

Abstract
Malignant Melanoma remains one of the most deadly human cancers with no effective cures for metastatic disease. The poor efficacy of current therapy in advanced melanoma highlights the need for better understanding of molecular mechanisms contributing to the disease. Recent work has shown that epigenetic changes, including aberrant DNA methylation, lead to alterations in gene expression and are as important in the development of malignant melanoma as the specific and well characterized genetic events. Reversion of these methylation patterns could thus lead to a more targeted therapy and are currently under clinical investigation. The purpose of this review is to compile recent information on aberrant DNA methylation of melanoma, to highlight key genes and molecular pathways in melanoma development that have found to be epigenetically altered and to provide insight as of how DNA methylation might serve as targeted treatment option as well as a molecular and prognostic marker in malignant melanoma.
Keywords: Melanoma, Hypermethylation, Epigenetics, decitabine
Introduction
Although Malignant Melanoma comprises only 4 percent of all dermatologic cancers, it accounts for more than 75 percent of deaths from skin cancer. Surgical resection remains the only effective cure for tumors that have not yet reached a depth of greater than 1mm. However prognosis rapidly declines with more advanced stages and the current 5-year survival averages at about 15% for metastatic melanoma [1,2]. Furthermore, melanoma is one of the most common causes of cancer deaths between the ages of 20-35 [3] resulting in an immense socioeconomic impact. Thus research is needed to define newer therapeutic targets as current treatment options for advanced malignant melanoma have failed to show satisfactory results. [1,2,3].
The stages of progression of normal melanocytes to malignant melanoma can be divided into 1) Benign Nevus 2) Dysplastic nevus 3) Radial growth phase 4) Vertical growth phase and 5) Metastatic Melanoma [4]. These histologic changes can be related to alterations of specific key genes [5,6]. Chromosomal deletions, mutations and amplifications play a major role in these transformations. However recent investigations has revealed that epigenetic events may be equally important in carcinogenesis.
DNA methylation is involved in epigenetic silencing of genes
The term Epigenetics refers to heritable changes in gene expression that are not caused by changes in the DNA nucleotide sequence, but rather result from modification in the DNA backbone and DNA packaging [7,8,9]. The best-studied epigenetic event so far is DNA methylation. DNA methylation occurs by covalent addition of a methyl group at the 5′ carbon of the cytosine ring, resulting in 5-methylcytosine. This process is catalyzed by DNA methyltransferases (DNMTs) and takes place only at cytosine bases located 5′ to a guanosine base in a CpG dinucleotide [7,8]. These CpGs can be clustered in small stretches of DNA termed “CpG islands” [10]. Approximately 70% of human gene promoters are associated with a CpG island [112]. Promoter CpG islands are normally unmethylated in Euchromatin (the part of the genome, that is actively transcribed and translated), but show a heavy methylation pattern in heterochromatin (part of genome where activity and expression are suppressed) to facilitate transcriptional silencing of noncoding regions [12]. Examples of such fully methylated CpG islands associated with silent genes can be found on the inactive X Chromosome in females and in silenced alleles of some imprinted genes (genes that are expressed only from either maternal or paternal allele) [10]. However, aberrant promoter methylation of genes that control cell cycle and apoptosis can lead disruption of normal cell division and can contribute to carcinogenesis.
Although most of the work in melanoma has focused on gene-silencing events caused by hypermethylation, it is important to mention that cytosine methylation can influence tumorigenicity by other mechanisms. It has been shown for example, that Histone deacetylation – another epigenetic event leading to transcriptional silencing can be stimulated by cellular machinery triggered by aberrant methylation [13-16]. There is also growing evidence that hypermethylation can actually predispose to mutational events as it has been shown in MLH1 and MGMT- two DNA repair genes where aberrant methylation resulted in microsatellite instability increasing the frequency of mutations [29-31]. 5-Methylcytosine itself has been shown to be mutagenic and can potentially undergo spontaneous hydrolytic deamination causing C→T transitions [32]. Additionally, in regards to Malignant Melanoma it is important to mention that the presence of methyl groups in CpG dinucloutides in coding regions of TP53 increases the rate of mutations induced by UV-light by shifting the UV absorption spectrum for cytosine to a region in spectrum that is prevalent in sunlight [33].
DNA Methyltransferases and their Inhibitors
As explained above DNMTs catalyze the covalent addition of methyl groups to cytosine residues in DNA. The most common functional DNMTs in mammalian cells include DNMT1, DNMT3a and DNMT3b [43,44]. The most abundant one – DNMT1 – appears to be responsible for maintaining established patterns of DNA methylation, while DNMT3a and DNMT3b seem to play a role in the establishment of new, de-novo, methylation [45, 46]. It has been shown however that DNMT1 in cancer cells was not alone responsible for maintaining abnormal methylation patterns suggesting that DNMT3s may cooperate for this function in malignancies [47,48]. While DNMT1 and DNMT3b priory were known to promote tumor growth and play important roles in cancer cell survival and tumorigenesis [46, 113], the role of DNMT3a in carcinogenesis remains mostly unknown. A recent study by Deng et al. now suggests that DNMT3a also has a significant impact on tumor growth and metastasis formation in malignant melanoma [113]. Several DNMT inhibitors have been employed in experimental and clinical trials, the most common one being 5-aza-2′-deoxycytidine, also known as DAC or Decitabine. As a Cytidine analog Decitabine is incorporated into newly synthesized DNA where it covalently links with DNMTs, which potentially induces cell death and cell damage by obstructing DNA synthesis and structural instability at the site of incorporation [50-52]. DNMTs are depleted by being bound to these agents and are thereby unavailable for methylation, resulting in significant demethylation after repeated replication [46,53]. DNMT Inhibitors are inactivated by deamination by cytidine deaminase [49].
Methylated Genes in Melanoma
Abnormal patterns of DNA methylation in melanoma have been recognized over the last few years and so far more than 70 aberrantly hypermethylated genes have been discovered (Table 1 and 2). These genes have been found to be hypermethylated either by direct examination of the methylated CpGs (Table 1) or indirectly by their activation upon treatment with demethylating agents (Table 2). Most of this work has been done in melanoma cell lines. Interestingly, cell lines of malignant tumors in general seem to have a higher level of CpG island hypermethylation than their primaries. Paz et al. who investigated 70 different cell lines from 12 different tumors – including malignant melanoma- compared the methylation status of the cultured cell lines to their primary tumors. Cultured cell lines showed 5′CpG hypermethylation in 26.6% vs. 17% in primary tumors (34). The studies reflected in this article seem to have an equal tendency towards increased promoter hypermethylation in cultured cell lines. Table 1 contains 34 studies where aberrant methylation was tested in melanoma cell lines as well as in melanoma tumor samples. 25 of the 34 studies showed increased methylation in Melanoma cell lines compared to their primary tumors, whereas two showed equal methylation patterns and seven showed increased methylation in the primary tumors as compared to the cell lines. In spite of these limitations, some genes have been consistently shown to be hypermethylated in melanoma and its cell lines and have even been implicated in the progression of mole to malignant melanoma. The role of these genes in pathobiology of melanoma is reviewed in greater detail.
Table 1.
| Gene | Reference | Samples | Frequency of Methylation |
|---|---|---|---|
|
3-OST-2 (3-O-sulfotransferase-2) |
Furuta et al. 2004 [67] | 25 Melanoma samples (4 primary, 21 metastatic) | 14/25 (56%) |
|
APC (Adenomatous polyposis coli gene) |
Worm et al. 2004 [96] | 40 Melanoma cell lines (incl. FM39, FM79, FM95, FM79 and FM95 otherwise unspecified) 54 melanoma biopsies - 15 primary tumors 39 metastases |
5/40 (13%) of the cell lines (FM39, FM79, FM95, FM79 and FM95). 1/ 15 (7%) primary melanomas, and 8/39 (21%) tumor metastases |
|
ASC/PYCARD (ASC/TMS1) (PYD, an N-terminal PYRIN-domain; and CARD, a C terminal caspase-recruitment domain) |
Guan et al. 2003 [97] | 12 human melanoma cell lines: C32TG, WM35, WM793, MeWo, VMRC-MELG, A2058, GAK, HMV-I, HMV-II, SK-Mel-28 and G361 32 Melanoma tissues (10 used for methylation assessment only) |
6/12 (50%) in HMV-I, C32TG, A2058, MeWo, WM35, G361 5 /10 (50%) |
|
BST 2 (Bone marrow stromal cell antigen) |
Muthusamy et al. 2006 [20] | 9 Melanoma cell lines (MelJuSo, UACC903, C8161, Neo6/C8161, WM1205 Lu, WM35, Roth, Carney, and WM455) 20 melanoma tumor samples |
8/9 (89%) in all but WM35 0/20 (50%) |
| Caspase 8 | Fulda et al. 2001 [98] | 6 Melanoma cell lines (Colo, IGR, HS, MEWO, MML, MRI) | 2/6 (33%) in MEWO, MML |
