

Abstract
A model binding site was used to investigate charge–charge interactions in molecular docking. This simple site, a small (180 Å3) engineered cavity in cyctochrome c peroxidase (CCP), is negatively charged and completely buried from solvent, allowing us to explore the balance between electrostatic energy and ligand desolvation energy in a system where many of the common approximations in docking do not apply. A database with about 5300 molecules was docked into this cavity. Retrospective testing with known ligands and decoys showed that overall the balance between electrostatic interaction and desolvation energy was captured. More interesting were prospective docking scre”ens that looked for novel ligands, especially those that might reveal problems with the docking and energy methods. Based on screens of the 5300 compound database, both high-scoring and low-scoring molecules were acquired and tested for binding. Out of 16 new, high-scoring compounds tested, 15 were observed to bind. All of these were small heterocyclic cations. Binding constants were measured for a few of these, they ranged between 20 μM and 60 μM. Crystal structures were determined for ten of these ligands in complex with the protein. The observed ligand geometry corresponded closely to that predicted by docking. Several low-scoring alkyl amino cations were also tested and found to bind. The low docking score of these molecules owed to the relatively high charge density of the charged amino group and the corresponding high desolvation penalty. When the complex structures of those ligands were determined, a bound water molecule was observed interacting with the amino group and a backbone carbonyl group of the cavity. This water molecule mitigates the desolvation penalty and improves the interaction energy relative to that of the “naked” site used in the docking screen. Finally, six low-scoring neutral molecules were also tested, with a view to looking for false negative predictions. Whereas most of these did not bind, two did (phenol and 3-fluorocatechol). Crystal structures for these two ligands in complex with the cavity site suggest reasons for their binding. That these neutral molecules do, in fact bind, contradicts previous results in this site and, along with the alkyl amines, provides instructive false negatives that help identify weaknesses in our scoring functions. Several improvements of these are considered.
Keywords: molecular docking, electrostatic, solvation, cyctochrome c peroxidase, X-ray crystallography
Introduction
Molecular docking is widely used to discover new ligands for biological targets with a known 3D structure.1,2 Notwithstanding important successes,1,3–10 docking screens remain hampered by the prediction of false positives and negatives.11–17 This is tolerated for two reasons: docking focuses on easily available compounds and hit rates are often higher than those obtained by random high throughput screening.18–20 Nevertheless, it is clear that improved scoring functions would have considerable impact. Isolating the effects of particular changes in scoring functions is difficult because of the entanglement of various energetic contributions in ligand–receptor binding. These include receptor and ligand desolvation, other entropic contributions, polar and non-polar interactions, the hydrophobic effect, and receptor flexibility, among others.21,22 Therefore, it would be useful to have model systems that are simple enough to allow one to separate the different energetic contributions and thereby isolate the effect of individual terms in a scoring function.
Examples of such simple systems are cavities engineered in the core of T4 lysozyme. The cavity created by the substitution Leu99→Ala is completely buried from solvent, uniformly hydrophobic and contains no ordered water molecules.23 The ligands that bind to this pocket are small hydrophobic compounds like benzene or indene.24 The cavity does not tolerate ligand polarity well: toluene binds to the cavity, but there is no evidence that phenol does. By the additional substitution Met102→Gln, a single polar atom was introduced in the wall of this cavity.25 This new cavity accommodates the hydrophobic ligands of the L99A mutant cavity but also polar compounds like phenol or 3,5-difluoroaniline. The simplicity of these sites, combined with well established binding assays and crystallization conditions, makes these pockets good model systems to test scoring functions both retro- and prospectively, and to guide their improvement.12,25–27 In recent work, Gilson and colleagues have taken this approach one step further using organic host–guest complexes as model systems to explore enthalpy–entropy compensation.28 The motivation behind each of these systems it to simplify molecular recognition to the point where individual driving forces can be isolated and studied.
The aspects of scoring functions and docking algorithms that can be probed in a model system are determined by its properties. The T4 lysozyme cavities provide systems to examine ligand binding in a hydrophobic and a slightly polar environment12,25 and to investigate limited receptor flexibility.26,27 To simulate other aspects of ligand binding, such as charge–charge interactions, new systems are needed.
A model site well-suited for this purpose is an engineered pocket in cyctochrome c peroxidase (CCP) that was created by the substitution Trp191→Gly (Figure 1).29 This substitution creates a small pocket that in some ways resembles those of the T4 lysozyme cavities. It has roughly the same volume as the lysozyme cavities (180 Å3 versus 150 Å3) and it, too, is completely buried from solvent. Unlike the lysozyme cavities, the CCP W191G cavity is negatively charged and “wet”, containing five ordered water molecules and a potassium ion. The charge owes to the presence of Asp235, and the water molecules and the potassium ion ligate both the carboxylate group of this residue and several exposed backbone carbonyl groups. Twenty-three ligands and 17 compounds that do not bind to this pocket are known.29–31 Most ligands are small heterocycles bearing a single positive charge (Table 1). For 18 of these, the X-ray crystal structures of the cavity-complexes were determined. Typically, non-ligands, which we will refer to as “decoys”,12 are small enough to fit in the pocket but have the wrong net charge (0 or +2). This model site was used previously for two retrospective studies related to inhibitor design. Brooks and colleagues tested their λ-dynamics approach to predict binding affinities.32,33 Olson and colleagues tested the ability of AutoDock34 to reproduce crystallographically observed binding modes and to predict binding affinities of the known ligands.31
Figure 1.
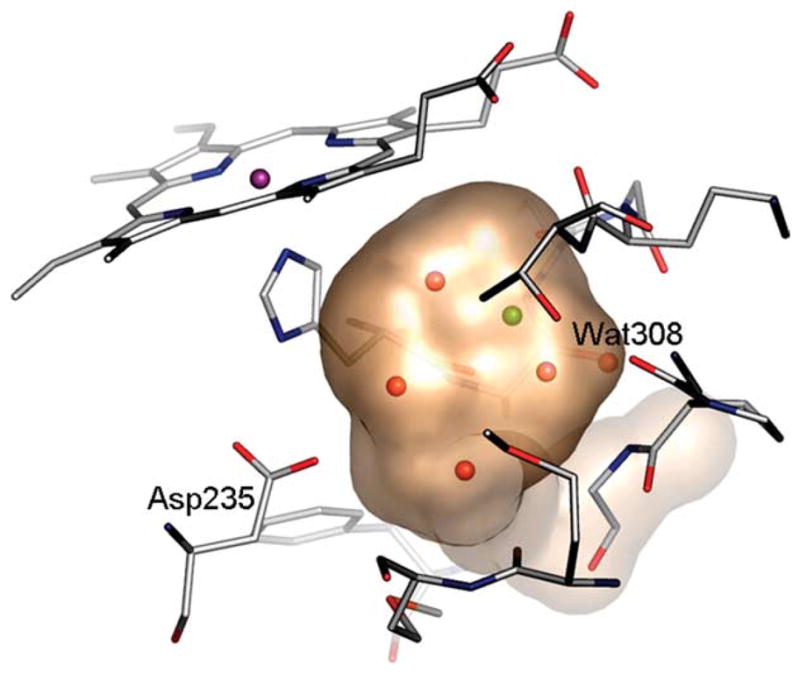
The cavity in CCP W191G. A transparent surface is displayed showing four ordered water molecules (red) and one potassium ion (green) in the cavity of the apo-structure. Water molecule 308 is conserved in all structures. (This Figure was made using PyMOL (www.pymol.org, as were Figures 4 and 5).)
Table 1.
rmsd for the top scoring docking pose compared to the previously determined crystallographic structure
| No | Structure | PDB code | rmsd (Å) |
|
|---|---|---|---|---|
| AMSOLa | Gaussianb | |||
| 1 | 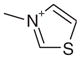 |
1AEB | 1.68 (12) | 0.44 |
| 2 | 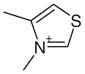 |
1AED | 3.01 (31) | 2.95 (5) |
| 3 | 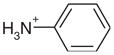 |
1AEE | 0.35 | 0.45 |
| 4 | 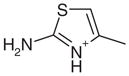 |
1AEH | 3.02 (29) | 3.52 (16) |
| 5 | 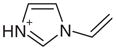 |
1AEJ | 0.54 | 0.63 |
| 6 | 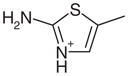 |
1AEN | 1.73 (5) | 1.74 (4) |
| 7 | 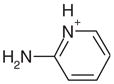 |
1AEO | 0.37 | 0.34 |
| 8 | 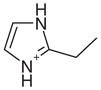 |
1AEQ | 0.61 | 0.35 |
| 9 | 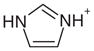 |
1AES | 0.61 | 0.7 |
| 10 | 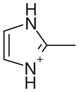 |
1AEU | 0.89 | 0.96 |
| 11 | 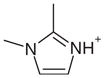 |
1CMP | 0.45 | 0.29 |
| 12 | 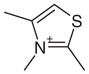 |
1AC4 | 2.46c | 2.46c |
| 13 | 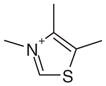 |
1AC8 | 2.4c | 2.32c |
Where the best scoring pose has an rmsd >1 Å, the best rank for a pose with an rmsd <1 Å is given in parentheses.
Using AMSOL to calculate ligand partial charges and desolvation energies.
Using Gaussian to calculate ligand partial charges and desolvation energies.
These ligands make a steric clash in the cavity.
