

Abstract
Motivation: Genome-wide association studies (GWASs) have been widely used to map loci contributing to variation in complex traits and risk of diseases in humans. Accurate specification of familial relationships is crucial for family-based GWAS, as well as in population-based GWAS with unknown (or unrecognized) family structure. The family structure in a GWAS should be routinely investigated using the SNP data prior to the analysis of population structure or phenotype. Existing algorithms for relationship inference have a major weakness of estimating allele frequencies at each SNP from the entire sample, under a strong assumption of homogeneous population structure. This assumption is often untenable.
Results: Here, we present a rapid algorithm for relationship inference using high-throughput genotype data typical of GWAS that allows the presence of unknown population substructure. The relationship of any pair of individuals can be precisely inferred by robust estimation of their kinship coefficient, independent of sample composition or population structure (sample invariance). We present simulation experiments to demonstrate that the algorithm has sufficient power to provide reliable inference on millions of unrelated pairs and thousands of relative pairs (up to 3rd-degree relationships). Application of our robust algorithm to HapMap and GWAS datasets demonstrates that it performs properly even under extreme population stratification, while algorithms assuming a homogeneous population give systematically biased results. Our extremely efficient implementation performs relationship inference on millions of pairs of individuals in a matter of minutes, dozens of times faster than the most efficient existing algorithm known to us.
Availability: Our robust relationship inference algorithm is implemented in a freely available software package, KING, available for download at http://people.virginia.edu/∼wc9c/KING.
Contact: wmchen@virginia.edu
Supplementary information: Supplementary data are available at Bioinformatics online.
1 INTRODUCTION
Genome-wide association studies (GWASs) have been widely used to identify common variants that contribute to variation in complex human phenotypes and diseases. Pedigree integrity is crucial to the performance of family-based GWA, as well as in population-based data with unknown family structure. High-throughput genotyping performed in a GWAS presents new opportunities for pedigree error detection using millions of SNPs to assess the degree of relationship between a pair of individuals. With these opportunities come the challenges of accounting for linkage disequilibrium among typed markers, while managing computational resources to analyze the large amount of genotype data. Compared to linkage studies, association studies also require consideration of population substructure, misreported race and ethnicity and unreported familial relationships among samples recruited as unrelated individuals.
One well-developed approach for relationship inference in linkage studies offers fully parametric methods for sib pairs (Boehnke and Cox, 1997) and extensions to general pedigrees (McPeek and Sun, 2000) using hidden Markov models (HMM) to calculate multipoint marker probabilities, incorporated into a likelihood framework to assess evidence in support of particular pair-wise relationships. In considering full multipoint marker probabilities, computational demands increase with the number of markers genotyped, making analysis of GWAS SNPs for all pairs of individuals prohibitive. A simple method, known as GRR (Graphical Representation of Relationship errors; Abecasis et al., 2001), uses clustering of readily available non-parametric estimates for mean and standard deviation (SD) of identical by state (IBS) statistics at a series of markers for each pair of relatives. GRR identifies outliers of clusters as relationship errors. Performance of the clustering algorithm used to classify relative pairs depends on the panel of genetic markers, the underlying allele frequencies of genetic markers for different individuals, and the number of individuals genotyped. If certain pairs of individuals do not cluster—either due to limitations in sample size or due to the different underlying allele frequencies between different pairs (e.g. in the presence of population structure)—GRR fails to detect the pedigree errors. One efficient implementation of relationship inference in GWAS data is available in a widely used software package, PLINK (Purcell et al., 2007). The identical-by-descent (IBD) statistics between each pair of individuals are estimated using the average of IBS and the estimation of sample-level allele frequencies at each SNP according to Hardy–Weinberg Equilibrium (HWE) assumptions.
All popular algorithms for relationship inference depend on reliable estimates of allele frequencies at each SNP, assuming a homogeneous population without stratification (Abecasis et al., 2001; Boehnke and Cox, 1997; Lynch and Ritland, 1999; McPeek and Sun, 2000; Purcell et al., 2007). Recent GWAS analytic advances for association mapping have incorporated the presence of unknown family and population structure (Choi et al., 2009; Kang et al., 2010; Thornton and McPeek, 2010; Yang et al., 2010; Zhang et al., 2010); however, algorithms to estimate family relationships remain based on the assumption of population homogeneity. In samples with undetected population substructure, this strong assumption of population homogeneity leads to biased results, systematically inflating the degree of relatedness among individuals of the same racial group.
