

Abstract
Intrauterine bacterial infections are a well-established cause of pregnancy complications. One key observation in a number of abnormal pregnancies is that placental apoptosis is significantly elevated. First trimester trophoblast cells are known to express TLR1 and TLR2 and to undergo apoptosis following exposure to Gram-positive bacterial peptidoglycan (PDG). Thus, the objectives of this study were to determine whether PDG-induced pregnancy complications are associated with placental apoptosis and to characterize the cellular mechanisms involved. We have demonstrated, using an animal model, that delivery of PDG to pregnant mice early in gestation resulted in highly elevated placental apoptosis, evidenced by trophoblast M-30 and active caspase 3 immunostaining. Using an in vitro model of human first trimester trophoblasts, apoptosis induced by PDG was found to be mediated by both TLR1 and TLR2 and that this could be blocked by the presence of TLR6. Furthermore, in the presence of TLR6, exposure to PDG resulted in trophoblast NF-κB activation and triggered these cells to secrete IL-8 and IL-6. The findings of this study suggest that a Gram-positive bacterial infection, through TLR2 and TLR1, may directly promote the elevated trophoblast cell death and that this may be the underlying mechanism of pregnancy complications, such as preterm delivery. Furthermore, the expression of TLR6 may be a key factor in determining whether the response to PDG would be apoptosis or inflammation.
Intrauterine bacterial infections can gain access to the placenta by either ascending into the uterus from the lower tract or via the maternal circulation and represent a significant threat to fetal well-being and pregnancy outcome (1). It has been postulated that through the expression of the innate immune TLRs, the placenta has the ability to control microorganisms that may injure the embryo/fetus, thereby protecting the pregnancy (2). However, intrauterine infections have been associated with pregnancy complications such as preterm labor, intrauterine growth restriction, and preeclampsia (1, 3–6), although the connection between bacterial infections and preterm labor is the most well established (1, 7, 8). Although the precise mechanisms by which an infection can lead to such pregnancy complications remain largely undefined, excessive inflammation and/or cell death at the maternal-fetal interface are thought to be key contributors in a compromised pregnancy (9–13). Therefore, an inappropriate or inadequately controlled TLR-mediated reaction to a pathogen by the placenta may provide the link between infection and adverse pregnancy outcome (2, 14).
TLRs allow both immune and nonimmune cells to recognize pathogen-associated molecular patterns expressed by microorganisms and subsequently mediate an immune response. To date, 10 functional human TLRs have been characterized. As a family, TLRs can sense the spectrum of microbes and viruses; however, each TLR has a distinct extracellular domain and, as a result, has distinct specificities (15). Intracellularly, however, all TLRs share a common Toll/IL-1R homology region (TIR)3 (16). Following ligation, the TLRs can signal through the intracellular signaling adapter protein MyD88 to activate the NF-κB pathway, which results in the production of cytokines, chemokines, and antimicrobial factors (17, 18). In addition to the classic TLR-induced inflammatory response, certain TLRs have been shown to trigger apoptosis. This TLR-mediated cell death appears to be highly dependent upon the receptor involved, the agonist used, and the cell type (19–26). Of the TLRs, the most common mediator of apoptosis is TLR2 (19, 21–23, 26). TLR2 is able to respond to Gram-positive bacterial peptidoglycan (PDG) and lipoteichoic acid (LTA) (27–30), meningococcal porins (31), bacterial lipopeptides and lipoproteins (32, 33), as well as fungal, parasitic, and viral pathogen-associated molecular patterns (34). This broad specificity is, in part, due to the unique ability of TLR2 to both homodimerize as well as form heterodimers with either TLR1 or TLR6 (35–39).
We have previously reported that first trimester trophoblast cells, which express TLR2 and TLR1, but lack TLR6, undergo apoptosis following exposure to the TLR2 ligands, PDG and LTA (40). Furthermore, PDG and LTA are known to trigger prematurity in vivo (41, 42). Therefore, the aims of this study were to determine whether PDG-induced pregnancy complications are associated with excessive placental apoptosis and to characterize the cellular mechanism by which TLR2 mediates PDG-induced apoptosis in first trimester trophoblast cells. In this study, we report, for the first time, that administration of PDG at low doses to pregnant wild-type mice results in extensive placental apoptosis. We have demonstrated that PDG-induced trophoblast apoptosis is mediated by both TLR1 and TLR2, and that this can be inhibited by the presence of TLR6. Moreover, in the presence of TLR6, PDG triggers trophoblast cells to secrete IL-8 and IL-6. These results suggest that a Gram-positive bacterial infection during pregnancy may promote trophoblast apoptosis via TLR2/TLR1 heterodimers. Expression of TLR6, however, may protect the trophoblast from PDG-induced apoptosis and instead may promote an inflammatory response, suggesting that apoptosis may not be the only TLR2-mediated pathway for adverse pregnancy outcome. These findings shed new light on the mechanisms by which Gram-positive bacterial infections may affect placental function and pregnancy outcome.
Materials and Methods
First trimester trophoblast cell lines
Two human first trimester trophoblast cell lines were used in this study. The SV40-transformed HTR8 cells, which were a gift from Dr. C. Graham (Queens University, Kingston, Ontario, Canada) (43), and the 3A cells, which were transformed by SV40 ts30 (40). Both cell lines were maintained at 37°C/5% CO2 in RPMI 1640 (Invitrogen) supplemented with 10% FBS (HyClone), 10 mM HEPES, 0.1 mM MEM nonessential amino acids, 1 mM sodium pyruvate, and 100 nm penicillin/streptomycin (Invitrogen).
Stably transfected trophoblast cell lines
3A cells were stably transfected with either the pZERO expression plasmid containing human TLR1ΔTIR, the pZERO expression plasmid containing human TLR2ΔTIR, or the pUNO expression plasmid containing full-length human TLR6 (InvivoGen). TLR1ΔTIR and TLR2ΔTIR act as dominant negatives (DN) because their TIR domains have been deleted. Thus, TLR1ΔTIR and TLR2ΔTIR can compete with endogenous TLR1 or TLR2, respectively, for ligand binding, but cannot transduce a signal. Briefly, cells were transfected for 18 h with 2 μg of DNA using Fugene 6 (Roche Diagnostics). Following transfection, cells were allowed to recover in growth medium for 24 h, after which selection for stable expression was performed. Cells expressing the TLR1ΔTIR (TLR1-DN) or the TLR2ΔTIR (TLR2-DN) were selected for using 200 μg/ml puromycin (InvivoGen), and cells expressing TLR6 (TLR6+) were selected for using 200 μg/ml blasticidin (InvivoGen). Untransfected cells served as the wild-type control.
Animal studies
C57BL/6 mice were obtained from The Jackson Laboratory. All mice were housed in a specific pathogen-free facility supervised by the Central Research Department of Rhode Island Hospital. All protocols were approved by the Lifespan Animal Welfare Committee and conducted according to its guidelines. Mice received i.p. injections of 100 μl of either saline or PDG at 25 μg/mouse on embryonic day 6.5. On embryonic day 12, animals were sacrificed and the uteri, placenta, and spleens were harvested as previously described (44). Tissues were either paraffin embedded, snap frozen and stored at −80°C, or analyzed immediately. Mouse placental tissue lysates were prepared by homogenization in 1 ml of lysis buffer/1 g weight containing 1% Nonidet P-40/0.1% SDS in PBS in addition to 0.2 μg/ml PMSF and a protease inhibitor mixture (Roche Applied Science). After a 30-min incubation on ice, samples were centrifuged for 60 min at 13,500 rpm/4°C. Supernatants were then collected and stored at −40°C until further analysis. Protein concentrations were determined using the BCA assay (Pierce).
Flow cytometry
Mouse uterine and splenic mononuclear cells were isolated as previously described (44). Cells were then washed in PBS and resuspended in PBS containing 0.1% sodium azide and 1% BSA. FITC-conjugated anti-CD45 (30-511), PE-conjugated anti-NK1.1 (PK136; BD Biosciences), or PE-conjugated anti-CD11b (M1/70; BD Biosciences) and PerCP-conjugated anti-CD3 (145-2C11; BD Biosciences) were then added simultaneously and allowed to incubate at 4°C for 30 min. Fluorochome-conjugated isotype Abs of irrelevant specificity were used as controls (44).
