

Abstract
Reactive oxygen species (ROS) are critical in a broad spectrum of cellular processes including signaling, tumor progression, and innate immunity. The essential nature of ROS signaling in the immune systems of Drosophila and zebrafish has been demonstrated; however, the role of ROS, if any, in mammalian adaptive immune system development and function remains unknown. The current work provides the first clear demonstration that thymus specific elevation of mitochondrial superoxide (O2·−) disrupts normal T-cell development to impair function of the mammalian adaptive immune system. To assess the effect of elevated mitochondrial superoxide in the developing thymus, we used a T-cell specific knockout of manganese superoxide dismutase (i.e. SOD2) and have thus established a murine model to examine the role of mitochondrial superoxide in T-cell development. Conditional loss of SOD2 led to increased superoxide, apoptosis, and developmental defects in the T-cell population resulting in immunodeficiency and susceptibility to influenza A virus (IAV), H1N1. This phenotype was rescued with mitochondrially targeted superoxide scavenging drugs. These new findings demonstrate that loss of regulated levels of mitochondrial superoxide lead to aberrant T-cell development and function, and further suggest that manipulations of mitochondrial superoxide levels may significantly alter clinical outcomes resulting from viral infection.
Keywords: Superoxide, Manganese Superoxide Dismutase, Cre/loxP, Transgenic, Mouse, Adaptive Immunity, Developmental Biology, Influenza, Anti-oxidants, Apoptosis, Immunodeficiency
Introduction
The term reactive oxygen species (ROS) describes a large group of free radical and non-free radical oxygen containing compounds (e.g. superoxide, O2·−; hydrogen peroxide, H2O2; peroxynitrite, ONOO−; hydroxyl radical, ·OH, etc) [1]. It is commonly accepted that ROS are byproducts of normal metabolism, and as such act to damage cellular components such as nucleic acid, proteins, or lipid [2–4]. Due to this, ROS have been implicated in many different diseases such as cancer, atherosclerosis, amyotrophic lateral sclerosis, Alzheimer’s disease, and many others [5–8]. One specific role of ROS is their ability to enhance the pathogenesis of infections, such as influenza [9, 10]. It has been demonstrated that during times of influenza infection ROS may damage lung parenchyma cells, but that this injury may be ameliorated by anti-oxidant supplementation [11–14]. Current theories propose the mechanism behind this benefit is attenuation of ROS produced by the innate immune system, but this is not commonly accepted and is still highly debated. More recently it has been shown that cells possess the ability to exploit ROS for signaling and functional purposes. For example, many transcription factor pathways are sensitive to oxidative stress, and as such are able to help cells adapt to large deviations in redox status [15–18]. Moreover, ROS are essential in the development of certain organ systems and even whole organisms [19, 20]. With this knowledge, the importance of ROS in biology is being elucidated, but many questions about tissue specific dependence, specific ROS functions, and ROS mechanisms of action remain unanswered.
One organ system in which ROS have been widely described is that of the immune system. The biological relevance of ROS was first depicted in this system as it was found that leukocytes depended upon ROS for the oxidative burst to neutralize pathogens [21]. Other studies have demonstrated the importance of ROS in the downstream intracellular signaling post-T-cell activation [22–24]. Furthermore, evidence has shown that hydrogen peroxide acts as an important chemoattractant to direct leukocytes to wound margins at sites of injury, which was pivotal in demonstrating immune cells are able to respond to exogenous ROS in addition to producing endogenous levels [25]. Recent studies have demonstrated the role of ROS in priming the development of the primitive immune system in Drosophila, illuminating the first described function of ROS in the development of this organ system in an invertebrate species [26]. Finally, it has been demonstrated that an intracellular pro-oxidant shift occurs prior to normal thymocyte apoptosis during selection [27], and numerous groups have shown increased ROS levels in pathologic T-cell systems [28–30]. On the contrary, few studies to date have focused on how pro- or anti-oxidants affect the normal mammalian adaptive immune system, and to our knowledge no published studies have directly addressed the role of excess mitochondrial superoxide in the growth or function of the adaptive immune system. With the understanding that ROS play a major part in intracellular signaling and cellular damage, we hypothesized that mitochondrial superoxide may play a distinct role in normal development and maintenance of the mammalian adaptive immune system, which could be central to intercellular communication between the innate and adaptive branches of the immune system.
While the conceptual framework underlying this hypothesis appears simple in nature, the availability of animal models with which to address this hypothesis is limited. Numerous constitutive anti-oxidant enzyme knock-out animals have demonstrated valuable information on the global developmental importance of these proteins [31–36], but these studies have not assessed the role of specific ROS in tissue-specific adaptive immunity. To address these deficits, we used a conditional T-cell manganese superoxide dismutase (i.e. SOD2) knock-out mouse to examine the role of increased steady-state levels of superoxide during mammalian adaptive immune system development [37]. The superoxide dismutase class of enzymes specifically scavenges superoxide in biological systems [38]. Mammals contain three variants of the enzymes: cytoplasmic Cu/Zn SOD, SOD1; mitochondrial MnSOD, SOD2; and extracellular SOD, SOD3. Since SOD2 has the explicit role of eliminating mitochondrial superoxide, tissue specific disruption of this activity should provide an excellent in vivo model under conditions where compartmentalized superoxide metabolism is disrupted. A constitutive SOD2 knock-out mouse has been created, but due to the post-natal developmental dependence of SOD2 the animal succumbs to numerous organ failures shortly after birth [39, 40]. Furthermore, due to the mouse’s limited lifespan no examination of the immune system was reported. Taken together, our model serves as the first described animal model of studying the effects of perturbing steady-state mitochondrial superoxide levels on the development and function of the mammalian T-cell adaptive immune system.
Materials and Methods
Mice
Mice homozygous for the floxed SOD2 allele (i.e. B6.Cg-Sod2tm1, shorthand SOD2L/L), in which exon 3 of the SOD2 gene is flanked by 2 loxP sequences, have been previous described [37]. B6.Cg-Tg-Lck-Cre548Jxm/J or Lck-Cre mice (Cre-recombinase is exogenously expressed under control of the proximal lymphocyte specific kinase, where Cre-recombinase becomes activated in αβ T-cells during the CD4−/CD8− to CD4+/CD8+ stage in development) and B6.Cg-Tg-Vav1-iCreA2Kio/J or Vav-iCre mice (Cre recombinase is exogenously expressed under control of the vav promoter, where Cre-recombinase becomes activated within the hematopoietic stem cell and affects all lymphoid, myeloid, and erythroid lineages) were generously donated by Dr. Adam Dupuy (Department of Anatomy and Cell Biology, The University of Iowa), and have been previously described[41, 42]. B6.Cg-Tg-AlbCre21Mgn/J or Albumin-Cre mice (Cre-recombinase is exogenously expressed under control of the albumin promoter, and as such expression is limited to liver) were generously donated by Dr. Curt Sigmund and have been previously described [43]. Finally, B6.129-Tg-MMTVCre4Mam/J or MMTV-Cre mice (Cre-recombinase is exogenously expressed under the mouse mammary tumor virus promoter, where expression is primarily limited to mammary tissue) were purchased from Jackson Laboratories, and have been previously described [44]. To obtain conditional T-cell SOD2 homozygous knock-out animals (i.e. SOD2−/−), parent strains of both the floxed SOD2 and Lck-Cre mice were bred to generate F1 heterozygotes (i.e. SOD2wt/−). The F1 generation was then bred back to the parent floxed SOD2 mice to create F2 homozygous knock-outs. Lck-Cre was only passed through male parents to limit non-specific oocyte expression. Mice used were of pure C57BL/6 background, and littermate floxed animals (i.e. SOD2L/L) served as controls. All work was performed under the approval of the Institutional Animal Care and Use Committee at the University of Iowa.
Tissue Isolation
For all experiments, fresh tissue was harvested and used from animals at 6-weeks of age unless otherwise noted. For thymocyte, splenocyte, and lymph node preps, organs were freshly harvested upon necropsy and placed in a solution of Hanks Buffered Salt Solution (HBSS) containing 10% fetal bovine serum (FBS). Tissues were dissociated by physical disruption using ground glass. Following this, lymphocytes were isolated by mouse optimized FicoLite-LM gradient (Atlanta Biologicals, Atlanta, GA). Remaining red blood cell contaminants were removed by red blood cell lysis buffer, and remaining lymphocytes were washed 2X with PBS before further analysis. Peripheral blood was isolated by retro-orbital bleeding into heparin coated capillary tubes.
Real-Time PCR
RNA was extracted using the Trizol method and was quantified by the use of a Nanodrop ND-1000. 1 μg from each sample was reverse transcribed using the ABI cDNA archive kit. Generated cDNA was then subjected to SYBR green quantitative real time PCR with primers specific to each individual transcript, and the 18S control (Supplementary Primer Sequences) under the following PCR parameters: 95°C for 10 min, followed by 40 cycles of 95°C for 15 s and 60°C for 1 min. A threshold in the linear range of PCR amplification was selected and the cycle threshold (Ct) determined. Levels of transcripts were then normalized to the 18S control, and compared relative to the SOD2L/L control using the ΔΔCt method.
