

Abstract
Hypersecretion of cortisol occurs in numerous patients with major depression and normalizes with clinical recovery during the course of chronic antidepressant treatment. These clinical data suggest that investigation of the effects of antidepressant treatments on the regulation of the brain-pituitary-adrenal axis may assist in elucidating the therapeutic basis of antidepressant actions. In the present investigation, both swim stress and acute fluoxetine challenge increased release of corticosterone and progesterone to reflect an activation of the brain pituitary-adrenal axis. The effects of chronic antidepressant treatment (21 days) on corticosterone and progesterone secretion induced by these challenges were investigated. Chronic fluoxetine treatment (5 mg/kg/day) completely blocked the increased secretion of corticosterone and progesterone in response to the acute fluoxetine challenge. Chronic treatment with desipramine, imipramine or amytriptyline (15 mg/kg/day) also markedly attenuated fluoxetine-induced corticosterone and progesterone secretion. However, chronic treatment with the monoamine oxidase inhibitors, phenelzine (5 mg/kg) and tranylcypromine (5 mg/kg), did not affect this hormonal response to acute fluoxetine challenge. Plasma levels of fluoxetine after acute challenge were not significantly different for the various chronic antidepressant treatment conditions from the chronic saline controls; therefore, an increase in the metabolism of fluoxetine can not explain the antagonism of the fluoxetine-induced hormonal response after chronic antidepressant treatment. In contrast to the effects of selected antidepressants on acute fluoxetine-induced steroid release, chronic treatment with imipramine (20 mg/kg/day), fluoxetine (5 mg/kg/day) or phenelzine (5 mg/kg) did not significantly alter this swim stress-induced corticosterone or progesterone secretion. Because chronic fluoxetine and tricyclic antidepressant drugs blocked the acute action of fluoxetine to increase adrenal cortical secretion, but did not alter swim stress-induced secretion of these steroids, we propose that distinct neurochemical mechanisms control fluoxetine and swim stress-induced steroid release. We speculate that the substantial adaptive response to those chronic antidepressant treatments, which minimize the effect of acute fluoxetine challenge to increase in corticosterone and progesterone secretion, may be relevant to the therapeutic actions of these drugs.
Several measures of brain-pituitary-adrenal axis function are elevated in major depressive illness (Ettigi and Brown, 1977; Holsboer and Barden, 1996). For example, a substantial portion of depressed patients have chronically elevated cortisol (Gibbons, 1964; Sachar, 1975) and also exhibit a subsensitive negative feedback system, as reflected by attenuated inhibition of cortisol secretion in response to dexamethasone (Stokes et al., 1975; Carroll et al., 1981). Additionally, in depressed patients, cerebral spinal fluid levels of CRF are elevated (Banki et al., 1987; Nemeroff et al., 1984) and CRF mRNA in the paraventricular nucleus of depressed suicide victims is markedly higher than in control subjects (Raadsheer et al., 1995). Depressed patients also exhibit a supersensitive adrenocorticotropin response to systemically administered CRF (Holsboer et al., 1987; Schmider et al., 1995). Because stress results in activation of the hypothalamic-pituitary-adrenal axis (Ganong and Forsham, 1960), the well-documented disturbances in the control of corticoid function in depression suggest that depressed patients may be in a state of chronic stress (Rubin et al., 1987). In the course of chronic antidepressant drug treatment, normalization of adrenal cortical secretion precedes or coincides with clinical recovery from depression (Linkowski et al., 1987; Steiger et al., 1989; Souetre et al., 1989).
In addition to stress, the brain-pituitary-adrenal axis is induced by acute treatment with serotonin uptake inhibitors and other drugs that increase the synaptic availability of serotonin, such as 5-hydroxytryptophan (Fuller and Snoddy, 1990). The magnitude of increased corticosterone secretion in response to acute fluoxetine challenge is similar to that observed by various physiological stressors (Fuller and Snoddy, 1990). The acute corticosterone response to an antidepressant drug like fluoxetine seems paradoxical, because normalization of cortisol levels occurs with clinical recovery after chronic antidepressant therapy (Linkowski et al., 1987; Steiger et al., 1989; Souetre et al., 1989).
The acute neuropharmacological properties of antidepressant drugs are well characterized. However, the immediate actions of these drugs on the function of the central nervous system can not account directly for their clinical efficacy, because chronic antidepressant administration is required for therapeutic effectiveness (Katz et al., 1987; Klein and Davis, 1969). This requirement for chronic antidepressant administration for therapeutic effectiveness suggests that drug-induced neural adaptation is responsible for the therapeutic actions of this drug class (Heninger and Charney, 1987; Duman et al., 1994). A common adaptive response produced by chronic treatment with several classes of antidepressant drugs, including tricyclics, MAO inhibitors and the selective serotonin uptake inhibitor fluoxetine, is a reduction in brain of levels of mRNA for CRF in animals (Brady et al., 1991, 1992). Thus, neurochemical control of the brain-pituitary-adrenal axis presents a biological substrate to explore the mechanism by which antidepressant action affects the elevated activity of the brain pituitary-adrenal axis in depressive illness.
Although progesterone has been considered only as a female reproductive hormone, elevated levels of plasma progesterone accompany the increase in corticosterone after stress in male rats (Nequin et al., 1975; Schaeffer and Aron, 1987; Deis et al., 1989; Purdy et al., 1991) and male humans (Breier and Buchanan, 1992). Stress-induced progesterone secretion in male and female rats is derived from the adrenal gland, because the response is abolished after adrenalectomy (Deis et al., 1989; Nequin et al., 1975; Purdy et al., 1991). The physiological function of progesterone secreted from the adrenals in males and females is presently unclear. However, it is well established that systemically circulating progesterone can enter the brain and serve as a precursor for the synthesis of neurosteriods, which are potent allosteric moduators of γ-aminobutyric acid receptors (Majewska et al., 1986; Morrow et al., 1987; Purdy et al., 1991; Paul and Purdy, 1992).
The purpose of the present investigation was to assess whether chronic antidepressant treatment would induce adaptive responses in the brain, such that corticosterone and progesterone responses to acute challenge with fluoxetine and swim stress would be attenuated. The swim stress protocol used was the forced swim test, a behavioral screen for antidepressant drug activity (Porsolt et al., 1978).
Materials and Methods
Animals and treatments
Male Charles River Sprague-Dawley (Raleigh, NC) rats were housed three per cage, given continuous access to food (Purina rat chow) and water,- and were on a 12-hr light-dark cycle with lights off at 1900 hr. For chronic treatments, rats were injected i.p. once a day for 21 days with drugs dissolved in 0.9% saline. Drugs were prepared at 4 mg/ml to minimize peritoneal irritation. At the termination of the experiments, when rats weighed 300 to 380 g, they were sacrificed by decapitation between 1300 and 1500 hr for collection of trunk blood.
