

Abstract
The adenomatous polyposis coli (APC) gene is known to act as a tumor suppressor gene in both sporadic and hereditary colorectal cancer by negatively regulating WNT signaling. Familial adenomatous polyposis (FAP) patients develop intestinal polyps due to the presence of a single germline mutation in APC. The severity of the FAP phenotype is a function of the position of the APC mutation, indicating a complex role for APC that extends beyond the canonical WNT pathway. APC encodes a large protein with multiple functional domains, including an armadillo repeat domain that has been linked to protein–protein interactions. To determine the effect of the armadillo repeat domain on intestinal tumorigenesis, we generated a congenic mouse line (ApcΔ242) carrying a gene trap cassette between exons 7 and 8 of the murine Apc gene. ApcΔ242/+ mice express a truncated Apc product lacking the armadillo repeat domain as part of a fusion protein with β-geo. Expression of the fusion product was confirmed by X-gal staining, ensuring that ApcΔ242 is not a null allele. In contrast, ApcMin/+ mice produce a truncated Apc product that contains an intact armadillo repeat domain. On the C57BL/6J background, ApcΔ242/+ mice develop more polyps than do ApcMin/+ mice along the entire length of the small intestine; however, polyps were significantly smaller in ApcΔ242/+ mice. In addition, polyp multiplicity in ApcΔ242/+ mice is affected by polymorphisms between inbred strains. These data suggest that the armadillo repeat domain of the Apc protein suppresses tumor initiation in the murine intestine while also promoting tumor growth.
Introduction
Colorectal cancer has the third highest mortality rate for cancers among men and women in the United States, accounting for 9–10% of total cancer-related deaths (www.cancer.org). Genetic evidence suggests that the APC gene acts as a classical tumor suppressor gene in both sporadic and hereditary colorectal cancers, with inactivation of both alleles resulting in tumor growth (Miyaki et al. 1994; Powell et al. 1992). Inactivation of APC results in nuclear localization of β-catenin and subsequent activation of the WNT signaling pathway (MacDonald et al. 2009). Familial adenomatous polyposis (FAP) is a dominantly inherited disorder in which individuals inherit one defective APC allele. The presence of a mutated APC allele makes FAP patients susceptible to the development of colorectal adenomas following inactivation of the remaining wild-type APC allele through somatic mutation and/or loss of heterozygosity (LOH) (Groden et al. 1991; Kinzler et al. 1991; Nishisho et al. 1991). As a result, FAP patients are diagnosed by the development of hundreds to thousands of benign adenomatous polyps throughout the intestinal tract.
The study of human diseases, such as FAP, has been greatly facilitated by the use of animal model systems. Mouse models provide a fixed genetic background and controlled environment in which to analyze the cellular processes leading to neoplasia (Thompson et al. 2004). Several models of intestinal tumorigenesis have been generated by introducing specific mutations into the murine ortholog of the APC gene (Apc), allowing investigation of its role in both polyp initiation and carcinoma progression (reviewed in Siracusa et al. 2004; Uronis and Threadgill 2009). The most extensively studied of these models is the multiple intestinal neoplasia (ApcMin) mouse, which was discovered through phenotypic screening following ENU mutagenesis (Moser et al. 1990). B6 mice carrying the ApcMin mutation develop an average of 106 benign adenomas throughout the length of the intestinal tract. Apc is a large, multidomain protein and the position of a mutation can affect polyp number, location, and stage in mice (Heyer et al. 1999; Taketo and Edelmann 2009). For example, ApcΔ716 mice develop over three times as many polyps as ApcMin/+ mice, while a more distal mutation, Apc1634N, results in greatly reduced tumor burden (Fodde et al. 1994; Oshima et al. 1995). These data recapitulate findings from FAP patients in which tumor multiplicity and secondary symptoms correlate with the location of the germline APC mutation (Burt and Jasperson 2008). Mouse lines carrying mutant Apc alleles have, therefore, provided valuable in vivo models for studying the diverse roles of Apc in intestinal tumorigenesis.
In this study we describe a new mouse model of FAP called ApcΔ242, generated by insertion of a gene trap cassette between exons 7 and 8 of the Apc gene. Mice carrying the ApcΔ242 allele express a fusion product consisting of the N-terminus of Apc and a functional β-geo protein. C57BL/6J (B6) mice carrying the mutant ApcΔ242 allele develop significantly more tumors throughout the length of the small intestine than do B6 ApcMin/+ mice. The ApcΔ242 mouse strain provides a model system for dissecting the roles of domains within the Apc protein during tumor initiation, growth, and progression.