Western Blot Analysis
Protein was extracted using standard RIPA buffer, and was quantified on the basis of the Bradford assay and a standard curve. Protein was run on a SDS-PAGE gel for separation of different sized proteins. The products were then transferred from the gel to a nitrocellulose membrane. Identification and quantification of amount of protein was performed by addition of specific primary antibody, and then the addition of a HRP-tagged secondary antibody to the first. Chemiluminescent substrate was added to the blot for exposure of products, and film was exposed and qualitatively quantified for protein amount. Loading errors were controlled by normalizing to beta-actin (ACTβ). Antibodies used for this study: SOD2 (Millipore, Billerica, MA), Aconitase (Abcam, Cambridge, MA), SDHB (Abcam, Cambridge, MA), LC3 (Cell Signaling Technology, Beverly, MA), p53 (Abcam, Cambridge, MA), Beta-Actin (Abcam, Cambridge, MA).
SOD2 Activity
To analyze the activity of the superoxide dismutase enzymes, the indirect competitive inhibition assay developed by Spitz and Oberley was used [45]. Briefly, superoxide is generated from xanthine by xanthine oxidase and detected by recording the reduction of nitroblue tetrazolium (NBT). SOD scavenges superoxide and competitively inhibits the reduction of NBT. One unit of SOD activity is defined as the amount of protein required to inhibit 50% of the maximal NBT reduction. To obtain the amount of MnSOD activity, sodium cyanide (5 mmol/L) is added to inhibit the CuZnSOD enzyme activity.
Dihydroethidium (DHE)/Dichlorfluorescein-diacetate (DCFH-DA) Staining
Superoxide specific dihydroethidium (DHE) analysis was performed as previously described [46]. Briefly, superoxide production was estimated using the fluorescent dye DHE from Molecular Probes (Eugene, Oregon). Cells were washed once with PBS and labeled in suspension at 37°C for 45 minutes in PBS (containing 5 mmol/L pyruvate) with DHE (10 μmol/L; in 1% DMSO). Samples were analyzed using a LSR flow cytometer (Superoxide specific excitation 405 nm, emission 585 nm band-pass filter; non-specific excitation 488 nm, emission 585 nm band-pass filter). The mean fluorescence intensity (MFI) of 10,000 cells was analyzed in each sample and corrected for autofluorescence from unlabeled cells. Dichlorfluorescein-diacetate(DCFH-DA) analysis was performed as previously described [47]. Briefly, steady-state levels of pro-oxidants were determined using the peroxide oxidation-sensitive (CDCFH2, 10 μg/mL) and oxidation-insensitive (CDCF, 10 μg/mL) fluorescent dyes (dissolved in 1% DMSO) obtained from Molecular Probes. The cells were washed once with PBS and labeled with the fluorescent dyes for 15 minutes at 37°C in PBS. Cells then were then resuspended in PBS, and analyzed using a LSR flow cytometer (Excitation 488 nm, emission 530 nm band-pass filter). The MFI of 10,000 cells was analyzed in each sample and corrected for autofluorescence from unlabeled cells.
Aconitase and Succinate Dehydrogenase (SDH) Activities
Aconitase activity assay was adapted from the previously described experiment [48]. Briefly, protein from freshly isolated thymocytes was isolated by sequential freeze/thaws (3X) in a Tris/MnCl2/Citrate buffer (50 mM Tris, 600 μM MnCl2, and 5 mM NaCitrate). Protein was then quantified by the Bradford assay. Following this, 200 μg protein was combined with NADP+ (200 μM) and isocitrate dehydrogenase (10 Units). Reaction was monitored at 340 nm for the appearance of NADPH formed every 5 minutes for 2 hours. Rates were determined from slopes determined by regression analysis of data. Succinate dehydrogenase activity was adapted from the previously described experiment [49]. Briefly, protein from freshly isolated thymocytes was isolated by sequential freeze/thaws (3X) in a phosphate buffer (20 mM). Protein was then quantified by the Bradford assay. Following this, 200 μg protein was combined with succinate, non-complex II inhibitors (Antimycin A, rotenone, and cyanide), CoQ, and 2, 6-Dichloroindophenol (DCIP). Reaction was monitored at 600 nm for the colorimetric change of DCIP accepting electrons every 5 minutes for 1 hour. Rates were determined from slopes determined by regression analysis of data.
Electron Microscopy
Isolated thymocytes were fixed overnight with 2.5% glutaraldehyde1 in 0.1 M cacodylate buffer. Post-fixation was carried out for 1 hour at room temperature with a buffered 1% osmium tetroxide solution reduced with 1.5% potassium ferrocyanide. Samples were en bloc stained with 2.5% uranyl acetate. Cells were then rinsed and dehydrated using gradually increasing concentrations of acetone to 100%. Infiltration of Spurr’s epoxy resin and acetone were carried out over several days to 100% resin and cured 48 hours in a 60°C oven. Sections of 90nm thickness were cut using an Ultracut E ultramicrotome (Reichert-Jung). Grids were then counterstained with 5% uranyl acetate for 2 minutes and Reynold’s lead citrate for 2 minutes. Samples were imaged using a JEOL 1230 transmission electron microscope at 120 KV. To perform quantitative morphometry, 100 random images of both SOD2L/L and SOD2−/− thymocytes were chosen and blindly analyzed using Image J (National Institutes of Health) for following parameters: cellular area, nuclear area, cytoplasmic area, number of mitochondria, mitochondrial perimeter, mitochondrial area, and mitochondrial density.
Flow Cytometry
T-cell specific marker analysis for development or post-influenza infection was performed as previously described [50]. After isolation of fresh cell preparations, cells were suspended in staining buffer (HBSS supplemented with 5% bovine calf serum and 0.1% NaN3) and incubated with conjugated monoclonal antibodies (mAb) in the presence of normal rat serum (to limit non-specific antibody binding). After incubation and washing, cells were suspended in fixative [1% formaldehyde in 1.25x phosphate buffered saline (PBS)]. Stained cells were run on a FACSVantage SE flow cytometer (Becton Dickinson & Co., Mountain View, CA) with a minimum of 30,000 events collected per sample. Antibodies were semi-purified from HB101 serum-free supernatants by 50% ammonium sulfate precipitation and conjugated to specific fluorescent dye by using standard procedures.
Apoptosis and Cell Cycle
For apoptosis, the Annexin V-FITC Apoptosis Detection Kit (Becton Dickinson) was used to assess Annexin V positive cells. Briefly, fresh cell preparations were incubated with 1X Annexin binding buffer and Annexin V-FITC (2.5 μg/ml) conjugated primary antibody for 15 minutes on ice. Following incubation, propidium iodide (10 μg/ml) was added to suspension and cells were analyzed by flow cytometry using a LSR flow cytometer. For cell cycle, analysis was adapted from the previously described report [51]. One million cells were centrifuged and suspended in 0.5 ml of Krishan reagent (0.1% Na citrate, 0.03% NP-40, 0.05 mg/ml PI, 0.02 mg/ml RNase A) prior to analysis. Analysis was performed by examining propidium iodide-stained nuclei on a LSR flow cytometer.
Influenza and Rescue
Mice (weighing approximately 20–25g) were infected with 1600 TCIU50 of mouse-adapted A/PR/8/34 Influenza virus A H1N1 by intranasal administration. For morbidity/mortality experiments, mice were observed and weighed daily for 14 days. For pulmonary CD8+ T cell analysis, mice were sacrificed at day 8 post-infection and T-cells isolated from lung tissue. IAV PA224 (H2D(b)/SSLENFRAYV) and NP366(H2D(b)/ASNENMETM) tetramers were obtained from National Institute of Allergy and Infectious Disease MHC Tetramer Core Facility (Atlanta, GA). For rescue experiments, Tempol was obtained from Sigma Aldrich Company. Mito-CTPO was graciously donated by Dr. Balaraman Kalyanaraman. Both Tempol and Mito-CTPO were dissolved in ethanol vehicle, and administered at either 25 mM or 250 nM respectively. Water bottles were supplemented with sucrose (4 g/100 mL) to offset taste of the antioxidants. Mice were treated from time of weaning until date of experimentation.
Statistics
Data were as expressed as mean and standard deviation. All experiments were performed at least 3 mice. For most experiments, comparisons between groups were analyzed by unpaired 2-tailed Student’s t-test. For Kaplan-Meier analysis, log-rank analysis was performed. A p-value of less than 0.01 was considered to be significant.