Chronic antidepressant treatment and hormonal responses to acute fluoxetine challenge
Rats were injected daily for 3 weeks with imipramine-HC1 (15 mg/kg), fluoxetine (5 mg/kg), desipramine-HC1 (15 mg/kg), amytriptyline (15 mg/kg), phenelzine (5 mg/kg) or tranylcypromine (7 mg/kg) before the hormonal response to a challenge dose of fluoxetine was assessed. The 5 mg/kg challenge dose of fluoxetine was chosen because preliminary studies demonstrated that this dose induced robust secretion of corticosterone and progesterone. Furthermore, previous work by Fuller and Snoddy (1990) had shown that this dose was slightly submaximal. Trunk blood was collected and stored on ice until centrifugation to allow collection of the serum. Previous work showed that peak levels of corticosterone are obtained 40 min after i.p. injection of fluoxetine and that the levels of the hormone remain high and constant from 40 to 60 min after injection (Fuller and Snoddy, 1990). Therefore, for the acute fluoxetine challenge, rats were injected with fluoxetine-HC1 (5 mg/kg), 20 to 24 hr after the final saline or drug injection in the 3-week drug treatment series, and sacrificed 40 min later.
Chronic antidepressant treatment and hormonal measurement in the forced swim test
Rats were injected daily i.p for 3 weeks with 0.9% saline, imipramine-HCl (20 mg/kg) or fluoxetine-HCl (5 mg/kg). The dose of 20 mg/kg imipramine considered the maximal dose of the drug for chronic administration and in previous work was demonstrated to antagonize swim stress-induced Fos (Duncan et al., 1996). Desipramine was not examined in this work with the swim test because imipramine is metabolized to desipramine and substantial levels of this demethylated metabolite are present in brain after administration of imipramine. After the chronic saline or antidepressant treatment, rats were processed in the forced swim test (Porsolt et al., 1978). For the conditioning swim on the first day of the test, the rats were placed in the swim tanks (40 × 19 cm Plexiglas cylinders) containing water (25°C) at a depth of 16 to 17 cm for 15 min. The final drug or saline injection was given after the conditioning swim. On the next day, for the test swim, the rats were placed in the tanks (25°C water) for 5 min and the duration of immobility measured. Rats were sacrificed 30, 60 or 120 min after the test swim, and trunk blood was collected and stored on ice until centrifugation.
Measurement of serum corticosterone and progesterone
Serum levels of corticosterone and progesterone were measured with radioimmunoassay kits (ICN Biomedicals, Inc. Costa Mesa, CA) according to the manufacturer’s instruction, with 125I-corticosterone and 125I-progesterone. Duplicate samples for each rat were assayed for each hormone. The approximate detection limits for the corticosterone and progesterone were 10 and 0.1 ng/ml, respectively. There was less than 1% cross-reactivity between corticosterone and progesterone for the respective antisera in the kits.
Measurement of serum fluoxetine
Serum fluoxetine and norfluoxetine metabolite were determined by a high-performance liquid chromatographic method. Fluoxetine and norfluoxetine were extracted from 100-μl serum samples with hexane/n-butylamine (9:1) after the addition of internal standard (imipramine) and pH adjustment with ammonium hydroxide. The organic phase was evaporated to dryness under a gentle stream of nitrogen at room temperature. After reconstitution with 75 μl of methanol, the sample was transferred to injection vials. Fluoxetine, norfluoxetine and internal standard were separated on a 3-μm (4.6 × 250 mm) Hypersil silica RP column (Keystone Scientific, Inc, Bellefonte, PA) and a mobile phase of acetonitrile/methanol/ammonium hydroxide (860:140:5) at 2.0 ml/min. Quantitation of each compound was performed with UV detection at 254 nm (Linear Instruments, Reno, NV) and Chrom Perfect chromatographic software (version 2.05, from Justice Innovations, Inc., Mountainview, CA). Standard curves were linear from 25 ng/ml, the lower limit of quantitation, up to 1000 ng/ml for both compounds. The within- and between-day coefficients of variation were <9%.
Statistics
Data were analyzed by two-way analysis of variance and post hoc comparisons were made with Tukey’s test.
Results
Effects of acute fluoxetine challenge on secretion of corticosterone and progesterone
In accord with previous results (Stark et al., 1985; Fuller and Snoddy 1990), administration of fluoxetine (5 mg/kg) induced a robust secretion of corticosterone (fig. 1) that was similar in magnitude to that induced by swim (compare with figs. 8–10). Additionally, just as with stress in male rats (Purdy et al., 1991), acute fluoxetine administration resulted in an increase in progesterone (fig. 1). To determine whether the increased secretion of these steroids involved activation of the brain-pituitary-adrenal axis, the synthetic glucocorticoid dexamethasone was administered 4 hr before fluoxetine injection. Dexamethasone is a potent synthetic glucocorticoid that suppresses adrenal cortical secretion by activating negative feedback mechanisms in the brain and pituitary gland (Meikle and Tyler, 1977). Pretreatment of rats with dexamethasone completely blocked fluoxetine-induced increases in corticosterone and progesterone secretion (fig. 1). These data indicate that fluoxetine induces the secretion of these steroids via an action on the brain and/or the pituitary gland.
Fig. 1.
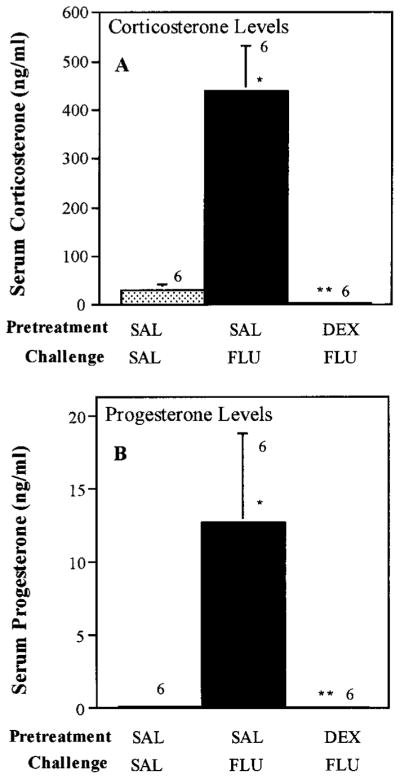
Acute fluoxetine-induced increase in corticosterone and progesterone secretion: Effect of dexamethasone. Rats were pretreated with saline (SAL) or dexamethasone (DEX, 0.5 mg/kg) 4 hr before challenge with saline or fluoxetine (FLU). Rats were sacrificed 40 min after the saline or fluoxetine challenge. Data are means ± S.E.M. with n = 6 for each group. *P < .01 compared with SAL; **P < .01 compared with SAL-FLU.
