.27E-06 |
| GOTERM_MF_FAT | GO:0004984∼olfactory receptor activity | 47 | 3.63 | 1.32E-06 |
| GOTERM_BP_FAT | GO:0007608∼sensory perception of smell | 48 | 3.71 | 1.51E-06 |
| GOTERM_BP_FAT | GO:0051252∼regulation of RNA metabolic process | 150 | 11.58 | 3.55E-06 |
| GOTERM_BP_FAT | GO:0045449∼regulation of transcription | 205 | 15.83 | 3.64E-06 |
| GOTERM_BP_FAT | GO:0006355∼regulation of transcription, DNA-dependent | 147 | 11.35 | 3.87E-06 |
| GOTERM_BP_FAT | GO:0006350∼transcription | 172 | 13.28 | 4.56E-06 |
| GOTERM_BP_FAT | GO:0007606∼sensory perception of chemical stimulus | 50 | 3.86 | 5.67E-06 |
| UP_SEQ_FEATURE | compositionally biased region:Poly-Pro | 50 | 3.86 | 8.67E-06 |
| GOTERM_MF_FAT | GO:0043565∼sequence-specific DNA binding | 61 | 4.71 | 2.56E-05 |
| SP_PIR_KEYWORDS | olfaction | 46 | 3.55 | 2.86E-05 |
All considered genes were used as the background set.
Fifteen categories with the lowest P-values are listed.
Table 5.
Gene set enrichment in genes with suboptimal context of sAUG and a dAUG present among codons 2–5 in human
| Category | Term | Count | % | P-value |
|---|---|---|---|---|
| GOTERM_BP_FAT | GO:0051254∼positive regulation of RNA metabolic process | 18 | 9 | 7.65E-04 |
| GOTERM_BP_FAT | GO:0045893∼positive regulation of transcription, DNA-dependent | 18 | 9 | 7.65E-04 |
| GOTERM_BP_FAT | GO:0045941∼positive regulation of transcription | 18 | 9 | 0.002729 |
| GOTERM_BP_FAT | GO:0045944∼positive regulation of transcription from RNA polymerase II promoter | 14 | 7 | 0.002746 |
| UP_SEQ_FEATURE | domain:EF-hand 1 | 7 | 3.5 | 0.003219 |
| UP_SEQ_FEATURE | domain:EF-hand 2 | 7 | 3.5 | 0.003219 |
| GOTERM_BP_FAT | GO:0010628∼positive regulation of gene expression | 18 | 9 | 0.003422 |
| INTERPRO | IPR011992:EF-Hand type | 8 | 4 | 0.00345 |
| GOTERM_CC_FAT | GO:0005886∼plasma membrane | 47 | 23.5 | 0.006591 |
| UP_SEQ_FEATURE | calcium-binding region:2 | 6 | 3 | 0.006667 |
| UP_SEQ_FEATURE | domain:EF-hand 4 | 5 | 2.5 | 0.006821 |
| UP_SEQ_FEATURE | domain:EF-hand 3 | 5 | 2.5 | 0.006821 |
| GOTERM_BP_FAT | GO:0045935∼positive regulation of nucleobase, nucleoside, nucleotide and nucleic acid metabolic process | 18 | 9 | 0.011303 |
| SP_PIR_KEYWORDS | Transcription | 39 | 19.5 | 0.011644 |
| GOTERM_CC_FAT | GO:0044459∼plasma membrane part | 26 | 13 | 0.013469 |
All genes with suboptimal context of sAUG were used as the background set.
Fifteen categories with the lowest P-values are listed.
Table 6.