|
CDH1 (Cadherin 1) |
Furuta et al. 2004 [67] | 13 Melanoma cell lines (MeWo, WM-266-4, WM-115, C32TG, MMAc, VMRC-MELG, COLO 679, GAK, A2058, SK-MEL- 28, G361, HMV-I, TK-Mel-1) |
6/13 (46%) in MeWo, WM-266-4, WM-115,C32TG, A2058, HMV-I) |
|
CDH8 (Cadherin 8) |
Muthusamy et al. 2006 [20] | 9 Melanoma cell lines (MelJuSo, UACC 903, C8161, Neo6/C8161, WM1205 Lu, WM35, Roth, Carney, and WM455) 20 melanoma tumor samples |
4/9 (44%) in C8161, Neo6/C8171, WM1205, Carney 2/20 (10%) |
|
CDKN1B (Cyclin Dependent Kinase Inhibitor 1B) |
Worm et al. 2000 [96] | 45 metastatic tumors 35 cell lines (unspecified) |
4/45 (9%) 3/35 (9%) in FM 9, FM 60, FM 70 |
|
CDKN1C Cyclin Dependent Kinase Inhibitor 1C) |
Muthusamy et al. 2006 [20] | 9 Melanoma cell lines (MelJuSo, UACC 903, C8161, Neo6/C8161, WM1205 Lu, WM35, Roth, Carney, and WM455) 20 melanoma tumor samples |
4/9 44%) in UACC903, C8161, WM1205, Carney 7/20 (35%) |
|
COL1A2 (Collagen, type 1, alpha2) |
Muthusamy et al. 2006 [20] | 9 Melanoma cell lines (MelJuSo, UACC 903, C8161, Neo6/C8161, WM1205 Lu, WM35, Roth, Carney, and WM455) 20 melanoma tumor samples |
8/9 (89%) in al but WM 455 16/20 (89%) |
| Koga et al. 2009 [112] | Normal skin n=7 and nevi n=2 Early stage melanomas n=8 Advanced stage melanomas n=16 |
0/9 90%0 4/8 (50%) 11/16 (69%) |
|
|
CXCR4 (C-X Chemokine receptor 4) |
Mori et al. 2005 [61] | 11 Melanoma Cell lines (MA-MK) | 4/11 (35%) in MA, MH, MC, MD |
| Cyclin D2 | Furuta et al. 2004 [67] | 13 Melanoma cell lines (MeWo, WM-266-4, WM-115, C32TG, MMAc, VMRC-MELG, COLO 679, GAK, A2058, SK-MEL- 28, G361, HMV-I, TK-Mel-1) |
2/13 (15%) in MMAc, HMV-I |
|
CYP1B1 Cytochrome P450, subfamily 1, polypeptid 1 |
Muthusamy et al. 2006 [20] | 9 Melanoma cell lines (MelJuSo, UACC 903, C8161, Neo6/C8161, WM1205 Lu, WM35, Roth, Carney, and WM455) 20 melanoma tumor samples |
8/9 (89%) in all but UACC903 20/20 (100%) |
|
DAL1 Differentially expressed in adenocarcinoma of the lung |
Muthusamy et al. 2006 [20] | 9 Melanoma cell lines (MelJuSo, UACC 903, C8161, Neo6/C8161, WM1205 Lu, WM35, Roth, Carney, and WM455) 20 melanoma tumor samples |
2/9 (22%) in C8162, NeoC/C8161 1/20 (5%) |
|
DAPK (Death associated Protein Kinase) |
Hoon et al. 2004 [90] | 15 Melanoma cell lines (MA-MO) 20 primary tumors 86 metastatic tumors |
0% 0% 16/86 (19%) |
|
DDIT4L (DNA-Damage-Inducible Transcript 4-like) |
Koga et al. 2009 [112] | Normal skin n=7 and nevi n=2 Early stage melanomas n=8 Advanced stage melanomas n=16 |
0/9 90%0 1/8 (13%) 6/16 (16%) |
|
DNAJC15 (DNAJ, E. coli homolog of subfamily C, member 15) |
Muthusamy et al. 2006 [20] | 9 Melanoma cell lines (MelJuSo, UACC 903, C8161, Neo6/C8161, WM1205 Lu, WM35, Roth, Carney, and WM455) 20 melanoma tumor samples |
6/9 (67%) in all but UACC903, WM1205, WM 455 10/20 (50%) |
| E-Cadherin | Tsutsumida et al. 2004 [99] | 8 Melanoma call lines (MeWo,, AKI, MMAc, SK-MEL-28, A375M, 9711 and MMIV | 2/8 (25%) in AKI and A375M |
|
ER (Human Estrogen Receptor) |
Furuta et al. 2004 [67] | 13 Melanoma cell lines (MeWo, WM-266-4, WM-115, C32TG, MMAc, VMRC-MELG, COLO 679, GAK, A2058, SK-MEL- 28, G361, HMV-I, TK-Mel-1) |
2/13 (15%) in MMAc, HMV-I |
|
GATA4 (GATA binding Protein 4) |
Tanemura et al. 2009 [60} | Stage I, Primary (18) Stage II, Primary (17) Stage III, Primary (17) Stage III, Metastatic (18) Stage IV, Metastatic (52) Overall 122 samples from 107 patients |
16.7% 8.3% 42.9% 22.2% 34% 27.5% |
|
GDF15 (Growth differentiation factor 15) |
Muthusamy et al. 2006 [20] | 9 Melanoma cell lines (MelJuSo, UACC 903, C8161, Neo6/C8161, WM1205 Lu, WM35, Roth, Carney, and WM455) 20 melanoma tumor samples |
4/9 (44%) in C8161, WM1205, WM35, Roth 15/20 (75%) |
|
HMW-MAA (Human high molecular weight melanoma associated antigen) |
Luo et al. 2006 [100] | 5 Melanoma cell lines (M14#5, 1520, SK-MEL-5, A375, M21) | 2/5 (40%) in M14#5 and 1520, weak expression in SK-MEL-5 |
|
HOXB13 (Homeo Box B13) |
Muthusamy et al. 2006 [20] | 9 Melanoma cell lines (MelJuSo, UACC 903, C8161, Neo6/C8161, WM1205 Lu, WM35, Roth, Carney, and WM455) 20 melanoma tumor samples |
3/9 (33%) in MelJuSo, C8161, Neo6/C8161 4/20 (20%) |
|
LOX (Lysil Oxidase) |
Liu et al. 2008 [101] | 20 Melanoma cell lines (A375-Mel, Lox, C8161.9 and 17 daughter cell lines) 40 Melanoma samples (8 primary, 32 metastasis) |
12/20 (60%) 18/40 (45%) |
|
LRRC1 (Leucine-rich repeat-containing protein1) |
Muthusamy et al. 2006 [20] | 9 Melanoma cell lines (MelJuSo, UACC 903, C8161, Neo6/C8161, WM1205 Lu, WM35, Roth, Carney, and WM455) 20 melanoma tumor samples |
4/9 (44%) in MelJuSo, C8161, Neo6/C8161, Roth 1/20 (5%) |
|
LXN (Latexin) |
Muthusamy et al. 2006 [20] | 9 Melanoma cell lines (MelJuSo, UACC 903, C8161, Neo6/C8161, WM1205 Lu, WM35, Roth, Carney, and WM455) 20 melanoma tumor samples |
8/9 (90%) in all but Neo6/C8161 19/20 (95%) |
|
Maspin/Serpin (Serin Protease inhibitor) |
Denk et al. 2007 [102] | 6 Melanoma cell lines (MelIm, MelWei, MelJu, MelHo, SkMel3, SKMel28, HTZ19d) | 6/6 (100%) |
|
MFAP2 (Microfibril-associated protein 2) |
Muthusamy et al. 2006 [20] | 9 Melanoma cell lines (MelJuSo, UACC 903, C8161, Neo6/C8161, WM1205 Lu, WM35, Roth, Carney, and WM455) 20 melanoma tumor samples |
6/9 (67%) in all but C8161, Neo6/C8161, WM 455 6/20 (30%) |
|
MGMT (O6-methylguanine DNA methyltransferase) methyltransferase) |
Hoon et al. 2004 [90] | 15 Melanoma cell lines (MA-MO) 20 primary tumors 86 metastatic tumors |
4/15 (27%) in MD, MH, MK and MO 2/20 (10%) primary tumors 29/86 (34%) metastatic tumors |
| Kohonen-Corish et all 2006 [92] |
84 Melanoma Metastases from 47 patients | 26/84 (31%) | |
| Furuta J et al. 2004 [67] | 13 Melanoma cell lines (MeWo, WM-266-4, WM-115, C32TG, MMAc, VMRC-MELG, COLO 679, GAK, A2058, SK-MEL- 28, G361, HMV-I, TK-Mel-1) |
0/13 (0%) | |
|
MINT 17 (Methylated in tumor loci 17) |
Tanemura et al. 2009 [60] | Stage I, Primary (18) Stage II, Primary (17) Stage III, Primary (17) Stage III, Metastatic (18) Stage IV, Metastatic (52) Overall 122 samples from 107 patients |
11.1% 17.6% 41.2% 52.9% 38.5% 34.2% |
|
MINT 31 (Methylated in tumor loci 31) |
Tanemura et al. 2009 [60] | Stage I, Primary (18) Stage II, Primary (17) Stage III, Primary (17) Stage III, Metastatic (18) Stage IV, Metastatic (52) Overall 122 samples from 107 patients |
5.6% 23.5% 35.3% 27.8% 36.5% 28.1% |
|
MT1G (Metallothionein) |
Koga et al. 2009 [112] | Normal skin n=7 and nevi n=2 Early stage melanomas n=8 Advanced stage melanomas n=16 8 Melanoma Cell strains WW165, YUGEN8, YULAC, YUMAC, YURIF, YUSAC2, YUSIT1, YUSTE |
0/9 (0%) 1/8 (13%) 4/16 (25%) Increased Methylation and decreased gene expression compared to new born and adult melanocytes in all cell lines except YUSIT 1. |
|
NPM 2 (Nucleoplasmin 2) |
Koga et al. 2009 [112] | Normal skin n=7 and nevi n=2 Early stage melanomas n=8 Advanced stage melanomas n=16 |
0/9 (0%) 3/8 (38%) 9/16 (56%) |
|
MTAP (Methylthioadenosine Phosphorylase) |
Behrmann et al. 2003 [103] | 9 Melanoma cell lines (Mel Im, Mel Ei, Mel Wei, Mel Ho, Mel Juso, Mel Ju, SK-Mel-28, HMB2, and HTZ19d) | 8/9 (89%) in all cell lines but HTZ19d |
| Freedberg et al. 2008 [104] | 60 melanoma metastases 9 melanoma cell lines (19, 94, 100, 103, 147, 173, 187, 192, 197) |
34/60 (57%) 3/9 (33%) in 173, 192, 197 |
|
| Furuta et al. 2004 [67] | 13 Melanoma cell lines (MeWo, WM-266-4, WM-115, C32TG, MMAc, VMRC-MELG, COLO 679, GAK, A2058, SK-MEL- 28, G361, HMV-I, TK-Mel-1) |
0/13 (0%) | |
| p16/ INK14 | Freedberg et al. 2008 [104] | 9 melanoma cell lines (19, 94, 100, 103, 147, 173, 187, 192, 197) | 1/9 (11%) in 192 |
|
CDKN2A (Cyclin-dependent kinase inhibitor 2A INK4A) |
60 melanoma metastases | 16/60 (27%) | |
| Gonzalgo et al. 1997 [105] | 12 Melanoma Cell lines (SK-MEL-21,-28, -37, -39, -61, -63, - 147, -196, -241, A 375, 526, 888) 30 metastatic Melanoma samples |
3/12 (25%) in SK-MEL- 196, -241 and 888 3/30 (10%) |
|
| Van der Velden et al. 2001 [106] |
12 uveal melanoma cell lines (92.1; OCM-1, -3, -8; Mel-270, -285, -290;, MEL-202; EOM-3; OMM-1, -1.3, -1.5) 22 primary oveal melanomas |
6/12 (50%) in 92.1; Mel-270, -285, OMM-1, -1.3, -1.5 7/22 (32%) |
|
| Furuta et al. 2004 [67] | 13 Melanoma cell lines (MeWo, WM-266-4, WM-115, C32TG, MMAc, VMRC-MELG, COLO 679, GAK, A2058, SK-MEL- 28, G361, HMV-I, TK-Mel-1) |
1/13 (8%) in GAK | |
|
PCSK (Proprotein convertase, subtilisin/kexin-type 1) |
Muthusamy et al. 2006 [20] | 9 Melanoma cell lines (MelJuSo, UACC 903, C8161, Neo6/C8161, WM1205 Lu, WM35, Roth, Carney, and WM455) 20 melanoma tumor samples |
9/9 (100%) 12/20 (60%) |
|
PRDX2 (Peroxiredoxin 2) |
Furuta J et al. 2006 [68] | 13 Melanoma cell lines (MeWo, VMRC-MELG, A2058, C32TG, CAK, G361, SK-MEL-28, HMV-1, COLO 679, MMAc, WM- 266-4, WM-115, TK-Mel-1) 36 surgical Melanoma specimen from primary and metastatic site, stage III-IV |
4/13 (31%) in MMac, VMRC-MELG, GAK, SK-MEL-28 3/36 (8%) |
|
PTEN (Phosphatase and tensin homologue) |
Mirmohammadsadegh et al. 2006 [58] |
11 peripheral blood specimens (circulating DNA) from patients with metastatic melanoma melanoma cell line SK-Mel-28 and MV3 |
62% 58-92% |
| Furuta et al. 2004 [67] | 13 Melanoma cell lines (MeWo, WM-266-4, WM-115, C32TG, MMAc, VMRC-MELG, COLO 679, GAK, A2058, SK-MEL- 28, G361, HMV-I, TK-Mel-1) |
3/13 (23%) in MeWo, MMAc and HMV-I | |
|
PTGS2 (Prostaglandin-endoperosid synthase 2) |
Muthusamy et al. 2006 [20] | 9 Melanoma cell lines (MelJuSo, UACC 903, C8161, Neo6/C8161, WM1205 Lu, WM35, Roth, Carney, and WM455) 20 melanoma tumor samples |
2/9 (22%) in UAC903, C8161 4/20 (20%) |
|
QPTC (Glutaminyl-peptide cyclotransferase) |