Here, we use the CCP W191G pocket for studying charge–charge and charge–polar interactions in docking screens of large compound databases. These electrostatic interactions are common in protein–ligand binding, but can be difficult to model using physics-based scoring functions, such as the one we use in this work.35 This scoring function, implemented in DOCK3.5.54,25,36 includes van der Waals (Evdw) and electrostatic terms (Eelec) and is corrected for ligand desolvation (ΔGsolv):
| (1) |
For charge–charge interactions, the large gain in electrostatic energy must be balanced against the corresponding large desolvation energy penalty. An additional complication is that the absolute error in calculating desolvation energies for charged compounds is usually higher than for neutral compounds.37,38
Running a virtual screening campaign against this relatively simple model system allowed us to address several questions that only emerge in database screens, when not only potential ligands, but also a vast number of decoy molecules are fit into the site and ranked. First, how well balanced are electrostatic and desolvation energy in the docking screen? Are molecules with the “right” overall charge picked out as likely ligands from among the decoys that dominate the database, or do either electrostatic or desolvation energy terms dominate? Second, can we discover any new chemotypes for this cavity? The known ligands were picked based on chemical intuition, and most resemble one-another. Screening a large database of compounds might allow us to find new classes of ligands. We docked a database with about 5300 neutral, single and double positively charged molecules, small enough to fit in the cavity, against this pocket, and tested high-ranking compounds. Third, we were curious as to why no neutral molecules were found as ligands for this cavity. Such neutral molecules can form a charged-dipole hydrogen bond with Asp235 and would be easier to desolvate relative to charged ligands. Fourth, we investigated how the docking predictions change when we used a higher level of theory for calculating partial charges and desolvation energies of the docked molecules, or when the value of the dielectric constant in the binding pocket is changed. To see how docking results for binding sites with different properties are affected by these changes, we included the T4 lysozyme cavities in the comparison study. Finally, we consider the false positive and negative predictions of the database screen against the CCP W191G cavity as a guide to future improvements of docking scoring functions.
Results
Retrospective tests
We began by evaluating the ability of the docking program to predict binding modes of known ligands and to recognize them as high scoring “hits” in retrospective database screens. Eighteen known ligands and 15 known decoys (test set) were seeded into a database of about 5300 neutral or positively charged molecules small enough to fit into the cavity in CCP W191G. Each database molecule was docked into the cavity in multiple orientations and conformations, scored for van der Waals and electrostatic complementarity and penalized for ligand desolvation energy. Because the CCP W191G cavity is small and completely buried, we did not consider differential receptor desolvation. The conformation of the cavity was held rigid, the potassium ion and all ordered water molecules except Wat308, which is conserved in all previous structures, were removed (Figure 1). Performance was evaluated based on the prediction of binding modes, enrichment of known ligands and downgrading of known decoys.
First, we checked the ability of the docking program to predict the binding modes for the ligands in the test set for which an unambiguous binding mode had been determined (Table 1).29,30,39 With AMSOL partial charges and desolvation energies for the small molecules (our standard procedure25), seven of 13 ligands had a binding mode close to that found in the crystal structure (rmsd <1 Å; Table 1). If the correct binding mode, i.e. having an rmsd <1 Å, among the top ten poses is considered success, eight correct predictions were made. For the ligands 12 and 13 no correct binding modes can be predicted. These ligands have van der Waals violations even when docked back into their own receptors, probably owing to lack of full refinement of the complex structures30, and should therefore be discounted.
We next turned to enrichment of ligands and downgrading of decoys. The test set was seeded into the 5300 compound database, docked into CCP W191G, and ranked by score. As expected, little correlation was observed when we compared the dock energies to the experimental binding constants (Figure S1; Supplementary Data). For docking, a less ambitious and more reasonable concern is the enrichment of known ligands among the top ranking docked molecules. Using AMSOL partial charges and desolvation energies, 72% of the ligands ranked in the top 2% of the database, an enrichment of 36, and no known decoys were found in the top 15% of the database (Figure 2(a)). The best scoring neutral molecule, 3,5-difluroaniline, ranked 147th; the best scoring dicationic compound, pyrimidine-2,4,5,6-tetraamine, ranked 3295th. The structure-based enrichment was much better than what would have been achieved based on simple chemical similarity to the known ligands (Figure 2(b)).
Figure 2.

Retrospective enrichment of previously known, “test set” ligands for the W191G cavity in CCP.29–31 (a) Using molecular docking, looking at enrichment of known ligands (continuous lines) and downgrading of known decoys (broken lines) with either AMSOL-based (blue curve) or Gaussian-based ligand partial charges and desolvation energies. (b) The hit-rate for finding the docking-derived novel ligands had we used chemical similarity to the previously known ligands. The enrichment for the docking-based enrichment against the same database is also shown for comparison.
Prospective predictions
A more compelling series of experiments involved prospective testing for new ligands and chemotypes. Twenty-four compounds from the database screen were picked for experimental testing of what we thought might be strengths and weaknesses of our scoring function (Table 2). Compounds 14–26 and 28–30 were chosen based on their high ranks and chemical diversity, i.e. we chose them based on standard docking criteria. The alkyl amines 33–35 were included to assess the limits between desolvation energy penalty and gain in electrostatic energy. These latter compounds ranked poorly in the screen because their desolvation energies are relatively high in magnitude. Consequently, the sums of their electrostatic and desolvation energies, i.e. the net electrostatic contribution to binding, average only −3.6 kcal/mol, whereas the average of those two terms is −14 kcal/mol for the known ligands in the test set. Therefore, it seemed likely to us that these were true negative predictions. Similarly, we also wanted to test neutral compounds such as 27, 31, 32, 36, and 37, which had a good steric fit with the pocket and would give us a chance to probe the previous finding that neutral compounds do not bind to this cavity.29
Table 2.
New docking-derived hits, tested for binding to CCP W191G
| No. | Structure | Conc. used in UV assay (mM)a | Crystal structure | Binding Kd (mM)b | Rank | rmsd (Å) |
|
|---|---|---|---|---|---|---|---|
| AMSOLc | AMSOLc | Gaussiand | |||||
| 14 | 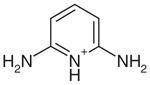 |
Yes | Yes (0.06) | 4 | 0.34 | 0.36 | |
| 15 | 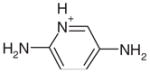 |
0.25 | Yes | Yes | 10 | 0.31 | 0.22 |
| 16 | 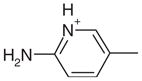 |
Yes | Yes (0.04) | 11 | 0.43 | 0.44 | |
| 17 | 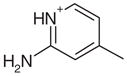 |
0.25 | Yes | Yes | 23 | 0.39 | 0.50 |
| 18 | 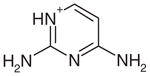 |
Yes | Yes (0.05) | 26 | 0.30 | 0.33 | |
| 19 | 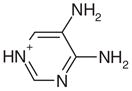 |
0.50 | No | Yes | 31 | NAe | NAe |
| 20 | 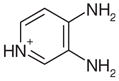 |
0.5 | No | Yes | 42 | NAe | NAe |
| 21 | 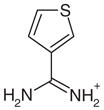 |
Yes | Yes (0.02) | 55 | 0.39 | 0.50 | |
| 22 | 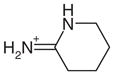 |
0.13 | Yes | Yes | 65 | 0.49 | 0.52 |
| 23 | 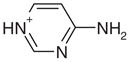 |
0.25 | No | Yes | 73 | NAe | NAe |
| 24 | 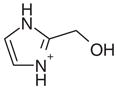 |
0.50 | Yes | Yes | 95 | 0.85 | 0.88 |
| 25 |  |
0.25 | Yes | Yes | 111 | 0.46 | 0.42 |
| 26 | 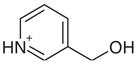 |
0.50 | No | Yes | 140 | NAe | NAe |
| 27 |  |
50.00 f | Yesg | No | 147 | NAe | NAe |
| 28 | 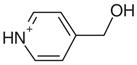 |
0.50 | Yes | Yes | 187 | 0.87 | 0.89 |
| 29 | 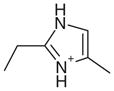 |
1.00 | No | Yes | 198 | NAe | NAe |
| 30 | 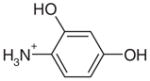 |
10.00 | Yesg | No | 351 | NAe | NAe |
| 31 | 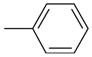 |
20.00 | No | No | 410 | NAe | NAe |
| 32 | 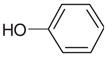 |
Yes | Yesh (4.10) | 420 | 2.60 | 2.58 | |
| 33 | 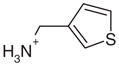 |
Yes | Yes (0.05) | 614 | 0.56 | 1.66 | |
| 34 | 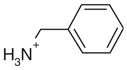 |
0.25 | Yes | Yes | 998 | 0.62 | 0.66 |
| 35 | 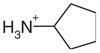 |
0.50 | Yes | Yes | 1122 | 0.64 | 0.66 |
| 36 | 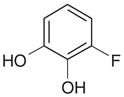 |
Yes | Yesh (7.70) | 1152 | 2.60 | 0.62 | |
| 37 | 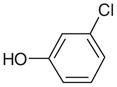 |
20.00 | No | No | 1518 | NAe | NAe |
Concentration is only given if no binding constant was determined.
The error of the binding constants is 30%.
Using AMSOL to calculate ligand partial charges and desolvation energies.
Using Gaussian to calculate ligand partial charges and desolvation energies.
Non-applicable, because no complex crystal structure was determined.
To assure the compound is neutral, the assay was done at pH 6.
No difference electron density for the ligand was obtained.
Binding of these compounds results in a blue shift.
All of the high-ranking charged compounds tested, except for compound 30, bind to CCP W191G when assayed at 0.5 mM or lower concentration (Table 2). To ensure that the compounds were protonated as modeled in the docking screen, the assay was performed at pH 4.5. Compound 30 gave no evidence of binding at 10 mM in the UV assay, and soaking CCP crystals at 50 mM did not reveal electron density for this compound. Therefore, we consider it to be a decoy. For selected ligands (14, 16, 18, and 21), we measured binding constants with full titration curves (Figure 3). These ranged from 20 μM to 60 μM. For ten of the new ligands (14–18, 21, 22, 24, 25, and 28), we determined crystal structures in complex with CCP W191G by X-ray crystallography. The resolution of these structures ranged from 1.12 to 1.70 Å(Table 3). All were extensively refined leading to Rcryst and Rfree values that ranged from 14.4 to 19.3 and from 15.2 to 22.6, respectively. The |Fo|−|Fc| omit electron density allowed us to position the ligands unambiguously (Figure 4). Typically, the docking predicted binding mode agreed well with the crystallographically determined one (<1 Å rmsd; Table 2).
Figure 3.

(a) Binding of cationic ligands induces a red shift in the Soret band (continuous line: spectra of the unbound, protein, broken line: spectra if a ligand is bound (here 18)). (b) Titration curve for 18. The continuous line represents the least-squares fit of the data according to the equation for single site binding described in Methods.