Materials and methods
Generation of the B6.129 ApcΔ242/+ colony
BayGenomics, Inc. (San Francisco, CA) generated gene trap clones by insertion of a β-geo cassette with a 5′ splice acceptor site into 129P2/OlaHsd embryonic stem (ES) cells via the pGTOpfs gene trap vector (Stryke et al. 2003). Neomycin selection was used to identify clones carrying the gene trap allele. 5′ RACE PCR was used to identify one clone, XA018, with an insertion between exons 7 and 8 of the Apc gene. XA018 ES cells were microinjected into C57BL/6J (B6) blastocysts by the KCC Transgenic/Knockout Mouse Facility to generate chimeric offspring. Genomic DNA was isolated from tail biopsies by DNeasy Kit (Qiagen), and mice were genotyped by PCR for the presence of the gene trap cassette (forward: GATGCCTGCTTGCCGAATATC; reverse: GAAGAACTCCAGCATGAGATCC). Offspring carrying the mutant allele were backcrossed to B6 mice from The Jackson Laboratory (Bar Harbor, ME) for ten generations to generate the congenic B6.129 ApcGt(XA018)Byg line, hereafter referred to as ApcΔ242. The colony was maintained in the AALAC-accredited TJU animal facility. B6.129 ApcΔ242/+ mice were mated to C3H/HeJ mice from The Jackson Laboratory to generate F1 offspring.
Confirmation of β-galactosidase expression
Male ApcΔ242/+ mice at 120 days of age were euthanized by CO2 asphyxiation followed by cervical dislocation. Distal small intestine and colon samples were washed in 1× PBS and fixed for 30 min in Formalde-Fresh 10% buffered formalin solution (Fisher Scientific). Fixed tissues were incubated at 37°C in a 1:40 dilution of X-gal Solution (Novagen) in BetaBlue Reaction Buffer (Novagen). Staining was visualized on a Nikon SMZ-U dissection scope at 22.5× magnification. Pictures were taken with a Nikon DXM1200F digital camera.
Evaluation of intestinal tumorigenesis in ApcΔ242/+ mice
The intestinal tract was washed with 1× PBS and separated into four segments: proximal small intestine (SI), middle SI, distal SI, and colon. Polyp multiplicity and average size were determined for each segment with a Nikon SMZ-U dissection microscope at 22.5× magnification. The diameter was measured at the widest point of the tumor. Tumor volume was estimated using the equation for the volume of a sphere [(4/3)πr3]. Tumor burden was calculated as the sum of the volumes of all tumors in an individual.
Histopathological analysis of tumors
Tissues were fixed in Formalde-Fresh 10% buffered formalin solution (Fisher Scientific) overnight at 4°C and stored in 70% EtOH. Fixed tissues were embedded in paraffin and tissues were cut and mounted on glass slides by the KCC Translational Research Core Facility. Tissues were stained with eosin and counterstained with hematoxylin. Staining was visualized on a Nikon Eclipse E600 microscope at either 4× or 20× magnification.
Results
Generation of a mouse strain carrying an Apc gene trap allele
The APC gene is mutated in both sporadic colorectal cancers and hereditary disorders such as FAP. To study the effects of the armadillo repeat domain of the Apc protein on intestinal tumorigenesis, a mouse model expressing a truncated Apc protein was generated. BayGenomics generated a library of ES cell clones using a randomly inserted gene trap vector (Stryke et al. 2003). The gene trap cassette carries a splice acceptor site, a sequence encoding the β-geo fusion protein, and a polyadenylation site (Fig. 1a). Insertion of the cassette into an intronic sequence results in the fusion of the N-terminus of the trapped gene to the β-geo protein. One clone (XA018) carried a cassette inserted between exons 7 and 8 of the Apc gene (Fig. 1a). The Apc gene consists of 16 exons, with the majority of the open reading frame (ORF) encoded by exon 16 (www.ensemble.org). Different Apc mutations result in different polyposis phenotypes in both FAP patients and mouse models (reviewed in Aoki and Taketo 2007). Therefore, we generated a congenic mouse strain carrying the ApcGt(XA018)Byg allele (ApcΔ242) to analyze the effect of this novel Apc mutation on intestinal tumorigenesis. XA018 ES cells were injected into C57BL/6J (B6) blastocysts. The resulting chimeric offspring were genotyped for the presence of the gene trap cassette (Fig. 1b) and mice carrying the ApcΔ242 allele were backcrossed to the B6 inbred strain for more than ten generations to generate a congenic line.
Fig. 1.