Muthusamy et al. 2006 [20] | 9 Melanoma cell lines (MelJuSo, UACC 903, C8161, Neo6/C8161, WM1205 Lu, WM35, Roth, Carney, and WM455) 20 melanoma tumor samples |
5/9 (56%) in all but WM 1205, MelJuSo, WM455, WM35 20/20 (100%) |
|
RAR-b2 (Retinoic acid receptor-b2) |
Hoon et al. 2004 [90] | 15 Melanoma cell lines (MA-MO) 20 primary tumors 86 metastatic tumors |
8/15 (53%) MD, ME, MH-MM 14/20 (70%) 60/86 (70%) |
| Furuta et al. 2004 [67] | 13 Melanoma cell lines (MeWo, WM-266-4, WM-115, C32TG, MMAc, VMRC-MELG, COLO 679, GAK, A2058, SK-MEL- 28, G361, HMV-I, TK-Mel-1) 25 Melanoma samples (4 primary, 21 metastatic) |
6/13 (46%) in MMAc, GAK, A2058, G361, HMV-1, SK-MEL-28 5/25 (20%) |
|
|
RASSF1A (RAS Association Domain Family Protein 1) |
Tanemura et al. 2009 [60] | Stage I, Primary (18) Stage II, Primary (17) Stage III, Primary (17) Stage III, Metastatic (18) Stage IV, Metastatic (52) Overall 122 samples from 107 patients |
58.3% 66.7% 64.3% 47.2% 56.3% 57.6% |
| Spugnardi et al 2003 [107] | 11 Melanoma cell lines (MA - MK) 44 Melanoma tumors AJCC stage III/IV |
9/11 (82%) Melanoma cell lines 24/44 (55%) |
|
| Hoon et al. 2004 [90] | 15 Melanoma cell lines (MA-MO) 20 primary tumors 86 metastatic tumors |
12/15 (80%) in MA, MC, ME-MK,MM-MO 3/15 (15%) 49/86 (57%) |
|
| Furuta et al. 2004 [67] | 13 Melanoma cell lines (MeWo, WM-266-4, WM-115, C32TG, MMAc, VMRC-MELG, COLO 679, GAK, A2058, SK-MEL- 28, G361, HMV-I, TK-Mel-1) 25 Melanoma samples (4 primary, 21 metastatic) |
6/13 (46%) in MMAc, GAK, A2058, SK-MEL-28, G361, HMV-I 9/25 (36%) |
|
| Tanemura et al. 2009 [60] | Stage I, Primary (18) Stage II, Primary (17) Stage III, Primary (17) Stage III, Metastatic (18) Stage IV, Metastatic (52) Overall 122 samples from 107 patients |
0% 0% 26.7% 27.8% 48.9% 28.6% |
|
|
RUNX3 (Runt-related transcription factor 3) |
Furuta et al. 2004 [67] | 13 Melanoma cell lines (MeWo, WM-266-4, WM-115, C32TG, MMAc, VMRC-MELG, COLO 679, GAK, A2058, SK-MEL- 28, G361, HMV-I, TK-Mel-1) |
3/13 (23%) in WM-266-4, WM-115, HMV-1 |
|
SOCS -1 (Suppressor of cytokine signaling 1) |
Liu et al. 2008 [101] | 20 Melanoma cell lines (A375-Mel, Lox, C8161.9 and 17 daughter cell lines) 40 Melanoma samples (8 primary, 32 metastasis) |
18/20 (90%) 30/40 (75%) |
| Tanemura et al. 2009 [60] | Stage I, Primary (18) Stage II, Primary (17) Stage III, Primary (17) Stage III, Metastatic (18) Stage IV, Metastatic (52) Overall 122 samples from 107 patients |
7.1% 23.1% 24% 23.5% 44.9% 31.5% |
|
|
SOCS-2 (Supressor of cytokine signaling 2) |
Liu et al. 2008 [101] | 20 Melanoma cell lines (A375-Mel, Lox, C8161.9 and 17 daughter cell lines) 40 Melanoma samples (8 primary, 32 metastasis) |
16/20 (80%) 30/40 (75%0 |
|
SYK (Spleen Tyrosine Kinase) |
Muthusamy et al. 2006 [20] | 9 Melanoma cell lines (MelJuSo, UACC 903, C8161, Neo6/C8161, WM1205 Lu, WM35, Roth, Carney, and WM455) 20 melanoma tumor samples |
8/9 (89%) in all cell lines but UACC903 6/20 (30%) |
|
TERC (Telomerase RNA component) |
Furuta et al. 2004 [67] | 13 Melanoma cell lines (MeWo, WM-266-4, WM-115, C32TG, MMAc, VMRC-MELG, COLO 679, GAK, A2058, SK-MEL- 28, G361, HMV-I, TK-Mel-1) |
3/13 (23%) in WM-266-4, WM-115, HMV-I |
|
TFPI-2 (Tissue factor pathway inhibitor-2) |
Nobeyama et al. 2007 | 12 Melanoma Cell lines (HMV-1, MeWo, WM-115, WM-216-4, VMRC-MELG, A2058, C32TG, GAK, G361, SK-MEL-28, MMAc) 37 Surgical Specimen (20 primary sites, 17 metastasis) |
4/12 (33%) in HMV-a, GAK, VMRC-MELG, G32TG 0% of primary sites, 5/12 (42%) of metastatic sites |
| Tanemura et al. 2009 [60] | Stage I, Primary (18) Stage II, Primary (17) Stage III, Primary (17) Stage III, Metastatic (18) Stage IV, Metastatic (52) Overall 122 samples from 107 patients |
0% 6.3% 17.6% 17.6% 44.9% 21.7% |
|
|
TNFRSF10C (DcR1) (Tumor necrosis factor receptor superfamily, member 10c, decoy without an intracellular domain) |
Liu et al. 2008 [101] | 20 Melanoma cell lines (A375-Mel, Lox, C8161.9 and 17 daughter cell lines) 40 Melanoma samples (8 primary, 32 metastasis) |
12/20 (60%) 23/40 (56%) |
|
TNFRSF10D (DcR2) (Tumor necrosis factor receptor superfamily, member 10d, decoy with truncated death domain) |
Liu et al. 2008 [101] | 20 Melanoma cell lines (A375-Mel, Lox, C8161.9 and 17 daughter cell lines) 40 Melanoma samples (8 primary, 32 metastasis) |
17/20 (85%) 32/40 (80%) |
|
TPM1 (Tropomyosin) |
Liu et al. 2008 [101] | 20 Melanoma cell lines (A375-Mel, Lox, C8161.9 and 17 daughter cell lines) 40 Melanoma samples (8 primary, 32 metastasis) |
4/12 (33%) 3/40 (7.5%) |
|
TSPY (Testis specific protein, Y linked) |
Gallaghar et al. 2005 [109] | 4 Melanoma cell lines (WM793, WM 793-P1, -P2; 1205 Lu 20 metastatic melanoma tumor samples (5male, 15 female) |
3/4 (75%) in all but WM793 5/20 (25%) in all male samples |
|
WIF1 (WNT inhibitory factor) |
Tanemura et al. 2009 [60] | Stage I, Primary (18) Stage II, Primary (17) Stage III, Primary (17) Stage III, Metastatic (18) Stage IV, Metastatic (52) Overall 122 samples from 107 patients |
5.6% 6.3% 31.3% 50% 44% 33.3% |
Table 2.
| Gene | Reference | Cell lines and tumor samples | More detailed explanation of re-expression |
|---|---|---|---|
|
APAF1 Apoptotoic protease activating factor 1 |
Soengas et al. 2001 [37] | 9 Melanoma Cell lines (SK-Mel 5,19, -28, -94, -103, -147, -197) | Recovery of APAF 1 expression after 5-Aza-d2C treatment |
|
APOD Apolipoprotein D |
Gallaghar et al. 2005 [109] | 4 Melanoma Cell lines WM 793, -P2, P2; 1205- Lu |
Increase in Gene expression and reduction in cell growth after 5- Aza-dC Treatment |
|
BST2 Bone marrow stromal cell antigen 2 |
Gallaghar et al. 2005 [109] | 4 Melanoma Cell lines WM 793, -P2, P2; 1205- Lu |
Increase in Gene expression and reduction in cell growth after 5- Aza-dC Treatment |
|
CAM Calmodulin |
Van der Velden et al. 2003 [106] | Primary Uveal Melanoma Cell line MEL-270 | 5 fold increase in expression after demythelation with 5-Aza-dC |
|
CCR-7 Chemokine receptor 7 |
Mori et al. 2005 [61] | 11 Melanoma Cell lines (MA-MK) | Upregulation of CCR-7 after treatment with 5-Aza-2dC and/or TSA |
|
CDH1 Cadherin 1 |
Paz et al. 2003 [34] | 18 Melanoma cell lines SK MEL2, -5, -19, -28, -29, -94, -100, -103, -147, -173, -187, -197, G-361, Malme-3M, M14-Mel, UACC-62, -257, LOX-LIVM |
Re-expression of CDH1 in 9/18 cell lines (50%), SK MEL19, -28, -103, -147, 1-73, -197, UACC62, LOX-LIVM |
|
CDKNA1 (p21) Cyclin-dependent Kinase Inhibitor 1A |
Halaban et al. 2009 [62] | 8 Melanoma cell strains YUMAC, YUSAC2, YULAC, YUSAIT, YUGEN8, WW 165, YURIF, 501 mel |
Significant increase in expression after treatment with 5-Aza-dC in all cell lines except YUSIT1 |
| Clu (Clusterin) | Halaban et al. 2009 [62] | 8 Melanoma cell strains YUMAC, YUSAC2, YULAC, YUSiIT1, YUGEN8, WW165, YURIF, 501 mel |
Significant increase in expression in all cell lines |
|
COL1A2 Collagen, type 1, alpha2 |
Koga et al. 2009 [112] | 2 Melanoma strains YUGEN8, YUMAC | > 400 fold increase in YUGEN8 and >500 fold increase in YUMAC after treatment with 5-Aza-dC |
|
CSK c-Src kinase |
Van der Velden et al. 2003 [106] | Primary Uveal Melanoma Cell line MEL-270 | 3 fold increase in expression after demythelation with 5-Aza-dC |
|
CSNK162 Casein kinase1 gamma2 |
Van der Velden et al. 2003 [106] | Primary Uveal Melanoma Cell line MEL-270 | 3 fold increase in expression after demythelation with 5-Aza-dC |
|
CSPCB Cartilage-specific proteoglycan core protein |
Van der Velden et al. 2003 [106] | Primary Uveal Melanoma Cell line MEL-270 | 3 fold increase in expression after demythelation with 5-Aza-dC |
|
CYBA Cytochrome B, alpha subunit |
Gallaghar et al. 2005 [109] | 4 Melanoma Cell lines WM 793, -P2, P2; 1205- Lu |
Increase in Gene expression and reduction in cell growth after 5- Aza-dC Treatment |
|
DDIT4L DNA-Damage-Inducible Transcript 4- like |
Koga et al. 2009 [112] | 2 Melanoma strains YUGEN8, YUMAC | >200 fold increase in YUGEN8 and >400 fold increase in YUMAC after treatment with 5-Aza-dC |
|
EBP1 Cell cycle protein p38-2G4 homolog |
Van der Velden et al. 2003 [106] | Primary Uveal Melanoma cell line MEL-270 | 3 fold increase in expression after demythelation with 5-Aza-dC |
|
EF1- alpha Elongation factor alpha |
Van der Velden et al. 2003 [106] | Primary Uveal Melanoma cell line MEL-270 | 3 fold increase in expression after demythelation with 5-Aza-dC |
|
ET2 Endothelin 2 |
Van der Velden et al. 2003 [106] | Primary Uveal Melanoma Cell line MEL-270 | 5 fold increase in expression after demythelation with 5-Aza-dC |
|
F6 ATP synthase coupling factor 6 mitochondrial |
Van der Velden et al. 2003 [106] | Primary Uveal Melanoma Cell line MEL-270 | 3 fold increase in expression after demythelation with 5-Aza-dC |
|
GARS Glycyl tRNA synthetase |
Van der Velden et al. 2003 [106] | Primary Uveal Melanoma Cell line MEL-270 | 3 fold increase in expression after demythelation with 5-Aza-dC |
|
GIP 3 Interferon-inducible peptide precursor |
Gallaghar et al. 2005 [109] | 4 Melanoma Cell lines WM 793, -P2, P2; 1205- Lu |
Increase in Gene expression and reduction in cell growth after 5- Aza-dC Treatment |
|
HSPB1 Heat-shock 27 KDA protein |
Gallaghar et al. 2005 [109] | 4 Melanoma Cell lines WM 793, -P2, P2; 1205- Lu |
Increase in Gene expression and reduction in cell growth after 5- Aza-dC Treatment |
|
HSBP6 Heat shock protein alpha-crystallin related B6 |
Koga et al. 2009 [112] | 2 Melanoma strains YUGEN8, YUMAC | >150 fold increase in YUGEN8 and >400 fold increase in YUMAC after treatment with 5-Aza-dC |
|
IFIT1 Interferon-induced protein with tetra- Tricopeptide repeats1 |
Gallaghar et al. 2005 [109] | 4 Melanoma Cell lines WM 793, -P2, P2; 1205- Lu |
Increase in Gene expression and reduction in cell growth after 5- Aza-dC Treatment |