Current approaches to relationship and population structure inference are somewhat circular. The relationship inference relies on correct specification of a homogeneous subpopulation (Purcell et al., 2007), while the detection of population structure relies on the correct identification of unrelated individuals (Zhu et al., 2008). In addition to the non-robustness to the population structure, existing approaches do not apply to small datasets, e.g. for comparison of a single pair of individuals, or relationship inference on a single pedigree.
We present a novel framework for relationship inference, Kinship-based INference for Genome-wide association studies (KING), together with a rapid algorithm for relationship inference appropriate for use on samples with thousands of individuals genotyped at millions of SNPs from autosomes, consistent with a scale typically achieved in a GWAS. Within this framework we present two methods: (i) KING-homo, derived under the assumption of population homogeneity and (ii) KING-robust that provides robust relationship inference in the presence of population substructure. The estimated pedigree information provided by KING (such as kinship coefficients) can be used to verify relationships, reconstruct pedigrees and conduct genetic association tests without relying on self-reported pedigree information. Our computationally efficient and flexible approach allows automated pedigree error detection, and is amenable to datasets involving a very small number of individuals, as encountered in forensic DNA analysis.
2 METHODS
Consider two individuals, indexed by i and j. Let ϕij denote the kinship coefficient, defined as the probability that two alleles sampled at random from two individuals are identical by descent, and π0ij, π1ij and π2ij denote the probability that the two individuals share zero, one and two alleles identical by descent, respectively. Table 1 lists values of ϕij and π0ij for relative pairs, including monozygotic twins, parent-offspring pairs, sibling pairs, 2nd-degree relative pairs (such as half-sibs, avuncular pairs and grandparent-grandchild pairs), 3rd-degree relative pairs (such as first cousins), and unrelated pairs. Note that the kinship coefficient is a function of IBD-sharing statistics with relationship 2ϕij = π1ij /2+π2ij. Inference criteria presented in Table 1 are derived using powers of 2, with the basis that this is the natural scale of the kinship and zero-IBD sharing statistics. In Section 3, we see that these inference criteria work well in practice.
Table 1.
Relationship inference criteria based on estimating kinship coefficients (ϕ) and probability of zero IBD sharing (π0)
| Relationship | ϕ | Inference criteria | π0 | Inference criteria |
|---|---|---|---|---|
| Monozygotic twin |  |
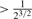 |
0 | <0.1 |
| Parent–offspring |  |
(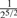 , ,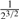
|
0 | <0.1 |
| Full sib |  |
(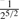 , ,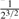
|
 |
(0.1,0.365) |
| 2nd Degree |  |
(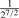 , ,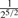
|
 |
(0.365,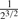
|
| 3rd Degree | 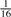 |
(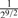 , ,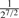
|
 |
(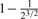 , ,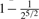
|
| Unrelated | 0 | 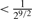 |
1 | 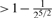 |
2.1 Relationship inference in a homogeneous population
We first summarize existing methods that allow relationship inference under the assumption of a homogeneous population. Assume p is the frequency of a reference allele (with label A) at a SNP, and the number of alleles IBS between individuals i and j is IBSij. Since only IBDij = 0 (not IBDij = 1 or 2) can result in IBSij = 0 (i.e. the pair of individuals has genotypes AA and aa), the expected proportion of SNPs with zero IBS can be specified assuming HWE:
| (1) |
This leads to the estimator
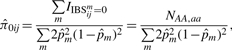 |
(2) |
where IIBSijm=0 is an indicator of whether the pair of individuals does not share any alleles at the m-th SNP, NAA,aa is the total number of SNPs at which the genotypes of the pair of individuals are different homozygotes, m indexes SNPs excluding those with missing genotypes in either individual of the pair, and allele frequency 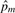 at the m-th SNP is estimated from the genotype frequencies in the entire sample as
at the m-th SNP is estimated from the genotype frequencies in the entire sample as
| (3) |
Note #AA, #Aa and #aa are the total number of individuals with genotype AA, Aa and aa, respectively, at the m-th SNP. The remaining two IBD statistics can be estimated based on NIBS=1, NIBS=2, 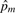 , and
, and 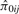 (Purcell et al., 2007). Since the sum of the three IBD statistics is unity, only two IBD statistics are needed to infer the relationship.