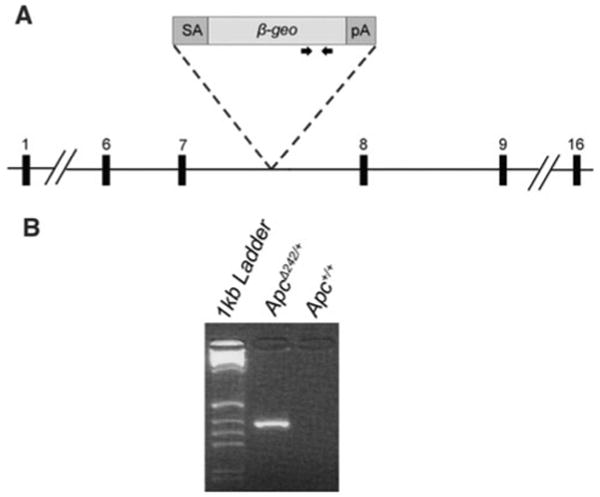
Generation of ApcΔ242/+ mice. a A gene trap cassette (BayGenomics) was inserted into mouse chromosome 18 between exons 7 and 8 of the Apc gene. The black numbered boxes indicate endogenous exons of murine Apc. Arrows represent the location of ApcΔ242 genotyping primers. b Genotyping of ApcΔ242/+ and Apc+/+ mice. A PCR product of ∼300 bp is produced
The ApcΔ242 allele generates an Apc fusion protein with β-galactosidase
Apc encodes a 2842-amino-acid protein containing multiple functional domains, including an N-terminal oligomerization domain and an armadillo repeat domain involved in protein–protein interactions (Fig. 2). The presence of the gene trap cassette in ApcΔ242/+ mice results in the splicing of the first seven exons of Apc to the β-geo gene. The ApcΔ242 allele generates a theoretical 172-kDa fusion product combining the first 241 residues of the Apc protein with the full-length β-geo protein (Fig. 2). The β-geo protein contains the functional domains of both the β-galactosidase enzyme and a neomycin resistant protein. To confirm expression of the mutant fusion protein, X-gal staining was performed on samples from the distal small intestine and distal colon of ApcΔ242/+ mice. Tissues from ApcΔ242/+ mice stained blue, while those of wild-type littermates did not stain blue, indicating the presence of functional β-galactosidase activity in the ApcΔ242/+ intestinal tract and expression of the mutant allele (Fig. 3).
Fig. 2.

ApcΔ242/+ mice lack the armadillo repeat domain of the Apc protein. Diagram comparing wild-type Apc protein with the truncated proteins generated by the ApcΔ242, ApcΔ474, ApcΔ580, and ApcMin mutations. The domains of the Apc protein are labeled on the wild-type protein. The ApcΔ474 and ApcΔ580 proteins have novel C-terminal residues as a result of frameshift mutations
Fig. 3.
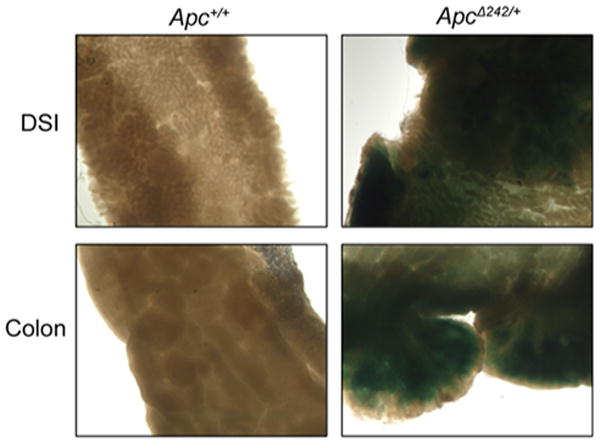
ApcΔ242/+ mice express an Apc fusion product with functional β-galactosidase. Distal small intestine and colon from Apc+/+ and ApcΔ242/+ mice after incubation with X-gal solution. Blue tissues indicate the presence of a functional β-galactosidase protein. Western blots probing intestinal tissue lysates from ApcΔ242/+ mice with β-galactosidase antibody indicated the presence of a fusion protein at the expected size of 172 kDa (data not shown)
ApcΔ242/+ mice develop adenomas in the small intestine and colon
The most prominent phenotype resulting from Apc mutations is intestinal tumorigenesis. To identify how the ApcΔ242 mutation affects polyposis, the intestinal tracts of ApcΔ242/+ mice were evaluated for the presence of polyps at 120 days of age. These mutant mice had an average of 177 ± 30 polyps throughout the length of the intestinal tract. The polyp multiplicity of ApcΔ242/+ mice is significantly higher than the polyp multiplicity of B6 ApcMin/+ mice (106 ± 28, P < 0.001). Analysis of tumor distribution indicates that this increased tumorigenesis occurs along the entire length of the small intestine, but not in the colon (Fig. 4a). ApcΔ242/+ mice had significantly smaller polyps in all areas of the small intestine (Fig. 4b). Both polyp multiplicity and polyp size contribute to overall tumor burden, a measurement that negatively correlates with mortality in mouse models of intestinal tumorigenesis (Guillen-Ahlers et al. 2010; Korsisaari et al. 2007). Despite the decrease in polyp size, ApcΔ242/+ mice have a significantly increased tumor burden in the distal small intestine compared to B6 ApcMin/+ controls (Fig. 4c).
