

Abstract
Oncogenic tyrosine kinases have proven to be promising targets for the development of highly effective anticancer drugs. However HER family tyrosine kinase inhibitors (TKIs) show only limited activity against HER2-driven cancers despite effective inhibition of EGFR and HER2 in vivo 1–8. The reasons for this are unclear. Signaling in trans is a key feature of this multimember family and the critically important PI3K/Akt pathway is driven predominantly through transphosphorylation of the kinase-inactive HER3 9,10. We report that HER3 and consequently PI3K/Akt signaling evade inhibition by current HER family TKIs in vitro and in tumors in vivo. This is due to a compensatory shift in HER3 phosphorylation-dephosphorylation equilibrium driven by increased membrane HER3 expression driving the phosphorylation reaction and reduced HER3 phosphatase activity impeding the dephosphorylation reaction. These compensatory changes are driven by Akt mediated negative feedback signaling. Although HER3 is not a direct target of TKIs, HER3 substrate resistance undermines their efficacy and has thus far gone undetected. The experimental abbrogation of HER3 resistance by siRNA knockdown restores potent pro-apoptotic effects to otherwise cytostatic HER TKIs, re-affirming the oncogene-addicted nature of HER2-driven tumors and the therapeutic promise of this oncoprotein target. However, since HER3 signaling is buffered against an incomplete inhibition of HER2 kinase, much more potent TKIs or combination strategies are required to effectively silence oncogenic HER2 signaling. The biologic marker to guide HER TKIs should be the transphosphorylation of HER3.
Selective inhibitors of Abl tyrosine kinase (TK) are effective in putting nearly all patients with bcr-abl driven leukemia in chronic phase into complete remission 11. This proof of concept sheds new hope for the treatment of other TK-driven cancers. Another important TK family is the human epidermal growth factor receptor (HER) family consisting of EGFR, HER2, HER3, and HER4. A subset of breast cancers are driven by overactive EGFR or HER2 TKs and an abundance of data from in vitro and mouse models suggests that continued activity of these TKs drives cancer progression 12–14. While the complexities of this multi-member TK family are not yet fully understood, their oncogenic signaling functions should, in theory, be amenable to silencing by TK inhibitors (TKIs). Several orally bioavailable HER family selective TKIs are in preclinical and clinical development. Although in in vitro biochemical assays these agents differ in their relative activities against individual HER kinase family members, in cell-based assays they are effective at inhibiting both EGFR and HER2 and equally effective at suppressing the growth of EGFR and HER2 driven tumor cells 15–19. They are also effective at inhibiting EGFR and HER2 phosphorylation in patients tissues and tumors 5–8. But these agents show very limited clinical anti-tumor activity 1–5. Their clinical development to this point has been driven largely by the detection of modest delays in tumor progression. The failure to reverse cancer progression despite an apparent inhibition of HER kinase function has created an enigma in the concept of TKI therapy of cancer that we have been exploring.
It is through heterodimerization and transphosphorylation that the HER family performs its signaling functions. Importantly, downstream PI3K/Akt pathway signaling is predominantly mediated through the transphosphorylation of the kinase-inactive member HER3 9,10. We have previously reported that sensitivity to HER family TKI therapy correlates with the inhibition of PI3K/Akt pathway signaling 15,20. We and others have also reported that failure to inhibit PI3K/Akt signaling leads to TK inhibitor resistance 20–22. In contrast to reports from in vitro models, Akt activity is not inhibited in most patients on HER TKI therapy 5,6,8. This discordancy has led us to look more closely at the inhibition of PI3K/Akt signaling.