Table 3.
Crystallographic data
| Complex with |
14 | 15 | 16 | 17 | 18 | 21 | 22 | 24 | 25 | 28 | 32 | 33 | 34 | 35 | 36 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| pH of soaking buffer | 4.5 | 7.0 | 6.0 | 6.0 | 6.0 | 7.0 | 7.0 | 6.0 | 7.0 | 6.0 | 4.5 | 6.0 | 6.0 | 7.0 | 4.5 |
| Resolution (Å) | 40.0–1.75 (1.81–1.75) | 10.0–1.35 (1.40–1.35) | 50.0–1.40 (1.45–1.40) | 10.0–1.12 (1.16–1.12) | 36.8–1.49 (1.54–1.49) | 10.0–1.55 (1.61–1.55) | 10.0–1.45 (1.50–1.45) | 50.0–1.45 (1.50–1.45) | 50.0–1.45 (1.50–1.45) | 50.0–1.39 (1.49–1.39) | 10.0–1.40 (1.45–1.40) | 50.0–1.30 (1.35–1.30) | 50.0–1.55 (1.61–1.55) | 10.0–1.45 (1.50–1.45) | 10.0–1.30 (1.35–1.30) |
| No. of unique reflections | 41,392 (410) | 90,551 (8687) | 62,656 (4696) | 124,366 (10,410) | 53,147 (3215) | 61,090 (5938) | 72,346 (6447) | 59,787 (5272) | 57,355 (4697) | 60,114 (9364) | 81,465 (7826) | 80,407 (6207) | 47,799 (3984) | 72,355 (6117) | 99,539 (7998) |
| Rmerge (%) | 6.4 (39.5)a | 3.3 (23.5) | 3.8 (33.5) | 7.2 (36.7) | 6.7 (31.7) | 4.1 (32.7) | 3.8 (28.1) | 4.2 (37.1) | 3.7 (28.3) | 6.4 (32.0) | 3.3 (36.1) | 3.5 (25.8) | 6.5 (37.1) | 2.8 (23.8) | 3.9 (31.5) |
| Completeness (%) | 96.9 (99.9) | 99.5 (96.7) | 94.2 (71.1) | 95.9 (81.6) | 94.2 (57.8) | 99.3 (98.0) | 98.6 (89.1) | 98.5 (87.9) | 96.0 (79.7) | 86.4 (68.4) | 99.4 (96.7) | 95.9 (74.9) | 94.6 (80.1) | 97.6 (83.8) | 97 (78.9) |
| I/σI b | 18.3 (3.5) | 28.4 (4.3) | 24.3 (2.4) | 36.4 (2.2) | 8.1 (1.5) | 23.2 (3.2) | 22.4 (3.3) | 21.4 (2.2) | 17.3 (2.2) | 15.8 (1.9) | 27.2 (2.6) | 26.2 (2.8) | 16.9 (2.5) | 30.1 (3.3) | 31.3 (3.1) |
| Rfree (%) | 19.9 | 16.5 | 17.1 | 15.2 | 22.6 | 20.0 | 17.7 | 16.1 | 18.1 | 17.1 | 18.4 | 16.6 | 17.8 | 18.3 | 13.8 |
| R-factor (%) | 18.1 | 13.4 | 14.8 | 14.3 | 19.3 | 15.2 | 13.7 | 14.5 | 15.2 | 14.7 | 14.6 | 14.6 | 14.8 | 14.2 | 17.5 |
| Average B-factor of protein atoms (Å2) | 16.4 | 16.3 | 17.0 | 12.4 | 20.3 | 15.3 | 17.0 | 15.7 | 14.0 | 14.8 | 18.8 | 11.8 | 11.5 | 16.4 | 19.3 |
| Average B-factor of ligand atoms (Å2) | 14.5 | 15.2 | 13.6 | 10.6 | 16.3 | 14.3 | 14.6 | 14.3 | 11.3 | 14.6 | 24.7 | 9.2 | 9.2 | 17.2 | 28.9 |
All crystals belong to the space group P212121.
Values in parentheses are for the highest resolution shell.
Rfree was calculated from a random selection of reflections constituting 5% of the data. The R factor was calculated with the remaining intensities.
Figure 4.




Crystal structures of selected ligands from Table 2 bound to CCP W191G. Left column: |Fo|−|Fc| omit map for the refined complexes, except for (a), (c), (k), (m), and (y) where the map of the unrefined complex is shown, contoured at 2.5σ (green) with the ligand left out of the calculation, but shown in the Figure for clarity. Right column: Superposition of the highest ranking dock pose (green carbon atoms) with the crystallographically determined binding mode (yellow carbon atoms). Hydrogen bonds are drawn as broken lines. (a) and (b) 14; (c) and (d) 15; (e) and (f) 16; (g) and (h) 17; (i) and (j) 18; (k) and (l) 21, the |Fo|−|Fc| map, contoured at 10σ (red) is also shown; (m) and (n) 22; (o) and(p) 24; (q) and (r) 25, the |Fo|−|Fc| map, contoured at 9σ (red) is also shown; (s) and (t) 28; (u) and (v) 33, the |Fo|−|Fc| map, contoured at 14σ (red) is also shown; (w) and (x) 34; (y) and (z) 35; (aa) and (bb) 36.
Complex structures for isosteric ligands
Ligands 14, 17, 18, and 25, as well as 15 and 16, have the same shape, but differ in charge distribution and spatial arrangement of hydrogen bond donors and hydrophobic groups. 2,4-Diaminopyrimidine (18) forms a double hydrogen bond to Asp235 (Figure 4(i) and (j)). In 2,6-diaminopyridine (14), a carbon atom replaces the ring nitrogen of 18, which in the complex structure interacts with Asp235. Interestingly, 2,6-diaminopyridine does not adopt a binding mode that would allow for a double hydrogen bond via its remaining ring nitrogen and an exocyclic amino group. Instead its binding mode resembles that of 2,4-diaminopyrimidine, allowing for only one hydrogen bond with Asp235 (Figure 4(a) and (b)). Despite the loss of this hydrogen bond, the binding constant of 2,6-diaminopyridine is similar to 2,4-diaminopyrimidine (0.05 versus 0.06 mM). In 2-amino-4-picoline (17, Figure 4(g) and (h)), the amino group of 2,6-diaminopyrimidine (18), which interacts with Leu177 and Wat308 (Figure 4(j)), is replaced by a methyl group. Superposition of both complexes reveals that this methyl group is further away from Leu177, resulting in the displacement of Lys179 and Thr180. 2,5-Diaminopyridine (15) and 2-amino-5-picoline (16) also differ only by the replacement of an amino group with a methyl group. Whereas in CCP W191G·15 the ligand forms a hydrogen bond with Leu177 (Figure 4(c) and (d)), in CCP W191G·16 (Figure 4(e) and (f)) the ligand is shifted away from Leu177. In this complex, in contrast to CCP W191G·17, Lys179 and Thr180 are not displaced. The binding constant of 16 is 0.02 mM. Due to high optical density, the binding constant of 15 could not be determined with the UV assay. Another isostere is 25, an N-methylated pyridinium in which the ring nitrogen is no longer available for direct hydrogen bonding. The position of 25 is defined unambiguously in the |Fo|−|Fc| electron density map with electron density for the pyridinium nitrogen still visible when contoured as high as 9σ (Figure 4(q) and (r)). The ligand does not interact with Asp235 via a hydrogen bond to Asp235 through its amino group, but via an ion-dipole interaction that some might classify as a CH–hydrogen bond (distance CH···O 3.2 Å, angle C–HO 152°).39–41
Complex structures for amidiniums
All previously discovered cyclic ligands are aromatic with their positive charge delocalized over the aromatic ring system (Table 1). The amidiniums 21 and 22 seemed interesting because they explore a new cationic functionality, and in the case of 22 the ligand is not even aromatic. Piperidinylideneamine (22) adopts a similar binding mode as 2-aminopyridine (7), forming two hydrogen bonds with Asp235 (Figure 4(m) and (n)). Thiopheneamidine (21) does not orient both nitrogen atoms of its charged group to Asp235 to form a double hydrogen bond, as do most ligands, but instead forms a double hydrogen bond with Met230 and only a single hydrogen bond with Asp235 (Figure 4(k) and (l)). Thiopheneamidine has the lowest (best) Kd value in the series of ligands measured for this paper (0.02 mM) and is among the better ligands discovered for this site to date.30
Complex structures for ligands with rotatable bonds
Most known ligands for CCP W191G are rigid (Table 1). Binding mode predictions for ligands with rotatable bonds are more challenging because of the increased search space. Therefore we selected two flexible ligands, imidazoylmethanol (24) and pyridinylmethanol (28), to test how well their binding mode is predicted (we note that these ligands are only slightly flexible, with one rotatable bond each, the cavity constraints tilt against much more flexible ligands). In the crystal structure the hydroxyl group of imidazoylmethanol orients towards Asp235 in agreement with the docking prediction (Figure 4(o) and (p)). In contrast, pyridinylmethanol hydrogen bonds with Asp235 with its ring nitrogen and its hydroxyl group interacts with the backbone carbonyl group of Leu177 (Figure 4(s) and (t)). Whereas the former interaction was predicted, the latter was not (Figure 4(t)). This result reflects the procedure used for preparing the database; the conformer found in the crystal structure was not generated. If the required conformer is added manually, the binding mode is predicted correctly. This is thus a failure of database preparation. Whereas database preparation is a critical challenge in virtual screening,35 this problem is not one of docking and scoring per se, the foci of this work.
The crystal structure of CCP W191G·24 revealed that an unmodeled water molecule mediates the contact between the ligand and the protein. This water molecule was also found in the previously determined CCP complex with 2-ethylimidazole42 and coincides with the position of the potassium ion in the apo-structure (Figure 4(p)). Despite the fact that this water molecule was not considered in the docking screen, imidazoylmethanol ranks in the top 3% of the database.