Results
The conditional loss of SOD2 increases superoxide specific oxidative stress
Due to the lack of a sufficient model to study ROS stress on in vivo development of the mammalian adaptive immune system, we reasoned that the creation of a T-cell specific SOD2 knock-out mouse would serve as an optimal platform to study the effects of mitochondrial superoxide on immune system development and function. Floxed SOD2 mice were bred with mice expressing Cre-recombinase under the control of the proximal promoter of the lymphocyte specific kinase (Lck) to the F2 generation to create homozygous T-cell SOD2 knock-outs. PCR analysis of tail sample DNA was used to confirm the proper genotype of animals used for studies (Supplemental Fig. 1, A–B). Following this, thymocytes were removed from animals to confirm the recombination of SOD2 in a tissue specific manner. Genomic DNA extracted from T-cells showed the specific excision product in SOD2−/− thymocytes, with no apparent Cre-recombination events in SOD2L/L animals (Fig. 1A). Furthermore, SOD2 specific mRNA transcript levels were significantly decreased in knock-out animals when compared to control animals (Fig. 1B). T-cell specific SOD2−/− mice were found to have minimal SOD2 protein and activity in thymus derived T-cells (Fig. 1, C–D), whereas no significant decreases were noted in any off-target tissues (e.g. heart, liver, brain, pancreas, or muscle) in either SOD2−/− or SOD2L/L mice (data not shown). In all assays utilized a small amount of residual SOD2 was detectable, suggesting the Cre-recombination may have been incompletely penetrant, or alternatively, that our thymocyte preparations may contain non-Lck-expressing cells (i.e. γδ T-cells, immature T-cell populations, or contaminating stromal cells) and thus do not possess Cre-recombinase. In either case, taken together, these data support the T-cell specificity and efficiency of the model of SOD2 knock-out.
Figure 1.
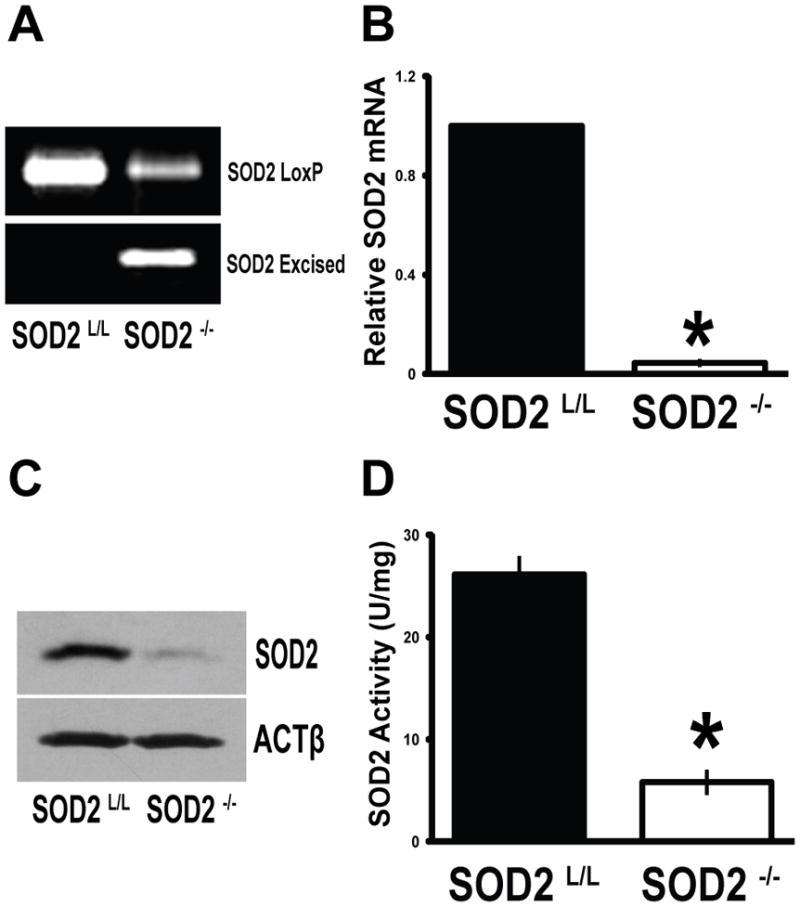
SOD2 is conditionally and effectively removed from T-cells. (A) Conventional PCR analysis of floxed or excised SOD2 products from genomic DNA extracted from 6-week old SOD2L/L or SOD2−/− thymocytes. (B) Quantitative real-time RT-PCR analysis of SOD2 mRNA extracted from 6-week old SOD2L/L or SOD2−/− thymocytes. Results shown as normalized to SOD2L/L. (C) Comparison of SOD2 protein in 6-week old SOD2L/L or SOD2−/− thymocytes. Beta-actin (ACTβ) shown as loading control. (D) Activity assay performed on protein extracted from 6-week old SOD2L/L or SOD2−/− thymocytes. At least 3 mice per experiment were analyzed; data are shown as mean and s.d. Where applicable, * = p<0.01 by Student’s t-test versus SOD2L/L.
To confirm that SOD2−/− mice did in fact harbor increased oxidative stress compared to their SOD2L/L littermates, we performed dihydroethidium (DHE) stain flow cytometry as a relative measure of superoxide content within the T-cell populations of these animals. It was observed that SOD2−/− demonstrated significantly increased steady-state levels of superoxide when compared to control SOD2L/L mice (Fig. 2A). Importantly, no change was noted in non-specific DHE oxidation or in the oxidation of a peroxide sensitive probe dihydrodichlorfluorescein-diacetate (DCFH-DA; Fig. 2B). We confirmed and extended these findings in two separate enzyme activity assays that are highly specific to disruption by superoxide (aconitase and succinate dehydrogenase), as both were shown to be significantly decreased within the SOD2−/− T-cells with no significant change in protein levels (Fig. 2, C–D). It was previously shown that reactive nitrogen species (RNS) may also inactivate these enzymes [52], but probing cellular proteins for damage by RNS showed non-detectable levels of nitrotyrosine (data not shown). Overall, it appeared that the conditional loss of SOD2 caused a specific and significant increase in steady-state levels of superoxide within the T-cell population of SOD2−/− mice, therefore these mice should serve as an effective model system for studying the effects of excess mitochondrial superoxide on the development and function of the mammalian T-cell adaptive immune system.
Figure 2.
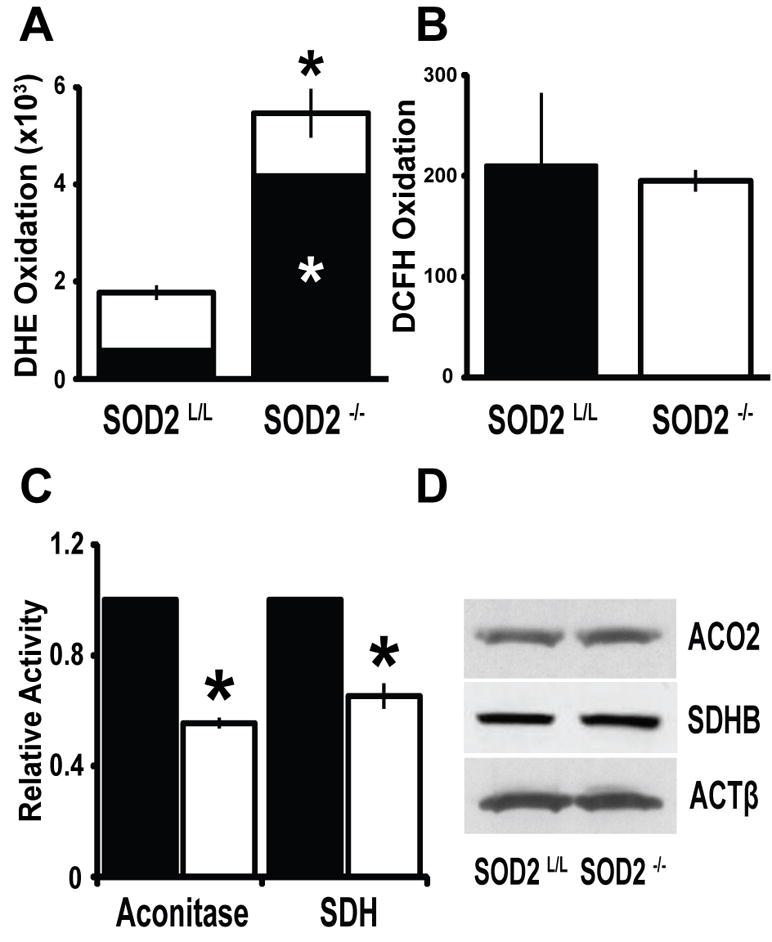
The loss of SOD2 causes superoxide specific oxidative stress in T-cells. (A) Quantification of total (entire bar) and superoxide specific (black section of bar) dihydroethidium (DHE) staining in 6-week old thymocytes. (B) The peroxide-sensitive probe dichlorofluorescein-diacetate (DCFH) demonstrated no significant change between SOD2L/L or SOD2−/− animals. (C) Quantification of total cellular aconitase and succinate dehydrogenase (SDH) activity in 6-week old thymocytes. Black bars indicate SOD2L/L and open bars indicate SOD2−/−. (D) Western blots of the iron-sulfur containing proteins aconitase 2 (ACO2) and succinate dehydrogenase B (SDHB) demonstrated no significant change in protein levels. For western blots, beta-actin (ACTβ) is displayed as loading control. At least 3 mice per experiment were analyzed; data are shown as mean and s.d. Where applicable, * = p<0.01 by Student’s t-test versus SOD2L/L.