Cytotoxicity assays
Uterine NK cell activity was measured using a standard chromium release assay as previously described (44). Briefly, target YAC-1 cells (5 × 103) were labeled with 0.15 mCi of Na2(51CrO4) (PerkinElmer) for 1 h at 37°C. Effector uterine mononuclear cells were added to target cells at descending half-fold E:T cell ratios of 50:1 to 1.65:1 in RPMI 1640 plus 10% FBS, and supernatants were harvested after an incubation of 5 h at 37°C in 10% CO2 and read on a gamma radiation counter. The percent lysis was calculated as: [(sample count − spontaneous release)/(maximal release − spontaneous release)] × 100. Spontaneous release was assessed by the radioactivity detected in the supernatant from target cells incubated alone, and maximal release was assessed by the radioactivity detected in the supernatant from target cells lysed with 1 N HCl.
Immunohistochemistry
Localization of caspase 3 and M30 expression in day 12 mouse placentas was performed as follows. Mouse placental samples were fixed with 4% paraformaldehyde and then paraffin embedded. Sections of placenta (5 μm) were adhered to glass slides, underwent deparaffinization, and then were blocked with mouse-on-mouse (MOM) blocking reagent (Vector Laboratories) for 1 h at room temperature. Following two washes with PBS, slides were incubated in MOM diluent for 5 min and then incubated for 30 min at room temperature with either the anti-active caspase 3 mAb at a 1/100 dilution (Calbiochem) or the M30 CytoDEATH mAb at a 1/25 dilution (Roche Applied Science). The M30 mAb detects caspase-cleaved cytokeratin 18 and is used to identify apoptosis in trophoblast and epithelial cells (45). MOM diluent served as negative controls. After two washes with PBS, specific staining was detected by incubating with a MOM biotinylated reagent (1/250 dilution) for 10 min followed by a 30- min incubation with streptavidin-HRP (Vector Laboratories). After three washes in 0.1 M Tris, slides were developed using diaminobenzidine substrate (Vector laboratories) and then counterstained with hematoxylin (Sigma-Aldrich) before dehydration with ethanol and Histosolve (Shandon). Slides were then mounted with Permount (Fisher Scientific) and visualized by light microscopy.
RT-PCR
Total RNA was isolated from cells using the RNeasy kit from Qiagen. Reverse transcription was performed on 5 μg of total RNA using the First Strand cDNA Synthesis kit from Amersham Biosciences according to the manufacturer’s directions. The primers and conditions used for amplification of human TLR6 and β-actin have been previously described (37, 40, 46).
Cell viability assay
The effects of PDG on wild-type and stably transfected trophoblast cell viability were determined using the CellTiter 96 viability assay (Pro-mega) as previously described (47). Cell lines were plated in wells of a 96-well plate at 5 × 103 cells/well in growth medium and cultured until 70% confluent. The medium was then replaced with serum-free Opti-MEM (Invitrogen) and cultured for another 4 h before treatment. Following treatment, with either medium or PDG isolated from Staphylococcus aureus (InvivoGen), the CellTiter 96 substrate was added to all wells and following a 1- to 4-h incubation at 37°C, ODs were read at 490 nm. All samples were assayed in triplicate and cell viability was presented as a percentage relative to the untreated control.
Caspase activity assay
The effects of PDG on caspase activity was determined using the Caspase-Glo assay (Promega) as previously described (47). Briefly, 10 μg of the whole cell lysates were incubated at room temperature in the dark for 1 h with either the caspase 3, caspase 8, or caspase 9 substrate. Following incubation, luminescence was measured using a TD-20/20 luminometer (Turner Designs). The amount of luminescence detected as relative light units (RLU) was proportional to caspase activity. All samples were assayed in triplicate.
Cytokine studies
The effect of PDG on trophoblast cytokine production was determined by Luminex technology. Trophoblast cells were treated with or without PDG. Following a culture of 48 h, the cell-free supernatants were collected by centrifugation at 400 × g for 10 min and stored at −80°C until analysis was performed. The concentrations of IL-8 and IL-6 were then evaluated using the Beadlyte Multiplex Assay with detection and analysis using the Luminex 100 IS system (Upstate Biotechnology) according to the manufacturer’s instructions and as recently described (48, 49). Mouse placental lysates collected from the animal studies were also evaluated for cytokine content by analyzing 62.5 μg of placental protein using the Bio-Plex Mouse Cytokine 23-Plex Panel (catalog no. 171-F11241) from Bio-Rad.
NF-κB activity assay
The effect of PDG on trophoblast cell NF-κB activity was determined using a luciferase reporter construct pBII-LUC containing two NF-κB sites before a Fos essential promoter (a gift from Dr. S. Ghosh, Yale University, New Haven, CT) (50). Trophoblast cells were transiently transfected with 2 μg of pBII-LUC using Fugene 6 (Roche Diagnostics) as previously described (51). Following transfection, cells were allowed to recover in growth medium for 24 h. The cells were then treated with or without PDG, lysed for protein, and luciferase activity was measured using the Luciferase Assay System (Promega) according to the manufacturer’s protocol. Briefly, 10 μg of protein in a total volume of 20 μl was mixed with 100 μl of the Luciferase Assay Reagent, and luminescence was measured using a TD 20/20 Luminometer (Turner Designs). Relative activity was calculated based on readings measured from untreated cells after subtracting blank values. Each sample was assayed in triplicate.
Statistical analysis
Data are expressed as mean ± SD. Statistical significance (p < 0.05) was determined using either t tests or, for multiple comparisons, one-way ANOVA followed by Bonferroni’s post test or multiple regression analyses. Unless stated otherwise, all experiments were performed in triplicate and a representative of at least three independent experiments is shown.
Results
PDG induces trophoblast apoptosis in vivo
Recently, Ilievski et al. (42) reported that i.p. administration of high doses of PDG to wild-type animals late in gestation induces preterm delivery. Because we previously reported that treatment with PDG induced first trimester trophoblast cells to undergo apoptosis (40), we sought to determine in an experimental model whether PDG-induced preterm labor is a result of PDG-induced apoptosis in placental cells. Thus, pregnant C57BL/6 mice were treated with either saline or 25 μg of PDG i.p. on embryonic day 6.5. This low dose of PDG, delivered early in gestation, did not induce preterm labor or fetal resorption (Table I) and, therefore, allowed us to fully evaluate the tissues. Following PDG administration, the fetoplacental units were collected on embryonic day 12 and evaluated for placental cytokine production, NK cell infiltration and cytotoxicity, and placental apoptosis. As shown in Fig. 1A, following treatment with PDG, high levels of active caspase 3 and M-30 expression were seen in both the placental trophoblast cells and in the fetal membranes. Placentas from animals treated with saline alone showed little, if any, active caspase 3 and M-30 expression (Fig. 1A). These results were confirmed by performing a caspase activity assay on the placental lysates. As shown in Fig. 1B, a significant increase in caspase 3, caspase 8, and caspase 9 activity was observed in placentas from mice treated with PDG when compared with the saline controls. No differences in placental cytokine expression levels were found between the saline-and PDG-treated mice (data not shown). Moreover, when compared with the saline control, administration of this low dose of PDG had no effect on the levels of uterine or splenic NK cells or myeloid cells (Fig. 1C), nor were there any differences in uterine NK cell cytotoxicity (Fig. 1D).
Table I.
Pregnancy outcome in mice treated i.p. on embryonic day 6.5 with 100 μl of either saline or PDG (25 μg)
| Treatment | Mean Time of Birth | No. of Resorptions |
|---|---|---|
| Saline | 19.57 ± 0.53 | 0/13 |
| PDG | 19.83 ± 0.41 | 0/9 |
FIGURE 1.