Complex structures with false negative alkyl amines
Surprisingly, the alkyl amines 33–35 also bind to this cavity. These alkyl amines rank not even in the top 10% of the database. Their low score is due to their localized charge which leads to a less favorable desolvation energy for these compounds compared to the desolvation energies of ligands with a delocalized charge (1–26 and 28–30). A good example of this is thiophenylmethylamine (33), whose localized charge makes it harder to desolvate then thiopheneamidinium (21), a close analog with a delocalized charge. Nevertheless, the Kd value of thiophenylmethylamine (33) is 0.05 mM, only slightly worse than that of thiopheneamidinium (21), which is 0.02 mM. Accordingly, the alkyl amines are clear false negatives. An explanation is provided by the complex structures; an unexpected water molecule mediates an additional contact between the alkyl amino group of the ligands and His175 (Figure 4(u), (w) and (y)). This water molecule was previously only observed in CCP W191·3. Since predicting the binding modes of most of the ligands in the test set (Table 1) was not possible, when this water molecule was present in the receptor we did not consider it for the database screen. In the docking screen, the correct orientation of the alkyl amino group with respect to Asp235 is not predicted correctly (Figure 4(v), (x) and (z)). When the alkyl amines are docked with the water molecule added to the receptor, the right orientation of the amino group is found for 33 and 34 (not shown). Also, the scores of these ligands improve by about 7 kcal/mol, which would result in rank 140 for thiophenyl-methylamine (33), 234 for benzylamine (34) and 306 for cyclopentylamine (35).
Complex structures with neutral ligands
Most of the neutral molecules did not bind, consistent with previous expectations.29 Surprisingly, two did, though not in the predicted geometry. As expected, the apolar and neutral molecule toluene (31) did not bind to CCP W191G when tested in the UV assay, nor did 3,5-difluoroaniline (27) and 3-chlorophenol (37). As a further test, we soaked CCP crystals in 50 mM 3,5-difluoroaniline in 25% methylpentanediol (MPD); no difference electron density for the compounds was observed. Soaking of phenol (32) at neutral pH was unsuccessful, but at pH 4.5 difference electron density suggested ligand binding and the presence of a partially occupied new water molecule (Wat308b; Figure 5(a)). Also observable in this structure at partial occupancy are the water molecules and the potassium ion that fill the apo cavity. The occupancy of phenol and Wat308b was refined to 65%, and the occupancies of the apo-water molecules and the potassium ion correspondingly to 35%. As a consequence of the displacement of Wat308 by phenol, part of a loop (Gly191 to Asn195) is also displaced (Figure 5(b)). Surprisingly, phenol does not hydrogen bond with Asp235 but rather with the carbonyl group of Leu177. The unsuccessful soaking at neutral pH, and the absence of a hydrogen bond between phenol and Asp235 suggests that Asp235 is protonated in the pH 4.5 complex. The binding constant of phenol is 4.1 mM at pH 4.5 and 3.3 mM at pH 6.0. Based on the crystal structures it is unclear why the binding constants of phenol at pH 6.0 and pH 4.5 are so similar.
Figure 5.

(a) |Fo|−|Fc| omit map of the refined phenol-CCP W191G complex contoured at 3.0σ, calculated with the ligand and the potassium ion and the cavity water molecules left out. The occupancy of the ligand was refined to 62%, that of Wat308b to 64%, that of Wat308a to 36%, and the occupancies of the remaining water molecules to 38%. (b) Superposition of the apo-structure (carbon atoms colored in cyan) with the phenol complex (carbon atoms colored in gray); water molecules which are not present when the ligand is bound are removed for clarity. In the complex the region from Gly191 to Asn195 is displaced relative to the apo-structure.
A second new neutral ligand that binds to the cavity in CCP W191G is 3-fluorocatechol (36). Like phenol, the binding constant is in the low millimolar range (7.7 mM). Soaking of this ligand was successful at neutral pH (electron density not shown) and pH 4.5 (Figure 4(aa)). The ligand is present at a partial occupancy of 77%. Also observed in this structure are the water molecules and the potassium ion associated with the apo cavity. As in the phenol complex, Wat308b is present, but at lower occupancy than the ligand. In contrast to the phenol complex, the conformation of part of a loop from Gly191 to Asn195 is unchanged relative to the apo-structure. The distance between Wat308b and Cβ of Asn195 is only 2.0 Å, and between Wat308a and Wat308b 1.9 Å. This suggests that Wat308b is present alternatively to Wat308a and the side-chain conformation of Asn195, as defined by the electron density. Refining the occupancies of this residue and the water molecules resulted in 77% for Asn195 and Wat308a and 23% for Wat308b.
Because 3-fluorocatechol is symmetric, if the atom types are not considered, there is some difficulty assigning the interactions in the complex unambiguously. At the resolution of the complex (1.3 Å), it is impossible to distinguish oxygen from fluorine atoms based on the electron density. In one binding mode that is consistent with the difference electron density, the ligand hydrogen bonds with Asp235 and Met235 (Figure 4(bb)). Due to the geometry of the hydrogen bond, Asp235 must be deprotonated (distance O3-fluorocatechol···OAsp 2.4 Å, angle O–H–OAsp 142°). This configuration seems the more likely to us, but we cannot rule out the possibility that there is an alternative binding mode in which the positions of the oxygen atom interacting with Asp235 and the fluorine atom are switched. In this binding mode, one oxygen atom of the ligand would be in close distance with Wat308 (2.6 Å) without being able to hydrogen bond with it for geometric reasons. It might therefore be the case that if the ligand adopts this binding mode, Wat308a is displaced and Wat308b is present. Based on the occupancies, the latter binding mode would be adopted in 23% of the unit cells, the former in 54% of the unit cells, and in the remaining unit cells the apo-water molecules and the potassium ion would be present. Neither of the possible binding modes was predicted by DOCK using AMSOL partial charges and desolvation energies (Figure 4(bb)).
Quantum mechanically calculated partial charges and desolvation energies
Both docked geometries and molecule rankings depend upon ligand partial atomic charges and desolvation energies. These were calculated by the semi-empirical quantum mechanical method AMSOL.43,44 This method had served us well in previous studies,25 but it seemed possible that in this charged cavity a higher level of theory would be appropriate. We recalculated the partial charges and desolvation energies for the entire database at the HF level using the 6-31G(d) basis set for neutral molecules and the 6-31+G(d) basis set for charged compounds, with the conductor-like polarizable continuum model (CPCM) as implemented in Gaussian. These combinations were chosen based on a recent benchmark study.37
There were no significant differences in binding mode predictions between ligands charged using AMSOL or those charged using Gaussian (Tables 1 and 2). With the Gaussian partial charges, the binding mode of 3-fluorocatechol (36) and the position of the sulfur atom of compound 1 (Table 1) is correctly predicted, unlike the predictions using AMSOL partial charges. The binding mode of 33 (Table 2) is only predicted correctly with the AMSOL partial charges. The overall enrichment of the compounds is also about the same (Figure 2(a)) with differences only in the ranking of individual compounds. Interestingly, the neutral compounds of the prospective test (27, 31, 32, 36, 37 in Table 2) all rank better with the Gaussian partial charges and desolvation energies, irrespective of whether they bind or not.
Probing the dielectric constant
There is no consensus on which value of the dielectric constant should be used for rigid protein binding sites; estimates vary from 1 to 20,45–47 and this range leads to large differences in predicted binding energies. In all calculations described above, we assumed a dielectric constant of 78 for the aqueous buffer and a dielectric constant of 2 for the protein. To test if a different dielectric constant would give us better results, we recalculated desolvation energies and partial charges of the small molecules in the database using dielectric constants ranging from 1.84 to 10.19 (these values were chosen based on defined solvent parameters for AMSOL). We then redocked the database against the cavities in CCP W191G, T4 lysozyme L99A and L99A/M102Q using the same dielectric constant for calculating the electrostatic potential of the receptor as used for calculating the properties of the small molecules. In all three systems, no significant change in the enrichment is obtained if the dielectric constant in the binding pocket is varied from 1.84 to 3.04, when judged by the number of ligands found in the top 2% of the database for the CCP W191G pocket and top 10% for the T4 lysozyme systems (Figure 6; for effects of AMSOL vs. Gaussian charges on rank, see Figure 7). If the dielectric constant is increased further, enrichment drops in all three systems. A worse enrichment can reflect two effects: either more decoys get enriched or unknown ligands show up in the top ranks. Based on previous results, only hydrophobic compounds can bind to the T4 lysozyme L99A cavity. If a dielectric of 10.19 is assumed for the binding pocket, 55 of the top 100 molecules contain nitrogen or oxygen atoms compared to 25 of the top 100 molecules for a dielectric constant of 2.02. This indicates that if the dielectric constant is increased, polar decoys are enriched. The same is true for the slightly polar cavity in T4 lysozyme L99A/M102Q. Only 59 out of the 100 top scoring molecules contain one or less nitrogen or oxygen atoms when a dielectric constant of 10.19 is assumed, compared to 85 for a dielectric constant of 2.02. For CCP W191G, all 100 top scoring molecules have a total charge of +1 when a dielectric constant of 2.02 is used. After increasing the dielectric constant to 10.19, one molecule in the top 100 has a charge of +2, and 12 have a total charge of 0. Most of these have no polar atoms, which makes it unlikely that they bind in this cavity. Taking these results together, increasing the dielectric constant to 10.19 led to enrichment of more decoys and consequently worse results in all three simple cavities.
Figure 6.

The variation of ligand enrichment (continuous lines) and decoy downgrading (broken lines) with protein dielectric constant when docking into: (a) the charged cavity of CCP W191G; (b) the hydrophobic cavity of T4 lysozyme L99A; (c) and(d) the slightly polar cavity of T4 lysozyme L99A/M102Q (for clarity, ligands and decoys are separated). For calculating the score, the dielectric constant in the pocket was varied from 1.84 to 10.19. The ligands and decoys are the corresponding “test set” compounds (see Methods) except for CCP W191G, where the test set was augmented with the newly-discovered docking hits (Table 2).
Figure 7.

Ranks of the CCP W191G cavity ligands (test set ligands and the new ligands in Table 2) scored using Gaussian charges and desolvation energies plotted against the ranks obtained using AMSOL charges and desolvation energies.