|
ISGF3G Interferon-stimulated transcription factor3, gamma |
Gallaghar et al. 2005 [109] | 4 Melanoma Cell lines WM 793, -P2, P2; 1205- Lu |
Increase in Gene expression and reduction in cell growth after 5- Aza-dC Treatment |
|
ITGAE Integrin alpha E |
Van der Velden et al. 2003 [106] | Primary Uveal Melanoma Cell line MEL-270 | 3 fold increase in expression after demythelation with 5-Aza-dC |
| ITGB8 Integrin beta 8 |
Van der Velden et al. 2003 [106] | Primary Uveal Melanoma Cell line MEL-270 | 3 fold increase in expression after demythelation with 5-Aza-dC |
| KIAA0324 | Van der Velden et al. 2003 [106] | Primary Uveal Melanoma Cell line MEL-270 | 3 fold increase in expression after demythelation with 5-Aza-dC |
|
LDHB L-Lactate dehydrogenase H subuint |
Van der Velden et al. 2003 [106] | Primary Uveal Melanoma Cell line MEL-270 | 3 fold increase in expression after demythelation with 5-Aza-dC |
|
MT1G Metallothionein |
Koga et al. 2009 [112] | 2 Melanoma strains YUGEN8, YUMAC | >50 fold increase in YUGEN8 and >200 fold increase in YUMAC after treatment with 5-Aza-dC |
|
MT2A Metallothionein 2A |
Gallaghar et al. 2005 [109] | 4 Melanoma Cell lines WM 793, -P2, P2; 1205- Lu |
Increase in Gene expression and reduction in cell growth after 5- Aza-dC Treatment |
|
MX1 Myxovirus resistance 1/interferon- inducible |
Gallaghar et al. 2005 [109] | 4 Melanoma Cell lines WM 793, -P2, P2; 1205- Lu |
Increase in Gene expression and reduction in cell growth after 5- Aza-dC Treatment |
|
NPM 2 Nucleoplasmin 2 |
Koga et al. 2009 [112] | 2 Melanoma strains YUGEN8, YUMAC | >150 fold increasein YUGEN8 and > 100 fold increase in YUMAC after treatment with 5-Aza-dC |
| P-Cadherin | Tsutsumida et al. 2004 [99] | Melanoma cell lines AKI and A375 | Re-expression after 5-Aza-dC treatment |
|
PCNA Proliferating cyclic nuclear antigen |
Van der Velden et al. 2003 | Primary Uveal Melanoma Cell line MEL-270 | 3 fold increase in expression after demythelation with 5-Aza-dC |
| PIRIN | Van der Velden et al. 2003 | Primary Uveal Melanoma Cell line MEL-270 | 3 fold increase in expression after demythelation with 5-Aza-dC |
|
RAR-b2 Retinoic acis receptor b2 |
Paz et al.2003 [34] | 18 Melanoma cell lines SK MEL2, -5, -19, -28, -29, -94, -100, -103, -147, -173, -187, -197, G-361, Malme-3M, M14-Mel, UACC-62, -257, LOX-LIVM |
Re-expression after 5 Aza-dC treatment in 10/18 cell lines (56%), SK MEL 2, -5, -19, -28 -29, - 100, -173, -187, Malme3M, UACC-257 |
|
RASSF1 Ras Association domain Family protein 1 |
Paz et al. 2003 [34] | 18 Melanoma cell lines SK MEL2, -5, -19, -28, -29, -94, -100, -103, -147, -173, -187, -197, G-361, Malme-3M, M14-Mel, UACC-62, -257, LOX-LIVM |
Re-expression of RASF 1 protein after treatment with 5-Aza-dC in all Cell lines (100%) |
|
RGS3 Regulator of G-protein signaling 3 |
Gallaghar et al. 2005 [109] | 4 Melanoma Cell lines WM 793, -P2, P2; 1205- Lu |
Increase in Gene expression and reduction in cell growth after 5- Aza-dC Treatment |
|
RPL37A Ribosomal protein L37A |
Gallaghar et al. 2005 [109] | 4 Melanoma Cell lines WM 793, -P2, P2; 1205- Lu |
Increase in Gene expression and reduction in cell growth after 5- Aza-dC Treatment |
|
S100A1 S100 Calcium-binding protein A1 |
Gallaghar et al. 2005 [109] | 4 Melanoma Cell lines WM 793, -P2, P2; 1205- Lu |
Increase in Gene expression and reduction in cell growth after 5- Aza-dC Treatment |
|
SIAT4A Sialyltransferase 4A |
Van der Velden et al. 2003 [106] | Primary Uveal Melanoma cell line MEL-270 | 3 fold increase in expression after demethylation with 5-Aza-dC |
|
TIMP 3 Tissue inhibitor of metalloproteinase 3 |
Van der Velden et al. 2003 [106] | Primary Uveal Melanoma cell line MEL-270 and metastatic uveal melanoma cell line OMM1.3 |
Methylation shown in both cell lines (100%), also 3 fold increase in expression after demythelation with 5-Aza-dC of MEL-270 |
|
TIMP1 Tissue inhibitor of metalloproteinase 1 |
Van der Velden et al. 2003 [106] | Primary Uveal Melanoma Cell line MEL-270 | 3 fold increase in expression after demethylation with 5-Aza-dC |
|
TGFBI (transforming growth factor, beta-induced) |
Halaban et al. 2009 [62] | 8 Melanoma cell strains YUMAC, YUSAC2, YULAC, YUSAIT, YUGEN8, WW 165, YURIF, 501 mel |
Significant increase in all cell lines except WW165 |
|
TNFRSF10A (DR4, TRAIL-1) Tumor necrosis factor receptor superfamily, member 10a |
Bae et al. 2008 [91] | SK-Mel-28, SK-Mel-3 and 8 additional Melanoma cell lines (unspecified) | >3 fold increase in re-expression after treatment with 5-Aza-dC |
|
TRP-1 Tyrosine-related protein1 |
Van der Velden et al. 2003 [106] | Primary Uveal Melanoma Cell line MEL-270 | 5 fold increase in expression after demethylation with 5-Aza-dC |
|
TSP1 Thrombospondin 1 |
Paz et al. 2003 [34] | 18 Melanoma cell lines SK MEL2, -5, -19, -28, -29, -94, -100, -103, -147, -173, -187, -197, G-361, Malme-3M, M14-Mel, UACC-62, -257, LOX-LIVM |
Re-expression of TSP1 after treatment with 5-Aza-dC in all Cell lines (100%) |
|
WFDC WAP 4-disulfide core domain 1 |
Muthusamy et al. 2006 [20] | 9 Melanoma cell lines (MelJuSo, UACC 903, C8161, Neo6/C8161, WM1205 Lu, WM35, Roth, Carney, and WM455) 20 melanoma tumor samples |
5 fold increase in expression after demethylation with 5-Aza-dC |
p16 (INK4A) is a tumor suppressor gene found on Chromosome 9p21 and is one of the two genes encoded by CDKN2A (Cyclin-dependent kinase inhibitor 2A)- the other one being p14 (ARF). 25-40% of familial melanomas have genetic alterations within the CDKN2A gene [22] and a majority of sporadic melanomas also exhibit a large amount of genetic/epigenetic events within this locus [25,26]. INK4A blocks the cell cycle at the G1-S checkpoint by inhibiting cyclin-D-dependent kinases, CDK4 and CKD6, thereby activating the tumor-suppressive effects of the retinoblastoma protein (RB) (5,6). Mice lacking INK4A are abnormally prone to the development of tumors, however development of melanoma in such mice requires additional epigenetic events in other regulatory genes [23,24].
p14 (ARF=alternative reading frame) derives its name from the use of an alternative reading frame of the exons it shares with p16INK4A. p14ARF plays a crucial role in maintaining p53 levels by controlling ubiquitination of the p53 protein. It acts through binding to and thereby neutralizing MDM2 (mouse double minute 2) protein, which triggers ubiquitination of p53 eventually leading to its destruction in the proteosome [27,28] (fig 2). Stress signals are transmitted to p14ARF in part through DAPK, a death-associated protein kinase. Activation of p14ARF eventually leads to increased p53 activity and cell apoptosis. Epigenetic silencing of p14 can thus lead to loss of p53 mediated apoptosis and tumor development. Interestingly, p53 itself has not shown to be grossly altered in malignant melanoma [37,38]. However, recent investigations have discovered another gene – Apaf1 (Apoptotic protease activating factor 1, Chromosome 12q22) – that acts with cytochrome c and caspase 9 to mediate p53 dependent apoptosis [36]. Activation of p53 leads to stimulation and release of cytochrome c in the mitochondria thereby activating Apaf1, which in turn stimulates caspase 9 eventually leading to cell death [38]. Furthermore it has been shown, that there exists a strong inverse correlation between Apaf1 expression and Melanoma progression [39,40]. Soengas et al. showed that treatment of Apaf1 negative Melanoma cell lines with 5-Aza-dC lead to re-expression of Apaf 1 and reduction of cell growth [37]. Paradoxically there could not be found a 5-CpG hypermethylation within the promoter region of the Apaf1 gene indicating possibly a different mechanism of Apaf1 silencing, which could be reversed with demethylating agents.
Fig 2.
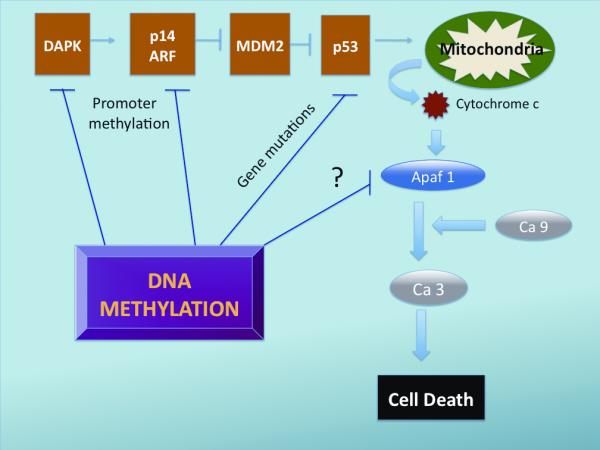
DNA Methylation and its effects on the apoptosis pathways. Promoter Methylation causes silencing of DAPK and p14 ARF decreasing thereby the inhibitory effect of p14ARF on MDM2 which in turn leads to increased ubiquitination and degradation of p53. p53 itself has not shown to be aberrantly methylated, however DNA methylation can predispose to gene mutations altering thereby functions of p53. Apaf 1, a newly recognized pro-apoptotic factor has shown to be underexpressed in cancer cells. Treatment with Decitabine can restore Apaf1 function, interestingly however the Apaf1 promoter was not found to be hypermethylated suggesting a different mechanism of gene silencing, which can be reversed with demethylating agents. Apaf1 activates in conjunction with Caspase 9 (Ca ) and Caspase 3 (Ca 3) leading ultimately to cell death.
PTEN (Phosphatase and tensin homologue) is a tumor suppressor gene that is located on Chromosome 10q23. It encodes a phosphatase that inhibits signaling by a variety of growth factors that use phosphatidylinositolphosphatate (PIP3) as intracellular signal. PIP3 becomes activated through the PI3K (Phosphatidylinositol-3-kinase) and in turn triggers stimulation of AKT (also known as Protein kinase B). Activated AKT inactivates proteins that suppress the cell cycle or stimulate apoptosis, thereby increasing proliferation and survival of cells. Mouse models have shown that suppression of AKT leads to reduced survival and growth of melanoma cells [41]; the amount of active AKT increases progressively during melanoma tumor progression with highest levels present in advanced-stage metastatic melanomas [42]. PTEN antagonizes PI3K activity by deactivating PIP3 thereby reducing AKT levels [5,6]. Epigenetic inactivation of PTEN thus can result in activation of the AKT pathway and lead to tumor development.