To investigate this discrepancy, we studied the durability of Akt inhibition by TKI with surprising results. Although as previously reported, gefitinib inhibits Akt signaling in HER2-driven cancer cells, this inhibition is not durable. Akt signaling resumes after a transient inhibition despite continued drug therapy (figures 1A,B). In light of this finding, we looked at the broader HER family signaling activities over a period of 96 hours following continuous exposure of BT474 breast cancer cells to gefitinib at concentrations that nonselectively inhibit EGFR and HER2. TKI treatment effects a sustained inhibition of EGFR and HER2 phosphorylation and a durable inhibition of downstream MAPK and JNK pathway signaling (figure 1A). However phosphorylation of the kinase-inactive family member HER3 is merely transient. HER3 signaling resumes and persists despite continued drug exposure and effective suppression of EGFR and HER2 (figure 1A,B). The reactivation of HER3 signaling explains the reactivation of Akt signaling since HER3 is the principal HER family member that binds PI3K and drives Akt signaling 9,10. TKI-refractory Akt signaling remains sensitive to PI3K inhibitors as expected (not shown). These time-dependent findings are not due to drug degradation since the drug is replenished daily in these studies and HER3/Akt signaling resumes despite repeatedly refreshing drug supply up to and beyond the point of resumption of Akt signaling (not shown). There is no significant expression of HER4 before or after drug treatment in these cells (data not shown). These findings are not unique to BT474 and SkBr3 cells and have been confirmed in other HER2 overexpressing breast cancer cells including MDA-453, AU565, MDA-361, HCC1954 (supplementary figure 1). These findings are not unique to gefitinib and are seen with other HER TKIs including agents with in vitro selectivity profiles favoring EGFR or HER2, such as erlotinib or AG825 (figure 1C,D). These findings are not artifacts of the in vitro models either. Treatment of mice bearing various HER2-driven xenograft tumors with gefitinib similarly fails to durably supress HER3 and Akt signaling, despite a transient suppression (figure 1E, and supplementary figure 2). This is not due to ineffective drug biodistribution, since in these models gefitinib was dosed three times higher than doses known to achieve sustained xenograft tumor concentrations above 2–4uM and averaging 6–10uM 23. Since we had previously established that inactivation of PI3K/Akt signaling is mechanistically linked to HER family TK inhibitor sensitivity in HER family driven cancers, we felt that the failure of these drugs to durably inactivate PI3K/Akt signaling is entirely consistent with their limited clinical activities. Therefore we set out to study the molecular basis by which HER3 evades TKI therapy.
Figure 1. HER TK inhibitors fail to induce sustained inhibition of HER3 signaling in HER2-driven breast cancer cells.

(A) BT474 cells were treated with 5 μM gefitinib for the indicated times and assayed for expression and phosphorylation of the indicated proteins. Lane 0* is an IgG immunoprecipitation control. (B) Data of interest shown from the identical experiment done in SkBr3 cells. (C) SKBr3 cells were treated with 5 μM erlotinib for the indicated times and lysates immunoblotted with the indicated antibodies. (D) SKBr3 cells were treated with 20 μM AG825 for the indicated times with drug being replenished 1 hr before the 72 hour harvest. (E) Mice bearing HCC1569 human breast cancer xenografts were treated with gefitinib at 150 mg/kg once daily. Animals were sacrificed at 2 hours after the first dose and 2 hours after the 4th dose (96 hours total treatment) and tumors rapidly harvested for immunoblotting with the indicated antibodies. The six lanes corresponding to each timepoint represent six different animals.
TKI-refractory HER3 phosphorylation is due to HER2, since it can be suppressed by anti-HER2 siRNA transfection (figure 2A). Although cross talk between receptor families can occur, we have found no evidence for non-HER family TKs mediating TKI refractory HER3 phosphorylation. The reactivation of HER3 signaling is not associated with the induction of any new TKs and remains resistant to the broad spectrum kinase inhibitor staurosporine (not shown). This apparent desensitization of HER3 signaling to TKIs is due to a forward shift in the equilibrium of the HER3 phosphorylation-dephosphorylation reactions establishing a new steady state HER3 phosphorylation despite significant inhibition of HER2 kinase and autophosphorylation activity by TKIs. This forward shift becomes clearly evident in the form of HER3 and Akt superphosphorylation when drug inhibition is withdrawn during the new steady state (figure 2B). HER3 phosphorylation remains suppressible by TKI in the new steady state, but much higher concentrations are required to completely dephosphorylate HER3 since the uninhibited HER3 phosphorylation state is significantly higher in the new steady state (figure 2C, compare left and right). Therefore drug-refractory HER3 phosphorylation is due to resistance at the level of the substrate HER3, and is driven by residual HER2 kinase activity. Similar characteristics apply to the more potent irreversible TKIs 24. The irreversible pan-HER family TKI PD168393, when used at partially or near-maximal inhibitory concentrations, induces a similar desensitization of HER3 to continued drug therapy (figure 2D, 0.1–0.2uM doses). However, at fully inactivating concentrations both reversible (figure 2C, 40uM dose) and irreversible TKI (Figure 2E) can durably suppress HER3 and Akt signaling.