Effect of PDG on placental apoptosis and immune cell recruitment in vivo. C57BL/6 pregnant mice were treated i.p. with either saline or PDG (25 μg/mouse) in 100 μl on embryonic day 6.5. On embryonic day 12, animals were sacrificed and the spleens and fetoplacental units were harvested. A, Paraffin-embedded placental tissue sections were immunostained for active caspase 3 and cell death using the apoptosis marker M-30. For each treatment (saline or PDG) and marker (caspase 3 and M-30), panels are labeled as: i, Placenta labyrinth (P) and fetal membranes (M) (original magnification, ×10); ii, placenta labyrinth and fetal membranes (original magnification, ×20); iii, placental labyrinth (original magnification, ×40); and iv, fetal membranes (original magnification, ×20–40). Insets show the Ab-negative controls (Neg). B, Placental tissue lysates were evaluated for activation of the caspase pathway using the Caspase-Glo assay. Bar graphs show caspase 3, caspase 8, and caspase 9 activities as RLU. Value of p < 0.05 relative to the saline controls. C, The proportion of uterine and splenic NK cells were evaluated using flow cytometry by first gating on the CD45+ cells and then by analyzing CD3, NK1.1, and CD11b expression. As shown on the scatter graphs, the proportion of uterine and splenic NK cells (NK1.1+CD3−) and myeloid cells (CD11b+CD3−) were similar between the mice treated with saline and those treated with PDG. C, Uterine NK cells were isolated and their cytotoxicity was tested against YAC-1 cells. D, Bar graph, Similar levels of uterine NK cell cytotoxicity from mice treated with either saline or PDG.
Kinetics of PDG-induced trophoblast apoptosis in vitro
Having confirmed that PDG was indeed inducing trophoblast apoptosis in vivo, we next sought to further characterize the molecular mechanism underlying this effect. For this, two first trimester trophoblast cell lines (HTR8 and 3A) were used as a model. The following experiments are described only for PDG, because we previously reported that LTA and PDG can both trigger trophoblast apoptosis in vitro and both are recognized by TLR2 (40). Thus, cells were treated for 48 h with either a low (20 μg/ml), moderate (40 μg/ml), or high (80 μg/ml) PDG concentration. As shown in Fig. 2A, trophoblast caspase 3 activity was significantly increased by PDG in a dose-dependent manner. Although the lowest dose only had an effect on the HTR8 cells, the moderate concentration of 40 μg/ml induced significant caspase 3 activation in both cell lines. Furthermore, following exposure to PDG, trophoblast caspase activation occurred in a time-dependent manner. No caspase 3 activation could be detected before 24 h of treatment (Fig. 2B). However, from 24 h onward, significant increases in caspase 3, caspase 8, and caspase 9 could be detected (Fig. 2C).
FIGURE 2.

Effect of PDG on trophoblast apoptosis in vitro. A, First trimester trophoblast cells (HTR8 and 3A) were incubated with PDG (0–80 μg/ml) for 48 h, after which cell lysates were prepared and caspase 3 activity was determined using the Caspase-Glo assay. Bar graphs show caspase activity in RLU. PDG significantly increased trophoblast cell caspase 3 activity in a dose-dependent manner. *, p < 0.001 relative to the untreated control (0). B, First trimester trophoblast cells (HTR8) were incubated with PDG (80 μg/ml) and, after 0, 6, 12, and 24 h, cell lysates were prepared and caspase 3 activity was evaluated. C, First trimester trophoblast cells (HTR8) were incubated with PDG (80 μg/ml). After 0, 24, 48, and 72 h, cell lysates were prepared and caspase 3, caspase 8, and caspase 9 activity was evaluated. PDG significantly increased trophoblast cell caspase activity in a time- dependent manner. *, p < 0.001 relative to time 0.
PDG triggers trophoblast apoptosis through TLR2 and TLR1
TLR2 can function as a homodimer (TLR2/TLR2) or as a heterodimer with either TLR1 or TLR6 (TLR2/TLR1 or TLR2/TLR6) (15). Since the HTR8 and 3A trophoblast cell lines express only TLR2 and TLR1 (40), we first evaluated which of these two TLRs were necessary for the induction of apoptosis. To inhibit TLR1 or TLR2, trophoblast cells were stably transfected to express either a TLR1-DN or a TLR2-DN and their responses to PDG were compared with the wild-type cells expressing only endogenous TLR1 and TLR2. The TLR1-DN and TLR2-DN both lack their TIR domains; therefore, they can compete with the endogenous receptors for ligand binding but cannot signal. As shown in Fig. 3, although PDG treatment significantly reduced wild-type trophoblast cell viability, expression of either the TLR1-DN or TLR2-DN significantly blocked this effect. The dependence of both TLR1 and TLR2 on PDG-mediated apoptosis was further confirmed by evaluating the effects of the dominant negatives on PDG-induced caspase activation. As shown in Fig. 4, when wild-type trophoblast cells were treated with the moderate concentration of PDG (40 μg/ml), a significant increase in caspase 3, caspase 8, and caspase 9 activity was observed. However, in the presence of either the TLR2-DN or the TLR1-DN, PDG-induced caspase activation was significantly reduced. These results suggest that both TLR2 and TLR1 are necessary for PDG-induced apoptosis in trophoblast cells.
FIGURE 3.
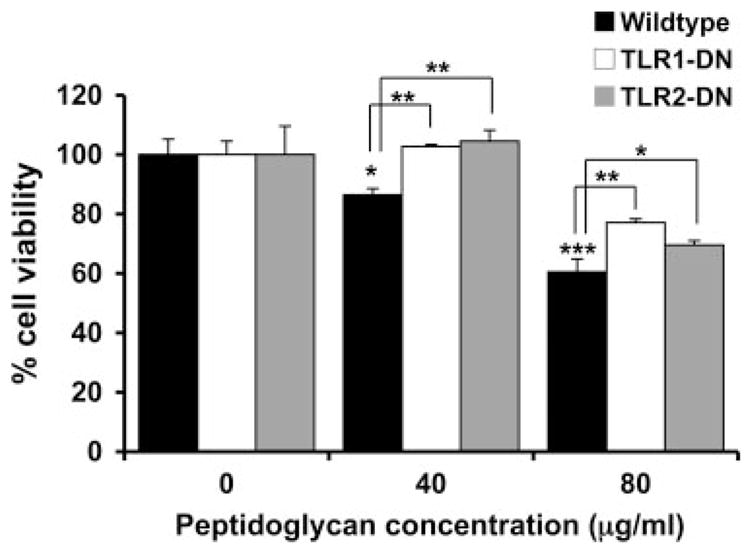
Expression of TLR1-DN or TLR2-DN inhibits PDG-induced trophoblast cell death. Wild-type or the trophoblast cell lines stably transfected with either TLR1-DN or TLR2-DN were incubated with PDG using 0, 40, or 80 μg/ml for 72 h. Cell viability was then determined using the CellTiter 96 assay. Bar graph, Percentage of cell viability relative to the untreated control (0). Treatment with PDG significantly reduced the wild-type trophoblast cell viability in a dose-dependent manner (*, p < 0.05; ***, p < 0.001). There was significantly less PDG-induced cell death in TLR1-DN- and TLR2-DN-expressing trophoblast cell lines when compared with the wild-type cells (*, p < 0.05; **, p < 0.01).
FIGURE 4.
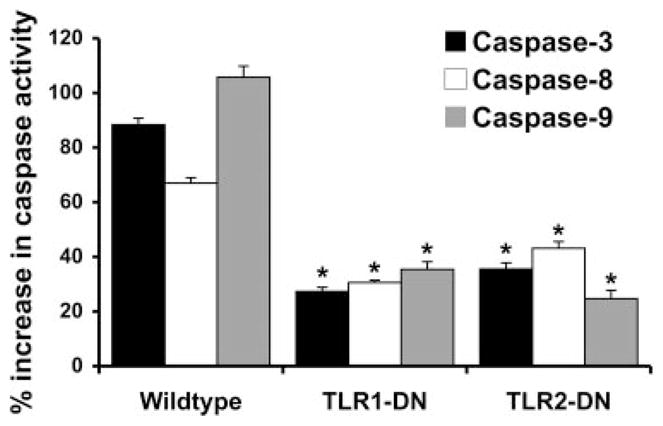
Expression of TLR1-DN or TLR2-DN inhibits PDG activation of the caspase pathway. Wild-type or the trophoblast cell lines stably transfected with either TLR1-DN or TLR2-DN were incubated with either no treatment or PDG at 40 μg/ml for 48 h, after which cell lysates were prepared and caspase 3, caspase 8, and caspase 9 activities were determined. Bar graph, The percent increase in caspase activity induced by PDG relative to the untreated control. Expression of either the TLR1-DN or TLR2-DN significantly reduced PDG-induced activation of caspase 3, caspase 8, and caspase 9 when compared with the response of the wild-type cells (*, p < 0.001).
