

Summary
The Merck STEP and the Thai RV144 human immunodeficiency virus (HIV) vaccine trials confirmed that we still have a long way to go before developing a prophylactic HIV vaccine. The main issue at hand is that we have yet to identify an immunological correlate of protection against HIV. While many question the T-cell-based approach towards vaccine development, it is likely that T cells will be a necessary part of any vaccine strategy. CD8+ T cells remain an attractive option because of their ability to specifically recognize and eliminate virally infected host cells. In this review, we recapitulate the evidence for CD8+ T cells as an immunological correlate against HIV, but more importantly, we assess the means by which we evaluate their antiviral capacity. To achieve a breakthrough in the domain of T-cell-based HIV vaccine development, it has become abundantly clear that we must overhaul our system of immune monitoring and come up with a ‘rational’ tactic to evaluate the efficacy of HIV-specific CD8+ T cells.
Keywords: T cells, AIDS, cytokines
Introduction
There continues to be a pressing need for a human immunodeficiency virus (HIV) vaccine, as acquired immunodeficiency syndrome (AIDS)-related illnesses remain one of the leading causes of death globally (1). Even though the overall incidence of HIV appears to be stabilizing, the HIV epidemic is continually evolving. For example, in Eastern Europe and Central Asia, where intravenous drug use was previously the main modality of HIV spread, there is a current outbreak of new infections that arose primarily via sexual transmission (1). HIV still accounts for 2 million deaths annually and is demonstrating the capacity to persist as a lethal pathogen (1). Thus, despite tremendous advances in therapeutic and prevention strategies, the development of an HIV vaccine remains of paramount importance.
The results of two recent HIV vaccine efficacy trials, the Merck STEP trial (2) and the Thai RV144 trial (3), necessitate an honest and thorough evaluation of the state of HIV vaccine development. The STEP trial was predicated upon substantial correlative evidence that virus-specific CD8+ T cells provide some aspect of viral load control within HIV-infected individuals (4, 5) and simian immunodeficiency virus (SIV)-infected non-human primates (6,7,8). In contrast, the RV144 trial was presumed to be an ill-fated combination of two previously failed HIV vaccine strategies designed to stimulate both a cellular and humoral immune response (9). While there are numerous caveats to the results and interpretations of the results from both trials, they have highlighted the glaring deficiency in the field of our collective inability to pinpoint a definitive correlate of protection against HIV. For the purposes of this review, we focus specifically upon adaptive cellular immune aspects of potential immune correlates of protection, with the acknowledgement that any successful HIV vaccine capable of generating broadly neutralizing antibody responses will trump any other vaccine strategy.
Defining a cellular correlate of protection against HIV is not a trivial issue, as several questions must first be answered before a correlate may be defined as protective. Which cell types are most relevant? Which function(s) mediate control or elimination of HIV-infected target cells? Is this particular functional profile correlated to a specific cellular phenotype? By what measure may we ascertain its biological relevance? Is there a gold standard towards which we may aspire? Is it possible to manipulate the correlate of protection for the purpose of vaccine design? How may we pre-empt HIV’s evasion mechanisms? These questions are framed in the context of HIV vaccine research but actually cast a huge spotlight onto the field of immunology as a whole. Although we have made great strides over the past ~30 years, paradigms, ideologies, rationales, and technologies must all breach new territory if we are to solve the HIV riddle.
Lessons from vaccine trials: routine immunoassays fail to predict vaccine efficacy
Although most licensed vaccines stimulate effective humoral immunity (10), HIV’s uncanny ability to evade its host’s neutralizing antibody response prompted HIV researchers to investigate CD8+ T cells as a potential correlate of protection. Several findings suggested an important role for CD8+ T cells in controlling HIV, including the temporal association between the appearance of HIV-specific CD8+ T cells and the decline in peak viremia during acute HIV infection (4, 5), the prevalence of specific human leukocyte antigen (HLA)-class I alleles among humans with non-progressive disease (11), and the rapid evolution of sequence mutations within immunodominant epitopes recognized by CTL in both humans (12, 13) and non-human primates (14, 15). CD8+ depletion studies in non-human primates, with a concomitant rise in SIV viremia, further promoted a significant role for CD8+ T cells in controlling HIV replication (6,7).
Based on these premises, the Merck STEP trial evaluated a vaccine platform designed specifically to stimulate an HIV-1 specific cell-mediated immune (CMI) response (8,16). The Merck test-of-concept trial used a mixture of recombinant Adenovirus type 5 (Ad5) vectors expressing the HIV-1 gag, pol, and nef genes, which are conserved across different clades of HIV-1 and encode for antigens that are frequently recognized by CD8+ T cells during natural infection (16). During phase I clinical testing, prototype Ad5 vaccines containing only the gag gene proved to be very immunogenic [particularly for CD8+ T cells, as measured by the IFN-γ enzyme-linked immunospot (ELISpot) assay], more than other commonly used CMI vaccine vectors such as DNA plasmids and poxvirus vectors (17–20). The Merck Ad5 Gag/Pol/Nef vaccine platform ultimately entered a test-of-concept trial, largely in an attempt to address the considerable uncertainty about how a CMI vaccine may control HIV viral replication (2). Random sampling of the study vaccinees for IFN-γ ELISpot responses at the week 8 timepoint revealed that 75% of the subjects receiving the vaccine responded to one or more HIV antigens with a geometric mean magnitude of over 200 SFC/million PBMCs (2). Despite this promising result, the Step trial was subsequently terminated immediately after interim analysis revealed the vaccine neither prevented HIV-1 infection nor lowered the viral load set points, and perhaps had the adverse effect of increasing HIV acquisition in Ad5-seropositive vaccinees (2).
While many researchers consider the Merck STEP trial a failure, the landmark study is of paramount importance, because we are able to glean substantial knowledge about CMI vaccine correlate(s) of protection. The obvious conclusion is that IFN-γ production by T cells is not a correlate of protection against HIV. In the STEP trial, despite positive IFN-γ ELISpot responses in 75% of the vaccinees at an early timepoint, the vaccine failed to protect against HIV acquisition compared to a placebo control. Clearly we are misinterpreting the IFN-γ ELISpot assay, as we are placing too much value in its results. IFN-γ does not directly inhibit HIV replication; it is a good indicator of the presence of a response but cannot be used to infer an anti-HIV property of T cells. Furthermore, while the ELISpot assay is extremely simple, rapid, amenable to high though-put analyses, and conducive to robust validation, we do not know what magnitude of response corresponds to biological relevance. The Merck Ad5 Gag/Pol/Nef vaccine elicited responses >200 SFC/million; is this frequency sufficient to achieve protection from or control of HIV infection? Such a measurement proved to be inadequate for IFN-γ; however, for another function like IL-2, it might be of the proper magnitude. We have no idea what threshold of immunogenicity must be crossed for vaccine efficacy; we need to determine what assay results correlate with in vivo relevance for every functional output of antigen-specific T cells.
Unlike the STEP trial, the Thai RV144 test-of-concept HIV vaccine trial evaluated a vaccine platform aimed at eliciting both humoral and cellular immunity; the vaccine consisted of the subtype B canarypox-HIV vector ALVAC-HIV (vCP1521) prime with a VaxGen AIDSVAX bivalent gp120 B/E boost (3, 21). Even though canarypox-based prime-boost regimens have historically induced poor CD8+ T-cell responses based on the IFN-γ ELISpot assay (22, 23) and a phase 3 trial of AIDSVAX B/E alone showed no effect on HIV-1 acquisition (24), the combinatorial vaccine strategy showed a marginal effect on reducing HIV acquisition early during the vaccine course (3). The mechanisms responsible for this are currently unknown, but the paucity of detectable HIV-specific CD8+ T cell responses in vaccine recipients has been interpreted to exclude a meaningful contribution of CD8+ T cells in preventing acquisition. During the RV144 trial, vaccine efficacy for T-cell induction was assessed using the IFN-γ ELISpot assay as well as by measuring IFN-γ and IL-2 production by intracellular cytokine staining (ICS)(3). The observed responses were extremely weak, as Gag and Env stimulation induced positive ELISpot T-cell responses in only 8.3% and 15.9% of vaccinees, respectively (3). CD8+ T-cell responses measured by ICS were worse, as Gag-specific cytokine production was detected in only 7.6% of vaccinees, whereas Env-specific responses were measured in only 11.1% (3). Gag stimulation resulted in a positive CD4+ T-cell cytokine response in only 2/144 vaccinees, whereas Env stimulation induced a positive CD4+ T-cell cytokine response in 34% of vaccinees compared to 3.6% of placebo recipients (p<0.001)(3). The data are consistent with a negligible contribution from T cells towards the observed protection.
In contrast, the phase 2 trial of the ALVAC-HIV prime/AIDSVAX boost vaccine immediately prior to RV144 made use of the chromium-release cytotoxic T lymphocyte (CTL) assay to measure CD8+ T-cell responses (21). The overall frequency of HIV-specific CTL responses in vaccinees was 24%, whereas none of the placebo recipients had such responses at any timepoint (21). Nine of the 22 responders (41%) displayed repeat positive CTL responses at multiple timepoints (21). Since the vaccine was deemed to be moderately successful in the RV144 trial (31% efficiency in low-risk subjects) and the phase 2 chromium-release assays demonstrated moderate activity, assaying for CTL cytotoxicity may be a more accurate predictor of vaccine efficacy than cytokine production from T cells.