Gene set enrichment in genes with a dAUG present among codons 2–5 in human and conserved in all other vertebrates
| Category | Term | Count | % | P-value |
|---|---|---|---|---|
| GOTERM_MF_FAT | GO:0043565∼sequence-specific DNA binding | 35 | 13.06 | 5.57E-10 |
| GOTERM_MF_FAT | GO:0030528∼transcription regulator activity | 54 | 20.15 | 1.36E-09 |
| GOTERM_MF_FAT | GO:0003700∼transcription factor activity | 43 | 16.04 | 6.70E-09 |
| SP_PIR_KEYWORDS | dna-binding | 59 | 22.01 | 6.49E-08 |
| GOTERM_MF_FAT | GO:0003677∼DNA binding | 65 | 24.25 | 3.04E-07 |
| GOTERM_BP_FAT | GO:0051252∼regulation of RNA metabolic process | 57 | 21.27 | 1.14E-06 |
| SP_PIR_KEYWORDS | Homeobox | 15 | 5.60 | 4.76E-06 |
| GOTERM_BP_FAT | GO:0006355∼regulation of transcription, DNA-dependent | 54 | 20.15 | 5.15E-06 |
| INTERPRO | IPR001356:Homeobox | 14 | 5.22 | 1.10E-05 |
| INTERPRO | IPR017970:Homeobox, conserved site | 14 | 5.22 | 1.10E-05 |
| GOTERM_BP_FAT | GO:0045449∼regulation of transcription | 70 | 26.12 | 2.05E-05 |
| SMART | SM00389:HOX | 14 | 5.22 | 2.73E-05 |
| UP_SEQ_FEATURE | DNA-binding region:Homeobox | 13 | 4.85 | 3.77E-05 |
| INTERPRO | IPR012287:Homeodomain-related | 14 | 5.22 | 4.26E-05 |
| SP_PIR_KEYWORDS | nucleus | 96 | 35.82 | 5.31E-05 |
Conservation of dAUG was required in all species were alignment was present.
All genes with dAUG present among codons 2–5 were used as the background set.
Fifteen categories with the lowest P-values are listed.
Genes with suboptimal context of sAUG carried substantially longer 5′ UTRs both in human and in D. melanogaster. In both species, length of 5′ UTRs was also increased in genes with dAUG, and among those genes, was very weakly positively correlated with conservation of dAUG (Table 7). Similarly, in D. melanogaster, genes with suboptimal context of sAUG and with dAUGs tended to encode longer proteins, and protein length was very weakly positively correlated with conservation of dAUG (Table 8). In humans, no such differences were observed.
Table 7.
Mean length in nucleotides of 5′UTR in genes with different patterns of TISs
| sAUG |
dAUG |
Correlation with dAUG conservation (Spearman’s R) | |||
|---|---|---|---|---|---|
| Optimal | Suboptimal | Absent | Present | ||
| H. sapiens | 205.03 | 261.79*** | 208.57 | 244.97*** | 0.09* |
| D. melanogaster | 207.67 | 242.50* | 208.99 | 244.49*** | 0.20*** |
Asterisks denote significance of difference between genes with optimal or suboptimal context of sAUG (Mann–Whitney U-test), between genes with dAUG present or absent among codons 2–5 (Mann–Whitney U-test), and significance of Spearman’s correlation: ***P < 0.001; **P < 0.01; *P < 0.05.
Table 8.
Mean length of proteins with different patterns of TISs
| sAUG |
dAUG |
Correlation with dAUG conservation (Spearman’s R) | |||
|---|---|---|---|---|---|
| Optimal | Suboptimal | Absent | Present | ||
| H. sapiens | 562.58 | 544.91 | 560.38 | 565.44 | 0.05 |
| D. melanogaster | 541.95 | 570.58*** | 542.64 | 581.72** | 0.08* |
Length of canonical protein isoforms was measured in amino acids.
Asterisks denote significance of difference between genes with optimal or suboptimal context of sAUG (Mann–Whitney U-test), between genes with dAUG present or absent among codons 2–5 (Mann–Whitney U-test), and significance of Spearman’s correlation: ***P < 0.001; **P < 0.01; *P < 0.05.
Finally, we asked whether the genes with alternative TISs are also more likely to be alternatively spliced. In D. melanogaster, the mean number of splice variants per gene was marginally higher in genes with a suboptimal context of sAUG, and somewhat higher in genes with a dAUG (Table 9). Although no such difference was observed in H. sapiens (Table 9), alternative splicing showed up as one of the characteristics associated with the suboptimal context of sAUG in gene set enrichment analysis in humans (Table 4). In both species, among genes with a dAUG, the number of isoforms was positively correlated with the conservation of dAUG, although the correlations were extremely weak (Table 9).