Partial charges
In all calculations described above, the partial charges for the molecules in the database were calculated in the medium of low dielectric. Intuitively, this might be the obvious way to proceed, because this is the same dielectric assumed for the cavity. However, the partial charges of the ligands might be polarized upon ligand binding. To simulate this process, we calculated the partial charges of the compounds in the database in water and redocked them in the cavities of the model systems. No change in enrichment was obtained in any system (Figure S2; Supplementary Data).
Discussion
Modeling charge–charge interactions in docking is challenging because the gain in electrostatic energy upon ligand binding has to be balanced against desolvation energies. Both values are high in magnitude, as are their errors in computer simulations. This study allowed us to probe charge–charge interactions in a controlled environment, a small pocket completely buried from solvent. If we are able to get this balance right anywhere, it should be in such a relatively simple site. Correspondingly, mispredictions are particularly informative because they come much less entangled by the approximations necessary in more complicated sites. Five points stand out from this study. First, overall electrostatic and desolvation energy appear to be balanced well in the physics-based scoring function. No neutral compound ranked among the top 100 molecules, and the first dicationic molecule scores poorly at rank 3295. Second, from a practical standpoint, virtual screening with this cavity was successful. Fifteen of 16 chemically diverse compounds, which ranked in the top 5% of the database, did actually bind to the site when tested experimentally. For all ten high-ranking ligands for which the crystal structures in complex with CCP W191G were determined, the binding modes were predicted within <1 Å rmsd. Third, neither using a higher level of theory for calculating partial charges and desolvation energies, nor changing the dielectric constant in the cavity, improves these results. Fourth, although the overall performance is good, problems exist for neutral compounds. The only neutral ligand found that interacts with the deprotonated Asp135 (36) ranks poorly (1152nd), whereas the best scoring neutral decoy 27 ranks 147th. Fifth, analyzing false negative predictions points to weaknesses in current docking protocols and can guide the improvement of scoring functions and docking algorithms. Examples of such instructive false negatives are the alkyl amines 33–35. Their poor ranking owes to an inadequate handling of explicit water molecules during docking. Similarly, the binding mode of phenol was not predicted correctly, because pKa shifts were not considered. We consider these points further below.
The physics-based scoring function used here (equation (1)) was surprisingly effective at enriching new ligands and predicting their binding geometries. We had expected the scoring function to have trouble balancing the interaction energy and desolvation terms, finding either more high-scoring neutral or dicationic hits than was warranted. Instead, the top scoring hits were dominated by singly charged cationic heterocycles, with the first neutral ligand ranked 147th (top 2.8% of the database) and the first dicationic molecule ranked 3295th (top 62.2% of the database). Of the 17 high-scoring molecules tested experimentally for binding, only two, 3,5-difluoroanline (27) and aminoresorcin (30) were not observed to bind (Table 2). It is debatable if aminoresorcin is really a false positive or rather a true negative prediction, since it does not even rank in the top 6% of the database. For four of the new high-ranking ligands binding constants were determined. They range from 20 μM to 60 μM putting them among the better ligands known for this cavity30 with a “ligand efficiency” for the best ligand close to the projected maximum.48–50
The geometric fidelity of the docking predictions was also high (Tables 1 and 2). At a first glance, predicting the correct pose might seem trivial, since most of the ligands are rigid and the pocket is small, but even in this simple system it can be a challenge. For instance, 2,6-diaminopyridine (14) does not form a double hydrogen bond with Asp235 via its ring and exocyclic nitrogen atoms as one might expect, and as it is actually observed for 2-aminopyridine,30 2,5-diaminopyridine (15; Figure 4(c) and (d)), 2-amino-5-picoline (16; Figure 4(e) and (f)), 2-amino-4-picoline (17; Figure 4(g) and (h)) and 2,4-diaminopyrimidine (18; Figure 4(i) and (j)). Instead 2,6-diaminopyridine only forms one hydrogen bond via the exocyclic amine group, and the protonated ring nitrogen does not have a hydrogen bonding partner at all (Figure 4(a) and (b)). Although this is not the binding mode we might intuitively predict for this ligand, it is correctly predicted in the docked geometry (Figure 4(b)). Also the binding mode of thiopheneamidinium (21) is predicted correctly, despite the absence of steric constraints to guide the position of the sulfur atom of the thiophene ring (Figure 4(k) and (l)). In summary, the quality of the docked geometries was typically high for the novel ligands, even in cases where distinguishing between the correct and incorrect pose involved a subtle balance of forces; even in a simple site, such balanced forces are often in play.
Along with the high hit rates came new and interesting chemotypes as ligands. Considering their small size, the enriched ligands are diverse and include disubstituted pyridines (14–17, 20, 25), pyrimidines (18, 19, 23), amidines (21, 22), alcohols (24, 26, 28), and non-aromatic ligands (22) (Table 2), none of which had previously been discovered. That said, all of these molecules are small and cationic; could they have been found by simpler methods, such as chemical similarity? Using Daylight fingerprints, only three of the 16 new ligands have a Tanimoto coefficient of 0.85 or better to the previously known ligands.29–31 Another way to pose this question is to ask how many of the supposedly novel docking hits would have been found by screening the database by similarity to the previously known ligands? Again using topological similarity as a metric, the enrichment of the docking-derived ligands from the similarity search was considerably lower than the structure-based docking enrichment (Figure 2(b)); most of the new chemotypes would not have been discovered solely by using a similarity search. Thus the docking-derived ligands seem genuinely novel, which is important for a model binding site as diverse ligands will avoid bias towards a particular type of chemotype when testing and improving scoring functions.
Taken together, the high enrichment of mono-cationic ligands and the high fidelity of the binding mode predictions suggest that the relatively simple, physics-based scoring function represented by equation (1) can at least separate likely from unlikely ligands, getting the overall balance between electrostatic interactions and desolvation correct. On closer inspection, however, problems with the predictions do emerge. Not all ligand interactions were correctly predicted (Figure 4(t)), one high-ranking docking hit did not bind (27), and two neutral ligands (32 and 36) were ranked poorly. What do these problems tell us about weaknesses in our scoring functions and how might they be overcome?
We had previously found, in the neutral lysozyme cavities, that docking could be improved by moving to a higher level of theory in modeling ligand desolvation and partial atomic charges.25 Here, we investigated moving one step further, from a semi-empirical quantum mechanical method to a fully quantum mechanical method to calculate ligand partial charges and desolvation energies. Overall, moving to higher theory had little effect, with changes only in the relative ranking of the ligands and decoys (Figures 2 and 6; Tables 1 and 2). The desolvation energies calculated by both methods can differ by several kcal/mol. The consequence for docking is that different ranks are predicted for specific ligands, without changing overall performance. Indeed, we may be reaching a limit on how well we can hope to do with even fairly sophisticated methods for calculating ligand desolvation. The error in the calculated energy for the transfer for a cation from water to vacuum with these methods is 3–4 kcal/mol.37,38 With our scoring function, a change of 3 kcal/mol can make a difference of about 200 rank units. To have a significant impact on molecular docking for virtual screening, a new method to calculate charges and desolvation energies must have a smaller error than this 3–4 kcal/mol uncertainty level that most of the current methods have for simple solvent transfer free energies.
There is no consensus as to what is the best dielectric constant to model electrostatics in a protein binding pocket.45–47 Based on strictly electronic effects, we used a dielectric constant of 2.25 This may be an extreme choice, given that we are docking to a rigid receptor. Also, changing the dielectric constant is a way to influence the weighting between the van der Waals term, ligand desolvation energy and electrostatic energy, and so, from a pragmatic standpoint, it seemed interesting to explore. We therefore repeated the docking screens using different dielectric constants for the protein binding site, leaving the external dielectric fixed at 78 (Figure 6). In addition to the negatively charged CCP W191G, we also docked the database against the hydrophobic cavity in T4 lysozyme L99A and the slightly more polar cavity in T4 lysozyme L99A/M102Q. In all three systems, the best enrichment is obtained for values between 1.84 and 3.04. We also compared the performance obtained when the partial charges are either calculated in water or cyclohexane (Figure S2; Supplementary Data). In all three systems, the enrichment is not influenced by these small changes. The similar behavior of these systems, in which the properties of the binding pocket range from completely hydrophobic to polar to charged, indicates that the physics-based scoring function used here is not grossly biased towards a particular type of interaction.
An attractive feature of model binding sites, such as CCP W191G, is that false positive and false negative predictions are often more informative than true predictions. We were thus almost disappointed by the high initial hit rate of the prospective docking screen. As we dug further, however, interesting problems did emerge. The neutral compound 3,5-difluoroaniline (27; Table 2) ranks well, but does not bind to the cavity, whereas another neutral compound, 3-fluorocatechol (36) scores badly but does bind. Also, the alkyl amines 33–35 rank poorly, but bind to the cavity. These ligands form one hydrogen bond to a water molecule, which was not considered during docking (Figure 4(v), (x) and (z)). If this water molecule is considered during docking, the scores of these alkyl amines improve, leading to a difference in more than 500 rank units. Unfortunately, simply adding a water molecule to the target is not a panacea. There are some molecules, like imidazoyl-methanol (24), that can dock with or without a water molecule despite the fact that it hydrogen bonds with it in the crystal structure (Figure 4(p)). Worse, most of the ligands will not bind with either of those water molecules present. To improve docking, algorithms are needed that treat the water structure flexibly, and that can balance the energetic costs and benefits of either binding or displacing ordered water molecules.51–54
Perhaps the most interesting mispredicted molecules are phenol (32) and 3-fluorocatechol (36), which are the first neutral ligands for this cavity (Table 2; Figures 4(a), (b) and 5). Neither of these molecules ranks well in the docking hit list. Admittedly, these neutral compounds are weaker ligands than many of the cationic ligands, though it is also true that compound 12 (Table 1) binds in the millimolar range (1.5 mM).30 The poor ranking of the neutral compared to the charged ligands points to weaknesses in the docking protocol. Most likely, binding of phenol is associated with a pKa shift of Asp235 (Figure 5). Such pKa shifts of either the ligand or the protein are not uncommon, but are not considered routinely in current docking protocols. Reliably modeling these changes would lead to better predictions. 3-Fluorocatechol interacts with the deprotonated Asp235 as modeled during docking, but still does not rank well. Thus, even though the docking screen performed well overall, there is room to improve the balance between electrostatic and desolvation energy; such an imbalance, obvious in this simple system, will become more deleterious in more complicated “drug-like” sites.