(Purcell et al., 2007). Since the sum of the three IBD statistics is unity, only two IBD statistics are needed to infer the relationship.
We propose an alternative framework to estimate the kinship coefficient between a pair of individuals. Suppose the frequency of a reference allele is p at a SNP for both individuals. The genotype score, defined by the number of the reference allele for individuals i, is X(i). We model genetic distance between a pair of individuals in terms of their kinship coefficient (derived under the assumption of HWE in the Supplementary Material) as
| (4) |
Let Ĥij/Mij be a consistent estimator of 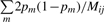 where Mij is the total number of non-missing markers for the pair of individuals. Now, we can estimate the kinship coefficient as
where Mij is the total number of non-missing markers for the pair of individuals. Now, we can estimate the kinship coefficient as
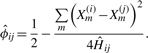 |
(5) |
Note only markers with genotype data for both individuals i and j are used in calculation of 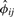 . When the sample of individuals is homogeneous, pm can be estimated by the observed allele frequency
. When the sample of individuals is homogeneous, pm can be estimated by the observed allele frequency 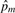 in (3). The plug-in estimator
in (3). The plug-in estimator
| (6) |
is consistent for 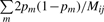 , and it follows that the estimator
, and it follows that the estimator 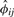 based on (5) and (6) is consistent for ϕij. We name the estimating method in Equations (5) and (6) as KING-homo. Together with the IBD estimator (2), all relationships presented in Table 1 can be determined uniquely. Note estimation of π1 and π2 can be derived easily according to equations
based on (5) and (6) is consistent for ϕij. We name the estimating method in Equations (5) and (6) as KING-homo. Together with the IBD estimator (2), all relationships presented in Table 1 can be determined uniquely. Note estimation of π1 and π2 can be derived easily according to equations 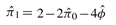 and
and 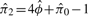 .
.
2.2 Analytical framework for efficient computation
We propose a general approach for computationally efficient relationship inference as follows. First, we derive an identity (details in the Supplementary Material) to represent the genetic distance between a pair of individuals in terms of their shared genotype counts
where IAa(i), IAa,Aa and IAA,aa indicate whether the i-th individual is heterozygous, whether both individuals are heterozygous, and whether the two individuals have different homozygotes, respectively. Now, we rewrite Equation (5) in terms of genotype counts
| (7) |
where NAa,Aa, NAa(i) and NAa(j) are the total numbers of SNPs at which both individuals of the pair are heterozygous, and the total number of heterozygotes for the i-th and j-th individual, respectively, excluding those SNPs with missing genotypes in either individual of the pair.
When each genotype is stored in two bits, NAa(i), NAa(j), NAa,Aa and NAA,aa can be computed using only bit operations (i.e. AND, OR, XOR and NOT), eliminating multiplication and division during the process of scanning the genome. For KING-homo, further computational savings is achieved by pre-calculating Ĥij at all SNPs prior to the pair-wise kinship coefficient estimation, and then updating to reflect the set of observed genotypes used in analysis of each pair of individuals.
2.3 Robust relationship inference in the presence of population substructure
A key assumption underlying KING-homo and other existing methods (e.g. Equation 2) is that genotypes for all individuals are representative of a common set of allele frequencies. Deviations from this assumption are expected in samples with population substructure. A simple approach to incorporate population stratification is a within-family adjustment, in which reported estimates of the kinship coefficient for each relative pair are adjusted by an inflation factor, representing the ratio of estimated-to-theoretical values of (1 − 2ϕ) averaged across all relative pairs for every family with three or more genotyped individuals. The rationale behind this adjustment is that inflation of allele frequencies measured by p(1 − p) (in Equation 4) should be identical across all individuals within each family, and larger than expected estimates (e.g. kinship coefficients for parent-offspring pairs greater than 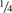 ) can indicate inflation of allele frequencies within this family. This approach results in more precise inference, particularly for larger families whose underlying allele frequencies differ from the overall values in the sample. While the family-specific adjustment performs well for large pedigrees, the approach may not improve inference in small families. Here, we present a general approach that is robust to population structure.
) can indicate inflation of allele frequencies within this family. This approach results in more precise inference, particularly for larger families whose underlying allele frequencies differ from the overall values in the sample. While the family-specific adjustment performs well for large pedigrees, the approach may not improve inference in small families. Here, we present a general approach that is robust to population structure.