Table 9.
Mean number of splice variants in genes with different patterns of TISs
| sAUG |
dAUG |
Correlation with dAUG conservation (Spearman’s R) | |||
|---|---|---|---|---|---|
| Optimal | Suboptimal | Absent | Present | ||
| H. sapiens | 2.95 | 2.86 | 2.95 | 2.92 | 0.07* |
| D. melanogaster | 1.53 | 1.59* | 1.52 | 1.76** | 0.11*** |
Asterisks denote significance of difference between genes with optimal or suboptimal context of sAUG (Mann–Whitney U-test), between genes with dAUG present or absent among codons 2–5 (Mann–Whitney U-test), and significance of Spearman’s correlation: ***P < 0.001; **P < 0.01; *P < 0.05.
DISCUSSION
It is commonly taken for granted that mature eukaryotic mRNAs contain a single start codon and encode a single protein. This assumption underlies most methods of eukaryotic gene structure prediction, and the vast majority of mRNAs annotated in GenBank have such a structure. However, it contradicts the experimental and bioinformatic data (9), which show that multiple start codons within a single gene can be used alternatively.
Ribosomes can initiate translation not only from the annotated start site, but also from the nearest downstream AUG codon(s) through leaky scanning (10). This can result in N-end truncated protein isoforms. If the truncation is minor, the protein isoforms may be isofunctional; otherwise, they may possess different functions. Therefore, alternative translation from a downstream AUG codon can be used either to increase the protein synthesis rate through additional production of an isofunctional variant, or to produce a protein variant with distinct properties (9).
The efficiency of an AUG codon as a TIS, and the propensity of scanning through it to be leaky, depends primarily on its nucleotide context. A considerable fraction of eukaryotic transcripts contains the annotated start AUG codon in a suboptimal context, and downstream start codons are more likely to be used for translation initiation in such genes. Comparison of the frequency and evolutionary conservation of downstream in-frame AUGs following sAUGs of different contexts can reveal the evolutionary pressure associated with the use of downstream translation start sites.
We showed that both the prevalence (Figure 1) and the conservation (Table 1, Figures 2 and 3) of dAUGs is elevated in genes with suboptimal sAUGs. Conceivably, this difference could be due to start codon annotation artifacts. Indeed, if a substantial fraction of the annotated suboptimal sAUGs are spurious, and translation in such genes is always initiated at the dAUGs, high conservation of the latter is to be expected. However, the context of the sAUG also affected conservation of dAUG in genes with proteomics-supported sAUGs (Table 2), in which translation starts at sAUG often enough for the peptide product to be detected experimentally. Therefore, the increased conservation of dAUGs in genes with suboptimal sAUGs is most plausibly explained by use of dAUGs as secondary TISs. Selection favoring such sites is strong enough to keep their frequency much higher than expected, and sufficiently long lasting to maintain them through hundreds of millions of years of evolution.
Our evidence of selection for alternative initiation is statistical, and does not allow us to pinpoint individual genes under selection with certainty. Nevertheless, we can study the characteristics of genes which plausibly employ it, i.e. those with conserved alternative TISs. We observe that the set of genes with suboptimal sAUGs is strongly enriched for transcription factors (Tables 4–6), in line with previous observations (9,23). Furthermore, within the genes with suboptimal sAUGs, dAUGs prevail and are particularly conserved among transcription factors. Regulatory proteins tend to be themselves subject to complex regulation; use of alternative TISs probably contributes to this complexity.
Different modes of selection may impose different constraints on the contexts of sAUGs and dAUGs. Selection for high expression may favor short 5′ UTR with optimal sAUG (23), and the context of dAUG in this case may be irrelevant, as it is seldom, if ever, used for initiation (10). A suboptimal dAUG, which follows a suboptimal sAUG, may result from selection for a generally low production of the protein (23); in this case, dAUG(s) further downstream have to be taken into account, which complicates the situation further. Finally, an optimal dAUG following a suboptimal sAUG may increase the overall production of the protein when the two isoforms are identical, or serve to produce a balanced amount of the N-truncated isoform with a distinct function.