Conclusions
The cavity site in CCP W191G is the third model system that we have studied for docking, adding a charge-dominated cavity to the hydrophobic and slightly polar sites represented by T4 lysozyme L99A and L99A/M101Q. CCP W191G allows us to explore the critical balance between electrostatic interaction energy and ligand desolvation in a site where many of the common approximations in docking do not apply. Docking was able to predict novel ligands at a surprisingly high hit rate, suggesting at least gross features of the desolvation–electrostatic balance were correct. That said, there were important and interesting failures, some neutral compounds rank low, but bind, others rank high, but do not bind, and the charged alkyl amines rank poorly, but also bind. The reasons for these failures are the same as observed in complex binding pockets: an inadequate handling of water molecules, neglect of pKa shifts and insufficient treatment of ligand desolvation energies. In this model system we can hope to study these problems in detail without the entanglement of other effects that occur in complex binding sites. We suspect that this charged cavity, in conjunction with the neutral cavity sites in T4 lysozyme, will provide illuminating models not only for docking methods but also for much more sophisticated theoretical techniques. The simplicity of these cavities, the dominance of particular terms in each of them, the atomic resolution structures available for multiple ligands and the ability to test new predictions prospectively, makes these sites interesting test cases for many molecular simulation methods.
Methods
Receptor preparation
Polar hydrogen atoms were added to CCP W191G (PDB code 1AC4) using MOLOC and their positions minimized using the MAB force field.55 Since water molecule 308 was observed in all complexes determined to date,30 it was kept as a rigid part of the receptor. All other water molecules in the pocket and the potassium ion were removed. AMBER charges56 were assigned to the protein atoms and to Wat308. Partial charges for the heme cofactor were calculated in cyclohexane using Jaguar (Schrödinger Inc.) with the 3-21G basis set for the Fe (III) atom and the 6-31+G(d) basis set for all other atoms. Grid-based excluded volume and van der Waals energy maps, the latter based on the AMBER potential function, were calculated for the cavity using the DOCK utilities DISTMAP and CHEMGRID. DelPhi57 was used to calculate an electrostatic potential for the receptor, with an internal dielectric of 2 and an external dielectric of 78, unless explicitly described otherwise in the text. To approximate the effect of ligand binding, the effective dielectric of the binding site was reduced by identifying the volume expected to be occupied by ligand atoms as a low dielectric region.25 Ligand atoms from the crystal structures, augmented with SPHGEN spheres,58 were used as receptor matching positions to dock molecules in the site. The cavities in T4 lysozyme L99A and L99A/M102Q were prepared as described.25
Test sets
The test set for the CCP pocket was composed of the ligands and decoys described previously.30 Several ligands do not interact directly with Asp235 but instead form a water-mediated contact; since we did not attempt to model explicit water molecules, we excluded these ligands from our test set. Two ligands, indoline and imidazo(1,2-a)pyridine, alter the protein conformation. They were therefore not considered, nor was quinoline, for which no complex structure exists, but which is even larger than these compounds. Including tautomers, there were 18 ligands and 15 decoys in the test set. The test set for the T4 lysozyme cavities was composed of previously published ligands and decoys.12,24,25 Since no attempt was made to model receptor flexibility, ligands which could not pass the DISTMAP filter for simple steric fit were not included. Altogether, there were 44 ligands and 31 decoys for the L99A cavity and 59 ligands and 18 decoys for L99A/M102Q cavity in the test sets. All of these are available free of charge from our laboratory site†.
Database preparation
With a python script based on OpenEye’s OE Chem library, duplicates in the Available Chemicals Directory (ACD) 2003 were removed and the remaining compounds filtered for molecules with a maximum of 15 heavy atoms and at least one ring. Subsequently, LigPrep (Schrödinger Inc.) was used to convert the molecules from 2D to 3D, enumerate stereoisomeres, tautomers and protonation states. In the latter step, a pH value of 5±2 was assumed resulting in all titrable groups with an assigned pKa value lower than 3.0 as deprotonated, above 7.0 as protonated, and both states were represented for the remaining groups. Conformations were sampled using Omega (OpenEye) and stored in a hierarchical flexibase.36 Partial atomic charges, desolvation energies and van der Waals parameters were calculated as described with one exception related to the treatment of the cavity terms in AMSOL.25,59 The desolvation energy in AMSOL is composed of two terms: the change in solute-electronic and solvent-polarization free energy (ΔGEP) and the cavity-dispersion-solvent-structure free energy (GCPS).38 The first term accounts for the electrostatic interactions of the solute molecule and the solvent, the second term accounts for forming a cavity in the solvent into which the solute is transferred. In our previous study on the T4 lysozyme systems, the desolvation penalty of the small molecules was calculated as:25
| (2) |
This was based on the assumption that the cavities in the apo-structure are preformed and free of solvent. Whereas this assumption is sensible, it might be problematic from a practical point of view. AMSOL is a parameterized semi-empirical method. During parameterization no attempt was made to get both terms correct, but only the overall desolvation energy. Thus, the GCPS term was also designed to make up for systematic deficiencies and intrinsic uncertainties in ΔGEP.25 Based on these considerations, the desolvation energy must be calculated as:
| (3) |
We therefore docked the small database (see below) with ligand desolvation energies calculated with both equations in the T4 lysozyme pockets (L99A and L99A/M102Q). Ligands were better enriched and decoys further downgraded in the top 10% of the database with a scoring function based on equation (3) (Figure S3; Supplementary Data). With the scoring function based on equation (2), all of the top scoring ligands contain several fluorine atoms together with polar groups (data not shown). In our experience, these molecules most likely do not bind to these rather hydrophobic pockets.24,25 In contrast, with the scoring function based on equation (3), fluorinated compounds are no longer enriched and the top scoring molecules closely resemble known ligands. Thus, in this study we calculated desolvation energies as the difference between the total desolvation calculated in water minus the total desolvation calculated in a solvent with lower dielectric constant.
To reduce the size of the database and to ensure that the compounds in the database have similar properties as the ligands and decoys in the test set,60 all molecules were docked into the CCP W191G pocket, and only those with a negative van der Waals score and a net charge of zero or higher were kept. Ligands of the test sets not present in the ACD were added manually. The final database contained about 5300 compounds, 131 of them were +2 charged, 996 were +1 charged and the remaining molecules are neutral. For these molecules, partial charges were also assigned according to the Merz-Singh-Kollman scheme,61 with desolvation energies for the transfer from water to cyclohexane calculated based on the CPCM method62,63 using GAUSSIAN 03(Gaussian Inc.) with the HF 6-31G(d) basis set for neutral molecules and HF 6-31+G(d) for charged molecules. If the dielectric constant was varied in the pocket, a solvent with the same dielectric constant was used for recalculating desolvation energies and partial charges with AMSOL.43,44
Docking protocol
DOCK3.5.5425,36 was used to dock a multi-conformer database of small molecules into the cavities. To sample ligand orientations, ligand, receptor and overlap bins were set to 0.2 Å; the distance tolerance for matching ligand atoms to receptor matching was set to 0.75 Å. Each docking pose was evaluated for steric fit. Compounds passing this filter were scored for electrostatic and van der Waals complementarity and corrected for desolvation.
Similarity search
A similarity search was performed with the test set of ligands as the reference structures, using Daylight fingerprints. Each ligand was compared to the full database used in the docking study. A Tanimoto-index of 0.85 was used as the cutoff for when two molecules were considered similar.64 The enrichment plot for the similarity search was made by using the test set ligands to search the full database with the Tanimoto-index threshold at zero. Then the top Tanimoto-coefficient for each compound in the database was used to rank the database as a whole by similarity to the known ligands. Ranks of the new binders from the similarity search were then compared to the ranks of the new binders from the docking run. Smiles strings for the ligands in the test set and the full database were generated using a python script based on OpenEye’s OE chem software version 1.3.4. Daylight fingerprints were built from the Smiles strings using the Fingerprint Toolkit in Daylight version 4.83 distributed by Chemical Information Systems, Inc (CIS Inc). The similarity search was performed utilizing a Tanimoto coefficient calculation derived from code in CACTVS subset 1.0 (CIS Inc).
Protein expression and purification
Ligand-binding measurements
Compound 25 was from Specs, 21, 24, and 33 were from Maybridge and all other compounds from Aldrich. Ligand binding was measured in 500 mM acetate buffer (pH 4.5), except 27, which was assayed at pH 6.0 to ensure that the compound was neutral. To avoid competition in ligand binding with small cations like potassium,29 the pH of the buffer was adjusted with Bis-Tris. The compounds were dissolved in either buffer or dimethyl sulfoxide (DMSO). Binding was monitored by the red shift and increase of absorbance of the heme Soret band, except for the neutral ligands where a blue shift was observed.29 Binding constants were obtained by plotting the difference in absorbance at 418 nM and fitting the data with GraFit (Erithacus Software Limited) to equation EL=(−(Lo +Eo +Kd) ± ((Lo +Eo +Kd)2−4EoLo)1/2)/2, where Eo is the total enzyme concentration, Lo is the total ligand concentration, EL is the concentration of the bound complex, which is proportional to the observed change in the Soret band, and Kd is the binding constant.