Figure 2. Forward shift in HER3 phosphorylation-dephosphorylation equilibrium following extended HER TKI treatment.

(A) BT474 cells were transfected with anti-HER2 (H) or control (C) siRNA and harvested 64 hours after transfection (lanes 1,2). Additional arms were treated with 48 hours of gefitinib untransfected (lanes 3,4) or following siRNA transfection (lanes 5,6). (B) SKBr3 cells were treated with 5μM gefitinib for 0,1, or 48 hours. Arm W was treated for 48 hours, washed, and incubated in drug-free media for one more hour. (C) SKBr3 cells were treated with the indicated concentrations of gefitinib for one hour (left side). Additional arms were treated with 5 μM gefitinib for 48 hours and subsequently treated with the indicated concentrations of gefitinib for one additional hour (right side). (D) SKBr3 cells were treated with the indicated concentrations and durations of PD168393. (E) SKBr3 cells were treated with 2 μM PD168393 for the indicated times. (F) SKBr3 cells were transfected with anti-HER3 (H) or control (C) siRNA and harvested 4 days after transfection (lanes 1,2). Additional arms were treated with gefitinib or control in untransfected cells (lanes 3,4) or following siRNA transfection (lanes 5,6). (G) In parallel with figure F, SkBr3 cells were either left untransfected, or transfected with anti-HER3 or control siRNA followed by 5uM gefitinib or control for 48 hours. Apoptotic cells were identified by their subG1 DNA content. (H) SKBr3 cells were treated as indicated for 48 hours. Apoptotic cells were identified by Annexin V expression.
The biological consequence of drug-refractory HER3 and Akt signaling is tumor cell survival. In fact the anti-proliferative activity of TKIs is reversible and tumor cells resume proliferative growth after drug withdrawal. If drug-refractory HER3 signaling is averted by anti-HER3 siRNA, TKI treatment of HER2-driven cancer cells leads to apoptotic cell death (figures 2F, 2G, and supplemental figure 4). This is the expected outcome of effective oncoprotein inactivation and recapitulates the apoptotic fate of oncogene withdrawal seen in reversible transgenic models of HER2 tumorigenesis 14. Sustained inhibition of HER3 signaling using TKIs at their fully inactivating doses (from figures 2C,2D,2E) also leads to apoptotic tumor cell death not seen with doses that allow HER3 escape (figure 2H).
The TKI-induced forward shift in the HER3 phosphorylation-dephosphorylation steady state is due to increased HER3 substrate concentration driving the forward reaction, and decreased phosphatase activity impeding the reverse reaction. Increased HER3 substrate concentration occurs through a significant increase in HER3 expression at the plasma membrane where the phosphorylation reaction occurs (figure 3A,B). Unlike HER2 which is predominantly localized to the plasma membrane, the HER3 pool is largely within intracellular compartments with some membrane expression 25. The TKI-induced forward shift in HER3 steady state phosphorylation is driven by HER3 relocalization to the plasma membrane and can be suppressed by inhibitors of vesicular trafficking (figure 3C,3D). The HER3 dephosphorylation rate is also slowed after 48 hours of TKI exposure (figure 3E). The retarded HER3 dephosphorylation rate may be due to reduced access to cytosolic protein tyrosine phosphatases (PTPs) due to altered endocytic trafficking, or it may be due to inhibition of PTPs. In support of the latter, TKI therapy leads to increased cellular reactive oxygen species (ROS) (figure 3F) which are known to inhibit PTPs and are emerging as an important regulator of PTP activity 26,27. Consistent with this, drug-refractory HER3 signaling can be suppressed by concomitant treatment with certain anti-oxidants (figure 3G).
Figure 3. Mechanism of HER3 reactivation following extended HER TKI treatment.