RASSF1A is a recently identified tumor suppressor gene, that has been studied with increasing interest since its first description in 2000 [76]. It is located in the 3p21.3 region of the human genome and has been found to be hypermethylated in various cancers [78-81]. Considering that RASSF1A also suffers point mutations in up to 15% of cancers, it is one of the most frequently inactivated proteins identified in neoplasms [77]. RASSF1a is a Ras effector that interacts with Ras GTPases promoting various cellular events including apoptosis, migration and mitosis [77, 84] (see figure 3). Mice whose RASSF1A gene has been knocked out exhibit increased tendency to develop spontaneous tumors [82, 83]. Re-expression of the RASSF1A gene in nude mice reduces tumorgenecity [84]. RASSF1A has been found to hypermethylated in upto 80% of samples in some studies. Interestingly the degree of methylation seems to increase with progressing tumor stage. Tanemura et al demonstrated that RASSF1A was not methylated in stage I and II melanoma, however aberrant methylation was present in almost 50% of stage IV melanoma, indicating that RASSF1A might play a role as marker of progression and prognosis in malignant melanoma.
Fig 3.
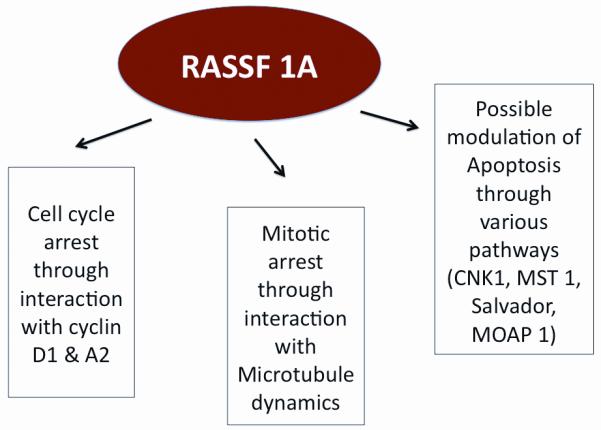
Multiple Effects of RASSF1A. Its functions as a tumor suppressor gene have shown to be secondary to cell cycle arrest in the G1 and G1/S phase through interaction with cyclin D1 & A2 and mitotic arrest by interacting with microtubule function. It is also thought that RASSF1A might play a role in the apoptosis pathway.
RAR-beta2 (Retinoic acid receptor β-2) silencing has been recently implicated in carcinogenesis as there is convincing evidence that it acts as putative tumor suppressor genes, however its exact mechanism in tumor suppression needs yet to be defined. Loss of RAR-b2 expression has been shown to be progressive in various premalignant and malignant tissues and increased expression correlates with clinical response [85, 86, 60]. Houle et al. demonstrated that lung carcinoma cells expressing transfected RAR-beta2 are less tumorigenic in nude mice [87] and transgenic mice expressing antisense RAR-beta2 develop lung cancer [88]. The precise molecular mechanisms responsible for RAR-beta2 mediated antitumor activity are not fully understood, but possibly include suppressed expression of EGFR, activating protein-1 (AP-1) and COX-2 and the phosphorylation of extracellular signal- regulated protein kinases 1 and 2 (ERK1/2) [85,89]. In malignant Melanoma the frequency of aberrant methylation and loss of expression of this protein are as high as 70% [90]. Bostick et al. demonstrated a significant correlation between hypermethylation of RAR-beta2 and increasing primary tumor Breslow thickness. Interestingly, however, the frequency of aberrant methylation seems to be similar in primary as well as in metastatic tumor [90,60] suggesting that RAR-b2 might play a role in initiation and progression of primary malignant melanoma, but not in metastasis formation.
Other genes, which have been shown to be hypermethylated in malignant melanoma, but whose role in tumorigenesis needs yet to be defined are SOCS, MGMT and TNFRSF10 receptors. Suppressor of cytokine signaling (SOCS) proteins have emerged as key physiological regulators of cytokine-mediated Jak-Stat signaling; including responses to Interferon, suggesting a role in the suppression of carcinogenesis [59]. O6-Methylguanine DNA Methyltransferase (MGMT) is a DNA repair protein involved in the reversal of DNA alkylation. Reduced MGMT expression as well as enzyme activity may result in an increased susceptibility to DNA alkylation and carcinogenesis [92]. TRAIL (TNF- related apoptosis-inducing ligand), also known as TNFRSF10, is a protein that belongs to the tumor necrosis factor (TNF) ligand family and preferentially induces apoptosis in transformed and tumor cells. TRAIL/TNFSF10 binds to at least 5 Receptors including R1 (DR4), R3 (DcR1) and R4 (DcR2). It is thought that underexpression of these receptors leads to reduced TRIAL/TNFSF10 mediated apoptosis.
Hypomethylation and gene activation in Malignant Melanoma
In spite of the increased prevalence of aberrant promotor hypermethylation in cancer, it has been shown paradoxically that tumor cells are globally hypomethylated when compared to wild-type cells [54,55]. There are suggestions that hypomethylation can contribute to carcinogenesis through activation of oncogenes and triggering of chromosome instability, as methylation of pericentromeric regions contributes to chromosome stability [10]. In malignant melanoma, hypomethylation has been shown to lead to activation of a number of Cancer testis (CT) genes, of which some examples include MAGE (Melanoma antigen genes), BAGE (B melanoma antigen), GAGE and NY-ESO-1 [56, 57, 116]. CT genes code for protein antigens with normal expression in adult testicular germ cells that can also be aberrantly activated and expressed in a range of human cancers. Since CT antigens are tumor specific and not expressed in somatic tissue, they have elicited increasing interest as a target for immunotherapy and vaccines [114, 115]. Though the biological function of CT genes in both the germ line and tumors remains poorly understood, it has been demonstrated that higher frequency of CT expression in tumors is often correlated with worse outcome and that CT antigens might play a role in tumorigenesis [114, 117]. Clinical trials to evaluate MAGE-A3 and NY-ESO-1 as therapeutic cancer vaccine targets in malignant melanoma patients have been conducted and have shown some promising results in inhibiting melanoma progression [118, 119]. A common feature of CT antigen gene expression is the induction by DNMT inhibitors like Decitabine, emphasizing hypomethylation as an important factor in activating CT gene expression [114]. Interestingly, when treating both, normal tissue and cancer cell lines with decitabine, Karpf et al. showed that MAGE-A1 was hypomethylated in normal as well as cancer cell lines, however the gene was only expressed in the cancer line suggesting that other transcriptional activators are required to express hypomethylated genes [56]. Further investigation is needed to define the role and mechanism of hypomethylation in cancer and to determine the impact of CT antigens in tumorigenesis and as target for immunotherapy.
Translational implications of aberrant DNA Methylation in melanoma
Methylation patterns could serve as diagnostic and prognostic biomarkers and be used in screening and risk assessment of cancer patients.
Use of aberrant DNA methylation marks in Diagnosis and Prognosis
De novo methylation of CpG islands occurs early in carcinogenesis and it has been demonstrated that these epigenetic changes can be detected in sputum, serum and urine samples [10]. Presumably, cancer cells release their DNA into the circulation after apoptosis or necrosis, after which it can be detected by simple blood tests. In malignant melanoma patients, Mirmohammadsadegh et al. detected aberrant methylated PTEN DNA in 62% of peripheral blood specimens [58]. Marini et al. were able to extract aberrant methylated SOCS 1 and 2, CDKN2a, RASSF1a and MGMT from peripheral sera of patients with malignant melanoma [59]. Paz et al analyzed different tumor cell lines and found that cell lines of a defined histological origin - including leukemia, melanoma, glioma, head and neck, thyroid and lymphoma cell lines - could be grouped according to their specific hypermethylation profiles by clustering these cell lines into independent terminal branches specific to their respective organ types [34]. These findings might lead to the development of molecular strategies for cancer detection and as a marker of recurrence after tumor excision. Furthermore, there is strong evidence that DNA methylation of selected genes increases quantitatively and qualitatively with tumor stages, implying that CpG Island methylator phenotypes predict progression of melanoma suggesting worse prognosis with increasing degrees of methylation [60]. Though these findings seem promising in using aberrant methylation as a parameter of diagnosis and prognosis more research is warranted to further determine the sensitivity and specificity of such testing.
Using methylation marks in Treatment and response to therapy
Current mainstay of therapy in advanced melanoma consists of chemotherapeutic agents in conjunction with immune modulators like Interferon or Interleukin-2, an approach also called biochemotherapy. Patient response to such treatment is however difficult to predict and tumor specific markers indicating the degree of response would be of tremendous clinical value [61]. Mori et al. investigated methylation patterns in responders of biochemotherapy to nonresponders and demonstrated that the frequency of circulating RASSF1A methylated DNA was significantly lower in responders than nonresponders [61]. The results imply that an increased degree of methylation does not only correlate with worse prognosis but also correlates with resistance to therapy. Consequently the use of demethylating agents has been implied in various clinical settings to improve outcome in cancer patients with aberrant methylation patterns [62, 75]. For example, decitabine is active in a variety of liquid cancers including myelodysplastic syndrome and acute myeloid leukemia (AML) [63-68], however clinical trials for solid tumors, such as melanoma, have yielded rather disappointing results when decitabine was used as a single agent [69-72, 111]. These findings are currently being re-evaluated using decitabine for potentiation of immune response or resensitization to chemotherapy. There is clinical evidence that Decitabine in combination with IL-2 might enhance activity of IL-2 in melanoma [73] or overcome resistance to Interferon induced Apoptosis [74] thus improving patient outcomes.
Conclusion and Perspective
The discovery of epigenetic changes in carcinogenesis over the last decades has opened the door to new strategies in treatment, diagnosis and prognosis in cancer. Unlike mutagenic events, epigenetic changes can be reversed and potentially restore the function of key control genes and pathways in premalignant and malignant cells. However the complexity of epigenetic events and interactions between epigenetic and mutagenic changes is not yet fully understood. Aberrant promoter methylation and loss of gene expression is found in an increasing number of genes, but for most of these genes it remains to be determined to what extent their loss of function contributes to tumorigenesis or whether their promoter methylation is just a natural process of aging. Also, there is a possibility that cells might have their own DNA-demethylating activities to protect themselves from aberrant methylation. Agius et al. were able to demonstrate that the plant Arabidopsis thaliana possesses an enzyme (glycosylase/lyase), which actively demethylates DNA [93] and it seems quite possible that human cells also might have enzymes with demethylating capacity. When used in clinical settings, demethylating agents act non-specifically on methylated DNA and can potentially re-express genes, that are intended to be silenced like proto-oncogenes, or can lead to hypomethylation which in turn causes chromosomal instability. Though such events have yet not been reported in clinical trials, there are studies showing that targeting DNMT-1 in mice is associated with an increased incidence of lymphoma [94-95]. Future investigation will certainly aim to precisely evaluate the molecular effects of demethylating agents in cancerous and normal tissue.
Fig 1.