Fig. 8.

Lack of effect of chronic fluoxetine treatment on swim-induced corticosterone or progesterone. Rats were injected daily with fluoxetine (5 mg/kg) for 21 days before processing in the forced swim test as described under “Materials and Methods.” After the test swim (20–24 hr after the final drug or saline injection), the rats were transferred to their home cages and sacrificed by decapitation at the indicated times after the swim for collection of trunk blood. Rats that received chronic injections of saline or fluoxetine and were not swum were sacrificed 20 to 24 hr after the final injection. The number of rats for each time and treatment are indicated above the bars. Data are expressed as the mean ± S.E.M. for each group.
Fig. 10.
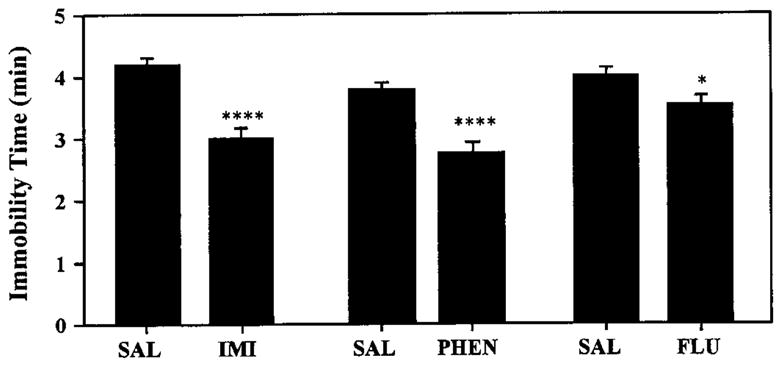
Immobility time for rats processed in the forced swim test after chronic antidepressant treatment. Rats were injected with saline (SAL), imipramine (IMI, 20 mg/kg) or fluoxetine (FLU, 5 mg/kg) and immobility time scored in the test swim (see “Materials and Methods”). ****P < .001,*P < .05 compared with saline.
Effects of chronic and subchronic treatment with fluoxetine on corticosterone and progesterone secretion in response to acute fluoxetine challenge
To evaluate the effect of chronic fluoxetine on the fluoxetine-induced increase in steroids, rats were challenged with 5 mg/kg fluoxetine after chronic administration of 0.9% saline or fluoxetine (5 mg/kg/day for 21 days). The fluoxetine challenge dose was given 20 to 24 hr after the final injection in the chronic fluoxetine treatment series. Chronic treatment with fluoxetine completely blocked the effects of the acute fluoxetine challenge on both corticosterone and progesterone secretion (fig. 2). In contrast to the effects of chronic fluoxetine treatment, rats injected with fluoxetine for 1 day, and challenged with 5 mg/kg fluoxetine the next day, did not exhibit diminished hormonal responses to the fluoxetine challenge (fig. 3).
Fig. 2.

Effect of chronic fluoxetine treatment on corticosterone and progesterone secretion in response to acute fluoxetine challenge. Rats were injected daily for 21 days with 0.9% saline or fluoxetine (5 mg/kg). Twenty-four hours after the final injection, a challenge dose of fluoxetine (5 mg/kg) or saline was administered and rats were sacrificed by decapitation 40 min later. Abbreviations: SAL, saline; FLU, fluoxetine. The number of rats in each group are given above the bars. Data are presented as the mean ± S.E.M. for each group. *P < .01 compared with SAL-SAL; ** P < .01 compared with SAL-FLU.
Fig. 3.

Lack of effect of 1-day fluoxetine treatment on corticosterone and progesterone secretion in response to fluoxetine challenge. Rats were treated with fluoxetine (FLU, 5 mg/kg) or saline (SAL) on day 1 and challenged with saline or fluoxetine (5 mg/kg) on day 2, as indicated. SAL-FLU levels of corticosterone and progesterone after SAL-FLU were not significantly different from levels in the FLU-FLU-treated rats.
Effects of chronic treatment with other antidepressant drugs on acute fluoxetine-induced corticosterone and progesterone secretion
To determine whether chronic treatment with antidepressants having pharmacological properties different from fluoxetine would antagonize the corticosterone and progesterone response to acute fluoxetine challenge, rats were treated chronically with various other clinically used antidepressants before an acute fluoxetine challenge. Antidepressants chosen for study were imipramine, desipramine, amytriptyline, tranylcypromine and phenelzine. Imipramine and amytriptyline inhibit the uptake of both serotonin and norepinephrine (Wood et al., 1986), whereas desipramine has predominant actions on norepinephrine uptake with little effect on serotonin uptake (Randrup and Braestrup, 1977). Desipramine also was chosen for study to determine whether an antidepressant drug, which does not directly affect serotonin function, would induce an adaptive response in serotonergic control of the brain-pituitary-adrenal axis, as assessed by the acute fluoxetine challenge. Chronic administration of imipramine, desipramine (fig. 4) or amytriptyline (fig. 5) markedly attenuated corticosterone and progesterone secretion induced by acute fluoxetine challenge. In contrast to the blockade by the tricyclic antidepressants on fluoxetine-induced steroid secretion, chronic treatment with the MAO inhibitors, tranylcypromine and phenelzine, did not affect the hormonal response to acute fluoxetine challenge (fig. 5).
Fig. 4.

Effect of chronic imipramine and desipramine treatment on fluoxetine-induced secretion of corticosterone and progesterone. Rats were injected daily for 21 days with 0.9% saline, imipramine or desipramine (15 mg/kg for each). Twenty-four hours after the final injection of desipramine and imipramine, a challenge dose of fluoxetine (5 mg/kg) was given and rats were sacrificed by decapitation 40 min later. The number of rats in each group are given above the bars. Data are presented as the mean ± S.E.M. for each group. Abbreviations: SAL, saline; IMI, imipramine; DMI, imipramine. *P < .01 compared with SAL-SAL; **P < .01 compared with SAL-FLU.
Fig. 5.