TLR6 expression protects trophoblast cells from peptidoglycan-induced apoptosis
In addition to TLR1, TLR2 is known to cooperate with TLR6 (15). Because both first trimester trophoblast cell lines used in this study lack TLR6 (40), we postulated that TLR6 might compete with TLR1 to prevent TLR2-mediated trophoblast apoptosis. Thus, trophoblast cells were stably transfected to express TLR6 (TLR6+; Fig. 5A), and their responses to PDG were compared with those of the wild-type cells. Expression of TLR6 significantly blocked the effect of PDG on trophoblast cell viability (Fig. 5B) and significantly inhibited PDG-induced caspase 3, caspase 8, and caspase 9 activation, when compared with the response of the wild-type cells (Fig. 6).
FIGURE 5.
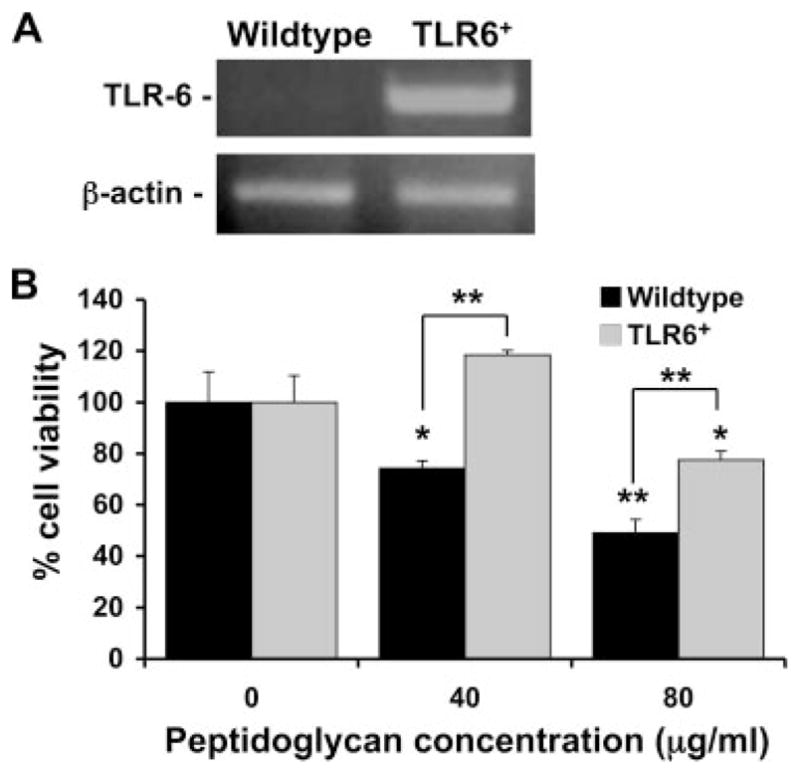
Expression of TLR6 inhibits PDG-induced trophoblast cell death. A, TLR6 mRNA expression was evaluated by RT-PCR in the wild-type trophoblast cells and in trophoblast cells stably transfected to express TLR6. Top panel, TLR6 expression (500 bp) and bottom panel, β-actin (620 bp). Cells were routinely monitored for TLR6 expression and only the stably transfected cells (TLR6+) were positive for this receptor. B, Wild-type or the TLR6+ trophoblast cells were incubated with PDG at 0, 40, or 80 μg/ml for 72 h. Cell viability was then determined using the CellTiter 96 assay. Bar graph, Percentage of cell viability relative to the untreated control (0). Treatment with PDG significantly reduced the wild-type trophoblast cell viability in a dose-dependent manner (*, p < 0.05; **, p < 0.001). Expression of TLR6 significantly inhibited the effect of PDG on trophoblast cell viability when compared with the responses of the wild-type cells (**, p < 0.001).
FIGURE 6.
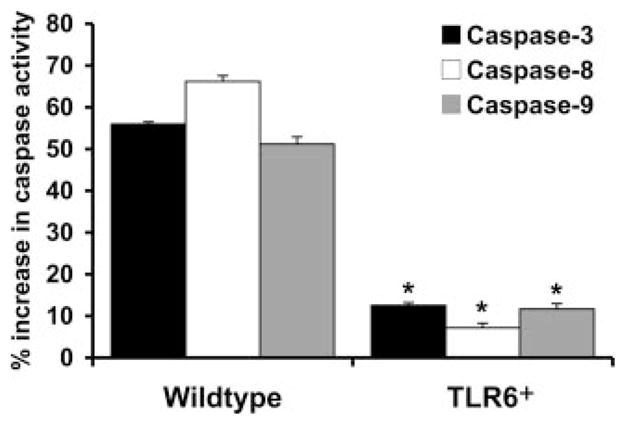
Expression of TLR6 inhibits PDG-induced trophoblast apoptosis. Wild-type trophoblast cells or TLR6+ trophoblast cells were incubated with either no treatment or PDG at 40 μg/ml for 48 h, after which cell lysates were prepared and caspase activity was determined. Bar graph, The percent increase in caspase 3, caspase 8, and caspase 9 activity induced by PDG relative to the untreated control. Expression of TLR6 significantly inhibited activation of caspase 3, caspase 8, and caspase 9 induced by PDG when compared with the response of the wild-type cells (*, p < 0.001).
TLR6 expression by trophoblast cells promotes NF-κB activation and cytokine production
Having established that TLR6 did indeed protect trophoblast cells from PDG-mediated apoptosis, we next determined whether the presence of TLR6 could fully reverse the effect of PDG on the trophoblast by triggering an inflammatory response. We have previously reported that after 12 h of treatment at a low dose of 10 μg/ml, PDG up-regulated the levels of IL-6 and IL-8 in the cell lysates (40). To further characterize the effects of PDG on cytokine and chemokine production, trophoblast cells (wild type and TLR6+) were treated with low to moderate concentrations of PDG (10–40 μg/ml) for 24 and 48 h and cytokine production was evaluated. As shown in Fig. 7, A and B, PDG significantly inhibited wild-type trophoblast content (lysates) and secretion (supernatants) of IL-8 and IL-6 in a dose- dependent manner at 24 and 48 h. However, in the presence of TLR6, this effect was reversed and we could observe a significant increase in the secreted levels of these two cytokines at 48 h (Fig. 7B). Since TLR-induced cytokine production is mediated through NF-κB activation (15), NF-κB activity in wild-type and TLR6+ trophoblast cells was monitored using a luciferase reporting system containing two NF-κB binding sites (50). As shown in Fig. 8, when wild-type trophoblast cells were treated with PDG, baseline NFκB activity was inhibited after 30 min. In contrast, treatment of the TLR6+ trophoblast cells with PDG resulted in a 12.5-fold increase in NF-κB activity after 30 min.
FIGURE 7.
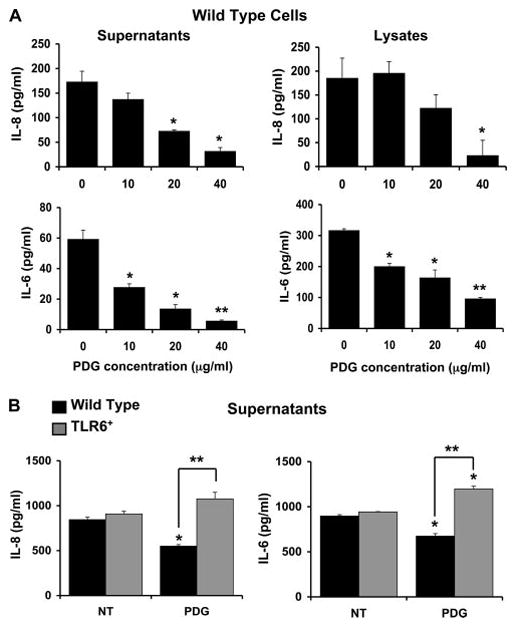
Expression of TLR6 reverses the inhibitory effect of PDG on trophoblast cytokine production. Wild-type first trimester trophoblast cells (HTR8 and 3A) and the trophoblast cells stably transfected with TLR6 (TLR6+) were treated with PDG at 0, 10, 20, or 40 μg/ml, after which cell-free supernatants were collected and assayed for IL-8 and IL-6 using the Beadlyte multiplex assay. A, Bar graphs show that in the HTR8 wild-type cells IL-8 and IL-6 content (lysates) and secretion (supernatants) was significantly reduced after 24 h of treatment with PDG in a dose-dependent manner. *, p < 0.05; **, p < 0.001 relative to the untreated control (0). B, After incubation with either no treatment (NT) or PDG (40 μg/ml) for 48 h, IL-8 and IL-6 secretion was inhibited in the wild-type 3A cells; however, in the TLR6+ cells, IL-8 and IL-6 secretion was significantly increased after PDG treatment (*, p < 0.05; **, p < 0.005).