Structure determination
Crystals were grown as described.30 Compounds 16–18, 24, 25, 28, 33, and 34 were soaked overnight by adding 1 μl of 100 mM stock solution dissolved in water to the mother liquor. Compounds 15, 21, 22, 25, and 27 were soaked for 1 h at a concentration of 50 mM in 25% MPD, and compounds 14 and 30 for 1 h at a concentration of 50 mM in 125 mM acetate buffer (pH 4.5) containing 25% MPD. Compounds 32 and 36 were soaked in both the MPD buffer and the acetate buffer. Diffraction data for the complex with 14 were collected at University of California San Francisco and for the complex with 18 at the Scripps Research Institute, San Diego, using a Rigaku X-ray generator equipped with a rotating copper anode and a Raxis IV image plate. Data for the complexes with 24 and 27 were collected on Beamline 5.0.1 of the Advanced Light Source (ALS) at Lawrence Berkley National Laboratory using an ADSC-CCD detector and for all remaining complexes on Beamline 8.3.1 of the ALS using an ADSC-CCD detector. All data sets were collected at 100 K. Data for the complex with 18 were reduced and scaled with Crystal Clearr and d*trek66 and for all other complexes with HKL2000.67 The complex with 14 was refined using CNS68 and the complexes with 15, 21, 22, 32, 35 and 36 were refined using SHELX.69 Parameters for these ligands were generated using PRODRG.70 The remaining complexes were refined using CNS and the CCP4 software package.71 Interactive model building was performed using O72 and Xtalview.73
Protein Data Bank accession code
The crystallographic coordinates for the complex structures presented in this work have been deposited with the RCSB Protein Data Bank† with accession codes 2ANZ, 2AQD, 2AS1, 2AS2, 2AS3, 2AS4, 2AS6, 2EUN, 2EUP, 2EUQ, 2EUO, 2EUR, 2EUS, 2EUT and 2EUU.
Supplementary Material
Acknowledgments
This work was supported by NIH grants GM59957 (to B.K.S.) and GM41049 (to D.B.G.). R.B. was supported by a fellowship from the Ernst Schering Research Foundation. We thank OpenEye Scientific Software (Santa Fe, NM) for the use of Omega, OEChem and other tools, and MDL (San Leandro, CA) for use of the ACD database and ISISBase. We thank Kerim Babaoglu for help in data collection, Michael Keiser for his help with the similarity search, BinQing Wei for many conversations and Alan Graves for reading the manuscript.
Abbreviations
- CCP
cyctochrome c peroxidase
- rmsd
root-mean-square deviation
- MPD
methylpentanediol
- CPCM
conductor-like polarizable continuum model
Footnotes
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.jmb.2006.01.034
References
- 1.Kitchen DB, Decornez H, Furr JR, Bajorath J. Docking and scoring in virtual screening for drug discovery: methods and applications. Nature Rev Drug Discov. 2004;3:935–949. doi: 10.1038/nrd1549. [DOI] [PubMed] [Google Scholar]
- 2.Shoichet BK. Virtual screening of chemical libraries. Nature. 2004;432:862–865. doi: 10.1038/nature03197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alvarez JC. High-throughput docking as a source of novel drug leads. Curr Opin Chem Biol. 2004;8:365–370. doi: 10.1016/j.cbpa.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 4.Abagyan R, Totrov M. High-throughput docking for lead generation. Curr Opin Chem Biol. 2001;5:375–382. doi: 10.1016/s1367-5931(00)00217-9. [DOI] [PubMed] [Google Scholar]
- 5.Huang D, Luthi U, Kolb P, Edler K, Cecchini M, Audetat S, et al. Discovery of cell-permeable non-peptide inhibitors of beta-secretase by high-throughput docking and continuum electrostatics calculations. J Med Chem. 2005;48:5108–5111. doi: 10.1021/jm050499d. [DOI] [PubMed] [Google Scholar]
- 6.Meiering S, Inhoff O, Mies J, Vincek A, Garcia G, Kramer B, et al. Inhibitors of Trypanosoma cruzi trypanothione reductase revealed by virtual screening and parallel synthesis. J Med Chem. 2005;48:4793–4802. doi: 10.1021/jm050027z. [DOI] [PubMed] [Google Scholar]
- 7.Carbone V, Ishikura S, Hara A, El-Kabbani O. Structure-based discovery of human L-xylulose reductase inhibitors from database screening and molecular docking. Bioorg Med Chem. 2005;13:301–312. doi: 10.1016/j.bmc.2004.10.030. [DOI] [PubMed] [Google Scholar]
- 8.Verras A, Kuntz ID, Ortiz de Montellano PR. Computer-assisted design of selective imidazole inhibitors for cytochrome p450 enzymes. J Med Chem. 2004;47:3572–3579. doi: 10.1021/jm030608t. [DOI] [PubMed] [Google Scholar]
- 9.Li C, Xu L, Wolan DW, Wilson IA, Olson AJ. Virtual screening of human 5-aminoimidazole-4-carboxamide ribonucleotide transformylase against the NCI diversity set by use of AutoDock to identify novel nonfolate inhibitors. J Med Chem. 2004;47:6681–6690. doi: 10.1021/jm049504o. [DOI] [PubMed] [Google Scholar]
- 10.Kraemer O, Hazemann I, Podjarny AD, Klebe G. Virtual screening for inhibitors of human aldose reductase. Proteins: Struct Funct Genet. 2004;55:814–823. doi: 10.1002/prot.20057. [DOI] [PubMed] [Google Scholar]
- 11.Cummings MD, DesJarlais RL, Gibbs AC, Mohan V, Jaeger EP. Comparison of automated docking programs as virtual screening tools. J Med Chem. 2005;48:962–976. doi: 10.1021/jm049798d. [DOI] [PubMed] [Google Scholar]
- 12.Graves AP, Brenk R, Shoichet BK. Decoys for docking. J Med Chem. 2005;48:3714–3728. doi: 10.1021/jm0491187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ferrara P, Gohlke H, Price DJ, Klebe G, Brooks CL., III Assessing scoring functions for protein-ligand interactions. J Med Chem. 2004;47:3032–3047. doi: 10.1021/jm030489h. [DOI] [PubMed] [Google Scholar]
- 14.Perola E, Walters WP, Charifson PS. A detailed comparison of current docking and scoring methods on systems of pharmaceutical relevance. Proteins: Struct Funct Genet. 2004;56:235–249. doi: 10.1002/prot.20088. [DOI] [PubMed] [Google Scholar]
- 15.Wang R, Lu Y, Fang X, Wang S. An extensive test of 14 scoring functions using the PDBbind refined set of 800 protein-ligand complexes. J Chem Inf Comput Sci. 2004;44:2114–2125. doi: 10.1021/ci049733j. [DOI] [PubMed] [Google Scholar]
- 16.Stahl M, Rarey M. Detailed analysis of scoring functions for virtual screening. J Med Chem. 2001;44:1035–1042. doi: 10.1021/jm0003992. [DOI] [PubMed] [Google Scholar]
- 17.Bissantz C, Folkers G, Rognan D. Protein-based virtual screening of chemical databases. 1 Evaluation of different docking/scoring combinations. J Med Chem. 2000;43:4759–4767. doi: 10.1021/jm001044l. [DOI] [PubMed] [Google Scholar]
- 18.Doman TN, McGovern SL, Witherbee BJ, Kasten TP, Kurumbail R, Stallings WC, et al. Molecular docking and high-throughput screening for novel inhibitors of protein tyrosine phosphatase-1B. J Med Chem. 2002;45:2213–2221. doi: 10.1021/jm010548w. [DOI] [PubMed] [Google Scholar]
- 19.Evensen E, Eksterowicz JE, Stanton RV, Oshiro C, Grootenhuis PD, Bradley EK. Comparing performance of computational tools for combinatorial library design. J Med Chem. 2003;46:5125–5128. doi: 10.1021/jm025618t. [DOI] [PubMed] [Google Scholar]
- 20.Paiva AM, Vanderwall DE, Blanchard JS, Kozarich JW, Williamson JM, Kelly TM. Inhibitors of dihydrodipicolinate reductase, a key enzyme of the diaminopimelate pathway of Mycobacterium tuberculosis. Biochim Biophys Acta. 2001;1545:67–77. doi: 10.1016/s0167-4838(00)00262-4. [DOI] [PubMed] [Google Scholar]
- 21.Zavodszky MI, Kuhn LA. Side-chain flexibility in protein-ligand binding: the minimal rotation hypothesis. Protein Sci. 2005;14:1104–1114. doi: 10.1110/ps.041153605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sims PA, Wong CF, Vuga D, McCammon JA, Sefton BM. Relative contributions of desolvation, inter- and intramolecular interactions to binding affinity in protein kinase systems. J Comput Chem. 2005;26:668–681. doi: 10.1002/jcc.20207. [DOI] [PubMed] [Google Scholar]
- 23.Eriksson AE, Baase WA, Wozniak JA, Matthews BW. A cavity-containing mutant of T4 lysozyme is stabilized by buried benzene. Nature. 1992;355:371–373. doi: 10.1038/355371a0. [DOI] [PubMed] [Google Scholar]
- 24.Morton A, Baase WA, Matthews BW. Energetic origins of specificity of ligand binding in an interior nonpolar cavity of {T4} lysozyme. Biochemistry. 1995;34:8564–8575. doi: 10.1021/bi00027a006. [DOI] [PubMed] [Google Scholar]
- 25.Wei BQ, Baase WA, Weaver LH, Matthews BW, Shoichet BK. A model binding site for testing scoring functions in molecular docking. J Mol Biol. 2002;322:339–355. doi: 10.1016/s0022-2836(02)00777-5. [DOI] [PubMed] [Google Scholar]
- 26.Wei BQ, Weaver LH, Ferrari AM, Matthews BW, Shoichet BK. Testing a flexible-receptor docking algorithm in a model binding site. J Mol Biol. 2004;337:1161–1182. doi: 10.1016/j.jmb.2004.02.015. [DOI] [PubMed] [Google Scholar]
- 27.Ferrari AM, Wei BQ, Costantino L, Shoichet BK. Soft docking and multiple receptor conformations in virtual screening. J Med Chem. 2004;47:5076–5084. doi: 10.1021/jm049756p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chang CE, Gilson MK. Free energy, entropy, and induced fit in host-guest recognition: calculations with the second-generation mining minima algorithm. J Am Chem Soc. 2004;126:13156–13164. doi: 10.1021/ja047115d. [DOI] [PubMed] [Google Scholar]
- 29.Fitzgerald MM, Churchill MJ, McRee DE, Goodin DB. Small molecule binding to an artificially created cavity at the active site of cytochrome c peroxidase. Biochemistry. 1994;33:3807–3818. [PubMed] [Google Scholar]