Effect of chronic amytriptyline (AMY), phenelzine (PHEN) and tranylcypromine (TRAN) treatment on fluoxetine-induced secretion of corticosterone and progesterone. Rats were injected daily for 21 days with either 0.9% saline, AMY (15 mg/kg), PHEN (5 mg/kg) or TRAN (5 mg/kg). Twenty-four hours after the final injection of these chronic exposures, a challenge dose of fluoxetine (FLU; 5 mg/kg) was given and rats were sacrificed by decapitation 40 min later. The number of rats in each group are given above the bars. Data are presented as the mean ± S.E.M. for each group. **P < .05 compared to SAL-FLU.
Effect of chronic antidepressant treatments on serum levels of fluoxetine
Because the reduced corticosterone and progesterone responses to acute fluoxetine challenge could have been caused by an altered metabolism of fluoxetine during the course of the chronic treatment of the antidepressants, plasma levels of fluoxetine were measured after chronic antidepressant treatments that antagonized the steroid response to fluoxetine. Serum fluoxetine levels after acute challenge were not significantly different in rats treated chronically with saline, fluoxetine, imipramine or desipramine (fig. 6). Serum norfluoxetine levels were significantly lower in rats treated chronically with desipramine and imipramine, but not fluoxetine (fig. 6). However, because fluoxetine levels were not reduced in the rats treated chronically with imipramine or desipramine, altered fluoxetine metabolism can not explain the attenuated hormonal responses to acute fluoxetine observed after chronic administration of these tricyclic antidepressants.
Fig. 6.

Lack of effect of chronic treatment with desipramine (DMI), imipramine (IMP) or fluoxetine (FLU) on serum levels of fluoxetine after acute challenge. Rats were treated as described in figures 2 and 4. Fluoxetine and norfluoxetine levels were measured by HPLC with UV detection in serum obtained 40 min after acute challenge with fluoxetine. The number of rats in each group are given above the bars. Data are presented as the mean ± S.E.M. for each group. The norfluoxetine levels were reduced significantly in the chronic DMI- and IMP-treated rats, but not in the FLU-treated rats. *P < .05 compared with SAL-FLU.
Effects of chronic treatment with fluoxetine, imipramine and phenelzine on swim stress-induced immobility and corticosterone and progesterone secretion
To determine whether the marked reduction in fluoxetine-induced corticosterone and progesterone secretion after chronic antidepressant treatment was caused by generalized desensitization of the brain-pituitary-adrenal axis, the effects of chronic treatment with imipramine and fluoxetine on swim stress-induced secretion of these hormones was examined. No significant difference in basal or swim-induced corticosterone or progesterone levels was observed between rats treated chronically (3 weeks) with imipramine (fig. 7), fluoxetine (fig. 8) or phenelzine (fig. 9) and those treated with saline.
Fig. 7.

Lack of effect of chronic imipramine treatment on swim-induced corticosterone or progesterone. Rats were injected daily with imipramine (20 mg/kg) for 21 days before processing in the forced swim test as described under “Materials and Methods.” After the test swim (20–24 hr after the final drug or saline injection), the rats were transferred to their home cages and sacrificed by decapitation at the indicated times after the swim for collection of trunk blood. Rats that received chronic injections of saline or imipramine and were not swum were sacrificed 20 to 24 hr after the final injection. The number of rats for each time and treatment are indicated above the bars. Data are expressed as the mean ± S.E.M. for each group.
Fig. 9.

Lack of effect of chronic phenelzine treatment on swim-induced corticosterone or progesterone. Rats were injected daily with phenelzine (PHEN, 5 mg/kg) for 21 days before processing in the forced swim test as described under “Materials and Methods.” After the test swim (20–24 hr after the final drug or saline injection), the rats were transferred to their home cages and sacrificed by decapitation at the indicated times after the swim for collection of trunk blood. Rats that received chronic injections of saline or phenelzine, and were not swum, were killed 20 to 24 hr after the final injection. The number of rats for each time and treatment are indicated above the bars. Data are expressed as the mean ± S.E.M. for each group.
Even though no effect of chronic antidepressant treatment was found on swim-induced steroid secretion, all three antidepressant drugs reduce the duration of immobility in the swim test (fig. 10; Porsolt et al., 1978). As reported previously (Detke et al., 1995), behavioral responses to fluoxetine were qualitatively distinct from the behavioral response obtained with imipramine and phenelzine. After treatment with imipramine and phenelzine, rats jumped from the bottom of the swim tanks and attempted to vigorously climb the walls of the tanks for much of the nonimmobile time. In contrast, such behavior was not exhibited by the fluoxetine-treated rats, which exhibited less vigorous “escape-directed” behavior, but instead would swim around the tank without jumping or climbing during the nonimmobile time. Thus, although a behavioral consequence could be elicited by the chronic administration of these antidepressants, their chronic treatment failed to affect the secretion of corticosterone or progesterone in response to swim stress.
Discussion
The present studies demonstrated that acute administration of fluoxetine, as well as swim stress, induced a robust increase in corticosterone and progesterone secretion. Because depressed patients can have chronically elevated cortisol levels (Gibbons, 1964; Sachar, 1975), it seems paradoxical that an acute response to an antidepressant (i.e., induction of corticosterone secretion by fluoxetine) is similar to a symptom manifested in some depressed patients. However, the recognized requirement for chronic antidepressant administration to achieve a therapeutic response in depressed patients, which includes a reduction in their hypersteroid secretion, demonstrates that acute pharmacological properties of fluoxetine do not account for the clinical efficacy of this drug class. One hypothesis tested in the present work was that chronic treatment with antidepressants would induce adaptive changes leading to an attenuation of the acute fluoxetine and stress-induced adrenal cortical activation. Our goal was to characterize a model system that could be used to explore antidepressant-induced neurochemical adaptation relevant to neural mechanism(s) by which antidepressants reduce the hypersecretion of cortisol in depressive illness.
Acute effects of fluoxetine and swim stress on corticosterone and progesterone secretion
Other investigators previously have observed that swim stress can increase progesterone as well as corticosterone secretion (Purdy et al., 1991). Although acute fluoxetine has been reported to increase corticosterone secretion (Stark et al., 1985; Fuller and Snoddy, 1990), our data are the first to demonstrate that acute fluoxetine administration also induces secretion of progesterone. Increased progesterone secretion during the menstrual cycle and administration of synthetic progesterone in oral contraceptives have been associated with depressive illness in some women (Backstrom, 1995; Crammer, 1986; Wagner and Berenson, 1994). However, many women do not experience depression while taking progestins in oral contraceptives, and anecdotal evidence suggests that the high levels of progesterone that exist during pregnancy are associated with a state of well-being. Thus, additional work is required to determine the psychophysiological consequences of alterations in progesterone levels in males and females. Little is known about the functional consequences of increased progesterone secretion in males, but plasma-derived progesterone can serve as a precursor in the brain for the synthesis of potent neuroactive steroids which can affect γ-aminobutyric acid-mediated neurotransmission (Majewska et al., 1986; Morrow et al., 1987; Purdy et al., 1991; Paul and Purdy, 1992). Such data indicate that adrenal-derived progesterone in males could be neurochemically relevant.