FIGURE 8.
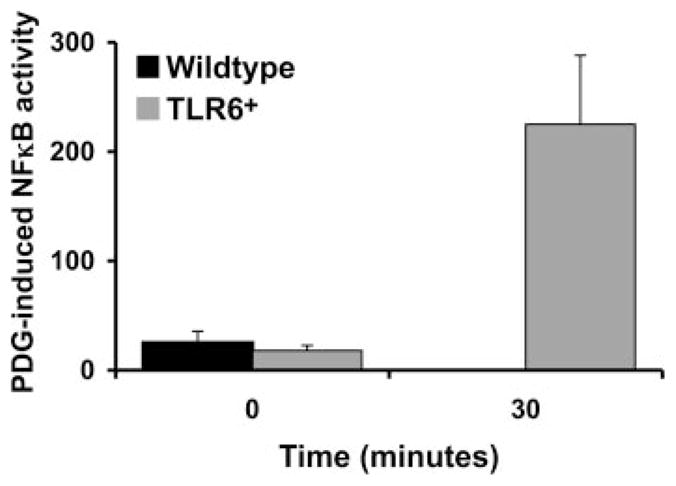
Expression of TLR6 results in PDG-induced NF-κB activation in trophoblast cells. Wild-type trophoblast cells or the TLR6+ trophoblast cells were transiently transfected with pBII-LUC. Cells were then treated with or without PDG (40 μg/ml) for 0 and 30 min, after which cell lysates were collected and assayed for NF-κB activity using the luciferase assay. Bar graph, PDG-induced NF-κB activity in RLU after normalization against the untreated controls.
Discussion
Intrauterine infections are a well-established cause of preterm labor and have been linked to other pregnancy complications, such as preeclampsia and intrauterine growth restriction (1, 3–8). However, the mechanisms involved are still largely undefined. A key observation in a number of abnormal pregnancies is that during early pregnancy, trophoblast cell apoptosis is significantly elevated (11–13). The findings of this current study suggest that an intra-uterine Gram-positive bacterial infection may cause the excessive trophoblast cell apoptosis observed in some abnormal pregnancies and that TLRs may provide the mechanism of pathogenesis. Specifically, our results demonstrate that Gram-positive bacterial PDG causes trophoblast apoptosis in vivo. Our in vitro studies implicate TLR1 and TLR2 in providing the direct mechanism of PDG-induced apoptosis which can be regulated by TLR6.
A number of animal models have been used to test the effects of infection on pregnancy. However, the majority of these studies have focused on Gram-negative bacteria or bacterial components (52). Two studies in mice have evaluated the effects of Gram-positive bacterial components on pregnancy outcome. Kajikawa et al. (41), found that i.p. administration of LTA on embryonic day 15 resulted in preterm delivery and trophoblast apoptosis. This is consistent with our previous in vitro observations that LTA can induce first trimester trophoblast apoptosis (40). In another study, i.p. administration of a high dose of PDG on embryonic day 14.5 was also found to trigger preterm delivery (42). However, contrary to another stimuli, such as bacterial LPS, which induces preterm delivery in association with elevated cytokine production and changes in intrauterine immune cell activation and distribution (44, 52), the effect of PDG was not associated with changes in cytokine production (42). We have also previously reported that PDG induces apoptosis in human first trimester trophoblast cells (40), suggesting that preterm labor observed in these animals may be mediated by PDG-induced trophoblast cell death. Since a high PDG dose of 1 mg per mouse given on day 14.5 of gestation (embryonic day 14.5) triggers rapid prematurity (42) and pregnancy complications are thought to be established early in pregnancy (1, 53, 54), a lower PDG concentration was selected and administered to mice early in gestation, so that prematurity would not be induced and the mechanisms involved could be assessed. Indeed, delivery of 25 μg of PDG on embryonic day 6.5 did not cause preterm delivery nor fetal resorption. In addition, there was no evidence of changes in uterine NK cell and myeloid cell levels or uterine NK cell cytotoxicity, as seen in other models (44). Instead, delivery of this low dose of PDG resulted in excessive trophoblast apoptosis, evidenced by M-30 and active caspase 3 immunostaining, as well as elevated placental caspase 3, caspase 8, and caspase 9 activities. Furthermore, high levels of apoptosis within the fetal membranes were observed. This latter observation may explain why in vivo, PDG causes preterm delivery rather than other pregnancy complications, and in humans this may manifest as preterm labor associated with membrane rupture (55). As mentioned above, unlike the LPS-induced models of preterm labor (44, 56–58), delivery of PDG failed to induce an inflammatory response at the maternal-fetal interface. There were no detectable differences in placental cytokine and chemokine levels, nor was there any significant decidual immune cell infiltrate. These findings suggest that placental apoptosis may provide the primary mechanism by which preterm labor is triggered in vivo by Gram-positive bacterial PDG.
Having established that PDG was indeed triggering trophoblast apoptosis in vivo, the next objective of this study was to determine the cellular mechanisms involved. As mentioned before, we have previously reported that first trimester trophoblast cells, which express TLR2 and TLR1, but lack TLR6, undergo apoptosis following exposure to the TLR2 ligands PDG and LTA (40). In contrast, the TLR4 ligand LPS does not induce trophoblast apoptosis, but instead triggers an inflammatory response (40, 59), indicating that this proapoptotic effect is specific for TLR2. Unlike other TLRs, TLR2 can cooperate with either TLR1 or TLR6 (35, 60, 61). This heterodimerization has been shown to allow TLR2 to discriminate between ligands, particularly with respect to lipoproteins and lipopeptides. For example, TLR2/TLR1 dimers can recognize tri-acylated lipopeptides, while TLR2/TLR6 dimers recognize diacylated lipopeptides (36, 38, 62), and differences in the extracellular domains of TLR1 and TLR6 are responsible for this ligand specificity (63). However, in some cases, usage of TLR1 and TLR6 appear to be more relevant to function rather than ligand specificity. TLR2 activation by S. aureus or its components is enhanced in the presence of TLR6 and inhibited by TLR1 (35, 64). Our results indicate that the type of TLR expressed by the trophoblast may determine the type of response generated. Thus, the proapoptotic effect appears to depend on the presence of TLR2 and TLR1, since inhibiting the signaling potential of either receptor decreases PDG-induced apoptosis. These results correlate with our previous findings where we demonstrated that PDG triggers first trimester trophoblast apoptosis through activation of the caspase pathway in a Fas-associated death domain protein-dependent manner (40). Similar results have been shown in other cell types, where bacterial lipoproteins are able to trigger TLR2-mediated apoptosis through the recruitment of Fas-associated death domain protein by MyD88 (22, 65). Furthermore, this induction of apoptosis is independent of the NF-κB pathway, since we did not observe either NF-κB activity or cytokine production. In a previous study, we evaluated the cytokine/chemokine content of the trophoblast (cell lysates) 12 h after treatment with a low PDG dose and found an increase in the intracellular levels of IL-8 and IL-6. For this we analyzed the trophoblast cell lysates after treatment with a low PDG dose (10 μg/ml) for 12 h (40). However, when we looked for changes in IL-8 and IL-6 at later times, we observed a different response. In the current study, we evaluated the cytokine content and secretion by the trophoblast following a 24- and 48-h treatment at low, moderate, and high PDG concentrations. Contrary to what we observed at 12 h, at these later time points, we saw both IL-8 and IL-6 content (lysates) and secretion (supernatants) inhibited, which correlates closely with the 24- to 48-h induction of caspase activation. We could hypothesize that the early increase in IL-8 and IL-6 content may represent an additional effort of the cell to prevent apoptosis, which at later time points becomes overridden by the proapoptotic event.