(A) SKBr3 cells were treated with 5 μM gefitinib or control for 48h and stained with anti-HER2 or anti-HER3 antibodies for immunofluorescence microscopy. (B) In addition, cell surface proteins in control or 48 hour pre-treated cells were biotinylated, precipitated, and immunoblotted as indicated. (C) Control or gefitinib pre-treated (5 μM, 48h) SKBr3 cells were treated with 20 μM monensin for the final 6h and analyzed by western blotting. (D) SkBr3 cells were treated with gefitinib for 48 hrs and with 20uM monensin for the final 6 hours. Membrane and total HER3 was immunoblotted from cell surface proteome pulldowns (above) or total lysates (below). (E) The dephosphorylation rate of p-HER3 following 48hrs of gefitinib treatment was determined immediately after initiation of fully inactivating concentrations of the irreversible HER TK inhibitor PD168393 (2 μM). (F) Reactive oxygen species were quantified in control (A) or gefitinib pre-treated (B) (5 μM, 48h)SKBr3 cells as described in methods. (G) SKBr3 cells were treated with 5 uM gefitinib or in combination with the indicated concentrations of the anti-oxidants Tempol or α-lipoic acid for 48 hours.
The changes in steady state HER3 signaling that evolve with TKI treatment are driven by the loss of Akt signaling and likely involve Akt-mediated negative feedback signaling. Consistent with this, HER3 signaling does not escape TKI treatment when a constitutively active Akt is transfected (figure 4A). Conversely, inhibition of Akt signaling by a PI3K inhibitor leads to a compensatory increase in HER3 phosphorylation (figure 4B). Complete inactivation of HER2 kinase with high doses of TKIs induces the maximum feedback signaling and HER3 redistribution, however due to the complete inactivation of HER2 kinase HER3 signaling cannot be restored and the feedback loop fails to rescue Akt activity.
Figure 4. Akt regulates HER3 signaling via negative feedback signaling.
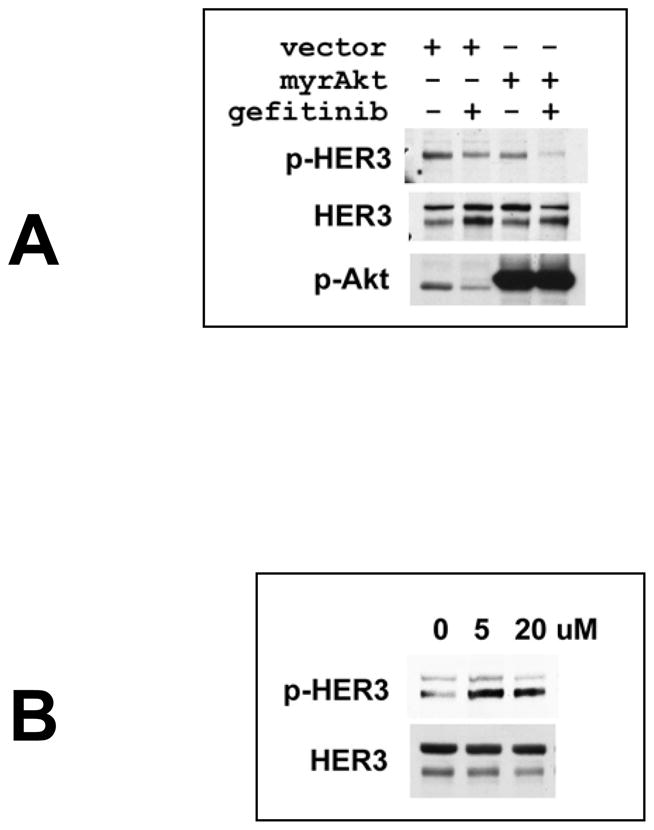
(A) 2×106 SKBr3 cells were transiently transfected with 5 μg of pcDNA3-myr-Akt plasmid or control pcDNA3 plasmid. The following day 5 μM gefitinib was added for an additional 48h where indicated. Lysates were immunoblotted as indicated. (B) SKBr3 cells were treated with 5 or 20 μM LY294002 for 8h and lysates immunoblotted as indicated.