Fig. 4.

Comparison of polyp location, size, and tumor burden in ApcMin/+ and ApcΔ242/+ mice at 120 days of age. a Average polyp numbers detected in the proximal, middle, and distal portions of the small intestine and colon. b Average polyp sizes detected in the proximal, middle, and distal portions of the small intestine and colon. Diameters of polyps were measured and reported in mm. c Tumor burden in the proximal, middle, and distal portions of the small intestine and colon. Tumor burden was calculated using the formula (4/3)πr3. *P < 0.05; **P < 0.005; ***P < 0.001. n = 15 for ApcMin/+ mice and n = 18 for ApcΔ242/+ mice. Error bars represent standard deviation
ApcΔ242/+ tumors are pathologically similar to ApcMin/+ tumors
While each polyp in individuals with FAP has only a small chance of progressing to adenocarcinoma, the large number of polyps in the intestinal tract guarantees the development of colorectal cancer in untreated patients. To determine if the increased intestinal tumorigenesis in ApcΔ242/+ mice results in the development of adenocarcinoma, tumors from 120-day-old ApcΔ242/+ mice were compared to tumors from age-matched ApcMin/+ mice. Intestinal tissue samples from ApcΔ242/+ and ApcMin/+ mice were stained with hematoxylin and eosin (H&E). H&E staining showed no invasion through the basement membrane in tumors from either model (Fig. 5a-d), further indicating that neither ApcΔ242/+ nor ApcMin/+ mice progress to adenocarcinoma by 120 days of age.
Fig. 5.

ApcΔ242/+ and ApcMin/+ polyps do not progress to adenocarcinoma. Intestinal polyp samples from 120-day-old ApcΔ242/+ and ApcMin/+ mice were stained with hematoxylin and eosin. Ten polyps were staged for each genotype. A representative distal small-intestine polyp from an ApcΔ242/+ mouse is shown at a 4× and b 20× magnification. A representative polyp from an ApcMin/+ mouse is also shown at c 4× and d 20× magnification. Black boxes indicate the areas shown in higher magnification
ApcΔ242-mediated tumorigenesis is altered by modifier genes on the C3H/HeJ genetic background
The ApcMin/+ mouse has been an invaluable model system for identifying modifier genes that affect intestinal tumorigenesis (reviewed in Siracusa et al. 2004). As a closely related mutation with a different phenotype, ApcΔ242 may provide a complementary model system for identifying genes and pathways relevant to colorectal cancer. The C3H/HeJ (C3H) genetic background is known to contain modifier loci that make the strain resistant to ApcMin-mediated polyposis (Koratkar et al. 2004). While B6 ApcMin/+ mice develop 106 ± 28 polyps, B6C3 F1ApcMin/+ offspring develop only 16 ± 6 polyps (Fig. 6). To determine if the ApcΔ242 phenotype is affected by genetic background, B6 ApcΔ242/+ mice were mated to C3H mice. B6C3 F1ApcΔ242/+ mice developed significantly fewer polyps (26 ± 13) than B6 ApcΔ242/+ mice in all sections of the small intestine, but not in the colon (Fig. 6). The percent decrease in polyp number due to the introduction of the C3H background was 86% in both ApcΔ242/+ and ApcMin/+ mice. These data indicate that the modifiers of ApcMin-mediated tumorigenesis found in the C3H inbred strain are also capable of modifying the phenotype of the ApcΔ242 allele.
Fig. 6.