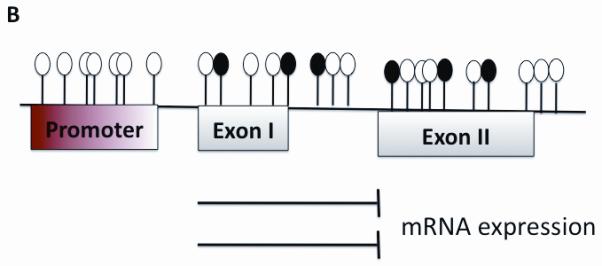
Mechanism of aberrant methylation.Transcription starts at the Promoter site and is only able to proceed when no aberrant methylation is found as seen in Part A. Part B shows multiple methylated CpGs in the promoter region. The methyl groups project into major grooves of DNA and effectively inhibit transcription and gene expression.
 unmethylated
unmethylated  methylated
methylated
Footnotes
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Cancer facts and figures 2007. American Cancer society; 2007. [Google Scholar]
- 2.Surveillance epidemiology and end results 2008. National Cancer Institute; 2008. [Google Scholar]
- 3.Houghton A, Polsky D. Focus on melanoma. Cancer Cell. 2002;2:275–278. doi: 10.1016/s1535-6108(02)00161-7. [DOI] [PubMed] [Google Scholar]
- 4.Clark WH, Jr, Elder DE, Guerry D, IV, Epstein MN, Greene MH, Van Horn M. A study of tumor progression: the precursor lesions of superficial spreading and nodular melanoma. Hum Pathol. 1984;15:1147–65. doi: 10.1016/s0046-8177(84)80310-x. [DOI] [PubMed] [Google Scholar]
- 5.Miller AJ, Mihm MC., Jr. Melanoma. N Engl J Med. 2006;355(1):51–65. doi: 10.1056/NEJMra052166. [DOI] [PubMed] [Google Scholar]
- 6.Dahl C, Guldenberg P. The Genome and Epigenome of malignant melanoma. APMIS. 2007;115:1161–1176. doi: 10.1111/j.1600-0463.2007.apm_855.xml.x. [DOI] [PubMed] [Google Scholar]
- 7.Baylin SB. DNA methylation and Gene silencing in cancer. Nature Clinical Practice Oncology. 2005;2:4–11. doi: 10.1038/ncponc0354. [DOI] [PubMed] [Google Scholar]
- 8.Jones PA, Baylin SB. The fundamental Role Of Epigenetic Events In Cancer. Nature Reviews Genetics. 2002;3:415–428. doi: 10.1038/nrg816. [DOI] [PubMed] [Google Scholar]
- 9.Rothammer T, Bosserhoff AK. Epigenetic events in malignant melanoma. Pigment Cel Res. 2007;20:92–111. doi: 10.1111/j.1600-0749.2007.00367.x. [DOI] [PubMed] [Google Scholar]
- 10.Herman JG, Baylin SB. Gene Silencing in Cancer in Association with Promoter Hypermethylation. N Engl J Med. 2003;349:2042–54. doi: 10.1056/NEJMra023075. [DOI] [PubMed] [Google Scholar]
- 12.Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16:6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- 13.Fuks F, Burgers WA, Alexander A, Hughes-Davies L Luke, Kouzarides T. DNA methyltransferase Dnmt1 associates with histonedeacetylase activity. Nature Genetics. 2000;24:88–91. doi: 10.1038/71750. [DOI] [PubMed] [Google Scholar]
- 14.Robertson KD, Ait-Si-Ali S, Yokochi T, Wade PA, Jones PL, Wolffe AP. DNMT1 forms a complex with Rb, E2F1 and HDAC1 and represses transcription from E2F-responsive promoters. Nature Genetics. 2000;25:338–342. doi: 10.1038/77124. [DOI] [PubMed] [Google Scholar]
- 15.Rountree MR, Bachman KE, Baylin SB. DNMT1 binds HDAC2 and a new co-repressor, DMAP1, to form a complex at replication foci. Nature Genetics. 2000;25:269–277. doi: 10.1038/77023. [DOI] [PubMed] [Google Scholar]
- 16.Fuks F, Burgers WA, Godin N, Kasai M, Kouzarides T. Dnmt3a binds deacetylases an is recruited by a sequence-specific repressor to silence transcription. The EMBO Journal. 2001;20(10):2536–2544. doi: 10.1093/emboj/20.10.2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liang G, Gonzales FA, Jones PA, Orntoft TF, Thykjaer T. Analysis of Gene Induction in Human Fibroblasts and Bladder Cancer Cells. Cancer Res. 2002;62(4):961–6. [PubMed] [Google Scholar]
- 18.Zardo G, Tiirikainen MI, Hong C, Misra A, Feuerstein BG, Volik S, et al. Integrated genomic and epigenomic analyses pinpoint biallelic gene inactivation in tumors. Nat Genet. 2002;32:453–8. doi: 10.1038/ng1007. [DOI] [PubMed] [Google Scholar]
- 19.Costello JF, Frühwald MC, Smiraglia DJ, Rush LJ, Robertson GP, Gao X, et al. Aberrant CpG-island methylation has non-random and tumor-type-specific patterns. Nat Genet. 2000;24:132–138. doi: 10.1038/72785. [DOI] [PubMed] [Google Scholar]
- 20.Muthusamy V, Duraisamy S, Bradbury M, Hobbs C, Curley DP, Nelson B, et al. Epigenetic Silencing of Novel Tumor Suppressors in Malignant Melanoma. Cancer Res. 2006;66(23):11187–11193. doi: 10.1158/0008-5472.CAN-06-1274. 21. [DOI] [PubMed] [Google Scholar]
- 21.Yi C, McCarty JH, Troutman SA, Eckman MS, Bronson RT, Kissil JL. Loss of the Putative Tumor Suppressor Band 4.1B/Dal1 Gene Is Dispensable for Normal Development and Does Not Predispose to Cancer. Molecular and Cellular Biology. 2005;25(22):10052–10059. doi: 10.1128/MCB.25.22.10052-10059.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thompson JF, Scolyer RA, Kefford RF. Cutaneous melanoma. Lancet. 2005;365:687–701. doi: 10.1016/S0140-6736(05)17951-3. [DOI] [PubMed] [Google Scholar]
- 23.Serrano M, Lee H, Chin L, Cordon-Cardo C, Beach D, DePinho RA. Role of the INK4a locus in tumor suppression and cell mortality. Cell. 2006;85:27–37. doi: 10.1016/s0092-8674(00)81079-x. [DOI] [PubMed] [Google Scholar]
- 24.Chin L, Pomerantz J, Polsky D, Jacobson M, Cohen C, Cordon-Cardo C, et al. Cooperative effects of INK4A and ras in melanoma susceptiblilty in vivo. Genes Dev. 1997;11:2822–34. doi: 10.1101/gad.11.21.2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Herman J0, Merlo A, Mao L, Lapidus RG, Issa JP, Davidson NE, et al. Inactivation of the CDKN2/p16/MTSJ gene is frequently associated with aberrant DNA methylation in all common human cancers. Cancer Res. 1995;55:4525–4530. [PubMed] [Google Scholar]
- 26.Walker GJ, Flores JF, Glendening JM, Lin AH, Markl ID, Fountain JW. Virtually 100% of melanoma cell lines harbor alterations at the DNA level within CDKN2A, CDKN2B, or one of their downstream targets. Genes Chromosomes Cancer. 1998;22:157–63. doi: 10.1002/(sici)1098-2264(199806)22:2<157::aid-gcc11>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 27.Pomerantz J, Schreiber-Agus N, Liégeois NJ, Silverman A, Alland L, Chin L, et al. The Ink4a tumor suppressor gene product, P19arf, interacts with MDM2 and neutralizes MDM2’s inhibition of p53. Cell. 1998;92:713–23. doi: 10.1016/s0092-8674(00)81400-2. [DOI] [PubMed] [Google Scholar]
- 28.Harris SL, Levine AJ. The p53 pathway: positive and negative feedback loops. Oncogene. 2005;24:2899–908. doi: 10.1038/sj.onc.1208615. [DOI] [PubMed] [Google Scholar]
- 29.Herman JG, Umar A, Polyak K, Graff JR, Ahuja N, Issa JP, et al. Incidence and functional consequences of hMLH1 promoter hypermethylation in colorectal carcinoma. Proc Natl Acad Sci USA. 1998;95:6870–6875. doi: 10.1073/pnas.95.12.6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kane MF, Loda M, Gaida GM, Lipman J, Mishra R, Goldman H, et al. Methylation of the hMLH1 promoter correlates with lack of expression of hMLH1 in sporadic colon tumors and mismatch repair-defective human tumor cell lines. Cancer Res. 1997;57:808–811. [PubMed] [Google Scholar]
- 31.Nakagawa H, Nuovo GJ, Zervos EE, Martin EW, Jr, Salovaara R, Aaltonen LA, et al. Age-related hypermethylation of the 5′ region of MLH1 in normal colonic mucosa is associated with microsatellite-unstable colorectal cancer development. Cancer Res. 2001;62:6991–6995. [PubMed] [Google Scholar]
- 32.Coulondre C, Miller JH, Farabaugh PJ, Gilbert W. Molecular basis of base substitution hotspots in Escherichia coli. Nature. 1978;274:775–780. doi: 10.1038/274775a0. [DOI] [PubMed] [Google Scholar]
- 33.Pfeifer GP, Tang M, Denissenko MF. Mutation hotspots and DNA methylation. Curr Tops Microbiol and Immunol. 2000;249:1–19. doi: 10.1007/978-3-642-59696-4_1. [DOI] [PubMed] [Google Scholar]
- 34.Paz MF, Fraga MF, Avila S, Guo M, Pollan M, Herman JG, et al. A systematic Profile of DNA Methylation in Human Cancer Cell Lines. Cancer Res. 2003;63:1114–1121. [PubMed] [Google Scholar]
- 35.Nelson-Rees WA, Daniels DW, Flandermeyer RR. Cross-contamination of cells in culture. Science. 1981;212(4493):446–52. doi: 10.1126/science.6451928. [DOI] [PubMed] [Google Scholar]
- 36.Soengas MS, Alarcon RM, Yoshida H, Giacca AJ, Hakem R, Mak TW, et al. Apaf-1 and caspase-9 inp53-dependent apoptosis and tumor inhibition. Science. 1999;284(5411):156–159. doi: 10.1126/science.284.5411.156. [DOI] [PubMed] [Google Scholar]
- 37.Soengas MS, Capodieci P, Polsky D, Mora J, Esteller M, Opitz-Araya X, et al. Inactivation of the apoptosis effector Apaf-1 in malignant melanoma. Nature. 2001;409:207–211. doi: 10.1038/35051606. [DOI] [PubMed] [Google Scholar]
- 38.Jones AJ. Death & Methylation. Nature. 2001;409:141–144. doi: 10.1038/35051677. [DOI] [PubMed] [Google Scholar]
- 39.Mustika R, Budiyanto A, Nishigori C, Ichihashi M, Ueda M. Decreased expression of Apaf-1 with progression of melanoma. Pigment Cell Res. 2004;18:59–62. doi: 10.1111/j.1600-0749.2004.00205.x. [DOI] [PubMed] [Google Scholar]
- 40.Niedojadło K, Łabedzka K, Łada E, Milewska A, Chwirot BW. Apaf-1 expression in human cutaneous melanoma progression and in pigmented nevi. Pigment Cell Res. 2005;19:43–50. doi: 10.1111/j.1600-0749.2005.00280.x. [DOI] [PubMed] [Google Scholar]
- 41.Stahl JM, Cheung M, Sharma A, Trivedi NR, Shanmugam S, Robertson GP. Loss of PTEN promotes tumor development in malignant melanoma. Cancer Res. 2003;63:2881–2890. [PubMed] [Google Scholar]
- 42.Stahl JM, Sharma A, Cheung M, Zimmerman M, Cheng JQ, Bosenberg MW, et al. Deregulated Akt3 activity promotes development of malignant melanoma. Cancer Res. 2004;64:7002–10. doi: 10.1158/0008-5472.CAN-04-1399. [DOI] [PubMed] [Google Scholar]
- 43.Bestor TH. The DNA methyltransferases of mammals. Hum Mol Genet. 2000;9:6870–6875. doi: 10.1093/hmg/9.16.2395. [DOI] [PubMed] [Google Scholar]
- 44.Robertson KD. DNA methylation, methyltransferases and cancer. Oncogene. 2001;20:3139–3155. doi: 10.1038/sj.onc.1204341. [DOI] [PubMed] [Google Scholar]