We observed that genes with suboptimal sAUGs tend to have longer 5′ UTR (Table 7). Furthermore, possible complexity of regulation, as evidenced by long 5′ UTRs, is correlated with the presence of dAUG and, when it is present, its conservation (Table 7). The weak positive correlation of alternative TIS use with protein length observed in D. melanogaster (Table 8) probably stems from the fact that short proteins are more highly expressed (24). Finally, alternative TIS use and conservation is correlated with alternative splicing (Table 9). Together, these results suggest that alternative TISs tend to be used by the highly regulated genes. The weakness of the observed statistical patterns should not come as a surprise, given the high number of factors which may affect the frequency and conservation of TISs.
Each mode of selection may favor the corresponding context-determining combination of nucleotides. Analysis of conservation patterns of such nucleotides may help elucidate selection favoring each of such combinations.
The optimal context of sAUGs, which is characteristic of the vast majority of genes, is conserved in evolution (Figures 4 and 5, left columns). Nevertheless, the prevalence (Figure 6) and conservation (Table 3) of the optimal context of dAUGs is higher in genes with the suboptimal sAUGs. Therefore, optimality of dAUGs is most important in genes where a suboptimal sAUG favors leaky scanning.
Why does a substantial fraction of genes have sAUGs in a suboptimal context? Is the suboptimal context of an sAUG advantageous, or is it simply the result of fixations of mildly deleterious mutations? Stronger selection for optimal dAUGs after suboptimal sAUGs is consistent with either hypothesis. Occasional leaky scanning of a suboptimal sAUG followed by initiation of translation from an optimal dAUG can provide balanced production of alternative isoforms from different AUG codons (25), contributing to functional diversity of proteins. In this case, the context of the sAUG has to remain suboptimal; otherwise, the downstream translation will be abolished. Alternatively, dAUGs may be selected as backups to produce a functionally identical isoform when a suboptimal sAUG is missed. Cases of identical as well as different roles of isoforms produced from different AUGs have been described (9), and it is not clear a priori which situation is more common.
We can distinguish between these models using the data on conservation of the suboptimal context of sAUGs. Indeed, the key nucleotides affecting translation efficiency are unlikely to evolve neutrally. Therefore, the pyrimidine at position −3 that corresponds to the suboptimal context of an sAUG may either be advantageous, or represent a deleterious mutation causing selection which is not strong enough to prevent its fixation. In the former case, pyrimidines at positions −3 are expected to be conserved. In the latter case, we expect conservation of such pyrimidines to be lower than that of the surrounding nucleotides.
Although the results are inconclusive, the suboptimal context of sAUGs seems to be conserved at least in vertebrates (Figure 4, right column). Conservation of suboptimal sAUGs is higher when a dAUG can provide an alternative TIS (Figure 5, right column), suggesting stronger selection for suboptimal sAUGs in such genes. Thus, at least in vertebrates, suboptimality of sAUG appears to be functional, rather than a byproduct of inability to prevent deleterious context-disrupting mutations. Evolutionary maintenance of suboptimal characters involved in gene regulation is relatively widespread. For example, suboptimality of transcription factor-binding sites (26), splicing sites (27) and codon usage (28) can be conserved.
Recent research into structural organization and coding potential of eukaryotic mRNAs revealed unexpected complexity. The AUG triplets located in 5′-UTR (uAUGs) are functionally significant (29–33), and the products of the short open reading frames (ORFs) starting with them may be translated (34,35). Analysis of evolutionary conservation of 5′-UTR and uAUGs also revealed the potential presence of hidden functional signals (29,36–38). Our results show that the pattern of conservation of multiple in-frame dAUG codons and their contexts is also indicative of their functional significance.