Possible mechanisms of acute fluoxetine-induced corticosterone and progesterone secretion
Inhibition of the serotonin transporter by fluoxetine results in an increased extracellular fluid level of serotonin (Fuller and Snoddy, 1990). Therefore, the most probable mechanism by which acute fluoxetine induces activation of the brain-pituitary-adrenal axis would be by increasing the synaptic concentration of serotonin, with consequent increased activation of a serotonin receptors. Nonetheless, the specific serotonergic receptor mechanism involved in the neuroendocrine activation by fluoxetine is unknown. Both 5HT-2 and 5HT-1A receptor agonists have been shown to increase corticosterone levels (Urban et al., 1986; Fuller et al., 1986; Koenig et al., 1987; Fuller and Snoddy, 1990) and chronic fluoxetine treatment reduces corticosterone secretion induced by the 5HT-1A agonists (Li et al., 1993,1994). However, 5-HT-1A and 5HT-2 receptor antagonists do not attenuate the acute fluoxetine-induced increase in corticosterone levels (Fuller and Snoddy, 1990). Furthermore, interactions between the two receptor subtypes can not explain the effects of fluoxetine on corticosterone secretion, because coadministration of 5HT-1A and 5HT-2 receptor antagonists also did not block this fluoxetine-induced endocrine response (Fuller and Snoddy, 1990). Consequently, if enhancement of synaptic availability of endogenous serotonin by the fluoxetine is responsible for the adrenal cortical activation, a serotonin receptor subtype other than 5HT-1A or 5HT-2 may be involved. Although one potential candidate is the recently cloned 5HT-7 receptor, which is enriched in the hypothalamus (To et al., 1995; Sleight et al., 1995), no direct information currently documents specific 5HT receptor subtypes in the control of fluoxetine-induced steroid release.
Effect of chronic antidepressant administration on fluoxetine-induced steroid secretion
Chronic administration of all antidepressant drug types which inhibit uptake of serotonin, including fluoxetine (Stark et al.; 1985), imipramine and amytriptyline (Randrup and Braestrup, 1977; Wood et al., 1986), blocked the increase in corticosterone and progesterone induced by the acute fluoxetine challenge, without affecting base-line levels of these steroids. Likewise, chronic administration of desipramine, which potently inhibits norepinephrine uptake with minimal effect on serotonin uptake (Randrup and Braestrup, 1977), also antagonized fluoxetine-induced steroid secretion. These findings suggest that chronic inhibition of either serotonin uptake or noradrenergic uptake results in adaptive changes that markedly attenuate the ability of fluoxetine to induce secretion of corticosterone and progesterone. Unlike the effects of chronic treatment with fluoxetine and tricyclic antidepressant drugs, chronic treatment with the MAO inhibitors, phenelzine and tranylcypromine, did not result in a significant attenuation of the corticosterone and progesterone responses to the acute challenge with fluoxetine. Thus, the attenuation of fluoxetine-induced steroid secretion is not a general feature of all antidepressant drugs.
Possible adaptive mechanisms responsible for the antagonism of the steroid release by chronic antidepressant treatments
After chronic fluoxetine treatment, in vivo microdialysis studies have shown that extracellular fluid levels of serotonin are markedly elevated after the last chronic fluoxetine treatment (Rutter et al., 1994). The increased extracellular level of serotonin induced by fluoxetine after chronic treatment was even greater than after an acute fluoxetine challenge (Rutter et al., 1994). Thus, a reduced effectiveness of fluoxetine to release serotonin after chronic administration does not appear to be responsible for the observed blockade of the steroid release after acute fluoxetine administration. Additionally, serum levels of fluoxetine after acute challenge were similar in groups treated chronically with saline or with the antidepressants that blocked the acute steroid response to fluoxetine challenge. Therefore, an increased metabolism of fluoxetine can not explain the antagonism of fluoxetine-induced steroid release by chronic treatment of selected antidepressant drugs.
Chronic treatment with antidepressant drugs which block serotonin or norepinephrine uptake sites must result in a substantial neurochemical adaptation in neural circuits which regulate fluoxetine-induced adrenal hormone secretion, as both types of drugs antagonized this response. Although chronic treatment with fluoxetine has been shown to reduce corticosterone secretion induced by 5HT-1A agonists, (Li et al., 1993), chronic treatment with desipramine, which also reduces the acute fluoxetine-induced increases in steroid secretion, does not modify 5HT-1A agonist-induced corticosterone secretion (Li et al., 1994). These data, considered with the data of Fuller and Snody (1990) discussed above, suggest that the antagonism of fluoxetine-induced adrenal hormone secretion after chronic antidepressant treatment can not be explained by a reduction in the sensitivity of 5HT-1A receptors.
Chronic desipramine treatment, which antagonizes fluoxetine-induced release of steroids, causes down-regulation of beta adrenergic receptor number and function (see Baker and Greenshaw, 1989). Although chronic fluoxetine treatment also antagonizes steroid release by acute fluoxetine administration, chronic fluoxetine does not cause a down-regulation of beta adrenergic receptors (Johnson, 1991). As observed for the tricyclic antidepressant drugs (Baker and Greenshaw, 1989), chronic administration of MAO inhibitors also induces an adaptive down-regulation of beta adrenergic receptors; however, this class of antidepressants does not have an effect on the acute fluoxetine-induced release of steroids. Collectively, these latter data suggest that antidepressant-induced adaptive changes in noradrenergic receptor function can not explain the present finding that selected antidepressant drugs block acute fluoxetine-induced release of steroids.
Chronic administration of fluoxetine, tricyclic antidepressants and MAO inhibitors has been shown to reduce the expression of mRNA for CRF in the paraventricular nucleus of the hypothalamus (Brady et al., 1991, 1992). However, this action is apparently not sufficient to explain the present results, because MAO inhibitors are ineffective in reducing fluoxetine-induced hormonal secretion. Chronic fluoxetine administration also has been shown to down-regulate the 5-HT-7 receptor subtype in hypothalamic membrane preparations after chronic fluoxetine treatment (Sleight et al., 1995); however, it is not known whether chronic desipramine affects this 5-HT receptor subtype. Thus, it can be concluded that the adaptive mechanism(s) by which chronic antidepressant treatment blocks the acute action of fluoxetine to stimulate corticosterone and progesterone secretion has yet to be identified.