Defining CD8+ T-cell correlates of protection in HIV infection: the dilemma of human research
The definitive mechanism by which CD8+ T cells actually control HIV has remained elusive, as unlike in immunological research involving murine models, studies in human populations are correlative, non-mechanistic, and inevitably yield exceptions to every model conceived. The absence of an animal model of protection emphasizes the need to look to human cohort studies to define protective mechanisms mediated by HIV-specific T cells. To this end, we and others have focused upon subjects who exhibit natural resistance to HIV disease progression, named long-term nonprogressors (LTNPs) or elite controllers (ECs), who exhibit stable normal CD4+ T-cell counts in the periphery and durably low to undetectable viral loads. Since no single functional output of HIV-specific CD8+ T cells has proven to correlate with protection (or even control), the prevailing paradigm in the field now is that the more functions a CD8+ T cell performs, the more antiviral it must be. Indeed, HIV LTNPs possess a greater proportion of HIV-specific CD8+ T cells that perform 5 functions simultaneously (IL-2, IFN-γ, TNF-α, CD107a, and MIP-1β) compared to HIV-infected subjects suffering from progressive disease (25). Further support of this concept comes from a mouse model of Leishmania major infection, in which polyfunctional CD4+ T cells exert protection from disease progression (26, 27). A similar phenomenon has not been demonstrated for CD8+ T cells; however, antigen-specific polyfunctional CD8+ T cells do produce more cytokine on a per cell basis than monofunctional CD8+ T cells (28). The ability of polyfunctional HIV-specific CD8+ T cells to suppress HIV appears to relate to high antigen sensitivity (29). In addition, HIV-specific CD8+ T cells from LTNPs exhibit a greater proliferative capacity and also a higher degree of cytotoxicity against autologous HIV-infected CD4+ T cells than those from progressor patients (30–32). The relatively high killing capacity of LTNP CD8+ T cells is thought to be a result of their ability to dramatically upregulate Granzyme B and perforin production after long-term culture (32, 33).
There are several caveats, however, to the LTNP studies, centered mainly on the issue of cause-and-effect. There is great debate as to whether HIV-specific CD8+ T-cell polyfunctionality actually drives a low viral load in patients or if a polyfunctional profile is merely a byproduct of a low antigenic presence. Given that a highly polyfunctional (capable of performing 5 functions simultaneously) HIV-specific CD8+ T-cell population only constitutes a very small proportion of the total CD8+ T-cell compartment and that many LTNP patients lack this functional subset, many argue it cannot be a significant immune correlate of protection. Furthermore, HIV-specific CD8+ T-cell polyfunctionality can be induced by antiretroviral therapy (ART) among chronic HIV subjects with progressive disease (34), although in most cases cessation of treatment inevitably results in virologic failure despite improved immune function (35–37). With regards to the mechanism of polyfunctionality, some literature suggests that polyfunctionality is actually characteristic of HIV-specific CD8+ T cells with low, not high, antigen sensitivity (38). Thus, CD8+ T-cell polyfunctionality remains an unqualified correlate of HIV control.
The phenomenon of LTNPs thus raises a critical question: are LTNP subjects an appropriate cohort from which to develop and model a putative HIV vaccine? LTNP cohorts have emerged as the standard for investigating immunological control of HIV, but is it warranted? While there is little doubt that this rare population of HIV-infected patients exhibits natural resistance to progression to AIDS, we must remember that every one of these subjects became infected with HIV. In fact, many LTNPs also lack detectable HIV-specific CD8+ T-cell responses (as well as other genetic factors that may confer resistance to progression)(39). Thus, one could argue whether anything we learn from LTNPs is applicable to the immunological assessments of a prophylactic vaccine. In theory, individuals who remain uninfected by HIV despite repeatedly engaging in activities that are high risk for HIV acquisition, such as unprotected sex and needle-sharing with HIV-infected partners, represent the ideal study cohort for immunological correlates important for HIV vaccine design. However, despite some studies reporting the presence of HIV-specific CD8+ T-cell (40–42) and NK cell activity (43, 44) among these persistently seronegative subjects, skepticism over the genuine exposure to HIV, the low magnitude and low frequency of the HIV-specific responses, and the durability of both the HIV-specific responses and the seronegative status of the subjects has stymied the field.
LTNPs have been anointed the gold standard in the field mainly due to their longevity and to the reliable detection of HIV-specific immune responses of robust magnitude and frequency. However, because the main criterion for LTNP status is length of time with stable CD4+ T-cell counts and/or low or undetectable viral load, most studies assay PBMC samples from these subjects during the chronic phase of infection, long after HIV infection has become well established and equilibrium between the host response and viral replication/latency dynamics has been reached (45). This context precludes a reasonable deduction of cause or effect. Thus, LTNP subjects may be ideally suited to study HIV non-progression and thus amenable to the design of a therapeutic vaccine but of debatable relevance for a prophylactic vaccine.
Debunking functional correlates of protection
The interplay between HIV replication immediately after infection and the host immune response, both innate and adaptive, determines the course of HIV disease. If indeed HIV-specific CD8+ T cells are responsible for the initial resolution of peak viremia during the first few weeks of infection, it follows that during this timeframe, the CD8+ T cells must be most antiviral. A detailed characterization of HIV-specific CD8+ T cells during acute HIV infection, therefore, affords the greatest likelihood of discovering how CD8+ T cells eliminate HIV-infected host cells before a thorough seeding of HIV reservoirs, CTL escape mutations, CD4 depletion, and T-cell exhaustion manifest themselves and confuse the assessment of CD8+ T-cell control over HIV.
What is the best way to characterize HIV-specific CD8+ T cells? The prevailing tactic in the field is to correlate the absolute number of functions an antigen-specific CD8+ T cell may perform simultaneously to its antiviral capacity. However, ‘polyfunctionality’ is an ideology that developed as a result of our inability to identify a specific functional correlate of protection from HIV. Since no single function emerged as a leading correlate, we surmised that multiple functions must work in concert if CD8+ T cells actually control HIV replication. Henceforth, a detailed characterization of the antiviral nature of HIV-specific CD8+ T cells must include an assessment of their capacity to be polyfunctional. However, this approach poses a conundrum: isn’t the polyfunctional nature of a CD8+ T cell simply a product of the number of functions for which are assayed? Are we not biasing our interpretation of the data by screening for 5, 6, or 7 functions? For example, just as a CD8+ T cell will never be considered polyfunctional if it is only tested in an IFN-γ ELISpot assay, surely a cell will be multi- functional if we assay it for 10 functions? Does that mean it is necessarily antiviral?
Consider the functional profile below (Fig. 1A). IFN-γ, TNF-α, and IL-2 are the most commonly measured T cell functions. Judging by the lack of polyfunctional subsets, one might conclude that these CD8+ T cells were not particularly antiviral. However, including CD107a and MIP-1β in our analysis alters the distribution of responses, resulting in an increase in functionality (Fig. 1B), thereby improving their perceived antiviral nature. It is not difficult to imagine that the more functions for which we screen the better the odds for polyfunctionality. As technology advances and the number of parameters we are able to quantify simultaneously expands, the concept of CD8+ T-cell polyfunctionality as an immune correlate of protection, defined simply as being able to perform more than 2 functions simultaneously, becomes increasingly ambiguous.
Fig. 1. The degree of polyfunctionality of a population of CD8+ T cells correlates to the number of functions for which are assayed.

(A). Human PBMCs were peptide- stimulated and screened for resulting IFN-γ, IL-2, and TNF-α. The 7 possible functional outcomes are grouped by color according to the number of functions: charcoal for 1, blue for 2, yellow for 3 functions. Note that none of the responding CD8+ T cells were capable of performing all 3 functions simultaneously. (B). When CD107a and MIP-1α were added to the screening panel, the same responding cells could now be considered polyfunctional, as there are yellow and red slices in the pie chart denoting positivity for 3 and 4 functions, respectively. The more functions for which are assayed, the greater the likelihood the test cells will turn out to be polyfunctional. Thus, when screening for HIV vaccine efficacy, it is not enough simply to assess the absolute number of functions the cell may perform simultaneously; we must evaluate how well the responding HIV-specific CD8+ T-cell population performs functions of consequence to HIV inhibition/clearance.
That brings us back to square one: trying to identify specific CD8+ T-cell functions that truly have a negative impact on HIV replication. The temporal association between the appearance of HIV-specific CTLs in the periphery and a sharp decline in HIV viremia resulted from an assessment of cytotoxic potential in vitro and formed the first premise for CD8+ T cells as a correlate of HIV control (4, 5). Since then, researchers have assayed for HIV-specific CD8+ T-cell responses by techniques that require much less labor, time, and manipulation of the cells. The ELISpot assay was popular for a long period of time (and still is), largely because it is amenable to strict validation procedures, but polychromatic flow cytometry has clearly become the assay-of-choice for measuring antigen-specific immune responses, because of its multi-dimensional nature. These techniques quantify mostly cytokines and other non-cytolytic proteins, which have dramatically expanded our characterization of antigen-specific T-cell responses in unprecedented detail.
In the case of HIV, however, it can be argued that the measurement of cytokines, such as IFN-γ and IL-2, has obfuscated our understanding of the host anti-HIV response. Whereas measurement of HIV-specific CD8+ T-cell frequency by MHC tetramers initially showed an inverse relationship to viral load (46), the measurement of HIV- specific IFN-γ production determined a proportional relationship between HIV-specific CD8+ T cells and HIV viral load (47). IFN-γ was presumed to be an antiviral marker, because CD8+ T-cell clones that produced IFN-γ early after stimulation were shown to develop into CTLs after further long-term culture (48, 49). Moreover, most research on HIV-specific CD8+ T cells during acute HIV infection reported weak responses in frequency, magnitude, and breadth, based on IFN-γ production (50–52), suggesting an immediate deficiency in anti-HIV-specific effector activity.
IL-2 secretion is considered by some to be indispensible for protection, since HIV-specific CD8+ T cells from chronically infected HIV progressors that upregulate IFN-γ failed to simultaneously produce IL-2, whereas this dual capability was preserved among CMV- and EBV-specific CD8+ T cells (53). Furthermore, IL-2 secretion by CD8+ T cells is correlated to proliferation (53, 54), which is preserved in HIV subjects with non-progressive infections (33). While IL-2 is certainly critical for the preservation of HIV-specific CD8+ T-cell function during chronic infection (31), its relevance during acute HIV infection is debatable. IL-2 does not itself have antiviral activity (although it may promote it in other cells) and, in the case of HIV, may be a driver of viral replication by augmenting the availability of activated target CD4+ T cells. IL-2 production is taken for granted as a necessary component of a protective immune response because of studies on CMV, EBV, and influenza, in which virus-specific CD8+ T cells often secrete IL-2 (38,53–55). Comparison of IL-2 production from CD8+ T cells specific for influenza, adenovirus, EBV, and CMV indicates differential production is likely linked to viral load (55). EBV and influenza infections generally do not result in significant chronic antigenic burdens. CMV remains active and establishes a constant antigenic presence; however, depending on the cycle, the viral burden may be high or low. Adenovirus-specific CD8+ T cells very likely undergo intermittent stimulation due to re-infection with alternate adenovirus serotypes (56) or adenovirus persistence (57), thus serving as a good model for CD8+ T-cell responses in the setting of frequent antigenic burden. Indeed, IL-2 production from EBV- and influenza-specific CD8+ T cells is typically high compared to that of CMV and adenovirus (55). Thus, when there is no circulating antigen or the level is low, IL-2 production by virus-specific CD8+ T cells is high, whereas in the presence of a constant viral burden IL-2 production is low or absent. This agrees with the murine LCMV model of CD8+ T-cell responsiveness, in which IL-2 producing CD8+ T cells are lost when clone 13 establishes chronic infection but not after resolution of acute infection by the Armstrong clone (58). A formal role for IL-2 in clearing primary viral infection has not been demonstrated. Thus, IL-2 should not be considered an important correlate of protection against viral infection and should rather be viewed as an indicator of response longevity in the absence of antigen.