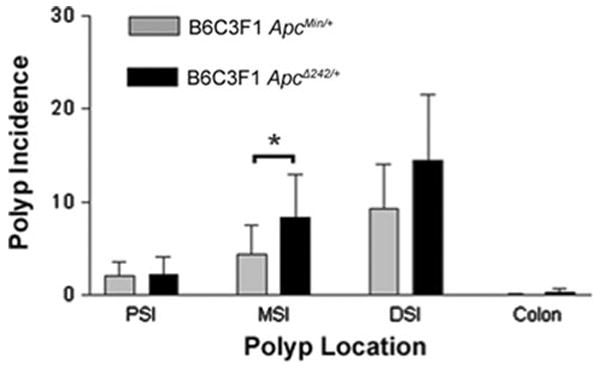
The C3H/HeJ background reduces polyp multiplicity in ApcMin/+ and ApcΔ242/+ mice. Average polyp numbers detected in the proximal, middle, and distal portions of the small intestine and colon of B6C3F1 ApcMin/+ mice and B6C3F1 ApcΔ242/+ mice at 120 days of age. *P < 0.05. n = 35 for B6C3F1 ApcMin/+ mice and n = 4 for B6C3F1 ApcΔ242/+ mice. Error bars represent standard deviation
Discussion
Numerous mouse models have been generated to study the role of the Apc gene in tumor initiation, growth, and progression in the intestinal tract. Many of these models carry Apc mutations that result in truncated proteins of varying sizes (Heyer et al. 1999; Taketo and Edelmann 2009). Comparison of polyp multiplicity, distribution, size, and stage between these Apc mutant mice can be used to dissect the functions of the various domains contained in Apc. ApcMin/+ mice, a common model of familial adenomatous polyposis (FAP), carry a point mutation in codon 850 that results in a premature stop codon and a truncated protein product (Fig. 2). The ApcΔ474/+ and ApcΔ580/+ mouse models harbor mutations further upstream than the ApcMin mutation, potentially elucidating the roles of the N-terminus of the Apc protein. ApcΔ580/+ mice, however, were generated on a mixed genetic background and were never made congenic on the B6 background (Kuraguchi et al. 2006). Different inbred backgrounds are known to harbor modifier genes that can alter the incidence, size, and stage of intestinal tumors (reviewed in Kwong and Dove 2009). Since ApcMin/+ mice are maintained on the B6 background, other mutant Apc alleles must also be present on the B6 background to provide a relevant comparison. ApcΔ474/+ mice are maintained on a B6 background and develop an average of 122 intestinal polyps; however, the truncated Apc protein expressed from the ApcΔ474 allele still carries a portion of the armadillo repeat domain (Fig. 2) (Sasai et al. 2000).
In this article we have described a new mouse model of intestinal tumorigenesis, ApcΔ242, which is maintained on the B6 background. Although the location and stage of tumors did not differ between B6 ApcΔ242/+ mice and B6 ApcMin/+ mice, B6 ApcΔ242/+ mice develop 70% more polyps than B6 ApcMin/+ mice. These polyps are significantly smaller in all areas of the small intestine. The ApcΔ242/+ and ApcMin/+ alleles can be used to identify some of the mechanisms by which different Apc mutations in FAP patients can result in different phenotypes.
The majority of APC mutations identified in patients fall within a region between codons 1286 and 1513, known as the mutation cluster region (MCR) (Aoki and Taketo 2007; Fearnhead et al. 2001; Miyoshi et al. 1992). MCR mutations result in C-terminal truncation proteins that are lacking the domains required for formation of the negative regulatory complex and degradation of β-catenin (Aoki and Taketo 2007). The presence of the MCR suggests a strong pressure selecting not only for loss of the C-terminus of Apc, but also retention of the N-terminus. Takacs et al. (2008) demonstrated that Apc proteins lacking the C-terminus upregulate signaling through the Wnt pathway in Drosophila melanogaster. Mutations in the MCR may, therefore, combine loss of the potent tumor suppressor function of Apc with the generation of an oncogenic truncated protein.
The disruption of the role of APC as a tumor suppressor in colorectal tumors has been extensively studied. The mechanism by which truncated APC may act as an oncogene, however, is still poorly understood. Truncated proteins resulting from MCR mutations retain the armadillo repeat and the oligomerization domains, both of which have potentially oncogenic functions. ApcMin/+ mice carry a mutation at codon 850 and produce a truncated Apc protein that also carries these two domains. The ApcΔ242 product, however, contains only the oligomerization domain (Fig. 2). ApcΔ242/+ mice develop significantly more polyps than ApcMin/+ mice and FAP patients with mutations similar to that of ApcΔ242 present with a severe form of the disease (Nugent et al. 1994). These data indicate that the ApcΔ242 mutation successfully mimics the human disease phenotype and suggest that loss of the armadillo repeats results in increased tumorigenesis. The molecular mechanism behind this increased tumorigenesis in both mice and humans remains unknown.