Increased mitochondrial superoxide levels impact proper development of murine T-cells
To understand the consequences of increased steady-state levels of mitochondrial superoxide during T-cell growth and selection, quantitative morphometry was performed upon transmission electron microscopy (TEM) images of developing thymocytes (Fig. 3A). It was observed that cellular cross sectional area was significantly increased within SOD2−/− T-cells, but no change was noted in nuclear area or mitochondrial size (Fig. 3B; Supplemental Fig. 2, A–B). Conversely, SOD2−/− thymocytes demonstrated an approximate doubling of the number of mitochondria per cell, but these mitochondria were found to be less electron dense (Fig. 3B) suggesting dysfunctional or degrading organelles, as this has been observed previously in oxidatively stressed systems [53]. These three mitochondrial parameters, decreased electron density, increased number per cell, and altered metabolism (Fig. 2B) all further support that the mitochondria within SOD2−/− T-cells are defective. To expand upon these observations, the role of mitochondrial autophagy was explored. It was shown that autophagy was in fact increased in the SOD2−/− T-cells as shown by increases in autophagic vesicles and the cleaved form of LC-3 protein (Fig. 3, C–D). Overall, the loss of SOD2 appears to have profound effects upon T-cell ultrastructure as well as metabolic function.
Figure 3.

Subcellular and mitochondrial alterations are consequences of T-cell specific loss of SOD2. (A) Representative transmission electron (TEM) images of SOD2L/L (left) and SOD2−/− (right) 6-week old thymocytes. Open arrowheads indicate mitochondria. (B) Quantitative morphometry of thymocyte cellular area (left), mitochondrial number (center), and mitochondrial density (right). Each “X” represents one data analysis point; clear bars represent means. (C) Representative transmission electron (TEM) images of two SOD2−/− 6-week old thymocytes. Solid arrowheads indicate apparent autophagic vesicles commonly seen in knockout animal thymocytes. (D) Western blot analysis comparing the increase of cleavage product (i.e. LC3B-II) of the autophagic marker LC3. At least 3 mice per experiment were analyzed. Where applicable, * = p<0.01 by Student’s t-test versus SOD2L/L.
Besides the morphological changes, decreases in thymocyte numbers as well as peripheral T-cells were noted (Fig. 4A). When total thymocytes were quantified at different ages throughout the mouse immune system development, an approximate 2-fold decrease was noted in SOD2−/− mice. In contrast to this, mice harboring a SOD2 knock-out from an earlier stage in development (i.e. vav-iCre mediated recombination) demonstrated no significant decrease in thymic cellularity (Fig. 4A, Supplemental Fig 3A). This finding suggests that various stages in T-cell development may be differentially sensitive to the presence of excess superoxide. Moreover, in contrast to the pan-organ failure noted in the aforementioned constitutive SOD2 knock-out mouse [40], our preliminary studies have revealed no overt phenotype in unstressed animals harboring either liver-specific or mammary-specific knockouts of SOD2 (Supplemental Fig 3A–C). Taken together, the identification of a cellular defect in the T-cell population due to the loss of SOD2 at a later (Lck-Cre mediated) rather than an earlier (vav-iCre mediated) stage of development suggests that superoxide mediated effects are tissue and time dependent, and not an abscopal phenomenon. Furthermore, the decrease in thymic cellularity of the Lck-Cre SOD2−/− mice was exacerbated by the fact that SOD2−/− mice also displayed reduced percentages of mature CD4+ and CD8+ thymocytes (Fig. 4B), a trend that was also observed in peripheral lymphoid organs as well (Supplemental Fig. 4B). On the contrary, the most immature T-cell populations in the thymus (i.e. CD4−, CD8−), γδ T-cells, as well as non-lymphocyte populations were shown to be static or increased within SOD2−/− mice (Fig. 4, B–C, Supplemental Fig. 4A). This is most likely attributed to the fact that none of these populations activates the lymphocyte specific kinase (Lck) promoter and thus they do not express the exogenous Cre-recombinase. Conversely, these populations may be exhibiting compensatory up-regulation due to the decrease in mature αβ T-cells, as this phenomenon has been described in other models of T-cell deficiencies [42, 54]. Interestingly, while examining peripheral T-cells it was observed that SOD2−/− lymphocytes expressed a greater proportion of Mel14−/CD44+ cells suggesting a more activated state among otherwise naïve T-cells (Fig. 4C). This finding was further supported by microarray analysis that demonstrated up-regulation of numerous genes involved in T-cell activation in the thymocytes of T-cell SOD2−/− mice (Supplemental Fig. 5, A–B), and a similar effect on gene regulation by mitochondrial superoxide has been recently reported [55]. An alternative hypothesis to explain this apparently activated T-cell phenotype could be due to the well-described phenomenon known as homeostatic expansion, a compensatory mechanism to replenish the global loss of T-cells throughout the SOD2−/− mice [56]. Taken together, these data suggest that increased mitochondrial derived superoxide alters intracellular signaling within the developing thymocytes, and may inappropriately mimic the activating conditions of an infection, thus inhibiting proper T-cell function during true infection.
Figure 4.

Significant T-cell developmental aberrations are observed in SOD2−/− animals. (A) Left, total thymic cellularity counts at different ages of T-cell development for SOD2L/L (blue circles) and SOD2−/− (red diamonds) animals. Clear bars show SOD2L/L averages, while solid bars show SOD2−/− averages. Middle, total cell counts for three peripheral lymphoid organs at 6-weeks of age. Right, total thymic cellularity counts for 6-week old mice that were created using the Vav-iCre promoter. No significant change in thymic numbers is noted when knocking-out SOD2 at a different stage in T-cell development. (B) Left, representative flow scatter diagrams of SOD2L/L (left) and SOD2−/− (right) 6-week old thymocytes for the developmental markers CD4 and CD8. Right, quantification of CD4+ and CD8+ 6-week old thymocytes. Colored arrowheads (left) correlate to respective colored quantification (right); Solid bars and arrowheads indicate SOD2L/L and open bars and arrowheads indicate SOD2−/−. (C) Left, Quantification of flow cytometric analysis of γδ+ T-cells, as well as for markers indicative of T-cell activation MEL14 and CD44 on splenic CD4+ cells (Middle) and CD8+ cells (Right). In all graphs, solid bars indicate SOD2L/L and open bars indicate SOD2−/−. At least 3 mice per experiment were analyzed; data are shown as mean and s.d. Where applicable, * = p<0.01 by Student’s t-test versus SOD2L/L.
The decrease in SOD2−/− T-cell numbers is due to increased apoptosis
During T-cell development apoptosis is the major selective mechanism for lymphocyte death [57]. With the understanding that apoptosis may be initiated through cytochrome c release from damaged mitochondria or through the non-caspase mediated p53 pathway [58], we postulated that increased apoptosis due to dysfunctional mitochondria may be the mechanism for decreased thymocytes in the SOD2−/− mice. When flow cytometry for annexin V/propidium iodide was performed, a 2-fold increase in the apoptotic fraction of thymocytes was found in the SOD2−/− mice (Fig. 5A). In addition, an approximate 2-fold increase in thymocyte sensitivity to oxygen toxicity in vitro as well as increased immunoreactive p53 was also noted, further suggesting increased susceptibility of SOD2−/− thymocytes to death by apoptosis (Supplemental Fig. 6, B–C). In addition, propidium iodide cell cycle analysis demonstrated a greater cycling population in SOD2−/− mice, which supports a role for compensatory proliferation in more primitive CD3−/CD4−/CD8− cells as previously discussed (Supplemental Fig. 6A). When mice were treated with small molecule superoxide scavengers (i.e. Tempol [59] or Mito-CTPO [60]) added to the water supply from the time of weaning, the anti-oxidant supplementation not only led to the complete rescue of the SOD2−/− phenotype, but also increased thymocyte numbers in control SOD2L/L animals by approximately 2-fold while decreasing their apoptotic fraction by 50% (Fig. 5B). There was a strong correlation between apoptotic fractions and total cell counts, suggesting that apoptosis is the primary mechanism for increased T-cell death in SOD2−/− animals (Fig. 5B). These data suggest a tight relationship between the regulation of steady-state mitochondrial superoxide levels within the normal developing thymus, and immune system aberrations.
Figure 5.