Having established the role of TLR1 and TLR2 in PDG-induced trophoblast apoptosis, we next questioned what effect the presence of TLR6 would have on the trophoblast response to PDG by introducing functional TLR6 into wild-type trophoblast cells. In the presence of TLR6, PDG-induced trophoblast cell death was inhibited. Furthermore, the presence of TLR6 was able to reverse the effect of PDG in terms of NF-κB activity and cytokine production. These data indicate that in response to PDG, TLR2/TLR1 dimers mediate trophoblast apoptosis, while TLR2/TLR6 dimers promote trophoblast survival, but instead triggers cytokine/chemokine production. In support of this, Nakao et al. (60), have demonstrated that PDG-induced cytokine production in monocytes is generated via TLR2 and TLR6, but not through TLR1 (60). TLR6 may have a greater affinity for PDG and, therefore, may be able to compete with TLR1 for ligand binding. Alternatively, the survival signals triggered by PDG through TLR2/TLR6 may outweigh the pro-apoptotic signals generate by TLR2/TLR1 (66).
Although the first trimester trophoblast cell lines used in these studies lack TLR6, term placental tissue has been shown to express TLR6 mRNA, albeit at low levels (67). However, the cellular localization of TLR6 is currently unclear, as is the expression levels and distribution earlier in gestation. Nonetheless, studies have shown that TLR6 expression in myeloid cells can be up-regulated by IL-6 and LPS and down-regulated following exposure to whole Gram-negative bacteria (67). Thus, the microenvironment at the implantation site my have a powerful influence on the expression of TLRs (68). In the case of TLR6, its expression by the trophoblast may determine whether there is an active immune response to the bacterial infection or placental cell death triggered by the same microbe. Furthermore, in the presence of TLR6, a change in the placenta’s response to PDG from apoptosis to NF-κB activity and inflammation may have significant physiologic consequences. Instead of cell death-triggered prematurity, the inflammatory response may act to facilitate resolution of the infection and protection of the pregnancy. However, if too strong, the TLR2/TLR6-mediated inflammation may itself trigger an adverse pregnancy outcome.
In summary, we have demonstrated, using an animal model, that PDG induces trophoblast apoptosis, suggesting that elevated placental cell death may provide the mechanism underlying PDG-induced prematurity. Using a human in vitro model, we have shown that PDG-induced trophoblast apoptosis is mediated by TLR2 and TLR1 and can be reversed by the presence of TLR6. Furthermore, TLR2/TLR6 responses to PDG results in trophoblast NF-κB activation and cytokine/chemokine production. The findings of this study suggest that a Gram-positive bacterial infection, through TLR2 and TLR1, may directly promote the elevated trophoblast cell death observed in a number of pregnancy complications. Together, these findings suggest that in the trophoblast the expression of TLR6 is a key factor determining whether the response to PDG would be apoptosis or inflammation.
Footnotes
This study was supported in part by Grant RO1HD049446-01 from the National Institute of Child Health and Human Development (to V.M.A.), Grants P20RR018728 and P42ES013660 from National Institutes of Health and National Institute on Environmental Health Sciences (to S.S.), and the Perinatology Research Branch, Division of Intramural Research of National Institute of Child Health and Human Development.
Abbreviations used in this paper: TIR, Toll/IL-1R homology region; LTA, lipoteichoic acid; DN, dominant negative; MOM, mouse-on-mouse; RLU, relative unit.
Disclosures
The authors have no financial conflict of interest.
References
- 1.Goncalves LF, Chaiworapongsa T, Romero R. Intrauterine infection and prematurity. Ment Retard Dev Disabil Res Rev. 2002;8:3–13. doi: 10.1002/mrdd.10008. [DOI] [PubMed] [Google Scholar]
- 2.Mor G, Romero R, Aldo PB, Abrahams VM. Is the trophoblast an immune regulator? The role of Toll-like receptors during pregnancy. Crit Rev Immunol. 2005;25:375–388. doi: 10.1615/critrevimmunol.v25.i5.30. [DOI] [PubMed] [Google Scholar]
- 3.von Dadelszen P, Magee LA. Could an infectious trigger explain the differential maternal response to the shared placental pathology of preeclampsia and normotensive intrauterine growth restriction? Acta Obstet Gynecol Scand. 2002;81:642–648. [PubMed] [Google Scholar]
- 4.Romero R, Chaiworapongsa T, Espinoza J. Micronutrients and intrauterine infection, preterm birth, and the fetal inflammatory response syndrome. J Nutr. 2003;133:1668S–1673S. doi: 10.1093/jn/133.5.1668S. [DOI] [PubMed] [Google Scholar]
- 5.Arechavaleta-Velasco F, Koi H, Strauss JF, III, Parry S. Viral infection of the trophoblast: time to take a serious look at its role in abnormal implantation and placentation? J Reprod Immunol. 2002;55:113–121. doi: 10.1016/s0165-0378(01)00143-7. [DOI] [PubMed] [Google Scholar]
- 6.Hsu CD, Witter FR. Urogenital infection in preeclampsia. Int J Gynaecol Obstet. 1995;49:271–275. doi: 10.1016/0020-7292(95)02373-k. [DOI] [PubMed] [Google Scholar]
- 7.Goldenberg RL, Hauth JC, Andrews WW. Intrauterine infection and preterm delivery. N Engl J Med. 2000;342:1500–1507. doi: 10.1056/NEJM200005183422007. [DOI] [PubMed] [Google Scholar]
- 8.Lamont RF. Infection in the prediction and antibiotics in the prevention of spontaneous preterm labour and preterm birth. BJOG. 2003;110(Suppl 20):71–75. doi: 10.1016/s1470-0328(03)00034-x. [DOI] [PubMed] [Google Scholar]
- 9.Romero R, Espinoza J, Goncalves LF, Kusanovic JP, Friel L, Hassan S. The role of inflammation and infection in preterm birth. Semin Reprod Med. 2007;25:21–39. doi: 10.1055/s-2006-956773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Matthiesen L, Berg G, Ernerudh J, Ekerfelt C, Jonsson Y, Sharma S. Immunology of preeclampsia. Chem Immunol Allergy. 2005;89:49–61. doi: 10.1159/000087912. [DOI] [PubMed] [Google Scholar]
- 11.Allaire AD, Ballenger KA, Wells SR, McMahon MJ, Lessey BA. Placental apoptosis in preeclampsia. Obstet Gynecol. 2000;96:271–276. doi: 10.1016/s0029-7844(00)00895-4. [DOI] [PubMed] [Google Scholar]
- 12.Kakinuma C, Kuwayama C, Kaga N, Futamura Y, Katsuki Y, Shibutani Y. Trophoblastic apoptosis in mice with preterm delivery and its suppression by urinary trypsin inhibitor. Obstet Gynecol. 1997;90:117–124. doi: 10.1016/S0029-7844(97)00176-2. [DOI] [PubMed] [Google Scholar]
- 13.Smith SC, Baker PN, Symonds EM. Increased placental apoptosis in intrauterine growth restriction. Am J Obstet Gynecol. 1997;177:1395–1401. doi: 10.1016/s0002-9378(97)70081-4. [DOI] [PubMed] [Google Scholar]
- 14.Abrahams VM, Mor G. Toll-like receptors and their role in the trophoblast. Placenta. 2005;26:540–547. doi: 10.1016/j.placenta.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 15.Kawai T, Akira S. TLR signaling. Cell Death Differ. 2006;13:816–825. doi: 10.1038/sj.cdd.4401850. [DOI] [PubMed] [Google Scholar]