Alternative use of multiple start codons maintained by selection may, in turn, facilitate evolution of novel traits which would otherwise impede previous function and/or whose origin requires multiple mutations. Indeed, alternative start codons allow selection to test novel N-terminal isoforms of existing proteins at low translation levels. For example, a purine-to-pyrimidine mutation at the −3 position of sAUG will direct a substantial fraction of ribosomes to initiate translation from the dAUG(s). This will still allow some synthesis of a full-length protein isoform, probably at a sufficient level to preserve its function. In addition, a novel N-truncated variant will be synthesized. If the new variant is advantageous, perhaps due to a different targeting of the isofunctional protein, it will be picked up by selection. Conversely, a mutation creating an in-frame AUG codon upstream of the existing start codon can lead to translation of a novel N-extended isoform. If the context of the new upstream AUG is suboptimal, this isoform would be initially translated at a low level, allowing selection to shape the sequence of the novel coding segment of DNA between the two start codons. This exact mechanism of origin of a novel N-end-extended isoform has recently been observed in evolution of glycophorin C (GYPC) gene in human lineage (39). On a genome scale, origin of new C-terminal coding sequence from 3′-UTR via shift of stop codons has been described in multiple genes in yeast (40). In the 5′-UTR, an analogous process of addition of sequence to the coding region may be further facilitated by balanced translation from different AUGs. Such additional synthesis of N-end-truncated or N-end-extended protein isoforms due to alternative translation could provide an evolutionary playground for shaping their sequence.
FUNDING
The Russian Foundation for Basic Research (08-04-01394-a); the ‘Molecular and Cellular Biology’ Program of the Russian Academy of Sciences; the Russian Science Support Foundation (to G.A.B.); the Russian Ministry of Science and Education (P916 to G.A.B.; 2.1.1/6382; 02.740.11.0705, and P128 to A.V.K.); and in the frame of the SD RAS Complex Integration Program (to A.V.K.). Funding for open access charge: Waived by Oxford University Press.
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
We thank Mikhail Gelfand for discussion, Alexey Kondrashov, Ekaterina Ermakova and two anonymous reviewers for valuable comments on the manuscript, and Dmitriy Vinogradov for help with data management. Analysis was partially performed using the KIAE supercomputing farm.
REFERENCES
- 1.Mironov AA, Fickett JW, Gelfand MS. Frequent alternative splicing of human genes. Genome Res. 1999;9:1288–1293. doi: 10.1101/gr.9.12.1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nilsen TW, Graveley BR. Expansion of the eukaryotic proteome by alternative splicing. Nature. 2010;463:457–463. doi: 10.1038/nature08909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Suzuki Y, Taira H, Tsunoda T, Mizushima-Sugano J, Sese J, Hata H, Ota T, Isogai T, Tanaka T, Morishita S. Diverse transcriptional initiation revealed by fine, large-scale mapping of mRNA start sites. EMBO Rep. 2001;2:388–393. doi: 10.1093/embo-reports/kve085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kitagawa N, Washio T, Kosugi S, Yamashita T, Higashi K, Yanagawa H, Higo K, Satoh K, Ohtomo Y, Sunako T, et al. Computational analysis suggests that alternative first exons are involved in tissue-specific transcription in rice (Oryza sativa) Bioinformatics. 2005;21:1758–1763. doi: 10.1093/bioinformatics/bti253. [DOI] [PubMed] [Google Scholar]
- 5.Sunderland PA, West CE, Waterworth WM, Bray CM. Choice of a start codon in a single transcript determines DNA ligase 1 isoform production and intracellular targeting in Arabidopsis thaliana. Biochem. Soc. Trans. 2004;32:614–616. doi: 10.1042/BST0320614. [DOI] [PubMed] [Google Scholar]
- 6.Gandre S, Bercovich Z, Kahana C. Mitochondrial localization of antizyme is determined by context-dependent alternative utilization of two AUG initiation codons. Mitochondrion. 2003;2:245–256. doi: 10.1016/S1567-7249(02)00105-8. [DOI] [PubMed] [Google Scholar]