The oligomerization domain allows Apc proteins to interact with one another. Initial studies on the role of Apc as an oncogene focused on potential dominant negative effects of truncated Apc proteins. Studies have demonstrated that truncated Apc mutants can sequester full-length Apc proteins and this interaction could potentially block formation of the Apc–Axin–GSK3β negative regulatory complex (reviewed in Aoki and Taketo 2007). In addition to preventing formation of the regulatory complex, sequestration of full-length Apc results in improper formation of the mitotic spindle and errors in chromosomal segregation (Green et al. 2005). These data may explain the presence of Apc mutations at the earliest stages of adenoma development. The presence of the mutant Apc product results in chromosomal instability and increases the probability of the cell acquiring further genetic lesions (Fodde et al. 2001). The ApcΔ242 protein should still be capable of acting as a dominant negative due to the presence of the oligomerization domain. The observed increase in tumor multiplicity compared to ApcMin/+ mice could be the result of increased binding affinity between full-length Apc and truncated Apc mutants in which the armadillo repeats are absent.
The increased polyposis in ApcΔ242/+ mice compared to ApcMin/+, ApcΔ474, and ApcΔ580 mice more likely results from the loss of the armadillo repeat domain (Fig. 2). These repeats are involved in protein–protein interactions and there is conflicting evidence regarding the role of this domain in tumorigenesis (Barth et al. 2002). The human APC protein associates with the ends of microtubules during active cell migration, suggesting a role for APC in regulating migration (Mogensen et al. 2002). APC binds both the guanine nucleotide exchange factor ASEF and the GTPase effector IQGAP1, which in turn decrease cell adhesion and increase migration (Kawasaki et al. 2000; Watanabe et al. 2004). These protein–protein interactions occur via the armadillo repeat domain; truncated APC generated by MCR mutations is still able to activate ASEF, suggesting an oncogenic potential for the armadillo repeats (Kawasaki et al. 2000, 2003). In vivo data, however, demonstrate that B6 ApcMin/+ mice have decreased migration rates of intestinal cells along the crypt–villus axis compared to wild-type B6 mice (Javid et al. 2005). Other experiments have shown that the armadillo repeats are required for normal cell migration along the crypt–villus axis and indicate that the domain may have a tumor suppressive role (Kawasaki et al. 2003). Maintenance of the crypt–villus axis is essential for homeostasis in the intestinal tract. Cells of the gut are exposed to carcinogens present in the intestinal lumen and frequent turnover of intestinal epithelial cells serves as a protective mechanism. Dysregulation of cell migration along this axis can cause the accumulation of cells carrying genetic lesions and result in tumor initiation (Mahmoud et al. 1999). If the armadillo repeat domain of Apc is required for proper cell migration, loss of this domain in ApcΔ242/+ mice may result in increased tumorigenesis by disrupting cell turnover in intestinal crypts.
The specific roles of the oligomerization and armadillo repeat domains in intestinal tumorigenesis have not been fully dissected in an in vivo system. Here we demonstrate in a novel mouse model that complete loss of the armadillo repeats results in increased tumorigenesis in all parts of the small intestine. Our data demonstrate that ApcΔ242/+ mice are a model of severe FAP, developing significantly more tumors than ApcMin/+ mice while showing no increases in average tumor size or stage. These data suggest that the loss of the armadillo repeats region results in increased tumor initiation, implying an unknown tumor suppressive function of the armadillo repeats. Further comparative analyses between ApcMin/+ and ApcΔ242/+ mice can dissect the respective roles of the oligomerization domain and the armadillo repeats. Future work must focus on integrating the in vivo and in vitro data to better understand the function of the armadillo repeats.
Acknowledgments
We thank Dr. Revati Koratkar for providing the B6C3F1 ApcMin/+ data and Stephanie Nnadi and Jennifer Talarico for critical reading of the manuscript. RCC was supported by NIH training grant T32-CA09678. JJR was supported by NIH training grant T32-CA09683. Research was supported in part by grants from the NCI to AMB and LDS.