Killing in the name of protection
Cytotoxicity is one functional attribute of human CD8+ T cells that unequivocally combats acute viral infections, including CMV (59–61), EBV (62), HBV (63, 64), and HCV (65, 66). CTLs clear virally infected target cells primarily by inducing apoptotic cascades through perforin-mediated delivery of granzymes, serine proteases that cleave caspases (67, 68). Granzyme B is the most prominent protease, and it commonly targets caspase-3 for cleavage, which in turn activates an enzyme that degrades DNA, thus inducing apoptosis (69). Granzyme B also cleaves Bid, which recruits Bax and Bak to change the membrane permeability of the mitochondria, thereby releasing other proteins that activate different apoptotic pathways (69). Perforin is stored within lytic granules and is required for delivery of granzyme B, as genetic mutation or deletion of perforin causes impaired cellular cytotoxicity and profound immunodeficiency (70, 71).
HIV-specific CTL activity was first described by Walker et al. (72) in 1987, where HIV-infected CD4+ T cells were shown to be killed by autologous CD8+ T-cell clones as well as CD8+ T cells directly ex vivo. Although this initial study defined that cytotoxic activity was present within HIV-infected subjects, many subsequent studies have found that HIV-specific killing function is deficient in chronically infected individuals (73–75) as well as acute infection (4, 76). These studies, however, have come full circle in more recent works, with a more detailed understanding of cytotoxic potential in chronic HIV infection (32, 33, 77–80).
Many assays have been developed to measure, either directly or indirectly, CD8+ T-cell-mediated cytotoxicity, including chromium release, CD107a degranulation, fluorescence-based target elimination, and caspase cleavage assays, with each bearing their own distinct advantages and disadvantages. The chromium-release assay, the historical gold standard for measurement of killing, involves radioactivity, is insensitive using CD8+ T cells directly ex vivo, and is prone to background issues. Similarly, fluorescence-based killing assays have background and sensitivity issues and are difficult to perform using human samples directly ex vivo. The caspase cleavage assays represent a quite powerful technique to detect granzyme-B-mediated cleavage within target cells as a means to directly quantify killing activity by flow cytometry. Perhaps the most significant disadvantage of the chromium-release, caspase cleavage, and fluorescence-based killing assays are that they uniformly only assess the killing of the target cell and provide no information about the characteristics of the CD8+ T cell that induced the killing. This represents a critical loss of information that is absolutely necessary to understand the nature of HIV-specific CD8+ T-cell killing and how to harness this activity for vaccine design and analysis. At minimum, an assay needs to be used that can identify the responding CD8+ T cell in the act of killing. For the long term, we need to develop an assay that characterizes both the killer and the target cell simultaneously.
In this regard, why not just measure the presence of perforin and granzyme B directly within HIV-specific CD8+ T cells? Both perforin and granzyme B can be readily measured within CD8+ T cells by flow cytometry, and this has been applied to the study of HIV-specific CD8+ T cells extensively. With the advent of MHC class I tetramer technology, it became possible to directly measure within HIV-specific CD8+ T cells expression of perforin and granzyme B (75). This initial study showed unequivocally that HIV-specific CD8+ T cells directly ex vivo were deficient in perforin expression compared to CD8+ T cells specific for other viruses such as CMV. There are, however, a number of issues with this finding, most notably that it remains unclear how much perforin is necessary to initiate granzyme B-mediated killing. Are HIV-specific CD8+ T cells ‘deficient’ in perforin expression incapable of killing?
Perforin upregulation after activation of HIV-specific CD8+ T cells
The first idea that our view of perforin expression in HIV-specific CD8+ T cells required re-evaluation came after the demonstration that HIV-specific CD8+ T cells from LTNPs could upregulate perforin after proliferation (33). In essence, it was shown that HIV-specific CD8+ T cells directly ex vivo may (or may not) express perforin to some degree, but after several rounds of proliferation became uniformly positive for perforin. Notably, this property was deficient in proliferating HIV-specific CD8+ T cells from chronic progressors, solidifying the concept of a killing defect in chronic HIV infection. More recent work in this vein has gone on to show that proliferation-induced perforin upregulation directly engenders HIV-specific CD8+ T cells from LTNPs with enhanced killing ability (32).
At about the same period of time, we were developing the use of CD107a as a marker for degranulation of CD8+ T cells (81). Having had difficulty in measuring perforin expression in activated CD8+ T cells directly ex vivo, we instead began to assess exposure of CD107a on the cell surface of activated antigen-specific CD8+ T cells as a surrogate marker for induction of killing. Currently, this assay remains one of few means to directly assess degranulation in human CD8+ T cells, but there are several caveats to the assay that have more recently allowed us to re-examine perforin expression in activated CD8+ T cells. First and foremost is that degranulation should never be equated directly to actual killing, unless one shows both events taking place. Degranulation occurs in most CD8+ T cells immediately upon TCR triggering, representing a first line of defense against viral infections (82); however, it is the content of the granules that determines the degree of cytotoxicity that ensues (83). Our initial results indicated that, as expected, intracellular perforin staining after antigenic stimulation declined concomitant with an increase of cell surface CD107a (84), suggesting that all perforin-containing granules are immediately released. We soon found, however, that the perforin antibody (δG9 clone) in common use was sensitive to the degranulation assay conditions, which require the use of monensin to neutralize intracellular pH (84).
A great deal is understood regarding perforin synthesis in CD8+ T cells; for the purposes of this discussion, the most important is the fact that perforin structure is modified extensively post-translation (85). Given our observations regarding the pH sensitivity of perforin to antibody recognition, this prompted us to re-evaluate whether perforin upregulation indeed required proliferation (33, 86, 87). Using a different perforin antibody (clone D48), we found that human CD8+ T cells rapidly upregulate perforin de novo after antigen-specific stimulation without the requirement for proliferation (84, 88), and this newly synthesized perforin can be immediately transported to the immunological synapse to potentiate cytotoxicity (88).
How does our new insight regarding cytotoxicity influence our perspective on HIV vaccine development? Since we have been unable to identify an immunological correlate of protection against HIV, the field has instead characterized immune responses against viruses from which we are protected, such as EBV, CMV, and influenza. The rationale for this approach being that protective immune responses against one virus should be transferable to other viral settings as well. We therefore incorporated rapid perforin upregulation into an assessment of the functional breadth of virus specific human CD8+ T cells ex vivo, including CMV, EBV, influenza (flu), and adenovirus (Ad), which as discussed earlier have distinct differences in viral burden and chronicity (55). Activated IFN-γ-producing EBV- and flu-specific CD8+ T cells often produced IL-2 but rarely upregulated perforin (55). Activated Ad- and CMV-specific CD8+ T cells, in contrast, often upregulated perforin but rarely upregulated IL-2(55). This difference was clearly associated with the memory phenotype of the responding cells, wherein perforin-producing cells generally fell within an effector or effector-memory like phenotype, and IL-2 producing cells appeared more memory like (55). This inverse relationship between IL-2 and perforin production ability was confirmed in a subsequent study that focused upon CMV-specific CD8+ T-cell responses (54). Given the cross-sectional nature of these studies and the absence of any viral measurements in the serum of the healthy subjects, there are two potential conclusions we may infer. If the functional characteristics of virus-specific CD8+ T cells are static, then we must conclude that every viral infection stimulates a unique CD8+ T-cell response. As a consequence, the functional profile of one virus cannot serve as a target after which an HIV vaccine may be designed. By this logic, previous work comparing HIV-specific T-cell responses to those of CMV or EBV may have misled us in our thinking of what a protective HIV-specific T-cell response should be.
However, if the functional descriptions for every virus-specific CD8+ T-cell population merely reflect various stages of CD8+ T-cell differentiation that depend on the antigenic burden at the time of sampling, then we must conclude that an effector response, driven in part by rapid perforin upregulation, is necessary to combat replicating virus, whereas a central memory-type profile, driven by IL-2 production, is necessary to maintain a healthy antiviral CD8+ T-cell population when viral levels in the periphery are low or non-existent (Fig. 2). In support of this latter conclusion, a longitudinal study that monitored vaccine induced yellow fever virus- and smallpox-specific CD8+ T cells demonstrated that both vaccines generated a primary virus-specific CD8+ T-cell response that passed through an obligate effector phase characterized by the abundant expression of perforin and granzyme B (89). The cells then differentiated into long-lived memory cells, maintaining the ability to proliferate and secrete effector cytokines in response to antigenic re-stimulation (89). Thus, the perforin and IL-2 functional subsets we describe likely mediate protective immunity at different stages of infection.
Fig. 2. Two possible mechanisms to explain the functional heterogeneity of virus-specific CD8+ T-cell responses.