- 16.Rock FL, Hardiman G, Timans JC, Kastelein RA, Bazan JF. A family of human receptors structurally related to Drosophila Toll. Proc Natl Acad Sci USA. 1998;95:588–593. doi: 10.1073/pnas.95.2.588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Medzhitov R, Janeway CA., Jr Decoding the patterns of self and nonself by the innate immune system. Science. 2002;296:298–300. doi: 10.1126/science.1068883. [DOI] [PubMed] [Google Scholar]
- 18.Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu Rev Immunol. 2003;21:335–376. doi: 10.1146/annurev.immunol.21.120601.141126. [DOI] [PubMed] [Google Scholar]
- 19.Aliprantis AO, Yang RB, Mark MR, Suggett S, Devaux B, Radolf JD, Klimpel GR, Godowski P, Zychlinsky A. Cell activation and apoptosis by bacterial lipoproteins through Toll-like receptor-2. Science. 1999;285:736–739. doi: 10.1126/science.285.5428.736. [DOI] [PubMed] [Google Scholar]
- 20.Bannerman DD, Tupper JC, Kelly JD, Winn RK, Harlan JM. The Fas-associated death domain protein suppresses activation of NF-κB by LPS and IL-1β. J Clin Invest. 2002;109:419–425. doi: 10.1172/JCI14774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lopez M, Sly LM, Luu Y, Young D, Cooper H, Reiner NE. The 19-kDa Mycobacterium tuberculosis protein induces macrophage apoptosis through Toll-like receptor-2. J Immunol. 2003;170:2409–2416. doi: 10.4049/jimmunol.170.5.2409. [DOI] [PubMed] [Google Scholar]
- 22.Into T, Kiura K, Yasuda M, Kataoka H, Inoue N, Hasebe A, Takeda K, Akira S, Shibata K. Stimulation of human Toll-like receptor (TLR) 2 and TLR6 with membrane lipoproteins of Mycoplasma fermentans induces apoptotic cell death after NF-κB activation. Cell Microbiol. 2004;6:187–199. doi: 10.1046/j.1462-5822.2003.00356.x. [DOI] [PubMed] [Google Scholar]
- 23.Chan G, Guilbert LJ. Ultraviolet-inactivated human cytomegalo-virus induces placental syncytiotrophoblast apoptosis in a Toll-like receptor-2 and tumour necrosis factor-α dependent manner. J Pathol. 2006;210:111–120. doi: 10.1002/path.2025. [DOI] [PubMed] [Google Scholar]
- 24.Equils O, Lu D, Gatter M, Witkin SS, Bertolotto C, Arditi M, McGregor JA, Simmons CF, Hobel CJ. Chlamydia heat shock protein 60 induces trophoblast apoptosis through TLR4. J Immunol. 2006;177:1257–1263. doi: 10.4049/jimmunol.177.2.1257. [DOI] [PubMed] [Google Scholar]
- 25.Salaun B, Coste I, Rissoan MC, Lebecque SJ, Renno T. TLR3 can directly trigger apoptosis in human cancer cells. J Immunol. 2006;176:4894–4901. doi: 10.4049/jimmunol.176.8.4894. [DOI] [PubMed] [Google Scholar]
- 26.Aravalli RN, Hu S, Lokensgard JR. Toll-like receptor 2 signaling is a mediator of apoptosis in herpes simplex virus-infected microglia. J Neuroinflammation. 2007;4:11. doi: 10.1186/1742-2094-4-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yoshimura A, Lien E, Ingalls RR, Tuomanen E, Dziarski R, Golenbock D. Cutting edge: recognition of Gram-positive bacterial cell wall components by the innate immune system occurs via Toll-like receptor 2. J Immunol. 1999;163:1–5. [PubMed] [Google Scholar]
- 28.Lien E, Sellati TJ, Yoshimura A, Flo TH, Rawadi G, Finberg RW, Carroll JD, Espevik T, Ingalls RR, Radolf JD, Golenbock DT. Toll-like receptor 2 functions as a pattern recognition receptor for diverse bacterial products. J Biol Chem. 1999;274:33419–33425. doi: 10.1074/jbc.274.47.33419. [DOI] [PubMed] [Google Scholar]
- 29.Schwandner R, Dziarski R, Wesche H, Rothe M, Kirschning CJ. Peptidoglycan- and lipoteichoic acid-induced cell activation is mediated by Toll-like receptor 2. J Biol Chem. 1999;274:17406–17409. doi: 10.1074/jbc.274.25.17406. [DOI] [PubMed] [Google Scholar]
- 30.Iwaki D, Mitsuzawa H, Murakami S, Sano H, Konishi M, Akino T, Kuroki Y. The extracellular Toll-like receptor 2 domain directly binds peptidoglycan derived from Staphylococcus aureus. J Biol Chem. 2002;277:24315–24320. doi: 10.1074/jbc.M107057200. [DOI] [PubMed] [Google Scholar]
- 31.Massari P, Henneke P, Ho Y, Latz E, Golenbock DT, Wetzler LM. Cutting edge: immune stimulation by neisserial porins is Toll-like receptor 2 and MyD88 dependent. J Immunol. 2002;168:1533–1537. doi: 10.4049/jimmunol.168.4.1533. [DOI] [PubMed] [Google Scholar]
- 32.Hirschfeld M, Kirschning CJ, Schwandner R, Wesche H, Weis JH, Wooten RM, Weis JJ. Cutting edge: inflammatory signaling by Borrelia burgdorferi lipoproteins is mediated by Toll-like receptor 2. J Immunol. 1999;163:2382–2386. [PubMed] [Google Scholar]
- 33.Takeuchi O, Kaufmann A, Grote K, Kawai T, Hoshino K, Morr M, Muhlradt PF, Akira S. Cutting edge: preferentially the R-stereoisomer of the mycoplasmal lipopeptide macrophage-activating lipopeptide-2 activates immune cells through a Toll-like receptor 2- and MyD88-dependent signaling pathway. J Immunol. 2000;164:554–557. doi: 10.4049/jimmunol.164.2.554. [DOI] [PubMed] [Google Scholar]
- 34.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 35.Ozinsky A, Underhill DM, Fontenot JD, Hajjar AM, Smith KD, Wilson CB, Schroeder L, Aderem A. The repertoire for pattern recognition of pathogens by the innate immune system is defined by cooperation between Toll-like receptors. Proc Natl Acad Sci USA. 2000;97:13766–13771. doi: 10.1073/pnas.250476497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Takeuchi O, Kawai T, Muhlradt PF, Morr M, Radolf JD, Zychlinsky A, Takeda K, Akira S. Discrimination of bacterial lipoproteins by Toll-like receptor 6. Int Immunol. 2001;13:933–940. doi: 10.1093/intimm/13.7.933. [DOI] [PubMed] [Google Scholar]
- 37.Bulut Y, Faure E, Thomas L, Equils O, Arditi M. Cooperation of Toll-like receptor 2 and 6 for cellular activation by soluble tuberculosis factor and Borrelia burgdorferi outer surface protein A lipoprotein: role of Toll-interacting protein and IL-1 receptor signaling molecules in Toll-like receptor 2 signaling. J Immunol. 2001;167:987–994. doi: 10.4049/jimmunol.167.2.987. [DOI] [PubMed] [Google Scholar]
- 38.Takeuchi O, Sato S, Horiuchi T, Hoshino K, Takeda K, Dong Z, Modlin RL, Akira S. Cutting edge: role of Toll-like receptor 1 in mediating immune response to microbial lipoproteins. J Immunol. 2002;169:10–14. doi: 10.4049/jimmunol.169.1.10. [DOI] [PubMed] [Google Scholar]
- 39.Han SH, Kim JH, Martin M, Michalek SM, Nahm MH. Pneumococcal lipoteichoic acid (LTA) is not as potent as staphylococcal LTA in stimulating Toll-like receptor 2. Infect Immun. 2003;71:5541–5548. doi: 10.1128/IAI.71.10.5541-5548.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Abrahams VM, Bole-Aldo P, Kim YM, Straszewski-Chavez SL, Chaiworapongsa T, Romero R, Mor G. Divergent trophoblast responses to bacterial products mediated by TLRs. J Immunol. 2004;173:4286–4296. doi: 10.4049/jimmunol.173.7.4286. [DOI] [PubMed] [Google Scholar]
- 41.Kajikawa S, Kaga N, Futamura Y, Kakinuma C, Shibutani Y. Lipoteichoic acid induces preterm delivery in mice. J Pharmacol Toxicol Methods. 1998;39:147–154. doi: 10.1016/s1056-8719(98)00015-x. [DOI] [PubMed] [Google Scholar]