Assume P is a random variable representing the allele frequency at a SNP that is randomly picked from the genotyped SNPs of an individual. P should follow the same probability distribution among individuals from the same subpopulation. In the presence of population stratification, P may vary across individuals. Equation (4) becomes
Let IAa denote an indicator of whether an individual has genotype Aa at the randomly picked SNP with allele frequency P. Assuming HWE across SNPs with the same underlying allele frequency P within an individual, i.e. Pr(Aa|P) = 2P(1 − P),
| (8) |
Thus, genome-wide average heterozygosity E(2P(1 − P)) for an individual can be estimated by NAa/Mij. For a pair of individuals i and j, since NAa(i) and NAa(j) are not necessarily equal, one empirical estimator for E(2P(1−P)) is Ĥij/Mij = (NAa(i) + NAa(j))/2Mij, and the robust estimator for the kinship coefficient is
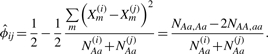 |
(9) |
Here, the genotype counts in the second representation of 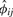 provide efficient computation, as described in Section 2.2. When the pair of individuals is sampled from the same population,
provide efficient computation, as described in Section 2.2. When the pair of individuals is sampled from the same population, 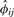 in Equation (9) is a consistent estimator of the kinship coefficient ϕij. When the pair of individuals is unrelated and from different populations (see details in the Supplementary Material),
in Equation (9) is a consistent estimator of the kinship coefficient ϕij. When the pair of individuals is unrelated and from different populations (see details in the Supplementary Material), 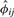 is a consistent estimator of a parameter with a negative value
is a consistent estimator of a parameter with a negative value
| (10) |
Thus, the robust estimator 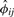 also can be used to determine the extent of population heterogeneity between the pair of individuals; an extreme negative value indicates the pair of individuals is drawn from two distinct populations. More rigorous derivation of an inference criterion for population heterogeneity using
also can be used to determine the extent of population heterogeneity between the pair of individuals; an extreme negative value indicates the pair of individuals is drawn from two distinct populations. More rigorous derivation of an inference criterion for population heterogeneity using 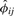 is the subject of future research.
is the subject of future research.
In most datasets, relative pairs are sampled from the same population, and pairs from different populations are unrelated. In both situations, the robust estimator given in Equation (9) is a consistent estimator (for either the kinship coefficient or a measure of population heterogeneity). It is possible that a pair of individuals is both related and from different populations, e.g. one or both individuals are mixed, in which case the robust estimator is no longer a consistent estimator of the kinship coefficient. In this scenario, the relationship inference within families could be less reliable; however, we have observed that this impact is rather small for the specification of relatives up to the 3rd-degree.
The only assumption required for our robust estimator of kinship coefficient (Equation 9) is HWE among SNPs with the same underlying allele frequencies. In practice, there is small proportion of individuals deviating from the HWE, due to reasons such as genotyping errors, recent admixture in a mixed population, or removing Mendelian errors in families. When the violation of HWE is in the direction of too little homozygosity (i.e. excessive heterozygosity), the robust estimator (Equation 9) can over-estimate the kinship coefficient. In order to guard against potential estimation inflation due to departure from individual-level HWE, we consider the smaller of the observed heterozygosity rates, min (NAa(i)/Mij, NAa(j)/Mij), as an alternative to E(2P(1 − P)). Without loss of generality, suppose the i-th individual has lower heterozygosity than the j-th individual. Then, the robust estimator is
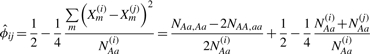 |
(11) |
The estimator above is no larger than the estimator in Equation (9), and both estimators are bounded above by 0.5.
We use estimator (Equation 9) for within-family relationship checking and estimator (Equation 11) for between-family relationship checking, naming this combined approach KING-robust. Using KING-robust, individuals of different ethnicities are less likely to be misspecified as relative pairs. All relationships in Table 1, except for the two types of 1st-degree relationships, can be uniquely specified through the kinship estimates provided by KING-robust. To further distinguish parent–offspring from full-sib pairs, we examine the observed IBS making use of the fact that IBS between a parent–offspring pair is always 1 or 2 at any SNP in the absence of genotyping errors. More advanced inference of pedigree structure can be carried out by simultaneously using the information from multiple pair-wise relationships.