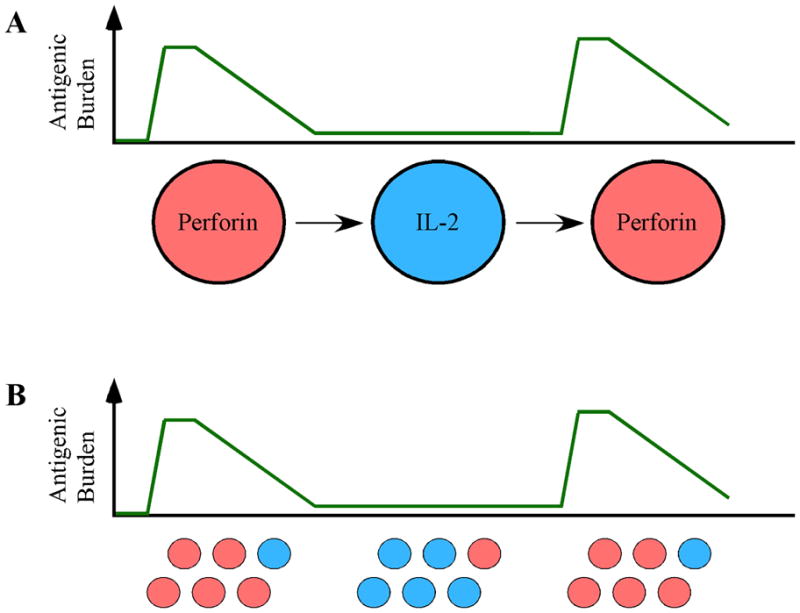
(A). During acute infection, when the viral load is high, the responding cells express perforin (red) in order to eliminate virally infected host cells. Upon resolution of infection, the responding CD8+ T cell differentiates into a resting memory cell, characterized by IL-2 secretion (blue), in order to maintain itself. During periods of viral re-activation, the cell then differentiates into an effector cell expressing perforin (red) once again, in an attempt to combat the virus anew. (B). Alternatively, both perforin-expressing (red) and IL-2-expressing (blue) CD8+ T-cell subpopulations are present in the host. During acute infection, when there is a high viral burden, the perforin-containing cells (red) proliferate in response to the viral insult. Upon viral clearance or latency, the perforin-expressing cells wane and the IL-2-expressing cells (blue) predominate. During viral re-challenge, the perforin-expressing subpopulation (red) prevails once again. For HIV vaccine design, it will be important to determine if vaccine-induced CD8+ T cells remain static or if they progress through various stages of differentiation. If it is the former, then an HIV-vaccine may need to induce both perforin-expressing and IL-2-expressing sub-populations.
Perforin upregulation and polyfunctionality in HIV
Work by others and us (25, 90) has determined that polyfunctionality is a correlate of control in HIV infection. Given the inverse relationship between perforin and IL-2 production in virus-specific CD8+ T cells, we first re-evaluated the role of polyfunctionality in HIV infection in a cohort of HIV elite controllers and chronic progressors (77). The findings of this study are important to our concept of immune correlates in HIV for a number of reasons. First, this study confirmed the relationship between polyfunctionality and control of viremia: HIV elite controllers have an increased frequency of highly functional HIV-specific CD8+ T cells compared to chronic progressors. Secondly, we found that HIV-specific CD8+ T cells from HIV elite controllers have a heightened ability to rapidly upregulate perforin directly ex vivo compared to chronic progressors. This directly implies that HIV-specific CD8+ T cells from HIV elite controllers should have the ability to directly eliminate HIV-infected target cells through cytotoxic mechanisms. Importantly, however, we did not find a direct link between polyfunctionality and perforin expression. Perforin was, in fact, more likely to be expressed in cells with less functionality, as measured by IFN-γ, IL-2, TNF-α, CD107a, or MIP-1α. The dominant perforin-expressing populations typically only otherwise degranulated (CD107a+) or produced MIP-1α. Thus, while highly polyfunctional HIV-specific CD8+ T cells are an immune correlate of control in HIV infection, so too are less functional cells, depending on the functional combinations they produce.
To extend these concepts further, we have begun to examine polyfunctionality and perforin upregulation in acute HIV infection (manuscript in preparation). We surmised that if polyfunctional CD8+ T cells were critical for ‘control’ of HIV disease progression, they should play a role in the early drop in viremia that occurs concomitant with the appearance of HIV-specific CD8+ T-cell responses during acute infection. Our preliminary findings indicate that polyfunctional CD8+ T-cell responses are quite rare during acute HIV infection. Instead, we have found that rapid perforin upregulation dominates the earliest response we can detect in nearly every subject’s CD8+ T-cell functional profile, with the most prevalent functional subset being perforin+CD107a+. Thus, a robust HIV-specific CD8+ T-cell response composed of rapid perforin upregulation with concomitant degranulation may be primarily responsible for T-cell-mediated clearance of acute viremia. This then suggests that the later appearance of IL-2+ polyfunctional CD8+ T cells in LTNPs is likely the result of effective control of viremia.
These data provide a compelling argument in support of a direct role for CD8+ cytotoxicity as an anti-HIV correlate of protection. Furthermore, rapid perforin upregulation should be considered a pre-eminent feature of any vaccine-induced anti-HIV CTL response, since a potent effector response should be the goal of any HIV vaccine.
The fork in the road
The field of HIV vaccine development must ask itself the following question: do we continue to build incrementally on the foundation we have laid out over the past 15 years or do we completely overhaul our system of immune monitoring? It is evident from the Merck and RV144 vaccine trials that we should re-evaluate our current ideology built with IFN-γ and IL-2 as the cornerstones of immune correlates of protection. As convenient and ingrained as our conventional immunoassays may be, we should adopt more recently developed assays and systems biology to serve as our primary means of assessing anti-HIV immunity. The ELISpot assay, while useful in defining the presence of a T-cell response, has proven irrelevant when it comes to determining a correlate of HIV protection. It may continue to serve as a useful output of T-cell activation but should only be used as a premise from which to proceed to more pertinent assays, not as an investigative endpoint. In addition, any CD8+ T-cell assay that involves extended periods of culture, including the cultured ELISpot assay, to detect a vaccine-induced response should be avoided (with the exception of proliferation assays). Stimulating antigen-specific CD8+ T cells for several days coerces the cells to divide and differentiate under artificial conditions, fundamentally altering the cell’s phenotypic and functional composition from its in vivo circumstance. While such cultured assays may yield results that agree with our pre-conceived notions about what an optimal CD8+ T-cell response should be, they more likely represent a great departure from their actual in vivo condition. It is simply not realistic to characterize a memory T-cell response after several days of in vitro stimulation and then extrapolate from those findings how an antigen-specific CD8+ T cell will react during a natural infection.
We must also scrutinize our selection of target cells for all immunoassays. Currently, incubating whole PBMCs with exogenously added peptide or peptide pools generates target cells for most immunological assays, but does this really portray what happens in vivo? HIV-specific CD8+ T cells may recognize peptide-loaded host cells but not necessarily HIV-infected host cells expressing the same epitopes. Consequently, the commonly used immunoassays may detect an HIV-specific CD8+ T-cell response that in reality does not mediate any antiviral effect on HIV in vivo. We need to perform a comprehensive study that compares the immunogenicity of peptide-loaded, protein-incubated, and whole virus-infected target cells. Most importantly, the study needs to determine which approach best recapitulates the natural situation. Infecting autologous CD4+ T cells with primary HIV strains is, of course, the ideal scenario for identifying CD8+ T cell correlates of protection against HIV; however, it is not a practical consideration for the standardization of immune monitoring during clinical trials.
The original studies assessing CD8+ T-cell polyfunctionality were designed in part based on convenience: the detection of IFN-γ, IL-2, TNF-α, CD107a, and MIP-1α/β by flow cytometry is well validated. To identify T-cell correlates of protection against HIV as well as for HIV vaccine immune monitoring, we must now adjust the design of polyfunctional assays to focus on T-cell parameters that most likely mediate an antiviral effect. With this frame of mind, assaying for several random functions will not translate into a more appropriate HIV-specific screening procedure, and certainly more functions may not signify enhanced protection against HIV. Instead, we need to determine the minimum set of immunological markers that will provide the best differential criteria for an HIV-specific T-cell response. Much like the recent call for ‘rational’ antibody-based HIV vaccine design (91), we call for ‘rational’ immunological analysis, based not upon convenience or historical precedence but rather on what may actually provide some component of an immune correlate of protection in HIV.
Rational immunological assessment of HIV vaccine responses
On which outputs of CD8+ T-cell function could ‘rational’ immunoassays focus? The bottom line is this: what type of T-cell response do we currently believe would provide optimal immunity to either clear or control HIV viremia after challenge? The simplest answer to this question is that we need, at minimum, high frequency effector CD8+ T cells present at the site of infection (the mucosa) with immediate cytolytic ability. Table 1 lists a plethora of T-cell markers that may, alone or in combination, correlate to protection against HIV and in part address whether this ‘optimal’ vaccine-induced CD8+ T-cell response is present or can be maintained. These markers fall into several different categories, including function, phenotype, transcriptional programming, homing, etc, and should be viewed merely as a beginning to what can be assessed on an HIV vaccine-induced T-cell response. While this is an extensive list, each category of markers bears potentially important characteristics to HIV-specific CD8+ T cells that might be induced by a vaccine. Importantly however, this list needs to be continually updated and incorporated into an evolving immunological assessment platform for optimal HIV vaccine development. Heretofore we have discussed in great deal various aspects of T-cell function; in the following section, we review some additional categories in the specific context of their relevance to HIV vaccine immunology.
Table 1.
Potential correlates of CD8+ T cell anti-HIV immunity
| T-cell Aspect | Markers |
|---|---|
| Memory Phenotype | CCR7, CD27, CD28, CD45RA/RO, CD57, CD62L |
| Cytokine Production | IFN-γ, TNF-α, IL-2, IL-4, IL-10, IL-12, IL-17, IL-23 |
| Cytotoxicity | Perforin (new and granule-associated), Granzyme A/B/K, Granulysin |
| Miscellaneous | Degranulation (CD107a exposure), MIP-1α/β, Proliferation (CFSE dilution) |
| Transcriptional Control | T-bet, Eomesodermin, BLIMP-1, GATA-3, ROR-γT, |
| Activation Status | HLA-DR, Ki67, bcl-2, CD38, CD69, CD95 |
| Exhaustion | PD-1, CD160, Lag-3, 2B4, TIM3 |
| Tissue Trafficking | α4β7, CCR5, CCR9, CCR10, CD161 |
| Regulatory Function | CTLA-4, Foxp3, GITR, CD25, CD39, CD73, TGF-β |
Memory phenotype
Several studies have related particular T-cell memory phenotypes to the control of certain viral infections, including CMV, EBV, HCV, and HBV (92–96). Assessment of memory phenotype can provide very useful information relating to the potential functional responses and response longevity, but differences in memory phenotype between various viral specificities should not be concluded as ‘superior’ or ‘deficient’. Memory phenotype properties most likely arise as a result of the antigenic history of the responding T cells. Another problem with phenotyping alone is that we cannot assume functionality of the virus-specific response without actually assaying for the different functions; there may be subtle differences in the functional quality of the responses between viruses that cannot be predicted by two or three marker phenotyping. For example, not every central memory CD8+ T cell can make IL-2. In addition, there is no consensus on what phenotype officially denotes a particular memory subset. Variations in memory subset nomenclature have confused more than clarified the differentiation process. Thus, memory markers are useful to measure but must be measured in context with other relevant functional markers or outputs to ascertain their importance.