Apoptosis explains the decrease in SOD2−/− thymocytes, which may be rescued by superoxide scavenger supplementation. (A) Representative annexin V and propidium iodide flow scatter diagrams of 6-week old thymocytes from vehicle treated SOD2L/L (left), vehicle treated SOD2−/− (middle), and Mito-CTPO treated SOD2−/− (right) mice. (B) Left, total thymic cellularity with and without pharmaceutical anti-oxidant supplementation. Middle, quantification of annexin V+ and propidium iodide+ thymocytes with and without pharmaceutical anti-oxidant supplementation. Colored quantification correlates to respective colored arrowheads in part (A). Right, regression analysis of total thymic cellularity versus apoptotic fraction. At least 3 mice per experiment were analyzed; data are shown as mean and s.d. Where applicable, * = p<0.01 by Student’s t-test versus vehicle treated SOD2L/L. Where applicable, ¥ = p<0.01 by Student’s t-test versus vehicle treated SOD2−/−.
Mitochondrial superoxide mediates susceptibility to influenza A, H1N1
The lack of proper T-cell development observed in the SOD2−/− mice strongly suggested an immunocompromised state. To test this hypothesis, mice were challenged with a sublethal dose of IAV, H1N1, a pathogen that causes a T-cell mediated immune response, and their ability to mount an immune response was observed. Surprisingly, 100% of SOD2−/− animals succumbed to the infection and died while only a small percentage of SOD2L/L animals exhibited mortality, and furthermore, SOD2−/− mice demonstrated increased morbidity (i.e. weight loss) with no recovery when compared to SOD2L/L mice (Fig. 6A). Moreover, treatment of SOD2−/− animals with the small molecule superoxide scavengers, Tempol (Supplemental Fig. 7) or Mito-CTPO (Fig. 6A), decreased this weight loss and rescued their ability to survive the influenza infection with delayed or no mortality noted. When examining immunological parameters of the infection, it was once again found that SOD2−/− mice manifest their immune defect with decreases in not only total T-cells, but also IAV-specific CD8+ IAV-peptide-MHC-tetramer+ cells, interferon gamma+ (IFNγ) cells, tumor necrosis factor alpha+ (TNFα) cells, and dual expressing IFNγ+ TNFα+ cells (Fig. 6B; Supplemental Fig. 8A). Interestingly, of the SOD2−/− T-cells that were present at the site of infection the quantity of IFNγ expression per cell was no less than control SOD2L/L T-cells, and TNFα expression actually increased within SOD2−/− T-cells (Supplemental Fig. 8B). These findings are consistent with other studies of immunodeficient states and sepsis in which T-cells compensate by over-producing pro-inflammatory markers in what is known as a “cytokine storm” [61] and may in part explain the apparently activated state of the naïve T-cells in the SOD2−/− mice. Overall, alterations in mitochondrial superoxide steady-state levels appear to significantly impact the outcomes associated the mammalian adaptive response to viral infection.
Figure 6.

SOD2−/− mice are immunologically susceptible to influenza virus, but may be rescued by superoxide scavenging pharmaceuticals. (A) Left, Kaplan-Meier analysis of mice succumbing to IAV, H1N1 infection during a two-week period after administration with and without Mito-CTPO administration. Right, relative weight loss of mice over a two-week period after infection with IAV with and without Mito-CTPO supplementation. Black squares/Solid line: Vehicle treated SOD2L/L, Black triangles/Dotted line: Vehicle treated SOD2−/−, Red circles/Dotted line: Mito-CTPO treated SOD2−/−. (B) Left, total lymphocyte counts harvested from lungs of both SOD2L/L and SOD2−/− animals 8 days post influenza virus infection. Also, flow cytometric analysis of CD8+ influenza-specific NP366 and PA224 MHC-tetramer positive cells (Middle) and analysis of NP366 and PA224-specific IFNγ+/TNFα+ (Right) CD8+ T-cells isolated from lungs 8 days post IAV, H1N1 infection. At least 6 mice per experiment were analyzed; data are shown as mean and s.d. For weights and graphs, * = p<0.01 by Student’s t-test versus SOD2L/L. For mortality, * = p<0.01 by log-rank analysis versus SOD2L/L.
Discussion
The current findings suggest an excess of mitochondrial superoxide at the stage of lymphocyte specific kinase (Lck) activation of development tilts the intracellular redox potential toward apoptosis, resulting in the decreased thymic cellularity observed. A relative increase in the normal steady-state levels of superoxide had severe consequences on the immunocompentency of the mammalian adaptive immune system. This novel finding complements the well established role of ROS in the innate immune system, and adds yet another effect of superoxide and its derived reactive oxygen intermediates in normal tissue growth and development. However, further studies are required to examine specific signaling pathways that are affected by increased mitochondrial superoxide. It would be predicted that redox-sensitive signaling cascades such as NFκB [17], AP-1 [15], or JAK/STAT [18] may be affected, and as such may in part explain the pseudo-activated state and microarray results observed in this study. Furthermore, the role of post-translational modifications on proteins such as the hypoxia inducible factor (HIF-1α) could also serve as valuable information on how ROS regulate cellular signaling [62]. Moreover, elucidation of how ROS affects peripheral lymphocyte mitosis, proliferation, and expansion in antigen unchallenged and challenged mice also warrants further examination. It may also be beneficial to understand the role of compartmentalization of superoxide (e.g. cytoplasmic, extracellular, etc), and as such these studies should be followed by similar investigations using SOD1 [63] or SOD3 [64] conditional knock-out animals. Due to the fact that both SOD1 and SOD3 constitutive knock-out animals are viable and have no gross pathologic phenotype as compared to the constitutive SOD2 knock-out mouse [31, 39, 40, 65], it would be postulated that superoxide compartmentalization may explain this phenomenon at least in part. We observed that elevated mitochondrial superoxide levels had severe effects on T-cell development, but it may be postulated that elevated superoxide in different cellular and tissue compartments may lead to a spectrum of developmental, functional, or disease (e.g. cancer) predispositions. Last, we have examined mitochondrial superoxide and the consequences of its excess on the development of the T-cell adaptive immune system. An in depth analysis of other reactive oxygen species (e.g. hydrogen peroxide, peroxynitrite, hydroxyl radical, etc) may prove highly informative to further understand the part each plays in development and function of organ systems. These data could serve as a platform for targeting specific immunodeficiencies that are currently not well understood and may be due to alterations in redox status, and tailor anti-oxidant therapies accordingly.
In this study, it was demonstrated that the loss of immunocompetence in a SOD2−/− background could be rescued using a mitochondria-targeted superoxide scavenger; an example of the aforementioned tailoring of specific therapies to immunodeficiencies. Serendipitously, it was shown that SOD2L/L mice with fully developed and functional adaptive immune systems also responded to the pharmaceutical intervention. These mice were shown to have significantly increased numbers of thymocytes compared to untreated control animals, and this appeared to be due to a decreased apoptotic fraction. At first glance, it appears that this increase in T-cells could potentially be a boost in immune system prowess, but SOD2L/L mice treated with superoxide scavengers showed no significant increase/decrease in the ability to recover from influenza A, H1N1 infection (data not shown). This finding is most likely attributed to the hypothesis that the increase in T-cells is due to an inhibition of proper apoptotic death of cells destined to die in the thymus (i.e. non-functional cells, cells that are self-recognizing, etc) as opposed to the expansion of pathogen specific immune cells. While in the short-term this had no observable consequence, it is speculated that the preserving of the cells predestined for death could lead to the potential for auto-immunity. Additional studies will be needed to confirm this potential role of superoxide in the development of autoimmune diseases in both mouse and humans. In this study, equal numbers of male and female mice were used for all assays, but anecdotally it was observed in many cases that females demonstrated more pronounced outcomes due to the effects of altering steady-state superoxide levels. With the understanding that females are more prone to auto-immune diseases as well as the recent observation of different anti-oxidant capacities in females and males [66], this preliminary observation may warrant further examination of this phenomenon in similar models of adaptive immune system anti-oxidant alterations.
In conclusion, we have used a previously undescribed model of T-cell specific superoxide scavenging deficiency to examine the development of the adaptive branch of the mammalian immune system and found that excess mitochondrial superoxide late in T-cell development is detrimental to normal T-cell maturation. In contrast, early developmental loss of SOD2 activity displayed no gross alteration in thymic cellularity, indicating the importance of the temporal nature of the loss of SOD2 function on thymic developmental processes. In addition, we have shown that the loss of SOD2 does not exert a similar damaging effect in development or maturation of other organs including liver and mammary glands, supporting previous findings by others using SOD2 floxed models [37, 67]. At this time the nature of the differences in cell type specificity and developmental timing of superoxide toxicity are poorly understood, but our findings warrant further investigation into possible mechanisms mediating these differential sensitivities.. Using the appropriate Cre-recombinase mouse, this model could be expanded to examining other elements of the immune system (e.g. B-Cells [68], Neutrophils [69], Macrophages [69], etc), different time points in development [41, 42, 70], or other organ systems all together [53, 71, 72]. In addition, due to the simplicity of the breeding scheme the floxed SOD2 mouse may be applied in combination with other protein deletions or additions (e.g. p53 null [73], myc over-expression [74, 75], etc) to examine the effect of superoxide oxidative stress in alternative immune model systems. Overall, this new model and these observations serve as an initial step in furthering the role of ROS in development and function of mammalian organ systems.