- 30.Musah RA, Jensen GM, Bunte SW, Rosenfeld RJ, Goodin DB. Artificial protein cavities as specific ligand-binding templates: characterization of an engineered heterocyclic cation-binding site that preserves the evolved specificity of the parent protein. J Mol Biol. 2002;315:845–857. doi: 10.1006/jmbi.2001.5287. [DOI] [PubMed] [Google Scholar]
- 31.Rosenfeld R, Goodsell D, Musah R, Garret M, Goodin D, Olson A. Automated docking of ligands to an artificial active site: augmenting crystallographic analysis with computer modeling. J Comput Aided Mol Des. 2003;17:525–536. doi: 10.1023/b:jcam.0000004604.87558.02. [DOI] [PubMed] [Google Scholar]
- 32.Banba S, Guo ZY, Brooks CL. Efficient sampling of ligand orientations and conformations in free energy calculations using the lambda-dynamics method. J Phys Chem ser B. 2000;104:6903–6910. [Google Scholar]
- 33.Banba S, Brooks CL. Free energy screening of small ligands binding to an artificial protein cavity. J Chem Phys. 2000;113:3423–3433. [Google Scholar]
- 34.Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, et al. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J Comput Chem. 1998;19:1639–1662. [Google Scholar]
- 35.Brenk R, Irwin J, Shoichet B. Here be dragons: docking and screening in an uncharted region of chemical space. J Biomo Screen. 2006;10:667–674. doi: 10.1177/1087057105281047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lorber DM, Shoichet BK. Hierarchical docking of databases of multiple ligand conformations. Curr Top Med Chem. 2005;5:739–749. doi: 10.2174/1568026054637683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Takano Y, Houk KN. Benchmarking the conductor-like Polarizable Continuum Model (CPCM) for aqueous solvation free energies of neutral and ionic organic molecules. J Chem Theory Comp. 2004;1:70–77. doi: 10.1021/ct049977a. [DOI] [PubMed] [Google Scholar]
- 38.Li JB, Zhu TH, Hawkins GD, Winget P, Liotard DA, Cramer CJ, et al. Extension of the platform of applicability of the SM5.42R universal solvation model. Theoret Chem Acc. 1999;103:9–63. [Google Scholar]
- 39.Musah RA, Jensen GM, Rosenfeld RJ, McRee DE, Goodin DB, Bunte SW. Variation in strength of an unconventional C-H to O hydrogen bond in an engineered protein cavity. J Am Chem Soc. 1997;119:9083–9084. [Google Scholar]
- 40.Taylor R, Kennard O. Crystallographic evidence for the existence of C–H···O, C–H···N, and C–H···C1 hydrogen-bonds. J Am Chem Soc. 1982;104:5063–5070. [Google Scholar]
- 41.Pierce AC, Sandretto KL, Bemis GW. Kinase inhibitors and the case for CH···O hydrogen bonds in protein–ligand binding. Proteins: Struct Funct Genet. 2002;49:567–576. doi: 10.1002/prot.10259. [DOI] [PubMed] [Google Scholar]
- 42.Fitzgerald MM, Musah RA, McRee DE, Goodin DB. A ligand-gated, hinged loop rearrangement opens a channel to a buried artificial protein cavity. Nature Struct Biol. 1996;3:626–631. doi: 10.1038/nsb0796-626. [DOI] [PubMed] [Google Scholar]
- 43.Li JB, Zhu TH, Cramer CJ, Truhlar DG. New class IV charge model for extracting accurate partial charges from wave functions. J Phys Chem ser A. 1998;102:1820–1831. [Google Scholar]
- 44.Chambers CC, Hawkins GD, Cramer CJ, Truhlar DG. Model for aqueous solvation based on class IV atomic charges and first solvation shell effects. J Phys Chem. 1996;100:16385–16398. [Google Scholar]
- 45.Schutz CN, Warshel A. What are the dielectric “constants” of proteins and how to validate electrostatic models? Proteins: Struct Funct Genet. 2001;44:400–417. doi: 10.1002/prot.1106. [DOI] [PubMed] [Google Scholar]
- 46.Honig B, Nicholls A. Classical electrostatics in biology and chemistry. Science. 1995;268:1144–1149. doi: 10.1126/science.7761829. [DOI] [PubMed] [Google Scholar]
- 47.Simonson T, Archontis G, Karplus M. A Poisson-Boltzmann study of charge insertion in an enzyme active site: the effect of dielectric relaxation. J Phys Chem ser B. 1999;193:6142–6156. [Google Scholar]
- 48.Kuntz ID, Chen K, Sharp KA, Kollman PA. The maximal affinity of ligands. Proc Natl Acad Sci USA. 1999;96:9997–10002. doi: 10.1073/pnas.96.18.9997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Carr RA, Congreve M, Murray CW, Rees DC. Fragment-based lead discovery: leads by design. Drug Discov Today. 2005;10:987–992. doi: 10.1016/S1359-6446(05)03511-7. [DOI] [PubMed] [Google Scholar]
- 50.Hopkins AL, Groom CR, Alex A. Ligand efficiency: a useful metric for lead selection. Drug Discov Today. 2004;9:430–431. doi: 10.1016/S1359-6446(04)03069-7. [DOI] [PubMed] [Google Scholar]
- 51.Minke WE, Diller DJ, Hol WG, Verlinde CL. The role of waters in docking strategies with incremental flexibility for carbohydrate derivatives: heat-labile enterotoxin, a multivalent test case. J Med Chem. 1999;42:1778–1788. doi: 10.1021/jm980472c. [DOI] [PubMed] [Google Scholar]
- 52.de Graaf C, Pospisil P, Pos W, Folkers G, Vermeulen NP. Binding mode prediction of cytochrome p450 and thymidine kinase protein-ligand complexes by consideration of water and rescoring in automated docking. J Med Chem. 2005;48:2308–2318. doi: 10.1021/jm049650u. [DOI] [PubMed] [Google Scholar]
- 53.Rarey M, Kramer B, Lengauer T. The particle concept: placing discrete water molecules during protein-ligand docking predictions. Proteins: Struct Funct Genet. 1999;34:17–28. [PubMed] [Google Scholar]
- 54.Osterberg F, Morris GM, Sanner MF, Olson AJ, Goodsell DS. Automated docking to multiple target structures: incorporation of protein mobility and structural water heterogeneity in AutoDock. Proteins: Struct Funct Genet. 2002;46:34–40. doi: 10.1002/prot.10028. [DOI] [PubMed] [Google Scholar]
- 55.Gerber PR. Charge distribution from a simple molecular orbital type calculation and non-bonding interaction terms in the force field MAB. J Comput Aided Mol Des. 1998;12:37–51. doi: 10.1023/a:1007902804814. [DOI] [PubMed] [Google Scholar]
- 56.Cornell WD, Cieplak P, Bayly CI, Kollman PA. Application of Resp charges to calculate conformational energies, hydrogen-bond energies, and free-energies of solvation. J Am Chem Soc. 1993;115:9620–9631. [Google Scholar]
- 57.Nicholls A, Honig B. A rapid finite-difference algorithm, utilizing successive over-relaxation to solve the Poisson-Boltzmann equation. J Comput Chem. 1991;12:435–445. [Google Scholar]
- 58.Kuntz ID, Blaney JM, Oatley SJ, Langridge R, Ferrin TE. A Geometric approach to macromolecule-ligand interactions. J Mol Biol. 1982;161:269–288. doi: 10.1016/0022-2836(82)90153-x. [DOI] [PubMed] [Google Scholar]
- 59.Meng EC, Shoichet BK, Kuntz ID. Automated docking with grid-based energy evaluating. J Comb Chem. 1992;13:505–524. [Google Scholar]
- 60.Verdonk ML, Berdini V, Hartshorn MJ, Mooij WT, Murray CW, Taylor RD, et al. Virtual screening using protein-ligand docking: avoiding artificial enrichment. J Chem Inf Comput Sci. 2004;44:793–806. doi: 10.1021/ci034289q. [DOI] [PubMed] [Google Scholar]
- 61.Besler BH, Merz KM, Kollman PA. Atomic charges derived from semiempirical methods. J Comput Chem. 1990;11:431–439. [Google Scholar]
- 62.Cossi M, Rega N, Scalmani G, Barone V. Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. J Comput Chem. 2003;24:669–681. doi: 10.1002/jcc.10189. [DOI] [PubMed] [Google Scholar]
- 63.Barone V, Cossi M. Quantum calculation of molecular energies and energy gradients in solution by a conductor solvent model. J Phys Chem ser A. 1998;102:1995–2001. [Google Scholar]
- 64.Martin YC, Kofron JL, Traphagen LM. Do structurally similar molecules have similar biological activity? J Med Chem. 2002;45:4350–4358. doi: 10.1021/jm020155c. [DOI] [PubMed] [Google Scholar]
- 65.Goodin DB, Davidson MG, Roe JA, Mauk AG, Smith M. Amino acid substitutions at tryptophan-51 of cytochrome c peroxidase: effects on coordination, species preference for cytochrome c, and electron transfer. Biochemistry. 1991;30:4953–4962. doi: 10.1021/bi00234a017. [DOI] [PubMed] [Google Scholar]
- 66.Pflugrath JW. The finer things in X-ray diffraction data collection. Acta Crystallog sect D. 1999;55:1718–1725. doi: 10.1107/s090744499900935x. [DOI] [PubMed] [Google Scholar]
- 67.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. In: Carter CWJ, Sweet RM, editors. Methods in Enzymology. Vol. 276. Academic Press; New York: 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- 68.Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, et al. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallog sect D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 69.Sheldrick G, Schneider T. SHELXL: high resolution refinement. Methods Enzymol. 1997;277:319–343. [PubMed] [Google Scholar]
- 70.Schuttelkopf AW, van Aalten DMF. PRODRG: a tool for high-throughput crystallography of protein–ligand complexes. Acta Crystallog sect D. 2004;60:1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- 71.Bailey S. The CCP4 suite—programs for protein crystallography. Acta Crystallog sect D. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 72.Jones TA, Kjeldgaard M. Electron-density map interpretation. Methods Enzymol. 1997;277:173–208. doi: 10.1016/s0076-6879(97)77012-5. [DOI] [PubMed] [Google Scholar]
- 73.McRee DE. XtalView/Xfit—a versatile program for manipulating atomic coordinates and electron density. J Struct Biol. 1999;125:156–165. doi: 10.1006/jsbi.1999.4094. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.