Activation markers
In terms of monitoring for a vaccine response, the measure of immune activation markers on T cells may be extremely important, especially if there are concerns of generalized immune activation (e.g. adenovirus platforms). Notably, the vast majority of vaccine clinical trials in humans do not collect peripheral blood samples at early time points after vaccination where a better picture of immune activation might be found (this also is especially important for studies of innate immune responses). The relevance of HLA-DR and Ki67 (the classically used indicators of immune activation) on HIV-specific CD8+ T cells is doubtful, however, as HIV-specific CD8+ T cells in chronic infection rarely express these markers despite widespread immune activation in the host at this stage of infection (97). It is important to start anew in this domain and to validate the precision with which each activation marker depicts the real activation state of the host. Expanding the repertoire of such markers to create an ‘activation profile’ may accomplish this goal.
Inhibitory markers
The meaning of inhibitory markers in HIV infection remains unclear. PD-1 is an inhibitory receptor that is highly expressed on HIV-specific CD8+ T cells (98–100). The consequent interpretation was that PD-1 expression on HIV-specific CD8+ T cells is a harbinger of their exhaustion and also that blockade of this pathway could recover CD8+ T-cell function. However, EBV-specific CD8+ T cells also express high levels of PD-1, yet they remain functionally competent (98–100), being highly polyfunctional with tremendous proliferative potential. Now there are four additional markers of exhaustion that have been identified (101). In the LCMV murine system, functional exhaustion of antigen-specific CD8+ T cells is proportional to the simultaneous expression of increasing numbers of these different inhibitory receptors (101). It remains to be seen if a similar phenomenon is at play during HIV infection. In addition, do these receptors impede all functional outlets of antigen-specific CD8+ T cells or just some? It will be important to assess the degree of inhibition these receptors exert on the CD8+ T-cell functions most critical for HIV control/prevention and then to account for this impact on a candidate HIV vaccine.
Homing properties
A previously under-appreciated topic of HIV T-cell immunology is tissue trafficking potential. Since the vast majority of HIV infections occur sexually and mucosal tissues are the primary sites of replication during both acute and chronic infection (102), any vaccine-induced CD8+ T cell will need to be present at the mucosa to combat HIV. To do this, the CD8+ T cell will need to express the proper cell-surface determinants. The interpretation of homing marker expression, however, can be problematic. For example, the expression of α4β7 is commonly used on peripheral blood T cells to indicate gut trafficking potential. However, what does the presence or absence of this marker on peripheral blood T cells mean? Presumably, α4β7-expressing T cells should traffic out of the peripheral blood and into the mucosa, so their absence in the blood cannot be interpreted as a deficiency. If high levels of α4β7-expressing CD8+ T cells are found in the blood, does this mean there is a failure in trafficking ability? The obvious fix for this problem is to assay directly from the relevant tissues themselves, instead of inferring trafficking potential from the wrong compartments. Moreover, once examining T cells directly from tissues, such as the gut for example, why bother to again assess expression of trafficking markers? The cells are being sampled from the relevant tissue directly, so obviously they have already trafficked to the site. Thus, understanding which receptors or which combination of receptors are most relevant in the context of HIV is of paramount importance for HIV vaccine development, but the data must be carefully interpreted and corroborated with sampling from the relevant tissue(s).
Into the Twilight Zone: CAF and viral inhibition assays
CD8+ T cells have long been known to possess the ability to directly interfere with HIV viral replication, both through cytolytic and non-cytolytic mechanisms. We have discussed cytolytic mechanisms extensively up to this point. At issue first is the mystery of the non-cytolytic mechanism of viral inhibition commonly referred to as CAF (CD8+ T-cell antiviral factor)(103). We have a superb idea of what CAF is not [basically any cytokine, chemokine, or cytolytic factor that we know of (104)] but little idea of what it is, beyond some basic characteristics (104,105). Notably, CAF appears to be a soluble factor that does not require direct cell-cell contact (103). It remains of great importance to determine the identity of CAF.
Recently the importance of measuring CD8+ T-cell cytokine production and cytolytic activity against HIV-infected targets has come into question, with some laboratories deeming HIV-specific ‘viral inhibition’ as the only important property of HIV-specific CD8+ T cells (106). Viral inhibition assays are similar to the CAF assay, wherein the readout is the absence (or loss) of HIV viral replication when HIV-specific CD8+ T cells are present (107). In general, CD4+ T-cell target cells are infected exogenously with a primary strain of HIV and then HIV replication is allowed to proceed for several days. Then, autologous effector CD8+ T cells are added to the target cells and any ensuing reduction in p24 content over the next several days in the culture supernatant represents the neutralizing activity of the CD8+ T cells. Differential abilities of HIV-specific CD8+ T cells to mediate viral inhibition based on various disease stages or progression status have been reported, as well as the possibility that certain HIV antigens represent better targets for viral inhibition than others (107). The major caveat to viral inhibition assays is the fact that they require extensive in vitro culture, which eliminates a direct connection to the in vivo state of the responding CD8+ T cells (for reasons described earlier) and allows for tremendous variability in the assay. Furthermore, the mechanism responsible for this activity is crucially important to understand but remains as yet undetermined. In the viral inhibition assay, MHC class I blockade abrogated CD8+ T-cell-mediated suppression of HIV replication, and physical separation of the effector CD8+ and target CD4+ T cells by Transwell chambers largely reverses the observed inhibition (107). These data strongly suggest that the CD8+ T cells do not interfere with HIV replication via soluble mediators such as cytokines (separating this function from ‘CAF’). Rather, the findings are consistent with the killing of target cells, though this remains to be formally demonstrated. It remains to be determined if this assay correlates with conventional measurements of rapid perforin expression, or proliferation induced perforin upregulation and acquisition of killing ability. Regardless of the caveats, viral inhibition assays do provide a useful readout of HIV-specific CD8+ T-cell ‘efficacy’ and should be considered as an integral component of a multi-pronged approach to defining the protective capacity of vaccine-induced HIV-specific CD8+ T cells.
We can develop an experimental framework for the deduction of the antiviral potential of HIV-specific CD8+ T cells. First, informed immunoassays may be employed to characterize a specific mechanism(s) by which HIV-specific CD8+ T cells mediate their protection against HIV. Then, a viral suppression assay may be used to evaluate the HIV neutralizing potential of the HIV-specific CD8+ T cells. Such a portfolio of immunological tests may be the best approach towards identifying CD8+ T-cell correlates of HIV protection, as well as for appraising candidate HIV vaccines.
The human league
The only way we will ever experience an HIV vaccine epiphany is by increasing the number of studies conducted in humans. This is not a slight to the non-human primate community, as we have learned a tremendous amount of information about SIV pathogenesis in non-natural hosts, where the infection is pathogenic, as well as in natural hosts, where infection appears to be largely non-pathogenic. This certainly has led us to fundamental insights into the immunology and virology of lentiviral infections. In terms of vaccine design, however, the monkey model has not been fruitful. Should the SIV/rhesus macaque model be used as a gatekeeper for HIV vaccine candidates? This is a topic of great debate and will continue to be so, until a definitive appropriate model of SIV infection can be agreed upon.
There have been several studies in which rhesus macaques were immunized with candidate HIV vaccines that elicited strong CD8+ T-cell responses (8, 17, 22, 108–112). When challenged with virus, the monkeys displayed relative protection from disease but in almost all cases the CTL response could not provide sterilizing immunity. The type of challenge virus used, as well as dose and route, in these studies is a huge issue in the field. SHIV89.6P was the initial standard choice but was summarily rejected because of its unusual coreceptor tropism and high sensitivity to the host adaptive immune response (113). Pathogenic SIVmac239 and SIVmac251 imitate natural HIV infection quite well; however, they always resulted in infection and never attested to sterilizing cellular immunity. The reason for this could be because high doses of virus have been used, in order to make sure the challenge was of sufficient strength. This approach, however, may have been too rigorous, thereby underestimating the protective capacity of the vaccine-induced CTL responses. Recently, repeated low dose challenge experiments have demonstrated promising results; however, infection still inevitably ensued (110,114–116). Thus, the SIV/non-human primate model of HIV should be revamped for it to be relevant for HIV vaccine design. Conventional concepts encompassing SIV challenge viruses, doses, vaccination protocols, and primary endpoints must all be re-examined and validated to best represent HIV infection.
What have we learned about immune correlates to HIV and SIV by studying natural hosts, such as the sooty mangabey and African Green monkey? Unfortunately not a great deal, although we have gained inroads into SIV pathogenesis and host adaptation in these models (117). Nor will natural hosts provide us with tangible information for prophylactic vaccine development, as for the most part natural hosts have simply evolved to thwart the virus rather than combat it directly with immune responses. It is more reasonable to believe that these models could provide potential strategies for therapeutic vaccination through genetic manipulation, rather than immunological enhancement.
With respect to the Merck STEP trial, non-human primate challenge studies demonstrated that SIV Ad5 prototype vaccines led to control of viremia in some but not all challenge models (8, 17, 110, 118). Sterilizing immunity had not been demonstrated, yet the decision was to move forward because the results from the monkey trials were among the most promising to date. Either the monkey model lied, or we misinterpreted the data through our rose-colored spectacles. Whatever the case, the end result is positive; we now have a veritable human vaccine trial, along with the RV144 trial, that may serve as a valuable comparator. We find ourselves in an unprecedented position of being able to integrate information on HIV-specific CD8+ T-cell immunity from the laboratory, the monkey model, and human clinical trials.
To identify true immunological correlates of protection against HIV, we need to continue to perform HIV-specific research in humans, be it in the laboratory or in clinical trials, so as to augment our human database. We need to remain critical of our own science to ensure that we are making the best use of irreplaceable human samples and research funding, such that we continue to advance the field rather than our own agenda. We need to harness systems biology to provide key information on potential immune correlates and better integrate data from appropriate viral infections in the mouse to assist in this process. Samples from the phase I trials must be made readily available for basic research purposes outside of prescribed vaccine networks, and future clinical trials must collect sufficient specimens for extended immunological analyses, rather than relying on simplistic, convenient, or out-dated immune assays. A human database of immunological assessments will enable us to hone our monkey models, our laboratory tools and systems, and our expectations to better reflect the human condition.