Supplementary Material
Acknowledgments
We would like to thank The University of Iowa Animal Pathology, Central Microscopy, and Flow Cytometry Facilities, especially David Meyerholz and Kathy Walters, for their help in tissue preparation and analysis. Microarray data were generated by the Genomics Core at the Arizona Cancer Center; supported by the Southwest Environmental Health Sciences Center. In addition, George Watts, Jose Munoz-Rodriguez, David Motto, Stephen Sangster, Joshua Madsen, Anna Johns, Melissa Teoh-Fitzgerald, Sean Martin, and C. Michael Knudson for their technical help or supplies. Last, Adam Dupuy, Curt Sigmund, Balaraman Kalyanaraman, Joy Joseph, and Takamune Takahashi for their generous donations. This work was supported by the following grants: NIH RO1 CA073612, DOD PC073831, NIAAA AA019438, NIAAA AA019437, NIAID AI071085, NIH P30 CA086862, and DOE-DE-SC0000830, NIEHS ES06694, NIH grant CA23074.
List of Abbreviations
- ROS
reactive oxygen species
- O2·−
superoxide
- SOD2
manganese superoxide dismutase
- IAV
influenza A virus
- H2O2
hydrogen peroxide
- ONOO−
peroxynitrite
- ·OH
hydroxyl radical
- SOD1
copper/zinc superoxide dismutase
- SOD3
extracellular superoxide dismutase
- SOD2−/−
conditional SOD2 knock-out mouse
- SOD2L/L
control loxP containing mouse
- DHE
dihydroethidium
- DCFH-DA
dihydrodichlorfluorescein-diacetate
- RNS
reactive nitrogen species
- TEM
transmission electron microscopy
- Lck
lymphocyte specific kinase
- INFγ
interferon gamma
- TNFα
tumor necrosis factor alpha
- HIF-1α
hypoxia inducible factor one alpha
- ACTβ
beta actin
- NBT
nitroblue tetrazolium
- MFI
mean fluorescence intensity
- HBSS
Hanks buffered salt solution
- PBS
phosphate buffered saline
Footnotes
The authors have no conflicts of interest to disclose on this research project
Author Contributions A.J.C performed or aided in all analyses; J.L.M. infected animals and aided in analysis of animals post-infection; L.T.T. prepared T-cells for developmental analysis; T.S. developed and provided SOD2 floxed animals; D.R.S., T.J.W., and K.L.L. were involved in study design as well as reagent donation. F.E.D. was principle investigator of study providing support, facilities, and supplies. A.J.C. and F.E.D. contributed equally in writing of the manuscript. All authors discussed the results and commented on the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.McCord JM, Fridovich I. The biology and pathology of oxygen radicals. Ann Intern Med. 1978;89:122–127. doi: 10.7326/0003-4819-89-1-122. [DOI] [PubMed] [Google Scholar]
- 2.Cerutti PA. Prooxidant states and tumor promotion. Science. 1985;227:375–381. doi: 10.1126/science.2981433. [DOI] [PubMed] [Google Scholar]
- 3.Fridovich I. Oxygen toxicity: a radical explanation. J Exp Biol. 1998;201:1203–1209. doi: 10.1242/jeb.201.8.1203. [DOI] [PubMed] [Google Scholar]
- 4.Leibovitz BE, Siegel BV. Aspects of free radical reactions in biological systems: aging. J Gerontol. 1980;35:45–56. doi: 10.1093/geronj/35.1.45. [DOI] [PubMed] [Google Scholar]
- 5.Kaneto H, Katakami N, Matsuhisa M, Matsuoka TA. Role of reactive oxygen species in the progression of type 2 diabetes and atherosclerosis. Mediators Inflamm. 2010:453892. doi: 10.1155/2010/453892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miao L, St Clair DK. Regulation of superoxide dismutase genes: implications in disease. Free Radic Biol Med. 2009;47:344–356. doi: 10.1016/j.freeradbiomed.2009.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nunomura A, Hofer T, Moreira PI, Castellani RJ, Smith MA, Perry G. RNA oxidation in Alzheimer disease and related neurodegenerative disorders. Acta Neuropathol. 2009;118:151–166. doi: 10.1007/s00401-009-0508-1. [DOI] [PubMed] [Google Scholar]
- 8.Oberley LW, Buettner GR. Role of superoxide dismutase in cancer: a review. Cancer Res. 1979;39:1141–1149. [PubMed] [Google Scholar]
- 9.Akaike T, Ando M, Oda T, Doi T, Ijiri S, Araki S, Maeda H. Dependence on O2− generation by xanthine oxidase of pathogenesis of influenza virus infection in mice. J Clin Invest. 1990;85:739–745. doi: 10.1172/JCI114499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oda T, Akaike T, Hamamoto T, Suzuki F, Hirano T, Maeda H. Oxygen radicals in influenza-induced pathogenesis and treatment with pyran polymer-conjugated SOD. Science. 1989;244:974–976. doi: 10.1126/science.2543070. [DOI] [PubMed] [Google Scholar]
- 11.Geiler J, Michaelis M, Naczk P, Leutz A, Langer K, Doerr HW, Cinatl J., Jr N-acetyl-l-cysteine (NAC) inhibits virus replication and expression of pro-inflammatory molecules in A549 cells infected with highly pathogenic H5N1 influenza A virus. Biochem Pharmacol. 2009 doi: 10.1016/j.bcp.2009.08.025. [DOI] [PubMed] [Google Scholar]
- 12.Jariwalla RJ, Roomi MW, Gangapurkar B, Kalinovsky T, Niedzwiecki A, Rath M. Suppression of influenza A virus nuclear antigen production and neuraminidase activity by a nutrient mixture containing ascorbic acid, green tea extract and amino acids. Biofactors. 2007;31:1–15. doi: 10.1002/biof.5520310101. [DOI] [PubMed] [Google Scholar]
- 13.Sidwell RW, Huffman JH, Bailey KW, Wong MH, Nimrod A, Panet A. Inhibitory effects of recombinant manganese superoxide dismutase on influenza virus infections in mice. Antimicrob Agents Chemother. 1996;40:2626–2631. doi: 10.1128/aac.40.11.2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Suliman HB, Ryan LK, Bishop L, Folz RJ. Prevention of influenza-induced lung injury in mice overexpressing extracellular superoxide dismutase. Am J Physiol Lung Cell Mol Physiol. 2001;280:L69–78. doi: 10.1152/ajplung.2001.280.1.L69. [DOI] [PubMed] [Google Scholar]
- 15.Gius D, Botero A, Shah S, Curry HA. Intracellular oxidation/reduction status in the regulation of transcription factors NF-kappaB and AP-1. Toxicol Lett. 1999;106:93–106. doi: 10.1016/s0378-4274(99)00024-7. [DOI] [PubMed] [Google Scholar]
- 16.Gius D, Spitz DR. Redox signaling in cancer biology. Antioxid Redox Signal. 2006;8:1249–1252. doi: 10.1089/ars.2006.8.1249. [DOI] [PubMed] [Google Scholar]
- 17.Schulze-Osthoff K, Los M, Baeuerle PA. Redox signalling by transcription factors NF-kappa B and AP-1 in lymphocytes. Biochem Pharmacol. 1995;50:735–741. doi: 10.1016/0006-2952(95)02011-z. [DOI] [PubMed] [Google Scholar]
- 18.Uckun FM, Qazi S, Ma H, Tuel-Ahlgren L, Ozer Z. STAT3 is a substrate of SYK tyrosine kinase in B-lineage leukemia/lymphoma cells exposed to oxidative stress. Proc Natl Acad Sci U S A. 2010;107:2902–2907. doi: 10.1073/pnas.0909086107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Allen RG, Balin AK. Oxidative influence on development and differentiation: an overview of a free radical theory of development. Free Radic Biol Med. 1989;6:631–661. doi: 10.1016/0891-5849(89)90071-3. [DOI] [PubMed] [Google Scholar]
- 20.Hitchler MJ, Domann FE. An epigenetic perspective on the free radical theory of development. Free Radic Biol Med. 2007;43:1023–1036. doi: 10.1016/j.freeradbiomed.2007.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tauber AI, Babior BM. Evidence for hydroxyl radical production by human neutrophils. J Clin Invest. 1977;60:374–379. doi: 10.1172/JCI108786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lu SP, Lin Feng MH, Huang HL, Huang YC, Tsou WI, Lai MZ. Reactive oxygen species promote raft formation in T lymphocytes. Free Radic Biol Med. 2007;42:936–944. doi: 10.1016/j.freeradbiomed.2006.11.027. [DOI] [PubMed] [Google Scholar]