Chronic antidepressant treatments on forced swim-induced immobility and corticosterone and progesterone secretion
The final aspect of our investigation was to determine whether chronic antidepressant treatment would antagonize the release of corticosterone and progesterone induced by swim stress (Purdy et al., 1991). This particular stressor was chosen because the protocol of forced swim is used to screen for antidepressant action (Porsolt et al., 1978). As expected from previous work (Duncan et al., 1985, 1996), chronic administration of the antidepressant drugs fluoxetine, imipramine and phenelzine resulted in a significant reduction in the immobility in the forced swim test paradigm. Even though chronic administration of the antidepressants produced a behavioral change induced by forced swim, swim stress-induced secretion of these steroids was not affected by such chronic antidepressant exposure. The inability of chronic treatment with fluoxetine and imipramine to modify adrenal cortical secretion in response to swim stress suggests that a generalized down-regulation of the brain-pituitary-adrenal axis was not induced by the antidepressant drug treatments.
In contrast to the present data showing no effect of chronic antidepressant treatment on swim-induced activation of the brain-pituitary-adrenal axis, Reul et al. (1993) found that chronic administration of amytriptyline reduced the corticosterone response induced by placing rats in a novel environment. Differences in the nature of these stressors and the possible activation of differing neural pathways by the stressors could explain the ability of chronic treatment to block the stress response induced by the novel environment but not affect the increase in corticosterone induced by the swim. On the other hand, the magnitude of the stress response could be a consideration, because swim stress could be expected to be more stressful than novelty stress. In this regard, swim stress typically increased corticosterone levels to 300 to 400 ng/ml, whereas in the study of Reul et al.(1993), the novelty stress condition increased corticosterone to about 250 ng/ml. Thus, one interpretation could be that chronic antidepressant treatment could not overcome the hormonal response of the more severe swim stress compared with the stress of novel environment. However, contrary to this interpretation, acute fluoxetine treatment increased steroids to a level as great as that observed with swim stress, yet this increase by the acute fluoxetine challenge was blocked completely by the chronic antidepressant treatments.
Therefore, the differential effects observed for the antidepressant treatments on swim stress-induced and fluoxetine-induced alterations in corticosterone and progesterone secretion may involve differing neural mechanisms associated with regulation of adrenal steroid secretion. In this regard, at least two distinct pathways controlling CRF release have been proposed previously, one originating from brainstem structures and the other involving projections from limbic regions of the forebrain (Herman and Cullinan, 1997; Sawchenko, 1990). Whether altered activity in these distinct pathways contributes to the differential effects of chronic antidepressant treatment on fluoxetine- and stress-induced adrenal steroid secretion deserves investigation.
Acknowledgments
The authors appreciate the supply of fluoxetine from Margaret Niedenthal at Lilly Laboratories, assistance in typing the manuscript by Doris Lee and the excellent technical assistance of Edna Titus.
ABBREVIATIONS
- CRF
corticotrophin releasing factor
- 5HT
serotonin
- MAO
monoamine oxidase
Footnotes
Supported by MH-39144, MH-33127, AA-00214 and HD-03110.
References
- Backstrom T. Symptoms related to the menopause and sex steroid treatments. Ciba Found Symp. 1995;191:171–180. doi: 10.1002/9780470514757.ch10. [DOI] [PubMed] [Google Scholar]
- Baker GB, Greenshaw AJ. Effects of long-term administration of antidepressants and neuroleptics on receptors in the central nervous system. Cell Mol Neurobiol. 1989;9:1–44. doi: 10.1007/BF00711441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banki CM, Bissette G, Arato M, O’Connor L, Nemeroff CB. CSF corticotropin-releasing factor-like immunoreactivity in depression and schizophrenia. Am J Psychiatry. 1987;144:873–877. doi: 10.1176/ajp.144.7.873. [DOI] [PubMed] [Google Scholar]
- Brady LS, Whitfield HJ, Jr, Fox RJ, Gold PW, Herkenham M. Long-term antidepressant administration alters corticotropin-releasing hormone, tyrosine hydroxylase, and mineralocorticoid receptor gene expression in rat brain. J Clin Invest. 1991;87:831–837. doi: 10.1172/JCI115086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady LS, Gold PW, Herkenham M, Lynn AB, Whitfield HJ., Jr The antidepressants fluoxetine, idazoxan and phenelzine alter corticotropin-releasing hormone and tyrosine hydroxylase mRNA levels in rat brain: therapeutic implications. Brain Res. 1992;572:117–125. doi: 10.1016/0006-8993(92)90459-m. [DOI] [PubMed] [Google Scholar]
- Breier A, Buchanan RW. The effects of metabolic stress on plasma progesterone in healthy volunteers and schizophrenic patients. Life Sci. 1992;51:1527–1534. doi: 10.1016/0024-3205(92)90563-5. [DOI] [PubMed] [Google Scholar]
- Carroll BJ, Feinberg M, Greden JF, Tarika J, Albala AA, Haskett RF, James NM, Kronfol Z, Lohr N, Steiner M, de Vigne JP, Young E. A specific laboratory test for the diagnosis of melancholia: Standardization, validation, and clinical utility. Arch Gen Psychiatry. 1981;38:15–22. doi: 10.1001/archpsyc.1981.01780260017001. [DOI] [PubMed] [Google Scholar]
- Crammer JL. Premenstrual depression, cortisol and estradiol treatment. Psychol Med. 1986;16:451–455. doi: 10.1017/s0033291700009284. [DOI] [PubMed] [Google Scholar]