The HWE assumption as in Equation (8) also allows estimation of the variance of allele frequencies in each individual as
| (12) |
Together with estimation of the allele frequency mean in each individual
| (13) |
the population structure in a GWAS dataset can be resolved, even in the presence of unspecified family structure. Thus, our approach (Equations 12 and 13) provides a useful tool for population structure analysis in the context of a family-based GWAS.
3 RESULTS
3.1 Resolution of relationship inference varies with genotyping density
We performed simulations to demonstrate the resolution of kinship coefficient estimation using high-throughput genotype data. We simulated 1000 three-generation pedigrees that contained 1st, 2nd and 3rd-degree relative pairs. The detailed algorithms of simulating pedigrees were shown previously (Chen and Deng, 2001; Chen et al., 2009). SNPs from 22 autosomes with varying densities (50k, 150k and 500k) were simulated, with minor allele frequencies ranging (randomly) from 0.1 to 0.5.
We first examined the distribution of actual or realized IBD sharing (versus the estimated distribution) between relative pairs that is defined as half of the actual proportion of the genome that is shared IBD between the pair of relatives The actual IBD sharing between a pair of relatives varies around its expectation except parent–offspring and monozygotic twin pairs (Visscher et al., 2008). This estimator is expected to provide an upper bound on precision for estimators of IBD-sharing statistics based on the same set of SNP data. Note that the realized IBD sharing of unrelated pairs is a constant, zero. We examined the distribution of the estimated kinship coefficients using the robust estimator (Equation 9). The distributions of realized IBD sharing with 150k SNPs, and estimated kinship coefficients with 150k SNPs, 5k SNPs and 500k SNPs are shown in Figure 1.
Fig. 1.

Distribution of kinship coefficient estimation. (A) Distribution of realized IBD-sharing with 150k SNPs; (B) distribution of kinship coefficient estimates with 150k SNPs; (C) distribution of kinship coefficient estimates with 5k SNPs; and (D) distribution of kinship coefficient estimates with 500K SNPs.
With 150k independent SNPs, the distribution of the realized IBD sharing and estimated kinship coefficients of relative pairs is rather similar, showing our robust kinship coefficient estimation achieves optimal power to classify relative pairs. However, even with the best possible estimation (e.g. the true value without estimation), pair-wise relationship misspecification can be still observed in GWAS data, especially for relative pairs of 3rd-degree and more distant. In all simulations, there is no distribution overlap between unrelated pairs and 3rd-degree relatives, and there is slight overlap between 3rd- and 2nd-degree relatives. The SDs for the estimated kinship coefficient of the 1st-, 2nd- and 3rd-degree relative pairs are 0.021, 0.017 and 0.012, respectively, in a GWAS with 150K SNPs. The SD for the estimated kinship coefficient of unrelated pairs is 0.002 (with 150K SNPs) or much less with denser SNPs. With a denser SNP panel, distributions between unrelated and related pairs are more separate, but its impact on the distribution for closely related pairs (up to 3rd-degree) is limited. In a linkage dataset with ∼5k SNPs, only closely related pairs (up to 2nd-degree) and unrelated pairs can be estimated reliably, and there is noticeable overlap of distributions between 3rd-degree and unrelated pairs. In linkage datasets, there could be millions of unrelated pairs and, therefore, it is not feasible to correctly distinguish 3rd-degree relatives from unrelated pairs. However, a linkage dataset is still valuable for detection of 1st- and 2nd-degree relative pairs among millions of unrelated pairs, frequently ignored in current analysis of linkage data. Dense SNP data were also simulated for over one million unrelated pairs (data not shown), and the robust estimate of the kinship coefficient never exceeded 0.022.
3.2 Robust relationship inference in the presence of population stratification
We illustrate our robust relationship inference through application to data from the 269 HapMap (International HapMap Consortium, 2005). The HapMap data used in this study consisted of 30 CEU trios, 30 YRI trios, 45 CHB samples and 44 JPT samples. Each individual is genotyped at ∼3 million SNPs in the consensus Phase II HapMap data with an average genotype missing rate 1.5% (note that ∼20% of SNPs are not polymorphic in each population). Potential pedigree errors can be viewed easily through graphical displays, in which the inferred kinship coefficients are plotted against the estimated probability of zero IBD (or proportion of zero IBS). Algorithms assuming a homogeneous population perform poorly to estimate the kinship coefficients (Fig. 2C–F), systematically inflating the degree of relatedness among individuals of the same racial group. The kinship coefficients (KING-homo) of unrelated CEU pairs within-families are estimated to be >0.044, so they are all incorrectly inferred to be 3rd-degree relative pairs.