- 23.Norell H, Martins da Palma T, Lesher A, Kaur N, Mehrotra M, Naga OS, Spivey N, Olafimihan S, Chakraborty NG, Voelkel-Johnson C, Nishimura MI, Mukherji B, Mehrotra S. Inhibition of superoxide generation upon T-cell receptor engagement rescues Mart-1(27–35)-reactive T cells from activation-induced cell death. Cancer Res. 2009;69:6282–6289. doi: 10.1158/0008-5472.CAN-09-1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sklavos MM, Tse HM, Piganelli JD. Redox modulation inhibits CD8 T cell effector function. Free Radic Biol Med. 2008;45:1477–1486. doi: 10.1016/j.freeradbiomed.2008.08.023. [DOI] [PubMed] [Google Scholar]
- 25.Niethammer P, Grabher C, Look AT, Mitchison TJ. A tissue-scale gradient of hydrogen peroxide mediates rapid wound detection in zebrafish. Nature. 2009;459:996–999. doi: 10.1038/nature08119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Owusu-Ansah E, Banerjee U. Reactive oxygen species prime Drosophila haematopoietic progenitors for differentiation. Nature. 2009;461:537–541. doi: 10.1038/nature08313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Macho A, Hirsch T, Marzo I, Marchetti P, Dallaporta B, Susin SA, Zamzami N, Kroemer G. Glutathione depletion is an early and calcium elevation is a late event of thymocyte apoptosis. J Immunol. 1997;158:4612–4619. [PubMed] [Google Scholar]
- 28.Choi J, Oh S, Lee D, Oh HJ, Park JY, Lee SB, Lim DS. Mst1-FoxO signaling protects Naive T lymphocytes from cellular oxidative stress in mice. PLoS One. 2009;4:e8011. doi: 10.1371/journal.pone.0008011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moulian N, Truffault F, Gaudry-Talarmain YM, Serraf A, Berrih-Aknin S. In vivo and in vitro apoptosis of human thymocytes are associated with nitrotyrosine formation. Blood. 2001;97:3521–3530. doi: 10.1182/blood.v97.11.3521. [DOI] [PubMed] [Google Scholar]
- 30.Peled-Kamar M, Lotem J, Okon E, Sachs L, Groner Y. Thymic abnormalities and enhanced apoptosis of thymocytes and bone marrow cells in transgenic mice overexpressing Cu/Zn-superoxide dismutase: implications for Down syndrome. EMBO J. 1995;14:4985–4993. doi: 10.1002/j.1460-2075.1995.tb00181.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Carlsson LM, Jonsson J, Edlund T, Marklund SL. Mice lacking extracellular superoxide dismutase are more sensitive to hyperoxia. Proc Natl Acad Sci U S A. 1995;92:6264–6268. doi: 10.1073/pnas.92.14.6264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Elchuri S, Oberley TD, Qi W, Eisenstein RS, Jackson Roberts L, Van Remmen H, Epstein CJ, Huang TT. CuZnSOD deficiency leads to persistent and widespread oxidative damage and hepatocarcinogenesis later in life. Oncogene. 2005;24:367–380. doi: 10.1038/sj.onc.1208207. [DOI] [PubMed] [Google Scholar]
- 33.Ho YS, Gargano M, Cao J, Bronson RT, Heimler I, Hutz RJ. Reduced fertility in female mice lacking copper-zinc superoxide dismutase. J Biol Chem. 1998;273:7765–7769. doi: 10.1074/jbc.273.13.7765. [DOI] [PubMed] [Google Scholar]
- 34.Ho YS, Magnenat JL, Bronson RT, Cao J, Gargano M, Sugawara M, Funk CD. Mice deficient in cellular glutathione peroxidase develop normally and show no increased sensitivity to hyperoxia. J Biol Chem. 1997;272:16644–16651. doi: 10.1074/jbc.272.26.16644. [DOI] [PubMed] [Google Scholar]
- 35.Ho YS, Xiong Y, Ma W, Spector A, Ho DS. Mice lacking catalase develop normally but show differential sensitivity to oxidant tissue injury. J Biol Chem. 2004;279:32804–32812. doi: 10.1074/jbc.M404800200. [DOI] [PubMed] [Google Scholar]
- 36.Sentman ML, Granstrom M, Jakobson H, Reaume A, Basu S, Marklund SL. Phenotypes of mice lacking extracellular superoxide dismutase and copper- and zinc-containing superoxide dismutase. J Biol Chem. 2006;281:6904–6909. doi: 10.1074/jbc.M510764200. [DOI] [PubMed] [Google Scholar]
- 37.Ikegami T, Suzuki Y, Shimizu T, Isono K, Koseki H, Shirasawa T. Model mice for tissue-specific deletion of the manganese superoxide dismutase (MnSOD) gene. Biochem Biophys Res Commun. 2002;296:729–736. doi: 10.1016/s0006-291x(02)00933-6. [DOI] [PubMed] [Google Scholar]
- 38.Fridovich I. The biology of oxygen radicals. Science. 1978;201:875–880. doi: 10.1126/science.210504. [DOI] [PubMed] [Google Scholar]
- 39.Lebovitz RM, Zhang H, Vogel H, Cartwright J, Jr, Dionne L, Lu N, Huang S, Matzuk MM. Neurodegeneration, myocardial injury, and perinatal death in mitochondrial superoxide dismutase-deficient mice. Proc Natl Acad Sci U S A. 1996;93:9782–9787. doi: 10.1073/pnas.93.18.9782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li Y, Huang TT, Carlson EJ, Melov S, Ursell PC, Olson JL, Noble LJ, Yoshimura MP, Berger C, Chan PH, Wallace DC, Epstein CJ. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat Genet. 1995;11:376–381. doi: 10.1038/ng1295-376. [DOI] [PubMed] [Google Scholar]
- 41.de Boer J, Williams A, Skavdis G, Harker N, Coles M, Tolaini M, Norton T, Williams K, Roderick K, Potocnik AJ, Kioussis D. Transgenic mice with hematopoietic and lymphoid specific expression of Cre. Eur J Immunol. 2003;33:314–325. doi: 10.1002/immu.200310005. [DOI] [PubMed] [Google Scholar]
- 42.Lee PP, Fitzpatrick DR, Beard C, Jessup HK, Lehar S, Makar KW, Perez-Melgosa M, Sweetser MT, Schlissel MS, Nguyen S, Cherry SR, Tsai JH, Tucker SM, Weaver WM, Kelso A, Jaenisch R, Wilson CB. A critical role for Dnmt1 and DNA methylation in T cell development, function, and survival. Immunity. 2001;15:763–774. doi: 10.1016/s1074-7613(01)00227-8. [DOI] [PubMed] [Google Scholar]
- 43.Postic C, Shiota M, Niswender KD, Jetton TL, Chen Y, Moates JM, Shelton KD, Lindner J, Cherrington AD, Magnuson MA. Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic beta cell-specific gene knock-outs using Cre recombinase. J Biol Chem. 1999;274:305–315. doi: 10.1074/jbc.274.1.305. [DOI] [PubMed] [Google Scholar]
- 44.Wagner KU, Wall RJ, St-Onge L, Gruss P, Wynshaw-Boris A, Garrett L, Li M, Furth PA, Hennighausen L. Cre-mediated gene deletion in the mammary gland. Nucleic Acids Res. 1997;25:4323–4330. doi: 10.1093/nar/25.21.4323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Spitz DR, Oberley LW. An assay for superoxide dismutase activity in mammalian tissue homogenates. Anal Biochem. 1989;179:8–18. doi: 10.1016/0003-2697(89)90192-9. [DOI] [PubMed] [Google Scholar]
- 46.Robinson KM, Janes MS, Pehar M, Monette JS, Ross MF, Hagen TM, Murphy MP, Beckman JS. Selective fluorescent imaging of superoxide in vivo using ethidium-based probes. Proc Natl Acad Sci U S A. 2006;103:15038–15043. doi: 10.1073/pnas.0601945103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Slane BG, Aykin-Burns N, Smith BJ, Kalen AL, Goswami PC, Domann FE, Spitz DR. Mutation of succinate dehydrogenase subunit C results in increased O2.−, oxidative stress, and genomic instability. Cancer Res. 2006;66:7615–7620. doi: 10.1158/0008-5472.CAN-06-0833. [DOI] [PubMed] [Google Scholar]
- 48.Morton RL, Ikle D, White CW. Loss of lung mitochondrial aconitase activity due to hyperoxia in bronchopulmonary dysplasia in primates. Am J Physiol. 1998;274:L127–133. doi: 10.1152/ajplung.1998.274.1.L127. [DOI] [PubMed] [Google Scholar]
- 49.Guzy RD, Sharma B, Bell E, Chandel NS, Schumacker PT. Loss of the SdhB, but Not the SdhA, subunit of complex II triggers reactive oxygen species-dependent hypoxia-inducible factor activation and tumorigenesis. Mol Cell Biol. 2008;28:718–731. doi: 10.1128/MCB.01338-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rijhsinghani AG, Bhatia SK, Tygrett LT, Waldschmidt TJ. Effect of pregnancy on thymic T cell development. Am J Reprod Immunol. 1996;35:523–528. doi: 10.1111/j.1600-0897.1996.tb00052.x. [DOI] [PubMed] [Google Scholar]