- 42.Ilievski V, Lu SJ, Hirsch E. Activation of Toll-like receptors 2 or 3 and preterm delivery in the mouse. Reprod Sci. 2007;14:315–320. doi: 10.1177/1933719107302959. [DOI] [PubMed] [Google Scholar]
- 43.Graham CH, Hawley TS, Hawley RG, MacDougall JR, Kerbel RS, Khoo N, Lala PK. Establishment and characterization of first trimester human trophoblast cells with extended lifespan. Exp Cell Res. 1993;206:204–211. doi: 10.1006/excr.1993.1139. [DOI] [PubMed] [Google Scholar]
- 44.Murphy SP, Fast LD, Hanna NN, Sharma S. Uterine NK cells mediate inflammation-induced fetal demise in IL-10-null mice. J Immunol. 2005;175:4084–4090. doi: 10.4049/jimmunol.175.6.4084. [DOI] [PubMed] [Google Scholar]
- 45.Kadyrov M, Kaufmann P, Huppertz B. Expression of a cytokeratin 18 neo-epitope is a specific marker for trophoblast apoptosis in human placenta. Placenta. 2001;22:44–48. doi: 10.1053/plac.2000.0616. [DOI] [PubMed] [Google Scholar]
- 46.Mor G, Sapi E, Abrahams VM, Rutherford T, Song J, Hao XY, Muzaffar S, Kohen F. Interaction of the estrogen receptors with the Fas ligand promoter in human monocytes. J Immunol. 2003;170:114–122. doi: 10.4049/jimmunol.170.1.114. [DOI] [PubMed] [Google Scholar]
- 47.Abrahams VM, Straszewski SL, Kamsteeg M, Hanczaruk B, Schwartz PE, Rutherford TJ, Mor G. Epithelial ovarian cancer cells secrete functional Fas ligand. Cancer Res. 2003;63:5573–5581. [PubMed] [Google Scholar]
- 48.Costello MJ, Joyce SK, Abrahams VM. NOD protein expression and function in first trimester trophoblast cells. Am J Reprod Immunol. 2007;57:67–80. doi: 10.1111/j.1600-0897.2006.00447.x. [DOI] [PubMed] [Google Scholar]
- 49.Fest S, Aldo PB, Abrahams VM, Visintin I, Alvero A, Chen R, Chavez SL, Romero R, Mor G. Trophoblast-macrophage interactions: a regulatory network for the protection of pregnancy. Am J Reprod Immunol. 2007;57:55–66. doi: 10.1111/j.1600-0897.2006.00446.x. [DOI] [PubMed] [Google Scholar]
- 50.Leung CH, Grill SP, Lam W, Gao W, Sun HD, Cheng YC. Eriocalyxin B inhibits nuclear factor-κB activation by interfering with the binding of both p65 and p50 to the response element in a noncompetitive manner. Mol Pharmacol. 2006;70:1946–1955. doi: 10.1124/mol.106.028480. [DOI] [PubMed] [Google Scholar]
- 51.Zhang M, Guller S, Huang Y. Method to enhance transfection efficiency of cell lines and placental fibroblasts. Placenta. 2007;28:779–782. doi: 10.1016/j.placenta.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 52.Elovitz MA, Mrinalini C. Animal models of preterm birth. Trends Endocrinol Metab. 2004;15:479–487. doi: 10.1016/j.tem.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 53.Roberts JM, Redman CW. Pre-eclampsia: more than pregnancy-induced hypertension. Lancet. 1993;341:1447–1451. doi: 10.1016/0140-6736(93)90889-o. [DOI] [PubMed] [Google Scholar]
- 54.Smith GC, Smith MF, McNay MB, Fleming JE. First-trimester growth and the risk of low birth weight. N Engl J Med. 1998;339:1817–1822. doi: 10.1056/NEJM199812173392504. [DOI] [PubMed] [Google Scholar]
- 55.Fortunato SJ, Menon R, Bryant C, Lombardi SJ. Programmed cell death (apoptosis) as a possible pathway to metalloproteinase activation and fetal membrane degradation in premature rupture of membranes. Am J Obstet Gynecol. 2000;182:1468–1476. doi: 10.1067/mob.2000.107330. [DOI] [PubMed] [Google Scholar]
- 56.Gendron RL, Nestel FP, Lapp WS, Baines MG. Lipopolysaccharide-induced fetal resorption in mice is associated with the intrauterine production of tumour necrosis factor-α. J Reprod Fertil. 1990;90:395–402. doi: 10.1530/jrf.0.0900395. [DOI] [PubMed] [Google Scholar]
- 57.Elovitz MA, Wang Z, Chien EK, Rychlik DF, Phillippe M. A new model for inflammation-induced preterm birth: the role of platelet-activating factor and Toll-like receptor-4. Am J Pathol. 2003;163:2103–2111. doi: 10.1016/S0002-9440(10)63567-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Diamond AK, Sweet LM, Oppenheimer KH, Bradley DF, Phillippe M. Modulation of monocyte chemotactic protein-1 expression during lipopolysaccharide-induced preterm delivery in the pregnant mouse. Re-prod Sci. 2007;14:548–559. doi: 10.1177/1933719107307792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Abrahams VM, Visintin I, Aldo PB, Guller S, Romero R, Mor G. A role for TLRs in the regulation of immune cell migration by first trimester trophoblast cells. J Immunol. 2005;175:8096–8104. doi: 10.4049/jimmunol.175.12.8096. [DOI] [PubMed] [Google Scholar]
- 60.Nakao Y, Funami K, Kikkawa S, Taniguchi M, Nishiguchi M, Fukumori Y, Seya T, Matsumoto M. Surface-expressed TLR6 participates in the recognition of diacylated lipopeptide and peptidoglycan in human cells. J Immunol. 2005;174:1566–1573. doi: 10.4049/jimmunol.174.3.1566. [DOI] [PubMed] [Google Scholar]
- 61.Sandor F, Latz E, Re F, Mandell L, Repik G, Golenbock DT, Espevik T, Kurt-Jones EA, Finberg RW. Importance of extra- and intracellular domains of TLR1 and TLR2 in NFκB signaling. J Cell Biol. 2003;162:1099–1110. doi: 10.1083/jcb.200304093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Buwitt-Beckmann U, Heine H, Wiesmuller KH, Jung G, Brock R, Akira S, Ulmer AJ. Toll-like receptor 6-independent signaling by diacylated lipopeptides. Eur J Immunol. 2005;35:282–289. doi: 10.1002/eji.200424955. [DOI] [PubMed] [Google Scholar]
- 63.Omueti KO, Beyer JM, Johnson CM, Lyle EA, Tapping RI. Domain exchange between human Toll-like receptors 1 and 6 reveals a region required for lipopeptide discrimination. J Biol Chem. 2005;280:36616–36625. doi: 10.1074/jbc.M504320200. [DOI] [PubMed] [Google Scholar]
- 64.Hajjar AM, O’Mahony DS, Ozinsky A, Underhill DM, Aderem A, Klebanoff SJ, Wilson CB. Cutting edge: functional interactions between toll-like receptor (TLR) 2 and TLR1 or TLR6 in response to phenol-soluble modulin. J Immunol. 2001;166:15–19. doi: 10.4049/jimmunol.166.1.15. [DOI] [PubMed] [Google Scholar]
- 65.Aliprantis AO, Yang RB, Weiss DS, Godowski P, Zychlinsky A. The apoptotic signaling pathway activated by Toll-like receptor-2. EMBO J. 2000;19:3325–3336. doi: 10.1093/emboj/19.13.3325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang CY, Mayo MW, Korneluk RG, Goeddel DV, Baldwin AS., Jr NF-κB antiapoptosis: induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science. 1998;281:1680–1683. doi: 10.1126/science.281.5383.1680. [DOI] [PubMed] [Google Scholar]
- 67.Zarember KA, Godowski PJ. Tissue expression of human Toll-like receptors and differential regulation of Toll-like receptor mRNAs in leukocytes in response to microbes, their products, and cytokines. J Immunol. 2002;168:554–561. doi: 10.4049/jimmunol.168.2.554. [DOI] [PubMed] [Google Scholar]
- 68.Kim YM, Romero R, Oh SY, Kim CJ, Kilburn BA, Armant DR, Nien JK, Gomez R, Mazor M, Saito S, et al. Toll-like receptor 4: a potential link between “danger signals,” the innate immune system, and pre-eclampsia? Am J Obstet Gynecol. 2005;193:921–927. doi: 10.1016/j.ajog.2005.07.076. [DOI] [PubMed] [Google Scholar]