References
- Aoki K, Taketo MM. Adenomatous polyposis coli (APC): a multi-functional tumor suppressor gene. J Cell Sci. 2007;120:3327–3335. doi: 10.1242/jcs.03485. [DOI] [PubMed] [Google Scholar]
- Barth AI, Siemers KA, Nelson WJ. Dissecting interactions between EB1, microtubules and APC in cortical clusters at the plasma membrane. J Cell Sci. 2002;115:1583–1590. doi: 10.1242/jcs.115.8.1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burt RW, Jasperson KW. APC - Associated Polyposis Conditions. In: Pagon RA, Bird TC, Dolan CR, Stephens K, editors. GeneReviews [Internet] Seattle (WA): University of Washington, Seattle; 2008. 1993-1998 Dec 18 [updated 24 July 2008] [PubMed] [Google Scholar]
- Fearnhead NS, Britton MP, Bodmer WF. The ABC of APC. Hum Mol Genet. 2001;10:721–733. doi: 10.1093/hmg/10.7.721. [DOI] [PubMed] [Google Scholar]
- Fodde R, Edelmann W, Yang K, van Leeuwen C, Carlson C, et al. A targeted chain-termination mutation in the mouse Apc gene results in multiple intestinal tumors. Proc Natl Acad Sci USA. 1994;91:8969–8973. doi: 10.1073/pnas.91.19.8969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fodde R, Kuipers J, Rosenberg C, Smits R, Kielman M, et al. Mutations in the APC tumour suppressor gene cause chromosomal instability. Nat Cell Biol. 2001;3:433–438. doi: 10.1038/35070129. [DOI] [PubMed] [Google Scholar]
- Green RA, Wollman R, Kaplan KB. APC and EB1 function together in mitosis to regulate spindle dynamics and chromosome alignment. Mol Biol Cell. 2005;16:4609–4622. doi: 10.1091/mbc.E05-03-0259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groden J, Thliveris A, Samowitz W, Carlson M, Gelbert L, et al. Identification and characterization of the familial adenomatous polyposis coli gene. Cell. 1991;66:589–600. doi: 10.1016/0092-8674(81)90021-0. [DOI] [PubMed] [Google Scholar]
- Guillen-Ahlers H, Suckow MA, Castellino FJ, Ploplis VA. Fas/CD95 deficiency in ApcMin/+ mice increases intestinal tumor burden. PLoS One. 2010;5:e9070. doi: 10.1371/journal.pone.0009070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyer J, Yang K, Lipkin M, Edelmann W, Kucherlapati R. Mouse models for colorectal cancer. Oncogene. 1999;18:5325–5333. doi: 10.1038/sj.onc.1203036. [DOI] [PubMed] [Google Scholar]
- Javid SH, Moran AE, Carothers AM, Redston M, Bertagnolli MM. Modulation of tumor formation and intestinal cell migration by estrogens in the Apc(Min/+) mouse model of colorectal cancer. Carcinogenesis. 2005;26:587–595. doi: 10.1093/carcin/bgh346. [DOI] [PubMed] [Google Scholar]
- Kawasaki Y, Senda T, Ishidate T, Koyama R, Morishita T, et al. Asef, a link between the tumor suppressor APC and G-protein signaling. Science. 2000;289:1194–1197. doi: 10.1126/science.289.5482.1194. [DOI] [PubMed] [Google Scholar]
- Kawasaki Y, Sato R, Akiyama T. Mutated APC and Asef are involved in the migration of colorectal tumour cells. Nat Cell Biol. 2003;5:211–215. doi: 10.1038/ncb937. [DOI] [PubMed] [Google Scholar]
- Kinzler KW, Nilbert MC, Vogelstein B, Bryan TM, Levy DB, et al. Identification of a gene located at chromosome 5q21 that is mutated in colorectal cancers. Science. 1991;251:1366–1370. doi: 10.1126/science.1848370. [DOI] [PubMed] [Google Scholar]
- Koratkar R, Silverman KA, Pequignot E, Hauck WW, Buchberg AM, et al. Analysis of reciprocal congenic lines reveals the C3H/HeJ genome to be highly resistant to ApcMin intestinal tumorigenesis. Genomics. 2004;84:844–852. doi: 10.1016/j.ygeno.2004.07.010. [DOI] [PubMed] [Google Scholar]
- Korsisaari N, Kasman IM, Forrest WF, Pal N, Bai W, et al. Inhibition of VEGF-A prevents the angiogenic switch and results in increased survival of Apc +/min mice. Proc Natl Acad Sci USA. 2007;104:10625–10630. doi: 10.1073/pnas.0704213104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuraguchi M, Wang XP, Bronson RT, Rothenberg R, Ohene-Baah NY, et al. Adenomatous polyposis coli (APC) is required for normal development of skin and thymus. PLoS Genet. 2006;2:e146. doi: 10.1371/journal.pgen.0020146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong LN, Dove WF. APC and its modifiers in colon cancer. Adv Exp Med Biol. 2009;656:85–106. doi: 10.1007/978-1-4419-1145-2_8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald BT, Tamai K, He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell. 2009;17:9–26. doi: 10.1016/j.devcel.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmoud NN, Bilinski RT, Churchill MR, Edelmann W, Kucherlapati R, et al. Genotype-phenotype correlation in murine Apc mutation: differences in enterocyte migration and response to sulindac. Cancer Res. 1999;59:353–359. [PubMed] [Google Scholar]