- 7.Leissring MA, Farris W, Wu X, Christodoulou DC, Haigis MC, Guarente L, Selkoe DJ. Alternative translation initiation generates a novel isoform of insulin-degrading enzyme targeted to mitochondria. Biochem. J. 2004;383:439–446. doi: 10.1042/BJ20041081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kaipio K, Kallio J, Pesonen U. Mitochondrial targeting signal in human neuropeptide Y gene. Biochem. Biophys. Res. Commun. 2005;337:633–640. doi: 10.1016/j.bbrc.2005.09.093. [DOI] [PubMed] [Google Scholar]
- 9.Kochetov AV. Alternative translation start sites and hidden coding potential of eukaryotic mRNAs. Bioessays. 2008;30:683–691. doi: 10.1002/bies.20771. [DOI] [PubMed] [Google Scholar]
- 10.Kozak M. Regulation of translation via mRNA structure in prokaryotes and eukaryotes. Gene. 2005;361:13–37. doi: 10.1016/j.gene.2005.06.037. [DOI] [PubMed] [Google Scholar]
- 11.Jackson RJ. Alternative mechanisms of initiating translation of mammalian mRNAs. Biochem. Soc. Trans. 2005;33:1231–1241. doi: 10.1042/BST0331231. [DOI] [PubMed] [Google Scholar]
- 12.Baird SD, Turcotte M, Korneluk RG, Holcik M. Searching for IRES. RNA. 2006;12:1755–1785. doi: 10.1261/rna.157806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pisarev AV, Kolupaeva VG, Pisareva VP, Merrick WC, Hellen CU, Pestova TV. Specific functional interactions of nucleotides at key -3 and +4 positions flanking the initiation codon with components of the mammalian 48S translation initiation complex. Genes Dev. 2006;20:624–36. doi: 10.1101/gad.1397906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kuhn RM, Karolchik D, Zweig AS, Wang T, Smith KE, Rosenbloom KR, Rhead B, Raney BJ, Pohl A, Pheasant M, et al. The UCSC Genome Browser Database: update 2009. Nucleic Acids Res. 2009;37:D755–761. doi: 10.1093/nar/gkn875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kellis M, Patterson N, Endrizzi M, Birren B, Lander ES. Sequencing and comparison of yeast species to identify genes and regulatory elements. Nature. 2003;423:241–254. doi: 10.1038/nature01644. [DOI] [PubMed] [Google Scholar]
- 16.Cliften P, Sudarsanam P, Desikan A, Fulton L, Fulton B, Majors J, Waterston R, Cohen BA, Johnston M. Finding functional features in Saccharomyces genomes by phylogenetic footprinting. Science. 2003;301:71–76. doi: 10.1126/science.1084337. [DOI] [PubMed] [Google Scholar]
- 17.Volkova OA, Kochetov AV. Interrelations between the nucleotide context of human start AUG codon, N-end amino acids of the encoded protein and initiation of translation. J. Biomol. Struct. Dyn. 2010;27:611–618. doi: 10.1080/07391102.2010.10508575. [DOI] [PubMed] [Google Scholar]
- 18.Yun DF, Laz TM, Clements JM, Sherman F. mRNA sequences influencing translation and the selection of AUG initiator codons in the yeast Saccharomyces cerevisiae. Mol. Microbiol. 1996;19:1225–1239. doi: 10.1111/j.1365-2958.1996.tb02468.x. [DOI] [PubMed] [Google Scholar]
- 19.Kochetov AV. AUG codons at the beginning of protein coding sequences are frequent in eukaryotic mRNAs with a suboptimal start codon context. Bioinformatics. 2005;21:837–840. doi: 10.1093/bioinformatics/bti136. [DOI] [PubMed] [Google Scholar]
- 20.Vizcaino JA, Cote R, Reisinger F, Foster JM, Mueller M, Rameseder J, Hermjakob H, Martens L. A guide to the proteomics identifications database proteomics data repository. Proteomics. 2009;9:4276–4283. doi: 10.1002/pmic.200900402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 22.Dennis G, Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: database for annotation, visualization, and integrated discovery. Genome Biol. 2003;4:P3. [PubMed] [Google Scholar]
- 23.Kochetov AV, Ischenko IV, Vorobiev DG, Kel AE, Babenko VN, Kisselev LL, Kolchanov NA. Eukaryotic mRNAs encoding abundant and scarce proteins are statistically dissimilar in many structural features. FEBS Lett. 1998;440:351–355. doi: 10.1016/s0014-5793(98)01482-3. [DOI] [PubMed] [Google Scholar]