Discussion
Since standard preclinical models have notoriously overestimated the clinical potential of HER TKIs, we challenged the traditional approach to evaluating TKIs in these models. Traditionally, signaling inhibitors are thought to have a continuous suppressive effect through rapid and sustained inhibition of their direct molecular targets and downstream signaling events. This notion of drug therapy may be too simplistic. Clearly, continuous exposure to a growth factor stimulus does not produce continuous high output downstream signaling. Rather, it leads to a sequelae of signaling events, programmed by negative and positive feedback signaling, until establishment of a new steady state in the presence of continued stimulus. We find here that continuous exposure to TKIs similarly leads to a sequence of signaling events that manifest over time, until a new steady state is reached. With this new perspective, we report that HER3/PI3K/Akt signaling is not effectively inhibited by current TKIs. In particular, the allocation of kinase and signaling functions to different members within the HER family allows the signaling substrate HER3 to restore signaling activity despite significant inhibition of HER2 kinase, in effect buffering HER3/PI3K/Akt signaling against an incomplete loss of HER2 kinase function. This inherent signal buffering capacity allows tumor cells to evade the pro-apoptotic effects of TKIs resulting in a significant loss of their anti-tumor activity. The highly effective treatment of HER-driven cancers may require drugs with much higher potency or drugs that completely inactivate HER kinase function. Irreversible TKIs, although more potent, are subject to similar limitations. Due to their reactive groups and reduced selectivity, many irreversible agents cannot be delivered at completely inactivating doses. Future highly selective irreversible inhibitors may turn out to be more effective. Until highly specific and fully inactivating drugs can be designed, combination treatment strategies designed to undermine the resiliency of HER family signaling may offer the most promising approach in the near future. In addition, inhibition of autophosphorylation activity deceptively overstates the efficacy of TKIs and is a poor in vivo biologic marker. The true biological marker of efficacy to guide future therapies should be HER3 transphosphorylation.
The signal buffering capacity endowed by the separation of kinase and signaling functions to different family members in the HER kinase family attests to an evolutionary advantage conferred by the loss of catalytic activity in the HER3 protein kinase. This can shed light on why approximately 10% of the human kinome appears to be catalytically inactive 28.
Brief Methods
Cell culture and reagents
PD168393 was synthesized as previously described 29. Commercially available gefitinib and erlotinib were purified for in vitro use. Reagent sources are detailed in supplementary materials. For immunofluorescence studies, cells grown on fibronectin coated cover slips were treated as indicated, fixed in 4% paraformaldehyde, permeabilized, and stained with the indicated primary antibodies and FITC conjugated secondary antibodies. Cells were visualized using a Zeiss Axioplan 2 fluorescence imaging microscope.
Apoptosis
Cells were seeded at 300,000–500,000 per well in 12-well or 6-well clusters. Apoptotic cells were identified and quantified by analysis of Annexin V binding using the Annexin V-FITC Apoptosis Detection Kit (Calbiochem) according to the manufacturer’s instructions, or by their sub-G1 DNA content and quantified by FACS analysis as previously described 30. All experimental arms were done in duplicate and displayed as averages with standard of deviation error bars.
Transfections
Cells were seeded at a density of 300,000 cells per well in 12-well plates and transfected the following day. For siRNA transfections 100–300nmol of siRNA (Dharmacon) was premixed with Lipofectemine2000 in Opti-MEM media and then added to each well. For plasmid transfections, 2 ug of plasmid DNA was premixed with Lipofectamine2000 in Opti-MEM media and added to wells for 6 hours.
Cell surface biotinylation
Cells were chilled on ice and rinsed twice with ice-cold PBS. Freshly prepared sulfo-NHS-SS-biotin was added to the final concentration of 0.5 mg/ml in PBS. Following 45min incubation at 4°C cells were lysed for immunoprecipitation.
Reactive Oxidation Species Assay
Cells were rinsed twice with PBS and incubated with 10 μM of freshly prepared H2DCFDA in phenol-red free media for 45 min at 37°C. Cells were then trypsinized and ROS levels were detected by flow cytometry.
Supplementary Material
Acknowledgments
This work was supported by the Susan Komen Foundation (MMM), the California Breast Cancer Research Program (MMM), and NIH AI-44009 (KMS). We thank David Stokoe and Frank McCormick for review of the manuscript.
Footnotes
Author contribution: All authors contributed to the experiments in this work. The studies were conceived by MMM with additional contributions from NS and KMS. The paper was written by NS and MMM.
Author information: The authors declare no competing financial interests.