- 51.Knudson CM, Johnson GM, Lin Y, Korsmeyer SJ. Bax accelerates tumorigenesis in p53-deficient mice. Cancer Res. 2001;61:659–665. [PubMed] [Google Scholar]
- 52.Tortora V, Quijano C, Freeman B, Radi R, Castro L. Mitochondrial aconitase reaction with nitric oxide, S-nitrosoglutathione, and peroxynitrite: mechanisms and relative contributions to aconitase inactivation. Free Radic Biol Med. 2007;42:1075–1088. doi: 10.1016/j.freeradbiomed.2007.01.007. [DOI] [PubMed] [Google Scholar]
- 53.Misawa H, Nakata K, Matsuura J, Moriwaki Y, Kawashima K, Shimizu T, Shirasawa T, Takahashi R. Conditional knockout of Mn superoxide dismutase in postnatal motor neurons reveals resistance to mitochondrial generated superoxide radicals. Neurobiol Dis. 2006;23:169–177. doi: 10.1016/j.nbd.2006.02.014. [DOI] [PubMed] [Google Scholar]
- 54.Molina TJ, Kishihara K, Siderovski DP, van Ewijk W, Narendran A, Timms E, Wakeham A, Paige CJ, Hartmann KU, Veillette A, et al. Profound block in thymocyte development in mice lacking p56lck. Nature. 1992;357:161–164. doi: 10.1038/357161a0. [DOI] [PubMed] [Google Scholar]
- 55.Kaminski MM, Sauer SW, Klemke CD, Suss D, Okun JG, Krammer PH, Gulow K. Mitochondrial reactive oxygen species control T cell activation by regulating IL-2 and IL-4 expression: mechanism of ciprofloxacin-mediated immunosuppression. J Immunol. 2010;184:4827–4841. doi: 10.4049/jimmunol.0901662. [DOI] [PubMed] [Google Scholar]
- 56.King C, Ilic A, Koelsch K, Sarvetnick N. Homeostatic expansion of T cells during immune insufficiency generates autoimmunity. Cell. 2004;117:265–277. doi: 10.1016/s0092-8674(04)00335-6. [DOI] [PubMed] [Google Scholar]
- 57.Williams GT. Role of apoptosis in the immune system. Biochem Cell Biol. 1994;72:447–450. doi: 10.1139/o94-059. [DOI] [PubMed] [Google Scholar]
- 58.Liu J, Cao L, Chen J, Song S, Lee IH, Quijano C, Liu H, Keyvanfar K, Chen H, Cao LY, Ahn BH, Kumar NG, Rovira II, Xu XL, van Lohuizen M, Motoyama N, Deng CX, Finkel T. Bmi1 regulates mitochondrial function and the DNA damage response pathway. Nature. 2009;459:387–392. doi: 10.1038/nature08040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mitchell JB, Xavier S, DeLuca AM, Sowers AL, Cook JA, Krishna MC, Hahn SM, Russo A. A low molecular weight antioxidant decreases weight and lowers tumor incidence. Free Radic Biol Med. 2003;34:93–102. doi: 10.1016/s0891-5849(02)01193-0. [DOI] [PubMed] [Google Scholar]
- 60.Dhanasekaran A, Kotamraju S, Karunakaran C, Kalivendi SV, Thomas S, Joseph J, Kalyanaraman B. Mitochondria superoxide dismutase mimetic inhibits peroxide-induced oxidative damage and apoptosis: role of mitochondrial superoxide. Free Radic Biol Med. 2005;39:567–583. doi: 10.1016/j.freeradbiomed.2005.04.016. [DOI] [PubMed] [Google Scholar]
- 61.Wang H, Ma S. The cytokine storm and factors determining the sequence and severity of organ dysfunction in multiple organ dysfunction syndrome. Am J Emerg Med. 2008;26:711–715. doi: 10.1016/j.ajem.2007.10.031. [DOI] [PubMed] [Google Scholar]
- 62.Kaewpila S, Venkataraman S, Buettner GR, Oberley LW. Manganese superoxide dismutase modulates hypoxia-inducible factor-1 alpha induction via superoxide. Cancer Res. 2008;68:2781–2788. doi: 10.1158/0008-5472.CAN-07-2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gong YH, Parsadanian AS, Andreeva A, Snider WD, Elliott JL. Restricted expression of G86R Cu/Zn superoxide dismutase in astrocytes results in astrocytosis but does not cause motoneuron degeneration. J Neurosci. 2000;20:660–665. doi: 10.1523/JNEUROSCI.20-02-00660.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gongora MC, Lob HE, Landmesser U, Guzik TJ, Martin WD, Ozumi K, Wall SM, Wilson DS, Murthy N, Gravanis M, Fukai T, Harrison DG. Loss of extracellular superoxide dismutase leads to acute lung damage in the presence of ambient air: a potential mechanism underlying adult respiratory distress syndrome. Am J Pathol. 2008;173:915–926. doi: 10.2353/ajpath.2008.080119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Reaume AG, Elliott JL, Hoffman EK, Kowall NW, Ferrante RJ, Siwek DF, Wilcox HM, Flood DG, Beal MF, Brown RH, Jr, Scott RW, Snider WD. Motor neurons in Cu/Zn superoxide dismutase-deficient mice develop normally but exhibit enhanced cell death after axonal injury. Nat Genet. 1996;13:43–47. doi: 10.1038/ng0596-43. [DOI] [PubMed] [Google Scholar]
- 66.Kim A, Joseph S, Khan A, Epstein CJ, Sobel R, Huang TT. Enhanced expression of mitochondrial superoxide dismutase leads to prolonged in vivo cell cycle progression and up-regulation of mitochondrial thioredoxin. Free Radic Biol Med. 2010;48:1501–1512. doi: 10.1016/j.freeradbiomed.2010.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shimizu T, Nojiri H, Kawakami S, Uchiyama S, Shirasawa T. Model mice for tissue-specific deletion of the manganese superoxide dismutase gene. Geriatr Gerontol Int. 2010;10(Suppl 1):S70–79. doi: 10.1111/j.1447-0594.2010.00604.x. [DOI] [PubMed] [Google Scholar]
- 68.Rickert RC, Roes J, Rajewsky K. B lymphocyte-specific, Cre-mediated mutagenesis in mice. Nucleic Acids Res. 1997;25:1317–1318. doi: 10.1093/nar/25.6.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Clausen BE, Burkhardt C, Reith W, Renkawitz R, Forster I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 1999;8:265–277. doi: 10.1023/a:1008942828960. [DOI] [PubMed] [Google Scholar]
- 70.Maekawa Y, Minato Y, Ishifune C, Kurihara T, Kitamura A, Kojima H, Yagita H, Sakata-Yanagimoto M, Saito T, Taniuchi I, Chiba S, Sone S, Yasutomo K. Notch2 integrates signaling by the transcription factors RBP-J and CREB1 to promote T cell cytotoxicity. Nat Immunol. 2008;9:1140–1147. doi: 10.1038/ni.1649. [DOI] [PubMed] [Google Scholar]
- 71.Lenart J, Dombrowski F, Gorlach A, Kietzmann T. Deficiency of manganese superoxide dismutase in hepatocytes disrupts zonated gene expression in mouse liver. Arch Biochem Biophys. 2007;462:238–244. doi: 10.1016/j.abb.2007.02.007. [DOI] [PubMed] [Google Scholar]
- 72.Lustgarten M, Jang Y, Liu Y, Muller F, Qi W, Steinhelper M, Brooks S, Larkin LM, Shimizu T, Shirasawa T, McManus L, Bhattacharya A, Richardson A, Van Remmen H. Conditional knockout of MnSOD targeted to type IIB skeletal muscle fibers increases oxidative stress and is sufficient to alter aerobic exercise capacity. Am J Physiol Cell Physiol. 2009 doi: 10.1152/ajpcell.00372.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jonkers J, Meuwissen R, van der Gulden H, Peterse H, van der Valk M, Berns A. Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nat Genet. 2001;29:418–425. doi: 10.1038/ng747. [DOI] [PubMed] [Google Scholar]
- 74.Park SS, Kim JS, Tessarollo L, Owens JD, Peng L, Han SS, Tae Chung S, Torrey TA, Cheung WC, Polakiewicz RD, McNeil N, Ried T, Mushinski JF, Morse HC, 3rd, Janz S. Insertion of c-Myc into Igh induces B-cell and plasma-cell neoplasms in mice. Cancer Res. 2005;65:1306–1315. doi: 10.1158/0008-5472.CAN-04-0268. [DOI] [PubMed] [Google Scholar]
- 75.Park SS, Shaffer AL, Kim JS, duBois W, Potter M, Staudt LM, Janz S. Insertion of Myc into Igh accelerates peritoneal plasmacytomas in mice. Cancer Res. 2005;65:7644–7652. doi: 10.1158/0008-5472.CAN-05-1222. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.