- 24.Lemos B, Bettencourt BR, Meiklejohn CD, Hartl DL. Evolution of proteins and gene expression levels are coupled in Drosophila and are independently associated with mRNA abundance, protein length, and number of protein–protein interactions. Mol. Biol. Evol. 2005;22:1345–1354. doi: 10.1093/molbev/msi122. [DOI] [PubMed] [Google Scholar]
- 25.Kozak M. Pushing the limits of the scanning mechanism for initiation of translation. Gene. 2002;299:1–34. doi: 10.1016/S0378-1119(02)01056-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kotelnikova EA, Makeev VJ, Gelfand MS. Evolution of transcription factor DNA binding sites. Gene. 2005;347:255–263. doi: 10.1016/j.gene.2004.12.013. [DOI] [PubMed] [Google Scholar]
- 27.Dewey CN, Rogozin IB, Koonin EV. Compensatory relationship between splice sites and exonic splicing signals depending on the length of vertebrate introns. BMC Genomics. 2006;7:311. doi: 10.1186/1471-2164-7-311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Neafsey DE, Galagan JE. Positive selection for unpreferred codon usage in eukaryotic genomes. BMC Evol. Biol. 2007;7:119. doi: 10.1186/1471-2148-7-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Churbanov A, Rogozin IB, Babenko VN, Ali H, Koonin EV. Evolutionary conservation suggests a regulatory function of AUG triplets in 5'-UTRs of eukaryotic genes. Nucleic Acids Res. 2005;33:5512–5520. doi: 10.1093/nar/gki847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Crowe ML, Wang XQ, Rothnagel JA. Evidence for conservation and selection of upstream open reading frames suggests probable encoding of bioactive peptides. BMC Genomics. 2006;7:16. doi: 10.1186/1471-2164-7-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hayden CA, Jorgensen RA. Identification of novel conserved peptide uORF homology groups in Arabidopsis and rice reveals ancient eukaryotic origin of select groups and preferential association with transcription factor-encoding genes. BMC Biol. 2007;5:32. doi: 10.1186/1741-7007-5-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hayden CA, Bosco G. Comparative genomic analysis of novel conserved peptide upstream open reading frames in Drosophila melanogaster and other dipteran species. BMC Genomics. 2008;9:61. doi: 10.1186/1471-2164-9-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tran MK, Schultz CJ, Baumann U. Conserved upstream open reading frames in higher plants. BMC Genomics. 2008;9:361. doi: 10.1186/1471-2164-9-361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oyama M, Kozuka-Hata H, Suzuki Y, Semba K, Yamamoto T, Sugano S. Diversity of translation start sites may define increased complexity of the human short ORFeome. Mol. Cell Proteomics. 2007;6:1000–1006. doi: 10.1074/mcp.M600297-MCP200. [DOI] [PubMed] [Google Scholar]
- 35.Ingolia NT, Ghaemmaghami S, Newman JR, Weissman JS. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009;324:218–223. doi: 10.1126/science.1168978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shabalina SA, Ogurtsov AY, Rogozin IB, Koonin EV, Lipman DJ. Comparative analysis of orthologous eukaryotic mRNAs: potential hidden functional signals. Nucleic Acids Res. 2004;32:1774–1782. doi: 10.1093/nar/gkh313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kovaleva GY, Bazykin GA, Brudno M, Gelfand MS. Comparative genomics of transcriptional regulation in yeasts and its application to identification of a candidate alpha-isopropylmalate transporter. J. Bioinform. Comput. Biol. 2006;4:981–998. doi: 10.1142/s0219720006002284. [DOI] [PubMed] [Google Scholar]
- 38.Resch AM, Ogurtsov AY, Rogozin IB, Shabalina SA, Koonin EV. Evolution of alternative and constitutive regions of mammalian 5'UTRs. BMC Genomics. 2009;10:162. doi: 10.1186/1471-2164-10-162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wilder JA, Hewett EK, Gansner ME. Molecular evolution of GYPC: evidence for recent structural innovation and positive selection in humans. Mol. Biol. Evol. 2009;26:2679–2687. doi: 10.1093/molbev/msp183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Giacomelli MG, Hancock AS, Masel J. The conversion of 3' UTRs into coding regions. Mol. Biol. Evol. 2007;24:457–464. doi: 10.1093/molbev/msl172. [DOI] [PMC free article] [PubMed] [Google Scholar]