Fig. 2.

Relationship checking in 269 HapMap samples (A), (C) and (E) are within-family relationship checking using three algorithms, and (B), (D) and (F) are between-family relationship checking using three algorithms. Negative kinship coefficient estimates are truncated to 0. Dashed lines indicate inference criteria as shown in Table 1. Solid lines follow the equation ϕ = (1 − π0)/4 that holds true for all relationships shown in Table 1, except for full sibs.
Estimation of between-family pairs is much worse. Many unrelated pairs between families are misspecified as 2nd-degree relatives using both algorithms (Fig. 2D and F), and a large proportion of unrelated pairs are misspecified as 1st-degree relatives in PLINK (Fig. 2F). In contrast, KING-robust gives clean results even in the presence of population stratification (Fig. 2A and B), with kinship coefficient estimates consistent with those from the stratified data (data not shown). All algorithms identify relatedness across three pairs of YRI trios; the closest relationships in these three pairs of families are 1st-, 2nd- and 3rd-degree relatives, respectively.
We compare the performance of algorithms in KING to identify the population structure with the PCA algorithm (Price et al., 2006; Zhu et al., 2008). Figure 3 demonstrates three clear clusters in the analyzed HapMap population, separating individuals of European (CEU), African (YRI) and Asian (CHB, JPT) ancestry. The robust kinship estimator identifies strong stratification across the distinct population groups, while individuals from the same population tend to have inferred kinship around 0 (Fig. 3A). The allele frequency statistics cluster the three populations (Fig. 3B), as does the principal component analysis (Fig. 3C). Relatedness between the three pairs of YRI families (reported above) produces the 3rd and 4th principal components (Fig. 3D).
Fig. 3.

Population structure in 269 HapMap samples. (A) Robust estimator of kinship coefficient as a tool for population structure discovery. Colored dots represent comparison of individuals from distinct populations. Within-population comparisons are shown in black; (B) mean and variance of allele frequencies at each individual; and (C) and (D) top four principal components from PCA.
We further investigated performance of our robust algorithm on a subset of 713 930 rare-variant SNPs with minor allele frequency <0.05. Results of the between-family relationship inference and population structure inference were very similar for this restricted set of SNPs (Supplementary Fig. S2) compared to the full HapMap SNP panel (Fig. 3A). These results demonstrate our algorithm is robust to the SNP panel used for relationship inference, providing a tool for both GWAS and studies of rare variants.
3.3 Robust relationship inference in a real GWAS
We further used the KING algorithms to screen pedigree errors in a GWAS of otitis media (Daly et al., 2004). This dataset includes 602 individuals from 143 families in which each is genotyped at 350K SNPs. The majority of individuals are Caucasian, one family of size four is Asian, one family of size two is Native American, and a few families have mixed ethnicity. Overall, we detect a higher degree of relatedness through analysis of genotype data compared to the relationships formally reported for the study. We detected 14 relationship errors within a family that are due to misspecification of one individual, and two disconnected families that are related (data not shown).
After fixing these two sets of errors, we display the inferred relationships in Figure 4A,B. We also applied the KING-homo and PLINK, both of which assume a homogeneous population (Fig. 4C–F). For this relatively homogenous dataset, different algorithms give similar results for the majority of pairs. By all three algorithms, 14 pairs of individuals from three unrelated sibships are estimated to be 3rd or 4th-degree relatives, and 2 out of 33 formally reported 3rd-degree relatives are misspecified as 4th-degree relatives (due to the limited power). Note the two pairs of 4th-degree relatives are correctly specified. In addition, PLINK reports six additional unrelated pairs as related (kinship > 0.022), while both KING algorithms clearly separate related pairs from unrelated pairs. KING-homo overestimates the kinship coefficient of a pair of unrelated Asian parents (Fig. 4C), which is expected given that Asians have a lower heterozygosity than other individuals (Note that 2Ĥij >NAa(i) + NAa(j) implies the kinship estimate in Equation 5 is larger than the one in Equation 9). The population structure of this data is shown in an allele frequency plot (Supplementary Fig. 1B) as well as in principal component plots (Supplementary Fig. 1C and D).