Reference List
- 1.Winer EP, Cobleigh M, Dickler M, Miller K, Fehrehbacher L, Jones CM, Anderson S, Eberhard D, Jones C. Phase II multicenter study to evaluate the efficacy and safety of Tarceva (Erlotinib HCl, OSI-774) in women with previously treated, locally advanced or metastatic breast cancer. San Antonio Br Can Symp. 2002:445. [Google Scholar]
- 2.Blackwell K, Kaplan EH, Franco SX, Marcom PK, Maleski MJ, Sorenson MS, Berger MS. A phase II, open-label, multicenter study of GW572016 in patients with trastuzumab-refractory metastatic breast cancer. Proc Amer Soc Clin Onc. 2004:3006. [Google Scholar]
- 3.Dees EC, Burris HA, Hurwitz H, Dowlati A, Smith D, Kock KM, Stead A, Mangum S, Harris JL, Spector N. Clinical summary of 67 heavily pre-treated patients with metastatic carcinomas treated with GW572016 in a phase Ib study. Proc Amer Soc Clin Onc. 2004:3188. [Google Scholar]
- 4.Campos SM, Seiden MV, Oza A, Plante M, Potkul R, Hamid O, Lenehan P, Kaldjian E, Jordan C, Hirte H. A phase 2, single agent study of CI-1033 administered at two doses in ovarian cancer patients who failed platinum therapy. Proc Amer Soc Clin Onc. 2004:5054. [Google Scholar]
- 5.Baselga J, et al. Phase II and tumor pharmacodynamic study of gefitinib in patients with advanced breast cancer. J Clin Oncol. 2005;23:5323–5333. doi: 10.1200/JCO.2005.08.326. [DOI] [PubMed] [Google Scholar]
- 6.Bacus SS, Beresford PJ, Yarden Y, Spector N, Smith B. The use of predicting factors and surrogate markers in patients’ cancer biopsies treated with targeted antibodies to erbB receptors and erbB tyrosine kinase inhibitors. Proc Amer Soc Clin Onc. 3408;22:2003. [Google Scholar]
- 7.Burris HA, Hurwitz H, Dees C, Dowlati A, Blackwell K, Ellis M, Overmoyer B, Jones S, Willcutt N, Smith DA, Harris JL, Spector NA. EGF10004: a randomized, multicenter, phase Ib study of the safety, biologic activity and clinical efficacy of the dual kinase inhibitor GW572016. San Antonio Br Can Symp. 2003:39. [Google Scholar]
- 8.Spector NL, et al. Study of the biologic effects of lapatinib, a reversible inhibitor of ErbB1 and ErbB2 tyrosine kinases, on tumor growth and survival pathways in patients with advanced malignancies. J Clin Oncol. 2005;23:2502–2512. doi: 10.1200/JCO.2005.12.157. [DOI] [PubMed] [Google Scholar]
- 9.Soltoff SP, Carraway KL, III, Prigent SA, Gullick WG, Cantley LC. ErbB3 is involved in activation of phosphatidylinositol 3-kinase by epidermal growth factor. Mol Cell Biol. 1994;14:3550–3558. doi: 10.1128/mcb.14.6.3550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim HH, Sierke SL, Koland JG. Epidermal growth factor-dependent association of phosphatidylinositol 3-kinase with the erbB3 gene product. J Biol Chem. 1994;269:24747–24755. [PubMed] [Google Scholar]
- 11.Kantarjian H, et al. Hematologic and cytogenetic responses to imatinib mesylate in chronic myelogenous leukemia. N Engl J Med. 2002;346:645–652. doi: 10.1056/NEJMoa011573. [DOI] [PubMed] [Google Scholar]
- 12.Muller WJ, Sinn E, Pattengale PK, Wallace R, Leder P. Single-step induction of mammary adenocarcinoma in transgenic mice bearing the activated c-neu oncogene. Cell. 1988;54:105–115. doi: 10.1016/0092-8674(88)90184-5. [DOI] [PubMed] [Google Scholar]
- 13.Slamon DJ, et al. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235:177–182. doi: 10.1126/science.3798106. [DOI] [PubMed] [Google Scholar]
- 14.Moody SE, et al. Conditional activation of Neu in the mammary epithelium of transgenic mice results in reversible pulmonary metastasis. Cancer Cell. 2002;2:451–461. doi: 10.1016/s1535-6108(02)00212-x. [DOI] [PubMed] [Google Scholar]
- 15.Moasser MM, Basso A, Averbuch SD, Rosen N. The tyrosine kinase inhibitor ZD1839 (“Iressa”) inhibits HER2-driven signaling and suppresses the growth of HER2-overexpressing tumor cells. Cancer Research. 2001;61:7184–7188. [PubMed] [Google Scholar]