Closing remark
To successfully engineer a prophylactic HIV vaccine, we need to identify the accurate immunological correlate(s) of protection. If CD8+ T cells are critical to this cause, in whole or in part, we must dramatically change our ideology. The use of IFN-γ and IL-2 as the dominant outputs of T-cell function in our research has stymied our progress, and, frankly, led us down dead-end avenues. A prophylactic T-cell based HIV vaccine must induce a response able to clear virally infected cells; it follows then that we must assess HIV-specific cytotoxicity as a primary function of vaccine-induced CD8+ T cells. That is not to say that other functions should not be measured; on the contrary, an immunological portfolio should be generated for the evaluation of vaccine efficacy, with perforin expression being amongst the foremost criteria for success. A multi-faceted evaluation, rather than relying on a single functional output, of HIV vaccine-specific CD8+ T cells offers the greatest odds of predicting vaccine efficacy. Furthermore, our vaccine ideology must integrate basic, non-human primate, and human research to hone our understanding of the process. This will only be accomplished by continued research in HIV-infected, HIV-exposed uninfected, and vaccinated humans, at the basic research level as well as within clinical trials.
References
- 1.UNAIDS. Report on the Global AIDS epidemic. Geneva: UNAIDS; 2009. [Google Scholar]
- 2.Buchbinder SP, et al. Efficacy assessment of a cell-mediated immunity HIV-1 vaccine (the Step Study): a double-blind, randomised, placebo-controlled, test-of-concept trial. Lancet. 2008;372:1881–1893. doi: 10.1016/S0140-6736(08)61591-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rerks-Ngarm S, et al. Vaccination with ALVAC and AIDSVAX to prevent HIV-1 infection in Thailand. N Engl J Med. 2009;361:2209–2220. doi: 10.1056/NEJMoa0908492. [DOI] [PubMed] [Google Scholar]
- 4.Koup RA, et al. Temporal association of cellular immune responses with the initial control of viremia in primary human immunodeficiency virus type 1 syndrome. J Virol. 1994;68:4650–4655. doi: 10.1128/jvi.68.7.4650-4655.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Borrow P, Lewicki H, Hahn BH, Shaw GM, Oldstone MB. Virus-specific CD8+ cytotoxic T-lymphocyte activity associated with control of viremia in primary human immunodeficiency virus type 1 infection. J Virol. 1994;68:6103–6110. doi: 10.1128/jvi.68.9.6103-6110.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jin X, et al. Dramatic rise in plasma viremia after CD8(+) T cell depletion in simian immunodeficiency virus-infected macaques. J Exp Med. 1999;189:991–998. doi: 10.1084/jem.189.6.991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schmitz JE, et al. Control of viremia in simian immunodeficiency virus infection by CD8+ lymphocytes. Science. 1999;283:857–860. doi: 10.1126/science.283.5403.857. [DOI] [PubMed] [Google Scholar]
- 8.Shiver JW, et al. Replication-incompetent adenoviral vaccine vector elicits effective anti-immunodeficiency-virus immunity. Nature. 2002;415:331–335. doi: 10.1038/415331a. [DOI] [PubMed] [Google Scholar]
- 9.Burton DR, et al. Public health. A sound rationale needed for phase III HIV-1 vaccine trials. Science. 2004;303:316. doi: 10.1126/science.1094620. [DOI] [PubMed] [Google Scholar]
- 10.Administration UFaD. Vaccines Licensed for Immunization and Distribution in the US with Supporting Documents. 2010;06/03/2010 Services USDoHH; [Google Scholar]
- 11.Carrington M, O’Brien SJ. The influence of HLA genotype on AIDS. Annu Rev Med. 2003;54:535–551. doi: 10.1146/annurev.med.54.101601.152346. [DOI] [PubMed] [Google Scholar]
- 12.Goonetilleke N, et al. The first T cell response to transmitted/founder virus contributes to the control of acute viremia in HIV-1 infection. J Exp Med. 2009;206:1253–1272. doi: 10.1084/jem.20090365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Salazar-Gonzalez JF, et al. Genetic identity, biological phenotype, and evolutionary pathways of transmitted/founder viruses in acute and early HIV-1 infection. J Exp Med. 2009;206:1273–1289. doi: 10.1084/jem.20090378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Allen TM, et al. Tat-specific cytotoxic T lymphocytes select for SIV escape variants during resolution of primary viraemia. Nature. 2000;407:386–390. doi: 10.1038/35030124. [DOI] [PubMed] [Google Scholar]
- 15.O’Connor DH, et al. Acute phase cytotoxic T lymphocyte escape is a hallmark of simian immunodeficiency virus infection. Nat Med. 2002;8:493–499. doi: 10.1038/nm0502-493. [DOI] [PubMed] [Google Scholar]
- 16.Priddy FH, et al. Safety and immunogenicity of a replication-incompetent adenovirus type 5 HIV-1 clade B gag/pol/nef vaccine in healthy adults. Clin Infect Dis. 2008;46:1769–1781. doi: 10.1086/587993. [DOI] [PubMed] [Google Scholar]
- 17.Casimiro DR, et al. Attenuation of simian immunodeficiency virus SIVmac239 infection by prophylactic immunization with dna and recombinant adenoviral vaccine vectors expressing Gag. J Virol. 2005;79:15547–15555. doi: 10.1128/JVI.79.24.15547-15555.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Catanzaro AT, et al. Phase 1 safety and immunogenicity evaluation of a multiclade HIV-1 candidate vaccine delivered by a replication-defective recombinant adenovirus vector. J Infect Dis. 2006;194:1638–1649. doi: 10.1086/509258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boyer JD, et al. Vaccination of seronegative volunteers with a human immunodeficiency virus type 1 env/rev DNA vaccine induces antigen-specific proliferation and lymphocyte production of beta-chemokines. J Infect Dis. 2000;181:476–483. doi: 10.1086/315229. [DOI] [PubMed] [Google Scholar]
- 20.Jaoko W, et al. Safety and immunogenicity of recombinant low-dosage HIV-1 A vaccine candidates vectored by plasmid pTHr DNA or modified vaccinia virus Ankara (MVA) in humans in East Africa. Vaccine. 2008;26:2788–2795. doi: 10.1016/j.vaccine.2008.02.071. [DOI] [PubMed] [Google Scholar]
- 21.Nitayaphan S, et al. Safety and immunogenicity of an HIV subtype B and E prime-boost vaccine combination in HIV-negative Thai adults. J Infect Dis. 2004;190:702–706. doi: 10.1086/422258. [DOI] [PubMed] [Google Scholar]
- 22.Belshe RB, et al. Induction of immune responses to HIV-1 by canarypox virus (ALVAC) HIV-1 and gp120 SF-2 recombinant vaccines in uninfected volunteers. NIAID AIDS Vaccine Evaluation Group. AIDS. 1998;12:2407–2415. doi: 10.1097/00002030-199818000-00009. [DOI] [PubMed] [Google Scholar]
- 23.AIDS Evaluation Group 022 Protocol Team. Cellular and humoral immune responses to a canarypox vaccine containing human immunodeficiency virus type 1 Env, Gag, and Pro in combination with rgp120. J Infect Dis. 2001;183:563–570. doi: 10.1086/318523. [DOI] [PubMed] [Google Scholar]
- 24.Russell ND, et al. Phase 2 study of an HIV-1 canarypox vaccine (vCP1452) alone and in combination with rgp120: negative results fail to trigger a phase 3 correlates trial. J Acquir Immune Defic Syndr. 2007;44:203–212. doi: 10.1097/01.qai.0000248356.48501.ff. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Betts MR, et al. HIV nonprogressors preferentially maintain highly functional HIV-specific CD8+ T cells. Blood. 2006;107:4781–4789. doi: 10.1182/blood-2005-12-4818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Darrah PA, et al. IL-10 production differentially influences the magnitude, quality, and protective capacity of Th1 responses depending on the vaccine platform. J Exp Med. 2010;207:1421–1433. doi: 10.1084/jem.20092532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Darrah PA, et al. Multifunctional TH1 cells define a correlate of vaccine-mediated protection against Leishmania major. Nat Med. 2007;13:843–850. doi: 10.1038/nm1592. [DOI] [PubMed] [Google Scholar]
- 28.Precopio ML, et al. Immunization with vaccinia virus induces polyfunctional and phenotypically distinctive CD8(+) T cell responses. J Exp Med. 2007;204:1405–1416. doi: 10.1084/jem.20062363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Almeida JR, et al. Antigen sensitivity is a major determinant of CD8+ T-cell polyfunctionality and HIV-suppressive activity. Blood. 2009;113:6351–6360. doi: 10.1182/blood-2009-02-206557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Horton H, et al. Preservation of T cell proliferation restricted by protective HLA alleles is critical for immune control of HIV-1 infection. J Immunol. 2006;177:7406–7415. doi: 10.4049/jimmunol.177.10.7406. [DOI] [PubMed] [Google Scholar]
- 31.Lichterfeld M, et al. Loss of HIV-1-specific CD8+ T cell proliferation after acute HIV-1 infection and restoration by vaccine-induced HIV-1-specific CD4+ T cells. J Exp Med. 2004;200:701–712. doi: 10.1084/jem.20041270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Migueles SA, et al. Lytic granule loading of CD8+ T cells is required for HIV-infected cell elimination associated with immune control. Immunity. 2008;29:1009–1021. doi: 10.1016/j.immuni.2008.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Migueles SA, et al. HIV-specific CD8+ T cell proliferation is coupled to perforin expression and is maintained in nonprogressors. Nat Immunol. 2002;3:1061–1068. doi: 10.1038/ni845. [DOI] [PubMed] [Google Scholar]