Fig. 4.

Relationship checking in OM GWAS data. (A), (C) and (E) are within-family relationship checking using three algorithms, and (B), (D) and (F) are between-family relationship checking using three algorithms. Negative kinship coefficient estimates are truncated to 0.
3.4 Computational efficiency: minutes rather than days
We compared the analysis time between the KING algorithms (both the robust algorithm and the one that assumes population homogeneity) with the algorithm implemented in PLINK, in the above two datasets as well as an additional GWAS dataset consisting of 2450 individuals (Table 2).
Table 2.
Computation time of two software implementations to estimate kinship coefficients in three sets of GWAS SNP data
| Summary of genome scan data |
Computing time |
||||
|---|---|---|---|---|---|
| Index | No. of SNPs | No. of samples | No. of pairs | KING | PLINK |
| 1 | 3 079 857 | 269 | 36 046 | 2 min | 2 h 9 min |
| 2 | 324 748 | 602 | 180 901 | 1 min | 1 h 13 min |
| 3 | 549 338 | 2 454 | 3 009 832 | 25 min | 28 h 30 min |
The computation time refers to the time to estimate kinship coefficients for all pairs of individuals, excluding overhead costs such as the time to load data into the computer memory. The two KING implementations (the robust algorithm and the algorithm assuming homogeneous samples) took a similar amount of computational time. This computation time can be estimated reliably as the analysis time for the entire data minus the analysis time for only the within-family data. The unit of computation time is in minutes hours. All computation was performed on and Intel Xeon with 3.20 GHz processor.
In all three datasets that we examined, the computational time of the KING implementation is in minutes; in contrast, it took hours to days to analyze the same datasets using PLINK on the same workstation. The computational saving of our implementation over PLINK is over 60-fold, and this result can be generalized to larger datasets. This computational efficiency makes our implementation particularly attractive for the analysis of large GWAS datasets that exceed 10 000 individuals (hours of computation time in KING, compared to a projected >1 month computation time using other software), making it feasible to perform the millions of pair-wise comparisons necessary for a comprehensive between-family analysis.
Note the reported computation time in Table 2 excludes time to load the data, which grows linearly as the sample size increases, in contrast to the exponential increase of analysis time. The time to load the data in KING was <30 s for all three scenarios when binary format genotypes were used as the input of the KING implementation, and was comparable to other software implementations (21 m, 6 m and 40 m, respectively) when the MERLIN (Abecasis et al., 2002) format genotypes were used as the input.
4 DISCUSSION
We have proposed a robust algorithm to infer relationships using high-density genotype data from a GWAS. Our approach to relationship inference incorporates simple estimates for key genetic parameters, reported with high precision due to the large number of SNPs typed by current high-throughput panels. The framework underlying the KING approach to relationship inference centers on modeling genetic distance between a pair of individuals as a function of their allele frequencies and kinship coefficient. In studies with homogeneous populations and relatively large sample sizes, allele frequencies at all SNPs can be estimated accurately from the given data, and used to inform the estimate of allelic heterogeneity needed to calculate the kinship coefficient in KING-homo. Under population stratification, a single set of allele frequencies for the given SNP panel is not appropriate for examination of the entire dataset, motivating our use of the robust estimator in KING-robust.
As demonstrated by our power analysis and application to the otitis media data, our approach based on estimation of the kinship-coefficient between any pair of individuals is sufficient to classify relative pairs as monozygotic twins, parent–offspring pairs, full sibs, 2nd- or 3rd-degree relatives. Unlike approaches that assume a homogeneous population, our robust approach classifies relative pairs correctly even under extreme population stratification seen in the pooled HapMap data. Our relationship inference is not impacted by the linkage disequilibrium structure among adjacent SNPs according to the large sample theory, and as demonstrated in the two GWAS analyses.
The robust algorithm in KING performs pair-wise relationship inference using only information from the two individuals under comparison. The inference is invariant to inclusion of any additional samples and to use of different SNP panels, producing reliable results using genotypes from GWAS or from studies of rare variants alone. The sample size of the data can be as small as two, and the analysis can be performed rapidly for a single pedigree or pair of individuals, with a wide range of applications, including forensic DNA analysis and paternity/maternity testing (assuming the current forensics technology transitions to high-density SNP genotyping). The ability to perform between-family relationship inference robust to population structure also allows population structure analysis without the worry of spurious principal components produced by undetected family structure. Ultimately, the combination of robust inference and rapid computation can be applied toward automated pedigree reconstruction and association mapping in the absence of any pre-specified pedigree or population structure (Chen and Abecasis, 2007; Chen et al., 2009; Choi et al., 2009; Kang et al., 2010; Thornton and McPeek, 2010; Zhang et al., 2010).