- 45.Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases DNMT3a and DNMT3b are essential for de novo methylation and mammalian development. Cell. 1999;9:247–257. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- 46.Baylin SB, Herman JG. DNA hypermethylation in tumorigenesis: epigenetics joins genetics. Trens Genet. 2000;16:168–174. doi: 10.1016/s0168-9525(99)01971-x. [DOI] [PubMed] [Google Scholar]
- 47.Goffin J, Eisenhauer E. DNA methyltransferase inhibitors – state of the art. Annals of Oncology. 2002;13:1699–1716. doi: 10.1093/annonc/mdf314. [DOI] [PubMed] [Google Scholar]
- 48.Rhee I, Jair KW, Yen RW, Lengauer C, Herman JG, Kinzler KW. CpG methylation is maintained in human cancer cells lacking DNMT1. Nature. 2000;404:1003–1007. doi: 10.1038/35010000. [DOI] [PubMed] [Google Scholar]
- 49.Lübbert M. DNA methylation inhibitors in the treatment of leukemias, myelodysplastic syndromes and hemoglobinopathies: clinical results and possible mechanisms of action. Curr Top Microbiol Immunol. 2000;249:135–164. doi: 10.1007/978-3-642-59696-4_9. [DOI] [PubMed] [Google Scholar]
- 50.Jüttermann R, Li E, Jaenisch R. Toxicity of 5-aza-2′-deosycytidine to mammalian cells is mediated primarily by covalent trapping of DNA methyltransferase rather than DNA demethylation. Proc Natl Acad Sci USA. 1994;91(25):11797–11801. doi: 10.1073/pnas.91.25.11797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Santi DV, Garrett CE, Barr PJ. On the mechanism of inhibition of DNA-cytosine methyltransferases by cytosine analogs. Cell. 1983;3:9–10. doi: 10.1016/0092-8674(83)90327-6. [DOI] [PubMed] [Google Scholar]
- 52.Bouchard J, Momparler RL. Incorporation of 5-aza-2′-deoxycytidine-5′-triphosphate into DNA. Interactions with mammalian DNA polymerase alpha and DNA methylase. Mol Pharmacol. 1983;24:109–114. [PubMed] [Google Scholar]
- 53.Santini V, Kantarjian HM, Issa JP. Changes in DNA methylation in neoplasia: pathophysiology and therapeutic implications. Ann Intern Med. 2001;134:573–586. doi: 10.7326/0003-4819-134-7-200104030-00011. [DOI] [PubMed] [Google Scholar]
- 54.Feinberg AP, Tycko B. The history of cancer epigenetics. Nature Reviews Cancer. 2004;4:143–153. doi: 10.1038/nrc1279. [DOI] [PubMed] [Google Scholar]
- 55.Feinberg AP, Vogelstein B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature. 1983;301:89–92. doi: 10.1038/301089a0. [DOI] [PubMed] [Google Scholar]
- 56.Karpf AR, Lasek AW, Ririe TO, Hanks AN, Grossman D, Jones DA. Limited Gene activation in tumor and normal epithelial cells treated with the DNA Methyltransferase Inhibitor 5-Aza-2′-deoxycytidine. Mol Pharmacol. 2004;65:18–27. doi: 10.1124/mol.65.1.18. [DOI] [PubMed] [Google Scholar]
- 57.DeSmet C, Loriot A, Boon T. Promoter-dependent mechanism leading to selective hypomethylation within the 5′ region of gene MAGE-A1 in tumor cells. Molecular and cellular Biology. 2004:4781–4790. doi: 10.1128/MCB.24.11.4781-4790.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mirmohammadsadegh A, Marini A, Nambiar S, Hassan M, Tannapfel A, Ruzicka T, Hengge UR. Epigenetic Silencing of the PTENGene in Melanoma. Cancer Res. 2006;66(13):6546–6552. doi: 10.1158/0008-5472.CAN-06-0384. [DOI] [PubMed] [Google Scholar]
- 59.Marini A, Mirmohammadsadegh A, Nambiar N, Gustrau A, Ruzicka T, Hengge UR. Epigenetic Inactivation of Tumor Suppressor Genesin Serum of Patients with Cutaneous Melanoma. Journal of Investigative Dermatology. 2006;126:422–431. doi: 10.1038/sj.jid.5700073. [DOI] [PubMed] [Google Scholar]
- 60.Tanemura A, Terando A, Sim MS, van Hoesel AQ, de Maat MF, Morton DL, et al. CpG Island Methylator Phenotype Predicts Progression of Malignant Melanoma. Clin Cancer Res. 2009;15(5):1801–1807. doi: 10.1158/1078-0432.CCR-08-1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mori T, O’Day SJ, Umetani N, Martinez SR, Kitago M, Koyanagi K, et al. Predicitve utility of Circulating Methylated DNA in Serum of Melanoma Patients Receiving Biochemotherapy. Journal of Clinical Oncology. 2005;23(36):9351–9358. doi: 10.1200/JCO.2005.02.9876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Halaban R, Krauthammer M, Pelizzola M, Cheng E, Kovacs D, Sznol M, et al. Integrative Analysis of Epigenetic Modulation in Melanoma Cell Response to Decitabine: Clinical Implications. PLoS ONE. 2009;4(2):e4563. doi: 10.1371/journal.pone.0004563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Momparler RL, Rivard GE, Gyger M. Clinical trial on 5-aza-2′-deoxycytidine in patients with acute leukemia. Pharmacol Ther. 1985;30:277–286. doi: 10.1016/0163-7258(85)90052-x. [DOI] [PubMed] [Google Scholar]
- 64.Rivard GE, Momparler RL, Demers J, Benoit P, Raymond R, Lin K, et al. Phase I study on 5-aza-2′-deoxycytidine in children with acute leukemia. Leuk Res. 1981;5:453–462. doi: 10.1016/0145-2126(81)90116-8. [DOI] [PubMed] [Google Scholar]
- 65.Wijermans P, Lubbert M, Verhoef G, Bosly A, Ravoet C, Andre M, et al. Low-dose 5-aza-2′-deoxycytidine, a DNA hypomethylating agent, for the treatment of high-risk myelodysplastic syndrome: a multicenter phase II study in elderly patients. J Clin Oncol. 2000;18:956–962. doi: 10.1200/JCO.2000.18.5.956. [DOI] [PubMed] [Google Scholar]
- 66.Pinto A, Zagonel V. 5-Aza-2′-deoxycytidine (decitabine) and 5-azacytidine in the treatment of acute myeloid leukemias and myelodysplastic syndromes: past, present and future trends. Leukemia. 1993;7(suppl 1):51–60. [PubMed] [Google Scholar]
- 67.Furuta J, Umebayashi Y, Miyamoto K, Kikuchi K, Otsuka F, Sugimura T, et al. Promoter methylation profiling of 30 genes in human malignant melanoma. Cancer Sci. 2004;95(12):962–8. doi: 10.1111/j.1349-7006.2004.tb03184.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Furuta J, Nobeyama Y, Umebayashi Y, Otsuka F, Kikuchi K, Ushijima T. Silencing of Peroxiredoxin 2 and aberrant methylation of 33 CpG islands in putative promoter regions in human malignant melanomas. Cancer Res. 2006;66(12):6080–6. doi: 10.1158/0008-5472.CAN-06-0157. [DOI] [PubMed] [Google Scholar]
- 69.Aparacio A, Eads CA, Leong LA, Laird PW, Newman EM, Synold TW, et al. Phase I trial of continuous infusion of 5-aza-2-deoxycytidine. Cancer Chemother Pharmacol. 2003;51:231–239. doi: 10.1007/s00280-002-0563-y. [DOI] [PubMed] [Google Scholar]
- 70.Momparler RL, Bouffard DY, Momparler LF, Dionne J, Belanger K, Ayoub J. Pilotphase I–II study on 5-aza-2′-deoxycytidine (decitabine) inpatients with metastatic lung cancer. Anticancer Drugs. 1997;8:2358–2368. doi: 10.1097/00001813-199704000-00008. [DOI] [PubMed] [Google Scholar]
- 71.Schwartsmann G, Schunemann H, Gorini CN, Filho AF, Gabrino C, Sabini G, et al. A phase I trial of cisplatin plus decitabine, a new DNA-hypomethylating agent, in patients with advanced solid tumors and a follow-up early phase II evaluation in patients within operable non-small cell lung cancer. Invest New Drugs. 2000;18:83–91. doi: 10.1023/a:1006388031954. [DOI] [PubMed] [Google Scholar]
- 72.Abele R, Clavel M, Dodion P, Bruntsch U, Gundersen S, Smyth J, et al. The EORTC Early Clinical Trials Cooperative Group experience with 5-aza-2′-deoxycytidine (NSC 127716) in patients with colo-rectal, head and neck, renal carcinomas and malignant melanomas. Eur J Cancer Clin Oncol. 1987;23:1921–1924. doi: 10.1016/0277-5379(87)90060-5. [DOI] [PubMed] [Google Scholar]
- 73.Gollob JA, Sciambi CJ, Peterson BL, Richmond T, Thoreson M, Moran K, et al. Phase I Trial of Sequential Low-Dose 5-Asa-2′Deoxycytidine Plus High-Dose Intravenous Bolus Interleukin-2 in Patients with Melanoma or Renal Cell Carcinoma. Clin Cancer Res. 2006;12(15):4619–4627. doi: 10.1158/1078-0432.CCR-06-0883. [DOI] [PubMed] [Google Scholar]
- 74.Reu FJ, Bae SI, Cherkassky L, Leaman DW, Lindner D, Beaulieu N, et al. Overcoming Resistance to Interferon-Induced Apoptosis of Renal Carcinoma and Melanoma Cells by DNA Demethylation. J Clin Oncol. 2006;24:3771–3779. doi: 10.1200/JCO.2005.03.4074. [DOI] [PubMed] [Google Scholar]
- 75.Jabbour E, Issa JP, Garcia-Manero G, Kantarjian H. Evolution of Decitabine Development. Accomplishments, ongoing investigations, and Future Strategies. Cancer. 2008;112(11):2341–2351. doi: 10.1002/cncr.23463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dammann R, Li C, Yoon J-H, Chin PL, Bates S, Pfeifer GP. Epigenetic inactivation of a RAS association domain family protein from lung tumour suppressor locus 3p21.3. Nat Genet. 2000;25:315–319. doi: 10.1038/77083. [DOI] [PubMed] [Google Scholar]
- 77.Donninger H, Vos MD, Clark GJ. The RASSF1A tumor suppressor. Journal of Cell Science. 2007;120:3163–3172. doi: 10.1242/jcs.010389. [DOI] [PubMed] [Google Scholar]
- 78.Agathanggelou A, Honorio S, Macartney DP, Martinez A, Dallol A, Rader J, et al. Methylation associated inactivation of RASSF1A from region 3p21.3 in lung, breast and ovarian tumors. Oncogene. 2001;20:1509–1518. doi: 10.1038/sj.onc.1204175. [DOI] [PubMed] [Google Scholar]
- 79.Chan MW, Chan LW, Tang NL, Lo KW, Tong JH, Chan AW, et al. Frequent hypermethylation of promoter region of RASSF1A in tumor tissues and voided urine of urinary bladder cancer patients. Int J Cancer. 2003;104:611–616. doi: 10.1002/ijc.10971. [DOI] [PubMed] [Google Scholar]
- 80.Tomizawa Y, Kohno T, Kondo H, Otsuka A, Nishioka M, Niki T, et al. Clinicopathological significance of epigenetic inactivation of RASSF1A at 3p21.3 in stage I lung adenocarcinoma. Clin Cacer Res. 2002;8:2362–2368. [PubMed] [Google Scholar]
- 81.Yu MY, Tong JH, Chan PK, Lee TL, Chan MW, Chan AW, et al. Hypermethylation of the tumor suppressor gene RASSF1A and frequent concomitatnt loss of heterozygosity at 3p21 in cervical cancers. Int J Cancer. 2003;105:204–205. doi: 10.1002/ijc.11051. [DOI] [PubMed] [Google Scholar]
- 82.Tommasi S, Damman R, Zhang Z, Wang Y, Liu L, Tsark WM, et al. Tumor susceptibility of RASSF1A knockout mice. Cancer Res. 2005;65:92–98. [PubMed] [Google Scholar]