- 34.Rehr M, et al. Emergence of polyfunctional CD8+ T cells after prolonged suppression of human immunodeficiency virus replication by antiretroviral therapy. J Virol. 2008;82:3391–3404. doi: 10.1128/JVI.02383-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ortiz GM, et al. Structured antiretroviral treatment interruptions in chronically HIV-1-infected subjects. Proc Natl Acad Sci USA. 2001;98:13288–13293. doi: 10.1073/pnas.221452198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Casazza JP, Betts MR, Picker LJ, Koup RA. Decay kinetics of human immunodeficiency virus-specific CD8+ T cells in peripheral blood after initiation of highly active antiretroviral therapy. J Virol. 2001;75:6508–6516. doi: 10.1128/JVI.75.14.6508-6516.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Davey RT, Jr, et al. HIV-1 and T cell dynamics after interruption of highly active antiretroviral therapy (HAART) in patients with a history of sustained viral suppression. Proc Natl Acad Sci USA. 1999;96:15109–15114. doi: 10.1073/pnas.96.26.15109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Harari A, et al. Skewed association of polyfunctional antigen-specific CD8 T cell populations with HLA-B genotype. Proc Natl Acad Sci USA. 2007;104:16233–16238. doi: 10.1073/pnas.0707570104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Emu B, et al. HLA class I-restricted T-cell responses may contribute to the control of human immunodeficiency virus infection, but such responses are not always necessary for long-term virus control. J Virol. 2008;82:5398–5407. doi: 10.1128/JVI.02176-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Makedonas G, et al. HIV-specific CD8 T-cell activity in uninfected injection drug users is associated with maintenance of seronegativity. AIDS. 2002;16:1595–1602. doi: 10.1097/00002030-200208160-00004. [DOI] [PubMed] [Google Scholar]
- 41.Kaul R, et al. CD8(+) lymphocytes respond to different HIV epitopes in seronegative and infected subjects. J Clin Invest. 2001;107:1303–1310. doi: 10.1172/JCI12433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rowland-Jones S, et al. HIV-specific cytotoxic T-cells in HIV-exposed but uninfected Gambian women. Nat Med. 1995;1:59–64. doi: 10.1038/nm0195-59. [DOI] [PubMed] [Google Scholar]
- 43.Scott-Algara D, et al. Cutting edge: increased NK cell activity in HIV-1-exposed but uninfected Vietnamese intravascular drug users. J Immunol. 2003;171:5663–5667. doi: 10.4049/jimmunol.171.11.5663. [DOI] [PubMed] [Google Scholar]
- 44.Boulet S, et al. Increased proportion of KIR3DS1 homozygotes in HIV-exposed uninfected individuals. AIDS. 2008;22:595–599. doi: 10.1097/QAD.0b013e3282f56b23. [DOI] [PubMed] [Google Scholar]
- 45.Miura T, et al. Impaired replication capacity of acute/early viruses in persons who become HIV controllers. J Virol. 2010;84:7581–7591. doi: 10.1128/JVI.00286-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ogg GS, et al. Quantitation of HIV-1-specific cytotoxic T lymphocytes and plasma load of viral RNA. Science. 1998;279:2103–2106. doi: 10.1126/science.279.5359.2103. [DOI] [PubMed] [Google Scholar]
- 47.Betts MR, et al. Analysis of total human immunodeficiency virus (HIV)-specific CD4(+) and CD8(+) T-cell responses: relationship to viral load in untreated HIV infection. J Virol. 2001;75:11983–11991. doi: 10.1128/JVI.75.24.11983-11991.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dunbar PR, et al. Cutting edge: rapid cloning of tumor-specific CTL suitable for adoptive immunotherapy of melanoma. J Immunol. 1999;162:6959–6962. [PubMed] [Google Scholar]
- 49.Dunbar PR, et al. Direct isolation, phenotyping and cloning of low-frequency antigen-specific cytotoxic T lymphocytes from peripheral blood. Curr Biol. 1998;8:413–416. doi: 10.1016/s0960-9822(98)70161-7. [DOI] [PubMed] [Google Scholar]
- 50.Dalod M, et al. Weak anti-HIV CD8(+) T-cell effector activity in HIV primary infection. J Clin Invest. 1999;104:1431–1439. doi: 10.1172/JCI7162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Alter G, et al. Human immunodeficiency virus (HIV)-specific effector CD8 T cell activity in patients with primary HIV infection. J Infect Dis. 2002;185:755–765. doi: 10.1086/339338. [DOI] [PubMed] [Google Scholar]
- 52.Altfeld M, et al. Cellular immune responses and viral diversity in individuals treated during acute and early HIV-1 infection. J Exp Med. 2001;193:169–180. doi: 10.1084/jem.193.2.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zimmerli SC, et al. HIV-1-specific IFN-gamma/IL-2-secreting CD8 T cells support CD4-independent proliferation of HIV-1-specific CD8 T cells. Proc Natl Acad Sci USA. 2005;102:7239–7244. doi: 10.1073/pnas.0502393102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cellerai C, et al. Proliferation capacity and cytotoxic activity are mediated by functionally and phenotypically distinct virus-specific CD8 T cells defined by interleukin-7R{alpha} (CD127) and perforin expression. J Virol. 2010;84:3868–3878. doi: 10.1128/JVI.02565-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Makedonas G, et al. Perforin and IL-2 upregulation define qualitative differences among highly functional virus-specific human CD8 T cells. PLoS Pathog. 2010;6:e1000798. doi: 10.1371/journal.ppat.1000798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.De Jong JC, et al. Adenoviruses from human immunodeficiency virus-infected individuals, including two strains that represent new candidate serotypes Ad50 and Ad51 of species B1 and D, respectively. J Clin Microbiol. 1999;37:3940–3945. doi: 10.1128/jcm.37.12.3940-3945.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Garnett CT, Erdman D, Xu W, Gooding LR. Prevalence and quantitation of species C adenovirus DNA in human mucosal lymphocytes. J Virol. 2002;76:10608–10616. doi: 10.1128/JVI.76.21.10608-10616.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wherry EJ, Ahmed R. Memory CD8 T-cell differentiation during viral infection. J Virol. 2004;78:5535–5545. doi: 10.1128/JVI.78.11.5535-5545.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Belz GT, Doherty PC. Virus-specific and bystander CD8+ T-cell proliferation in the acute and persistent phases of a gammaherpesvirus infection. J Virol. 2001;75:4435–4438. doi: 10.1128/JVI.75.9.4435-4438.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Doherty PC, Christensen JP, Belz GT, Stevenson PG, Sangster MY. Dissecting the host response to a gamma-herpesvirus. Philos Trans R Soc Lond B Biol Sci. 2001;356:581–593. doi: 10.1098/rstb.2000.0786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gillespie GM, et al. Functional heterogeneity and high frequencies of cytomegalovirus-specific CD8(+) T lymphocytes in healthy seropositive donors. J Virol. 2000;74:8140–8150. doi: 10.1128/jvi.74.17.8140-8150.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Callan MF. The evolution of antigen-specific CD8+ T cell responses after natural primary infection of humans with Epstein-Barr virus. Viral Immunol. 2003;16:3–16. doi: 10.1089/088282403763635401. [DOI] [PubMed] [Google Scholar]
- 63.Guidotti LG, et al. Intracellular inactivation of the hepatitis B virus by cytotoxic T lymphocytes. Immunity. 1996;4:25–36. doi: 10.1016/s1074-7613(00)80295-2. [DOI] [PubMed] [Google Scholar]
- 64.Moriyama T, et al. Immunobiology and pathogenesis of hepatocellular injury in hepatitis B virus transgenic mice. Science. 1990;248:361–364. doi: 10.1126/science.1691527. [DOI] [PubMed] [Google Scholar]
- 65.Lechner F, et al. Analysis of successful immune responses in persons infected with hepatitis C virus. J Exp Med. 2000;191:1499–1512. doi: 10.1084/jem.191.9.1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Thimme R, et al. Viral and immunological determinants of hepatitis C virus clearance, persistence, and disease. Proc Natl Acad Sci USA. 2002;99:15661–15668. doi: 10.1073/pnas.202608299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Heusel JW, Wesselschmidt RL, Shresta S, Russell JH, Ley TJ. Cytotoxic lymphocytes require granzyme B for the rapid induction of DNA fragmentation and apoptosis in allogeneic target cells. Cell. 1994;76:977–987. doi: 10.1016/0092-8674(94)90376-x. [DOI] [PubMed] [Google Scholar]
- 68.Peters PJ, et al. Cytotoxic T lymphocyte granules are secretory lysosomes, containing both perforin and granzymes. J Exp Med. 1991;173:1099–1109. doi: 10.1084/jem.173.5.1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bots M, Medema JP. Granzymes at a glance. J Cell Sci. 2006;119:5011–5014. doi: 10.1242/jcs.03239. [DOI] [PubMed] [Google Scholar]
- 70.Bolitho P, Voskoboinik I, Trapani JA, Smyth MJ. Apoptosis induced by the lymphocyte effector molecule perforin. Curr Opin Immunol. 2007;19:339–347. doi: 10.1016/j.coi.2007.04.007. [DOI] [PubMed] [Google Scholar]
- 71.Voskoboinik I, Smyth MJ, Trapani JA. Perforin-mediated target-cell death and immune homeostasis. Nat Rev Immunol. 2006;6:940–952. doi: 10.1038/nri1983. [DOI] [PubMed] [Google Scholar]
- 72.Walker BD, et al. HIV-specific cytotoxic T lymphocytes in seropositive individuals. Nature. 1987;328:345–348. doi: 10.1038/328345a0. [DOI] [PubMed] [Google Scholar]
- 73.Andersson J, et al. Perforin is not co-expressed with granzyme A within cytotoxic granules in CD8 T lymphocytes present in lymphoid tissue during chronic HIV infection. AIDS. 1999;13:1295–1303. doi: 10.1097/00002030-199907300-00005. [DOI] [PubMed] [Google Scholar]
- 74.Zhang D, et al. Most antiviral CD8 T cells during chronic viral infection do not express high levels of perforin and are not directly cytotoxic. Blood. 2003;101:226–235. doi: 10.1182/blood-2002-03-0791. [DOI] [PubMed] [Google Scholar]