The KING algorithms (robust and homo) for relationship inference have been implemented in a user-friendly software package. KING is able to process large-scale GWAS data consisting of thousands of individuals (a few minutes to check all pair-wise relationships for millions of pairs of individuals). Tools to detect population structure in the presence of genetic relatedness, including a modified PCA algorithm (Zhu et al., 2008) and allele frequency statistics (Equations 12 and 13) have also been implemented in KING to facilitate the analysis of GWAS data. Future toolsets include relationship inference between two groups of individuals (rather than two individuals), clustering samples in families and reconstructing pedigrees, automatic pedigree error fixing, robust PCA structure analysis and genome-wide association analysis in the presence of unknown genetic relatedness in the sample.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Gonçalo Abecasis for sharing C++ source code for the KING implementation, Xuanlin Hou for assistance in preparing the Otitis Media GWAS data, and three anonymous reviewers for valuable input that improved the article.
Funding: Research grant DC003166 (to K.D., in part).
Conflict of Interest: none declared.
REFERENCES
- Abecasis GR, et al. GRR: graphical representation of relationship errors. Bioinformatics. 2001;17:742–743. doi: 10.1093/bioinformatics/17.8.742. [DOI] [PubMed] [Google Scholar]
- Abecasis GR, et al. Merlin–rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- Boehnke M, Cox NJ. Accurate inference of relationships in sib-pair linkage studies. Am. J. Hum. Genet. 1997;61:423–429. doi: 10.1086/514862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen WM, Abecasis GR. Family-based association tests for genomewide association scans. Am. J. Hum. Genet. 2007;81:913–926. doi: 10.1086/521580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen WM, Deng HW. A general and accurate approach for computing the statistical power of the transmission disequilibrium test for complex disease genes. Genet. Epidemiol. 2001;21:53–67. doi: 10.1002/gepi.1018. [DOI] [PubMed] [Google Scholar]
- Chen WM, et al. A generalized family-based association test for dichotomous traits. Am. J. Hum. Genet. 2009;85:364–376. doi: 10.1016/j.ajhg.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi Y, et al. Case-control association testing in the presence of unknown relationships. Genet. Epidemiol. 2009;33:668–678. doi: 10.1002/gepi.20418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daly KA, et al. Chronic and recurrent otitis media: a genome scan for susceptibility loci. Am. J. Hum. Genet. 2004;75:988–997. doi: 10.1086/426061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International HapMap Consortium. A haplotype map of the human genome. Nature. 2005;437:1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang HM, et al. Variance component model to account for sample structure in genome-wide association studies. Nat. Genet. 2010;42:348–354. doi: 10.1038/ng.548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M, Ritland K. Estimation of pairwise relatedness with molecular markers. Genetics. 1999;152:1753–1766. doi: 10.1093/genetics/152.4.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPeek MS, Sun L. Statistical tests for detection of misspecified relationships by use of genome-screen data. Am. J. Hum. Genet. 2000;66:1076–1094. doi: 10.1086/302800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price AL, et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- Purcell S, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton T, McPeek MS. ROADTRIPS: case-control association testing with partially or completely unknown population and pedigree structure. Am. J. Hum. Genet. 2010;86:172–184. doi: 10.1016/j.ajhg.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visscher PM, et al. Heritability in the genomics era—concepts and misconceptions. Nat. Rev. Genet. 2008;9:255–266. doi: 10.1038/nrg2322. [DOI] [PubMed] [Google Scholar]
- Yang J, et al. Common SNPs explain a large proportion of the heritability for human height. Nat. Genet. 2010;42:565–569. doi: 10.1038/ng.608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, et al. Mixed linear model approach adapted for genome-wide association studies. Nat. Genet. 2010;42:355–360. doi: 10.1038/ng.546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, et al. A unified association analysis approach for family and unrelated samples correcting for stratification. Am. J. Hum. Genet. 2008;82:352–365. doi: 10.1016/j.ajhg.2007.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.