- 83.Van der Weyden L, Tachibana KK, Gonzalez MA, Adams DJ, Ng BL, Petty R, et al. The RASSF1A isoform of RASSF1 promotes microtubule stability and suppresses tumorigenesis. Mol Cell Biol. 2005;25:8356–8367. doi: 10.1128/MCB.25.18.8356-8367.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Katz ME, Mc Cornick F. Signal transduction from multiple Ras effectors. Curr Opin Genet Dev. 1997;7:75–79. doi: 10.1016/s0959-437x(97)80112-8. [DOI] [PubMed] [Google Scholar]
- 85.Xu XC. Tumor-suppressve activity of retinoic acid receptor- beta in cancer. Cancer Lett. 2007;253(1):14–24. doi: 10.1016/j.canlet.2006.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lotan R, Xu XC, Lippman SM, Ro JY, Lee JS, Lee JJ, et al. Suppression of retinoic acid receptor-beta in premalignant oral lesions and its up-regulation by isotretinoin. N Engl J Med. 1995;332:1405–1410. doi: 10.1056/NEJM199505253322103. [DOI] [PubMed] [Google Scholar]
- 87.Houle B, Rochette-Egly C, Bradley WE. Tumor-suppressive effect of the retinoic acid receptor beta in human epidermoid lung cancer cells. Proc Natl Acad Sci USA. 1993;90:985–989. doi: 10.1073/pnas.90.3.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Berard J, Laboune F, Mukuna M, Masse S, Kothoary R, Bradley WE. Lung tumors in mice expressing an antisense RARbeta2 transgene. FASEB J. 1996;10:1092–1097. doi: 10.1096/fasebj.10.9.8801172. [DOI] [PubMed] [Google Scholar]
- 89.Song S, Lippman SM, Zou Y, Ye X, Xu X-C. Induction of cyclooxygenase-2 by benzo[a]pyrenediolepoxide through inhibition of retinoic acid receptor-beta2 expression. Oncogene. 2005;24:8268–8276. doi: 10.1038/sj.onc.1208992. [DOI] [PubMed] [Google Scholar]
- 90.Hoon DS, Spugnardi M, Kuo C, Huang SK, Morton DL, Taback B. Profiling epigenetic inactivation of tumor suppressor genes in tumors and plasma from cutaneous melanoma patients. Oncogene. 2004;23(22):4014–22. doi: 10.1038/sj.onc.1207505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bae SI, Cheriyath V, Jacobs BS, Reu F, Borden EC. Reversal of methylation silencing of Apo2L/TRAIL receptor 1(DR4) expression overcomes resistance of SK-MEL-3 and SK-MEL-28. Oncogene. 2008;27:490–498. doi: 10.1038/sj.onc.1210655. [DOI] [PubMed] [Google Scholar]
- 92.Kohonen-Corish MR, Cooper WA, Saab J, Thompson JF, Trent RJ, Millward MJ. Promoter hypermethylation of the O(6)-methylguanine DNA methyltransferase gene and microsatellite instability in metastatic melanoma. J Invest Dermatol. 2006;126(1):167–71. doi: 10.1038/sj.jid.5700005. [DOI] [PubMed] [Google Scholar]
- 93.Agius F, Kappor A, Zhu JK. Role of the Arabidopsis DNA glycosylase/lyase ROS1 in active DNA demethylation. Pros Natl Acad Sci USA. 2006;103:11796–11801. doi: 10.1073/pnas.0603563103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Eden A, Gaudet F, Waghmare A, Jaenisch F. Chromosomal instability and tumors promoted by DNA hypomethylation. Science. 2003;300(5618):455. doi: 10.1126/science.1083557. [DOI] [PubMed] [Google Scholar]
- 95.Trinh BN, Long TI, Nickel AE, Shibta D, Laird PW. DNA methyltransferase deficiency modifies cancer susceptibility in mice lacking DNA mismatch repair. Mol Cell Ciol. 2002;22:2906–2917. doi: 10.1128/MCB.22.9.2906-2917.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Worm J, Christensen C, Grønbaek K, Tulchinsky E, Guldberg P. Genetic and epigenetic alterations of the APC gene in malignant melanoma. Oncogene. 2004;23(30):5215–26. doi: 10.1038/sj.onc.1207647. [DOI] [PubMed] [Google Scholar]
- 97.Guan X, Sagara J, Yokoyama T, Koganehira Y, Oguchi M, Saida T, et al. ASC/TMS1, a caspase-1 activating adaptor, is downregulated by aberrant methylation in human melanoma. Int J Cancer. 2003;107(2):202–8. doi: 10.1002/ijc.11376. [DOI] [PubMed] [Google Scholar]
- 98.Fulda F, Kufer MU, Meyer E, van Valen F. Dockhorn-Dworniczak B and Debatin KM. Sensitization for death receptor or drug induced apoptosis by re expression of caspase-8 through demethylation or gene transfer. Oncogene. 2001;20:5865–5877. doi: 10.1038/sj.onc.1204750. [DOI] [PubMed] [Google Scholar]
- 99.Tsutsumida A, Hamada J, Tada M, Aoyama T, Furuuchi K, Kawai Y, et al. Epigenetic silencing of E- and P-cadherin gene expression in human melanoma cell lines. Int J Oncol. 2004;25(5):1415–21. [PubMed] [Google Scholar]
- 100.Luo W, Wang X, Kageshita T, Wakasugi S, Karpf AR, Ferrone S. Regulation of high molecular weight-melanoma associated antigen (HMW-MAA) gene expression by promoter DNA methylation inhuman melanoma cells. Oncogene. 2006;25:2873–2884. doi: 10.1038/sj.onc.1209319. [DOI] [PubMed] [Google Scholar]
- 101.Liu S, Ren S, Howell P, Fodstad O, Riker AI. Identification of novel epigenetically modified genes in human melanoma via promoter methylation gene profiling. Pigment Cell Melanoma Res. 2008;21(5):545–58. doi: 10.1111/j.1755-148X.2008.00484.x. [DOI] [PubMed] [Google Scholar]
- 102.Denk AE, Bettstetter M, Wild PJ, Hoek K, Bataille F, Dietmaier W, Bosserhoff AK. Loss of maspin expression contributes to a more invasive potential in malignant melanoma. Pigment Cell Res. 2007;20(2):112–9. doi: 10.1111/j.1600-0749.2007.00363.x. [DOI] [PubMed] [Google Scholar]
- 103.Behrmann I, Wallner S, Komyod W, Heinrich PC, Schuierer M, Buettner R, Bosserhoff AK. Characterization of methyl thio adenosin phosphorylase (MTAP) expression in malignant melanoma. Am J Pathol. 2003;163(2):683–90. doi: 10.1016/S0002-9440(10)63695-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Freedberg DE, Rigas SH, Russak J, Gai W, Kaplow M, Osman I, Turner F, Randerson-Moor JA, Houghton A, Busam K, Bishop D Timothy, Bastian BC, Newton-Bishop JA, Polsky D. Frequent p16-independent inactivation of p14ARF in human melanoma. J Natl Cancer Inst. 2008;100(11):784–95. doi: 10.1093/jnci/djn157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Gonzalgo ML, Bender CM, E H, Glendening JM, Flores JF, Walker GJ, Hayward NK, Jones PA, Fountain JW. Low Frequency of pl6/CDKN2A Methylation in Sporadic Melanoma: Comparative Approaches for Methylation Analysis of Primary Tumors. Cancer Research. 1997;57:5336–5347. [PubMed] [Google Scholar]
- 106.van der Velden PA, Zuidervaart W, Hurks MH, Pavey S, Ksander BR, Krijgsman E, Frants RR, Tensen CP, Willemze R, Jager MJ, Gruis NA. Expression profiling reveals that methylation of TIMP3 is involved in uveal melanoma development. Int J Cancer. 2003;106(4):472–9. doi: 10.1002/ijc.11262. [DOI] [PubMed] [Google Scholar]
- 107.Spugnardi M, Tommasi S, Dammann R, Pfeifer GP, Hoon DS. Epigenetic inactivation of RAS association domain family protein 1 (RASSF1A) in malignant cutaneous melanoma. Cancer Res. 2003;63(7):1639–43. [PubMed] [Google Scholar]
- 108.Nobeyama Y, Okochi-Takada E, Furuta J, Miyagi Y, Kikuchi K, Yamamoto A, et al. Silencing of tissue factor pathway inhibitor-2 gene in malignant melanomas. Int J Cancer. 2007;121(2):301–7. doi: 10.1002/ijc.22637. [DOI] [PubMed] [Google Scholar]
- 109.Gallagher WM, Bergin OE, Rafferty M, Kelly ZD, Nolan IM, Fox EJ, et al. Multiple markers for melanoma progression regulated by DNA methylation: insights from transcriptomic studies. Carcinogenesis. 2005;26(11):1856–67. doi: 10.1093/carcin/bgi152. [DOI] [PubMed] [Google Scholar]
- 110.van der Velden PA, Metzelaar-Blok JA, Bergman W, Monique H, Hurks H, Frants RR, et al. Promoter hypermethylation: a common cause of reduced p16(INK4a) expression in uveal melanoma. Cancer Res. 2001;61(13):5303–6. [PubMed] [Google Scholar]
- 111.Samlowski WE, Leachman SA, Wade M, Cassidy P, Porter-Gill P, Busby L, et al. Evaluation ofa 7-day continuous intravenous infusion of decitabine: inhibition of promoter-specific and global genomic DNAmethylation. J Clin Oncol. 2005;23:3897–3905. doi: 10.1200/JCO.2005.06.118. [DOI] [PubMed] [Google Scholar]
- 112.Saxonov S, Berg P, Brutlag DL. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc Natl Acad Sci U S A. 2006 Jan 31;103(5):1412–7. doi: 10.1073/pnas.0510310103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Deng T, Kuang Y, Wang L, Li J, Wang Z, Fei J. An essential role for DNA methyltransferase 3a in melanoma tumorigenesis. Biochem Biophys Res Commun. 2009;387(3):611–6. doi: 10.1016/j.bbrc.2009.07.093. 25. [DOI] [PubMed] [Google Scholar]
- 114.Caballero OL, Chen YT. Cancer/testis (CT) antigens: potential targets for immunotherapy. Cancer Sci. 2009;100(11):2014–21. doi: 10.1111/j.1349-7006.2009.01303.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Simpson AJ, Caballero OL, Jungbluth A, Chen YT, Old LJ. Cancer/testis antigens, gametogenesis and cancer. Nat Rev Cancer. 2005;5:615–25. doi: 10.1038/nrc1669. [DOI] [PubMed] [Google Scholar]
- 116.Zendman AJ, de Wit NJ, van Kraats AA, Weidle UH, Ruiter DJ, van Muijen GN. Expression profile of genes coding for melanoma differentiation antigens and cancer/testis antigens in metastatic lesions of human cutaneous melanoma. Melanoma Res. 2001;11(5):451–9. doi: 10.1097/00008390-200110000-00003. [DOI] [PubMed] [Google Scholar]
- 117.Brasseur F, Rimoldi D, Lienard D, et al. Expression of MAGE genes in primary and metastatic cutaneous melanoma. Int J Cancer. 1995;63:375–80. doi: 10.1002/ijc.2910630313. [DOI] [PubMed] [Google Scholar]
- 118.Lehmann FLJ, Gaulis S, Gruselle O, Brichard V. Clinical Response to the MAGE-A3 Immunotherapeutic in Metastatic Melanoma Patients is associated with a Specific Gene Profile Present Prior to Treatment. XVIth meeting in the Cancer Research Institute International Cancer Immunotherapy Symposium Series and the 2008 Meeting of the Cancer Vaccine Consortium; New York City. 2008.p. S25. [Google Scholar]
- 119.Davis ID, Chen W, Jackson H, et al. Recombinant NY-ESO-1 protein with ISCOMATRIX adjuvant induces broad integrated antibody and CD4(+) and CD8(+) T cell responses in humans. Proc Natl Acad Sci U S A. 2004;101:10697–702. doi: 10.1073/pnas.0403572101. [DOI] [PMC free article] [PubMed] [Google Scholar]