- Miyaki M, Konishi M, Kikuchi-Yanoshita R, Enomoto M, Igari T, et al. Characteristics of somatic mutation of the adenomatous polyposis coli gene in colorectal tumors. Cancer Res. 1994;54:3011–3020. [PubMed] [Google Scholar]
- Miyoshi Y, Nagase H, Ando H, Horii A, Ichii S, et al. Somatic mutations of the APC gene in colorectal tumors: mutation cluster region in the APC gene. Hum Mol Genet. 1992;1:229–233. doi: 10.1093/hmg/1.4.229. [DOI] [PubMed] [Google Scholar]
- Mogensen MM, Tucker JB, Mackie JB, Prescott AR, Nathke IS. The adenomatous polyposis coli protein unambiguously localizes to microtubule plus ends and is involved in establishing parallel arrays of microtubule bundles in highly polarized epithelial cells. J Cell Biol. 2002;157:1041–1048. doi: 10.1083/jcb.200203001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser AR, Pitot HC, Dove WF. A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science. 1990;247:322–324. doi: 10.1126/science.2296722. [DOI] [PubMed] [Google Scholar]
- Nishisho I, Nakamura Y, Miyoshi Y, Miki Y, Ando H, et al. Mutations of chromosome 5q21 genes in FAP and colorectal cancer patients. Science. 1991;253:665–669. doi: 10.1126/science.1651563. [DOI] [PubMed] [Google Scholar]
- Nugent KP, Phillips RK, Hodgson SV, Cottrell S, Smith-Ravin J, et al. Phenotypic expression in familial adenomatous polyposis: partial prediction by mutation analysis. Gut. 1994;35:1622–1623. doi: 10.1136/gut.35.11.1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshima M, Oshima H, Kitagawa K, Kobayashi M, Itakura C, et al. Loss of Apc heterozygosity and abnormal tissue building in nascent intestinal polyps in mice carrying a truncated Apc gene. Proc Natl Acad Sci USA. 1995;92:4482–4486. doi: 10.1073/pnas.92.10.4482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell SM, Zilz N, Beazer-Barclay Y, Bryan TM, Hamilton SR, et al. APC mutations occur early during colorectal tumorigenesis. Nature. 1992;359:235–237. doi: 10.1038/359235a0. [DOI] [PubMed] [Google Scholar]
- Sasai H, Masaki M, Wakitani K. Suppression of polypogenesis in a new mouse strain with a truncated Apc(Delta474) by a novel COX-2 inhibitor, JTE-522. Carcinogenesis. 2000;21:953–958. doi: 10.1093/carcin/21.5.953. [DOI] [PubMed] [Google Scholar]
- Siracusa LD, Silverman KA, Koratkar R, Markova M, Buchberg AM. Genome-wide modifier screens: How the genetics of cancer penetrance may shape the future of prevention and treatment. In: Brenner C, Duggan D, editors. Oncogenomics: molecular approaches to cancer. Wiley; New York: 2004. pp. 255–290. [Google Scholar]
- Stryke D, Kawamoto M, Huang CC, Johns SJ, King LA, et al. BayGenomics: a resource of insertional mutations in mouse embryonic stem cells. Nucleic Acids Res. 2003;31:278–281. doi: 10.1093/nar/gkg064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takacs CM, Baird JR, Hughes EG, Kent SS, Benchabane H, Paik R, Ahmed Y. Dual positive and negative regulation of wingless signaling by adenomatous polyposis coli. Science. 2008;319:333–336. doi: 10.1126/science.1151232. [DOI] [PubMed] [Google Scholar]
- Taketo MM, Edelmann W. Mouse models of colon cancer. Gastroenterology. 2009;136:780–798. doi: 10.1053/j.gastro.2008.12.049. [DOI] [PubMed] [Google Scholar]
- Thompson DM, van der Weyden L, Biggs PJ, Chung YJ, Bradley A. Mouse models of cancer. In: Brenner C, Duggan D, editors. Oncogenomics: Molecular Approaches to Cancer. Wiley; New York: 2004. pp. 199–254. [Google Scholar]
- Uronis JM, Threadgill DW. Murine models of colorectal cancer. Mamm Genome. 2009;20:261–268. doi: 10.1007/s00335-009-9186-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe T, Wang S, Noritake J, Sato K, Fukata M, et al. Interaction with IQGAP1 links APC to Rac1, Cdc42, and actin filaments during cell polarization and migration. Dev Cell. 2004;7:871–883. doi: 10.1016/j.devcel.2004.10.017. [DOI] [PubMed] [Google Scholar]