- 16.Moulder SL, et al. Epidermal growth factor receptor (HER1) tyrosine kinase inhibitor ZD1839 (Iressa) inhibits HER2/neu (erbB2)-overexpressing breast cancer cells in vitro and in vivo. Cancer Research. 2001;61:8887–8895. [PubMed] [Google Scholar]
- 17.Anderson NG, Ahmad T, Chan K, Dobson R, Bundred NJ. ZD1839 (Iressa), a novel epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor, potently inhibits the growth of EGFR-positive cancer cell lines with or without erbB2 overexpression. International Journal of Cancer. 2001;94:774–782. doi: 10.1002/ijc.1557. [DOI] [PubMed] [Google Scholar]
- 18.Campiglio M, et al. Inhibition of proliferation and induction of apoptosis in breast cancer cells by the epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor ZD1839 (‘Iressa’) is independent of EGFR expression level. Journal of Cellular Physiology. 2004;198:259–268. doi: 10.1002/jcp.10411. [DOI] [PubMed] [Google Scholar]
- 19.Akita RW, Sliwkowski MX. Preclinical studies with Erlotinib (Tarceva) Seminars in Oncology 30. 2003;3 (suppl 7):15–24. [PubMed] [Google Scholar]
- 20.She Q, Solit D, Basso A, Moasser MM. Resistance to gefitinib (ZD1839, Iressa) in PTEN null HER overexpressing tumor cells can be overcome through restoration of PTEN function or pharmacologic modulation of constitutive PI3K/Akt pathway signaling. Clinical Cancer Research. 2003;9:4340–4346. [PubMed] [Google Scholar]
- 21.Bianco R, et al. Loss of PTEN/MMAC1/TEP in EGF receptor-expressing tumor cells counteracts the antitumor action of EGFR tyrosine kinase inhibitors. Oncogene. 2003;22:2812–2822. doi: 10.1038/sj.onc.1206388. [DOI] [PubMed] [Google Scholar]
- 22.Haas-Kogan DA, et al. Epidermal growth factor receptor, protein kinase B/Akt, and glioma response to erlotinib. J Natl Cancer Inst. 2005;97:880–887. doi: 10.1093/jnci/dji161. [DOI] [PubMed] [Google Scholar]
- 23.McKillop D, et al. Tumor penetration of gefitinib (Iressa), an epidermal growth factor receptor tyrosine kinase inhibitor. Mol Cancer Ther. 2005;4:641–649. doi: 10.1158/1535-7163.MCT-04-0329. [DOI] [PubMed] [Google Scholar]
- 24.Fry DW, et al. Specific, irreversible inactivation of the epidermal growth factor receptor and erbB2, by a new class of tyrosine kinase inhibitor. Proc Natl Acad Sci USA. 1998;95:12022–12027. doi: 10.1073/pnas.95.20.12022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Offterdinger M, Schofer C, Weipoltshammer K, Grunt TW. c-erbB-3: a nuclear protein in mammary epithelial cells. J Cell Biol. 2002;157:929–939. doi: 10.1083/jcb.200109033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Meng TC, Fukada T, Tonks NK. Reversible oxidation and inactivation of protein tyrosine phosphatases in vivo. Molecular Cell. 2002;9:387–399. doi: 10.1016/s1097-2765(02)00445-8. [DOI] [PubMed] [Google Scholar]
- 27.Tonks NK. Redox Redux: Revisiting PTPs and the control of cell signaling. Cell. 2005;121:667–670. doi: 10.1016/j.cell.2005.05.016. [DOI] [PubMed] [Google Scholar]
- 28.Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 29.Tsou HR, et al. 6-Substituted-4-(3-bromophenylamino)quinazolines as putative irreversible inhibitors of the epidermal growth factor receptor (EGFR) and human epidermal growth factor receptor (HER-2) tyrosine kinases with enhanced antitumor activity. J Med Chem. 2001;44:2719–2734. doi: 10.1021/jm0005555. [DOI] [PubMed] [Google Scholar]
- 30.Huron DR, et al. A novel pyridopyrimidine inhibitor of abl kinase is a picomolar inhibitor of Bcr-abl-driven K562 cells and is effective against STI571-resistant Bcr-abl mutants. Clin Cancer Res. 2003;9:1267–1273. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.