- Deis RP, Leguizamon E, Jahn GA. Feedback regulation by progesterone of stress-induced prolactin release in rats. J Endocrinol. 1989;120:37–43. doi: 10.1677/joe.0.1200037. [DOI] [PubMed] [Google Scholar]
- Detke MJ, Rickels M, Lucki I. Active behaviors in the rat forced swimming test differentially produced by serotonergic and noradrenergic antidepressants. Psychopharmacology. 1995;121:66–72. doi: 10.1007/BF02245592. [DOI] [PubMed] [Google Scholar]
- Duman RS, Heninger GR, Nestler EJ. Adaptations of receptor-coupled signal transduction pathways underlying stress- and drug-induced neural plasticity. J Nerv Ment Dis. 1994;182:692–700. doi: 10.1097/00005053-199412000-00003. [DOI] [PubMed] [Google Scholar]
- Duncan GE, Knapp DJ, Johnson KB, Breese GR. Functional Classification of antidepressants based upon antagonism of swim stress-induced Fos-like immunoreactivity. J Pharmacol Exp Ther. 1996;277:1076–1089. [PubMed] [Google Scholar]
- Duncan GE, Paul IA, Harden JK, Mueller RA, Stumpf W, Breese GR. Rapid down regulation of β-adrenergic receptors by combining antidepressant drugs with forced swim: A model of antidepressant-induced neural adaptation. J Pharmacol Exp Ther. 1985;234:402–408. [PubMed] [Google Scholar]
- Ettigi PG, Brown GM. Psychoneuroendocrinology of affective disorders: An overview. Am J Psychiatry. 1977;134:493–501. doi: 10.1176/ajp.134.5.493. [DOI] [PubMed] [Google Scholar]
- Fuller RW, Snoddy HD. Serotonin receptor subtypes involved in the elevation of serum corticosterone concentration in rats by direct- and indirect-aging serotonin agonists. Neuroendocrinology. 1990;52:206–211. doi: 10.1159/000125586. [DOI] [PubMed] [Google Scholar]
- Fuller RW, Snoddy HD, Molloy BB. Central serotonin agonist actions of LY 165163, 1-(m-trifluoromethylpheny)-4(p-aminophenylethyl)-piperazine, in rats. J Pharmacol Exp Ther. 1986;239:454–459. [PubMed] [Google Scholar]
- Ganong WF, Forsham PH. Adenohypophysis and adrenal cortex. Annu Rev Physiol. 1960;22:579–614. doi: 10.1146/annurev.ph.22.030160.003051. [DOI] [PubMed] [Google Scholar]
- Gibbons JL. Cortisol secretion rate in depressive illness. Arch Gen Psychiatry. 1964;10:572–575. doi: 10.1001/archpsyc.1964.01720240026004. [DOI] [PubMed] [Google Scholar]
- Heninger GR, Charney DS. Mechanisms of action of antidepressant treatments: implications for the etiology and treatment of depressive disorders. In: Meltzer HY, editor. Psychopharmacology: The Third Generation of Progress. Raven; New York: 1987. pp. 535–544. [Google Scholar]
- Herman JP, Cullinan WE. Neurocircuitry of stress: Central control of the hypothalamo-pituitary-adrenocortical axis. Trends Neurosci. 1997;20:78–84. doi: 10.1016/s0166-2236(96)10069-2. [DOI] [PubMed] [Google Scholar]
- Holsboer F, Barden N. Antidepressants and hypothalamic-pituitary-adrenocortical regulation. Endocr Rev. 1996;17:187–205. doi: 10.1210/edrv-17-2-187. [DOI] [PubMed] [Google Scholar]
- Holsboer F, von Bardeleben U, Wiedemann K, Muller OA, Stalla GK. Serial assessment of corticotropin-releasing hormone response after dexamethasone in depression-Implications for pathophysiology of DST non-suppression. Biol Psychiatry. 1987;22:228–234. doi: 10.1016/0006-3223(87)90237-x. [DOI] [PubMed] [Google Scholar]
- Johnson AM. The comparative pharmacological properties of selective serotonin re-uptake inhibitors in animals. In: Feighner JP, Boyer WF, editors. Perspectives in Psychiatry. vol 1: Selective Serotonin Re-uptake Inhibitors. John Wiley and Sons; New York: 1991. pp. 37–70. [Google Scholar]
- Katz MM, Koslow SH, Maas J, Frazer A, Secunda S, Bowden CL, Casper RC, Croughan J, Kocsis J, Redmond E. The timing, specificity, and clinical prediction of tricyclic drug effects in depression. Psychol Med. 1987;17:297–309. doi: 10.1017/s0033291700024831. [DOI] [PubMed] [Google Scholar]
- Klein DF, Davis JM. Diagnosis and Drug Treatment in Psychiatric Disorders. Williams & Wilkins Co; Baltimore: 1969. [Google Scholar]
- Koenig JI, Gudelsky GA, Meltzer HY. Stimulation of corticosterone and β-endorphin secretion in the rat by selective 5-HT receptor subtype activation. Eur J Pharmacol. 1987;45:305–310. doi: 10.1016/0014-2999(87)90175-0. [DOI] [PubMed] [Google Scholar]
- Li Q, Levy AD, Cabrere TM, Brownfield MS, Battaglia G, Van de Kar LD. Long-term fluoxetine, but not desipramine, inhibits the ACTH and oxytocin responses to the 5-HT1A agonist, 8-OH-DPAT, in male rats. Brain Res. 1993;630:148–156. doi: 10.1016/0006-8993(93)90652-4. [DOI] [PubMed] [Google Scholar]
- Li Q, Brownfield MS, Levy AD, Battaglia G, Cabrera TM, Van de Kar LD. Attenuation of hormone responses to the 5-HT1A agonist isapirone by long-term treatment with fluoxetine, but not desipramine, in male rats. Biol Psychiatry. 1994;36:300–308. doi: 10.1016/0006-3223(94)90627-0. [DOI] [PubMed] [Google Scholar]
- Linkowski P, Mendlewicz J, Kerkhofs M, Leclercq R, Goldstein J, Brasseur M, Copinschi G, Van Cauter E. 24-hour profiles of adrenocorticotropin, cortisol, and growth hormone in major depressive illness: Effect of antidepressant treatment. J Clin Endocrinol Metab. 1987;65:141–152. doi: 10.1210/jcem-65-1-141. [DOI] [PubMed] [Google Scholar]
- Majewska MD, Harrison NL, Schwartz RD, Barker JL, Paul SM. Steroid hormone metabolites and barbiturate-like modulators of the GABA receptor. Science. 1986;232:1004–1007. doi: 10.1126/science.2422758. [DOI] [PubMed] [Google Scholar]
- Meikle AW, Tyler FH. Potency and duration of action of glucocorticoids. Am J Med. 1977;63:200–207. doi: 10.1016/0002-9343(77)90233-9. [DOI] [PubMed] [Google Scholar]
- Morrow AL, Suzdak PD, Paul SM. Steroid hormone metabolites potentate GABA receptor-mediated chloride ion flux with nanomolar potency. Eur J Pharmacol. 1987;142:483–485. doi: 10.1016/0014-2999(87)90094-x. [DOI] [PubMed] [Google Scholar]