- 75.Appay V, et al. HIV-specific CD8(+) T cells produce antiviral cytokines but are impaired in cytolytic function. J Exp Med. 2000;192:63–75. doi: 10.1084/jem.192.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Safrit JT, Andrews CA, Zhu T, Ho DD, Koup RA. Characterization of human immunodeficiency virus type 1-specific cytotoxic T lymphocyte clones isolated during acute seroconversion: recognition of autologous virus sequences within a conserved immunodominant epitope. J Exp Med. 1994;179:463–472. doi: 10.1084/jem.179.2.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hersperger AR, et al. Perforin expression directly ex vivo by HIV-specific CD8 T-cells is a correlate of HIV elite control. PLoS Pathog. 2010;6:e1000917. doi: 10.1371/journal.ppat.1000917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Migueles SA, et al. Defective human immunodeficiency virus-specific CD8+ T-cell polyfunctionality, proliferation, and cytotoxicity are not restored by antiretroviral therapy. J Virol. 2009;83:11876–11889. doi: 10.1128/JVI.01153-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Saez-Cirion A, et al. HIV controllers exhibit potent CD8 T cell capacity to suppress HIV infection ex vivo and peculiar cytotoxic T lymphocyte activation phenotype. Proc Natl Acad Sci USA. 2007;104:6776–6781. doi: 10.1073/pnas.0611244104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rinaldo C, et al. High levels of anti-human immunodeficiency virus type 1 (HIV-1) memory cytotoxic T-lymphocyte activity and low viral load are associated with lack of disease in HIV-1-infected long-term nonprogressors. J Virol. 1995;69:5838–5842. doi: 10.1128/jvi.69.9.5838-5842.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Betts MR, et al. Sensitive and viable identification of antigen-specific CD8+ T cells by a flow cytometric assay for degranulation. J Immunol Methods. 2003;281:65–78. doi: 10.1016/s0022-1759(03)00265-5. [DOI] [PubMed] [Google Scholar]
- 82.Trambas CM, Griffiths GM. Delivering the kiss of death. Nat Immunol. 2003;4:399–403. doi: 10.1038/ni0503-399. [DOI] [PubMed] [Google Scholar]
- 83.Wolint P, Betts MR, Koup RA, Oxenius A. Immediate cytotoxicity but not degranulation distinguishes effector and memory subsets of CD8+ T cells. J Exp Med. 2004;199:925–936. doi: 10.1084/jem.20031799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hersperger AR, Makedonas G, Betts MR. Flow cytometric detection of perforin upregulation in human CD8 T cells. Cytometry A. 2008;73:1050–1057. doi: 10.1002/cyto.a.20596. [DOI] [PubMed] [Google Scholar]
- 85.Uellner R, et al. Perforin is activated by a proteolytic cleavage during biosynthesis which reveals a phospholipid-binding C2 domain. EMBO J. 1997;16:7287–7296. doi: 10.1093/emboj/16.24.7287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.White L, et al. Differential effects of IL-21 and IL-15 on perforin expression, lysosomal degranulation, and proliferation in CD8 T cells of patients with human immunodeficiency virus-1 (HIV) Blood. 2007;109:3873–3880. doi: 10.1182/blood-2006-09-045278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Meng Y, Harlin H, O’Keefe JP, Gajewski TF. Induction of cytotoxic granules in human memory CD8+ T cell subsets requires cell cycle progression. J Immunol. 2006;177:1981–1987. doi: 10.4049/jimmunol.177.3.1981. [DOI] [PubMed] [Google Scholar]
- 88.Makedonas G, et al. Rapid up-regulation and granule-independent transport of perforin to the immunological synapse define a novel mechanism of antigen-specific CD8+ T cell cytotoxic activity. J Immunol. 2009;182:5560–5569. doi: 10.4049/jimmunol.0803945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Miller JD, et al. Human effector and memory CD8+ T cell responses to smallpox and yellow fever vaccines. Immunity. 2008;28:710–722. doi: 10.1016/j.immuni.2008.02.020. [DOI] [PubMed] [Google Scholar]
- 90.Almeida JR, et al. Superior control of HIV-1 replication by CD8+ T cells is reflected by their avidity, polyfunctionality, and clonal turnover. J Exp Med. 2007;204:2473–2485. doi: 10.1084/jem.20070784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Walker LM, Burton DR. Rational antibody-based HIV-1 vaccine design: current approaches and future directions. Curr Opin Immunol. 2010;22:358–366. doi: 10.1016/j.coi.2010.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Appay V, et al. Memory CD8+ T cells vary in differentiation phenotype in different persistent virus infections. Nat Med. 2002;8:379–385. doi: 10.1038/nm0402-379. [DOI] [PubMed] [Google Scholar]
- 93.Gamadia LE, et al. Primary immune responses to human CMV: a critical role for IFN-gamma-producing CD4+ T cells in protection against CMV disease. Blood. 2003;101:2686–2692. doi: 10.1182/blood-2002-08-2502. [DOI] [PubMed] [Google Scholar]
- 94.Faint JM, et al. Memory T cells constitute a subset of the human CD8+CD45RA+ pool with distinct phenotypic and migratory characteristics. J Immunol. 2001;167:212–220. doi: 10.4049/jimmunol.167.1.212. [DOI] [PubMed] [Google Scholar]
- 95.Wills MR, et al. Identification of naive or antigen-experienced human CD8(+) T cells by expression of costimulation and chemokine receptors: analysis of the human cytomegalovirus-specific CD8(+) T cell response. J Immunol. 2002;168:5455–5464. doi: 10.4049/jimmunol.168.11.5455. [DOI] [PubMed] [Google Scholar]
- 96.Roos MT, et al. Changes in the composition of circulating CD8+ T cell subsets during acute epstein-barr and human immunodeficiency virus infections in humans. J Infect Dis. 2000;182:451–458. doi: 10.1086/315737. [DOI] [PubMed] [Google Scholar]
- 97.Papagno L, et al. Immune activation and CD8+ T-cell differentiation towards senescence in HIV-1 infection. PLoS Biol. 2004;2:E20. doi: 10.1371/journal.pbio.0020020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Petrovas C, et al. PD-1 is a regulator of virus-specific CD8+ T cell survival in HIV infection. J Exp Med. 2006;203:2281–2292. doi: 10.1084/jem.20061496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Day CL, et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature. 2006;443:350–354. doi: 10.1038/nature05115. [DOI] [PubMed] [Google Scholar]
- 100.Trautmann L, et al. Upregulation of PD-1 expression on HIV-specific CD8+ T cells leads to reversible immune dysfunction. Nat Med. 2006;12:1198–1202. doi: 10.1038/nm1482. [DOI] [PubMed] [Google Scholar]
- 101.Blackburn SD, et al. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat Immunol. 2009;10:29–37. doi: 10.1038/ni.1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Brenchley JM, Douek DC. The mucosal barrier and immune activation in HIV pathogenesis. Curr Opin HIV AIDS. 2008;3:356–361. doi: 10.1097/COH.0b013e3282f9ae9c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Levy JA. The search for the CD8+ cell anti-HIV factor (CAF) Trends Immunol. 2003;24:628–632. doi: 10.1016/j.it.2003.10.005. [DOI] [PubMed] [Google Scholar]
- 104.Mackewicz CE, Ortega H, Levy JA. Effect of cytokines on HIV replication in CD4+ lymphocytes: lack of identity with the CD8+ cell antiviral factor. Cell Immunol. 1994;153:329–343. doi: 10.1006/cimm.1994.1032. [DOI] [PubMed] [Google Scholar]
- 105.Mackewicz CE, et al. Lack of the CD8+ cell anti-HIV factor in CD8+ cell granules. Blood. 2003;102:180–183. doi: 10.1182/blood-2002-10-3034. [DOI] [PubMed] [Google Scholar]
- 106.Vojnov L, et al. Effective simian immunodeficiency virus-specific CD8+ T cells lack an easily detectable, shared characteristic. J Virol. 2010;84:753–764. doi: 10.1128/JVI.01596-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Spentzou A, et al. Viral inhibition assay: a CD8 T cell neutralization assay for use in clinical trials of HIV-1 vaccine candidates. J Infect Dis. 2010;201:720–729. doi: 10.1086/650492. [DOI] [PubMed] [Google Scholar]
- 108.Barouch DH, et al. Control of viremia and prevention of clinical AIDS in rhesus monkeys by cytokine-augmented DNA vaccination. Science. 2000;290:486–492. doi: 10.1126/science.290.5491.486. [DOI] [PubMed] [Google Scholar]
- 109.Hansen SG, et al. Effector memory T cell responses are associated with protection of rhesus monkeys from mucosal simian immunodeficiency virus challenge. Nat Med. 2009;15:293–299. doi: 10.1038/nm.1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wilson NA, et al. Vaccine-induced cellular immune responses reduce plasma viral concentrations after repeated low-dose challenge with pathogenic simian immunodeficiency virus SIVmac239. J Virol. 2006;80:5875–5885. doi: 10.1128/JVI.00171-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Liu J, et al. Immune control of an SIV challenge by a T-cell-based vaccine in rhesus monkeys. Nature. 2009;457:87–91. doi: 10.1038/nature07469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Evans DT, et al. Immunization of macaques with single-cycle simian immunodeficiency virus (SIV) stimulates diverse virus-specific immune responses and reduces viral loads after challenge with SIVmac239. J Virol. 2005;79:7707–7720. doi: 10.1128/JVI.79.12.7707-7720.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Feinberg MB, Moore JP. AIDS vaccine models: challenging challenge viruses. Nat Med. 2002;8:207–210. doi: 10.1038/nm0302-207. [DOI] [PubMed] [Google Scholar]
- 114.Keele BF, et al. Low-dose rectal inoculation of rhesus macaques by SIVsmE660 or SIVmac251 recapitulates human mucosal infection by HIV-1. J Exp Med. 2009;206:1117–1134. doi: 10.1084/jem.20082831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Reynolds MR, et al. Macaques vaccinated with SIVmac239Δnef delay Acquisition and Control Replication after Repeated Low Dose Heterologous SIV Challenge. J Virol. 2010;84:9190–9199. doi: 10.1128/JVI.00041-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Willer DO, et al. Multi-low-dose mucosal simian immunodeficiency virus SIVmac239 challenge of cynomolgus macaques immunized with “hyperattenuated” SIV constructs. J Virol. 2010;84:2304–2317. doi: 10.1128/JVI.01995-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Sodora DL, et al. Toward an AIDS vaccine: lessons from natural simian immunodeficiency virus infections of African nonhuman primate hosts. Nat Med. 2009;15:861–865. doi: 10.1038/nm.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Liang X, et al. Vectored Gag and Env but not Tat show efficacy against simian-human immunodeficiency virus 89.6P challenge in Mamu-A*01-negative rhesus monkeys. J Virol. 2005;79:12321–12331. doi: 10.1128/JVI.79.19.12321-12331.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]