- Nemeroff CB, Widerlow E, Bissette G, Walleus H, Karlsson I, Eklund K, Klits CD, Loosen PT, Vale W. Elevated concentrations of CSF corticotropin-releasing actor-like immunoreactivity in depressed patients. Science. 1984;226:1342–1344. doi: 10.1126/science.6334362. [DOI] [PubMed] [Google Scholar]
- Nequin LG, Alvarez JA, Campbell CS. Alterations in steroid and gonadotropin release resulting from surgical stress during the morning of proestrus in 5-day cyclic rats. Endocrinology. 1975;97:718–724. doi: 10.1210/endo-97-3-718. [DOI] [PubMed] [Google Scholar]
- Paul SM, Purdy RH. Neuroactive steroids. FASEB J. 1992;6:2311–2322. [PubMed] [Google Scholar]
- Porsolt RD, Anton A, Blavet N, Jalfre M. Behavioral despair in rats: A new model sensitive to antidepressant treatments. Eur J Pharmacol. 1978;47:379–391. doi: 10.1016/0014-2999(78)90118-8. [DOI] [PubMed] [Google Scholar]
- Purdy RH, Morrow AL, Moore PH, Jr, Paul SM. Stress-induced elevations of γ-aminobutyric acid type A receptor-active steroids in the rat brain. Proc Natl Acad Sci USA. 1991;88:4553–4557. doi: 10.1073/pnas.88.10.4553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raadsheer FC, van Heerikhuize JJ, Lucassen PJ, Hoogendijk WJG, Tilders FJH, Swaab DF. Corticotropin-releasing hormone mRNA levels in the paraventricular nucleus of patients with Alzheimer’s disease and depression. Am J Psychiatry. 1995;152:1372–1376. doi: 10.1176/ajp.152.9.1372. [DOI] [PubMed] [Google Scholar]
- Randrup A, Braestrup C. Uptake inhibition of biogenic amines by newer antidepressant drugs. Relevance to the dopamine hypothesis of depression. Psychopharmacology. 1977;52:308–314. doi: 10.1007/BF00492370. [DOI] [PubMed] [Google Scholar]
- Reul JMHM, Stec I, Sodr M, Holsboer F. Chronic treatment of rats with the antidepressant amytriptyline attenuates the activity of the hypothalamic-pituitary-adrenocortical system. Endocrinology. 1993;133:312–320. doi: 10.1210/endo.133.1.8391426. [DOI] [PubMed] [Google Scholar]
- Rubin RT, Poland RE, Lesser IM, Winston RA, Boldgett ALN. Neuroendocrine aspects of primary endogenous depression. Arch Gen Psychiatry. 1987;44:238–336. doi: 10.1001/archpsyc.1987.01800160032006. [DOI] [PubMed] [Google Scholar]
- Rutter JJ, Gundlah C, Auerbach SB. Increase in extracellular serotonin produced by uptake inhibitors is enhanced after chronic fluoxetine treatment. Neurosci Lett. 1994;171:183–186. doi: 10.1016/0304-3940(94)90635-1. [DOI] [PubMed] [Google Scholar]
- Sachar EJ. Neuroendocrine abnormalities in depressive illness. In: Sachar EJ, editor. Topics in Psychoendocrinology. Grune and Stratton; New York: 1975. pp. 256–298. [Google Scholar]
- Sawchenko PE. The final common path: Issues concerning the organization of central mechanisms controlling corticotropin secretion. In: Brown MR, Koob GF, Rivier C, editors. Stress Neurobiology and Neuroendocrinology. New York: Marcel Dekker, publisher; 1990. pp. 55–71. [Google Scholar]
- Schaeffer C, Aron CL. Stress-related effects on the secretion of progesterone by the adrenals in castrated male rats presented to stimulus males. Involvement of estrogen. Acta Endocrinol. 1987;114:440–445. doi: 10.1530/acta.0.1140440. [DOI] [PubMed] [Google Scholar]
- Schmider J, Lammers C-H, Gotthardt U, Dettling M, Holsboer F, Heuser IJE. Combined dexamethasone/corticotropin releasing hormone test in acute and remitted manic patients, in acute depression, and in normal controls. Biol Psychiatry. 1995;38:792–802. doi: 10.1016/0006-3223(95)00064-X. [DOI] [PubMed] [Google Scholar]
- Sleight AJ, Carolo C, Petit N, Zwingelstein C, Bourson A. Identification of 5-hydroxytryptamine7 receptor binding sites in rat hypothalamus: Sensitivity to chronic antidepressant treatment. Mol Pharmacol. 1995;47:99–103. [PubMed] [Google Scholar]
- Souetre E, Salvati E, Belugou JL, Pringuey D, Candito M, Krebbs B, Ardisson JL, Darcourt G. Circadian rhythms in depression and recovery: Evidence for blunted amplitude as the main chronobiological abnormality. Psychiatry Res. 1989;28:263–278. doi: 10.1016/0165-1781(89)90207-2. [DOI] [PubMed] [Google Scholar]
- Stark P, Fuller RW, Wong DT. The pharmacologic profile of fluoxetine. J Clin Psychiatry. 1985;46:7–13. [PubMed] [Google Scholar]
- Steiger A, Von Bardeleben V, Herth T, Holsboer F. Sleep electroencephalogram and nocturnal secretion of cortisol and growth hormone in male patients with endogenous depression before treatment and after recovery. J Affect Dis. 1989;16:189–195. doi: 10.1016/0165-0327(89)90073-6. [DOI] [PubMed] [Google Scholar]
- Stokes PE, Pick GR, Stoll PM, Nunn WD. Pituitary-adrenal function in depressed patients: Resistance to dexamethasone suppression. J Psychiatr Res. 1975;12:271–281. [Google Scholar]
- To ZP, Bonhaus DW, Elgen RM, Jakeman LB. Characterization and distribution of putative 5-ht7 receptors in guinea-pig brain. Br J Pharmacol. 1995;115:107–116. doi: 10.1111/j.1476-5381.1995.tb16327.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban JH, Van de Kar LD, Lorens SA, Bethea CL. Effect of the anxiolytic drug buspirone on prolactin and corticosterone secretion in stressed and unstressed rats. Pharmacol Biochem Behav. 1986;25:457–462. doi: 10.1016/0091-3057(86)90023-7. [DOI] [PubMed] [Google Scholar]
- Wagner KD, Berenson AB. Norplant-associated major depression and panic disorder. J Clin Psychiatry. 1994;55:478–480. [PubMed] [Google Scholar]
- Wood MD, Broadhurst AM, Wylie MG. Examination of the relationship between the uptake system for 5-hydroxytryptamine and the high affinity 3H-imipramine binding site. Neuropharmacology. 1986;25:519–525. doi: 10.1016/0028-3908(86)90178-4. [DOI] [PubMed] [Google Scholar]