

Abstract
Pleotropic drug resistant protein 5 (Pdr5p) is a plasma membrane ATP-binding cassette (ABC) transporter and the major drug efflux pump in Saccharomyces cerevisiae. The Pdr5p family of fungal transporters possesses a number of structural features significantly different from other modeled or crystallized ABC transporters, which include a reverse topology, an atypical ATP-binding site, a very low sequence similarity in the transmembrane section and long linkers between domains. These features present a considerable hurdle in molecular modeling studies of these important transporters. Here, we report the creation of an atomic model of Pdr5p based on a combination of homology modeling and ab initio methods, incorporating information from consensus transmembrane segment prediction, residue lipophilicity, and sequence entropy. Reported mutations in the transmembrane substrate-binding pocket that altered drug-resistance were used to validate the model, and one mutation that changed the communication pattern between transmembrane and nucleotide-binding domains was used in model improvement. The predictive power of the model was demonstrated experimentally by the increased sensitivity of yeast mutants to clotrimazole having alanine substitutions for Thr1213 and Gln1253, which are predicted to be in the substrate-binding pocket, without reducing the amount of Pdr5p in the plasma membrane. The quality and reliability of our model are discussed in the context of various approaches used for modeling different parts of the structure.
Keywords: ABC transporter, homology modeling, membrane protein, nucleotide-binding domain, transmembrane domain, substrate specificity
INTRODUCTION
ATP-binding cassette (ABC) transporters are transmembrane proteins that can be divided into two broad groups: importers and exporters. Both types couple ATP hydrolysis to the translocation of substrates across a membrane. The ABC importers are found primarily in bacteria and move nutrients into the cell. In contrast, the ABC exporters, also called efflux transporters, remove a variety of often structurally unrelated substances from the cell including ions, sex hormones, wastes, and toxins (Davidson and Chen, 2004). The efflux transporters are found in all organisms and alterations in human transporters or in their cellular expression are associated with a number of diseases including cystic fibrosis, adrenoleukodystrophy and multidrug resistant tumors (Gottesman and Ambudkar, 2001).
ABC transporters typically have four major structural domains: two transmembrane (TMD) and two nucleotide-binding domains (NBD). Half-transporters have only one of each (Rea, 1999), but are believed to function as either homo- or hetero-dimers (Davidson and Chen, 2004), Some ABC proteins even have supernumerary subunits that improve substrate import or regulate transporter activity (Davidson and Chen, 2004; Higgins, 2001). Numerous ABC transporters have their four major domains fused into a single polypeptide, although for a number of them each of these domains is a separate polypeptide chain. In a polypeptide chain of single or half transporter, the order of the TMDs and NBDs in the sequence determines the topology of the protein. In the forward topology the TMDs precede the NBDs, whereas the NBDs come first in the reverse topology (Rea, 1999). Each TMD of an ABC protein is composed of six or more membrane-spanning α-helices (Higgins, 2001), which forms one half of the transport channel for substrate binding and translocation (Rea, 1999). Conceivably, residues facing the transport channel play a major role in substrate recognition, binding, and transport.
The NBDs are the domains of ABC transporters that bind and hydrolyze ATP, driving substrate translocation (Higgins, 2001). Each NBD consists of two subdomains: a large catalytic domain and a small signaling domain. The catalytic domain has an α/β folding motif and contains well-conserved sequence elements such as the Walker A and B motifs, the D-loop, and the switch region. The signaling domain is composed of α-helices and contains the Q-loop as well as the characteristic ABC transporter signature motif known as the C-loop (Schmitt and Tampe, 2002). Together these structural elements are arranged into two ATP-binding sites in an NBD dimer. The first ATP site is composed of Walker A and B from the first and the C-loop and D-loop from the second NBD. The second ATP site is made up of Walker A and B from the second NBD and the C-loop and D-loop from the first NBD (Davidson and Chen, 2004; Higgins, 2001). Hydrolysis of ATP is thought to be communicated to the TMDs by interaction between the signaling domain and cytoplasmic extensions of the transmembrane helices (TMHs).
An important ABC transporter in the yeast Saccharomyces cerevisiae (S. cerevisiae) is Pdr5p. It is a 170 kDa plasma membrane protein with 1511 amino acids in a single polypeptide chain. It effluxes a wide range of structurally and functionally diverse compounds, such as rhodamine 6-G, tetrapropyltin, cycloheximide, tritylimidazole, and clotrimazole (Golin et al., 2003; Golin et al., 2007; Hanson et al., 2005; Kolaczkowski et al., 1996; Leppert et al., 1990; Rogers et al., 2001). Loss-of-function mutations in the pdr5 gene cause profound drug hypersensitivity, while overexpression creates multidrug hyperresistance (Meyers et al., 1992). Among the many human ABC transporters responsible for multidrug resistance (MDR) are P-glycoprotein (P-gp), MRP1p, and ABCG2p (Sharom, 2008), which are functional homologues of the S. cerevisiae Pdr5p transporter. Multidrug resistance conferred by ABC transporters is also prevalent in pathogenic organisms. In the U-genome database (Galperin, 2008), the ABC transporter CGR1p, from the opportunistic fungus Candida glabrata, has been linked to MDR. CGR1p has a 71% sequence identity and 85% sequence similarity with Pdr5p (supplementary materials, Fig. S1). Similarly, in Candida albicans, the protein CDR1p makes a significant contribution to MDR (Mishra et al., 2007). CDR1p has a 55% sequence identity and 76% sequence similarity with Pdr5p (supplementary materials, Fig. S1). The well established genetic background of S. cerevisiae, the tractability of the organism, and the close sequence and functional similarity of Pdr5p to clinically relevant ABC proteins makes it an important model transporter in examining the mechanisms of drug resistance (Sipos and Kuchler, 2006).
Modeling of Pdr5p becomes necessary since the methods for the experimental determination of structures at atomic resolution of this membrane protein have not yet been successful or fully explored. In general, the number of known membrane proteins far exceeds the number of experimentally determined structures. While every effort is made to elucidate the details of membrane protein structures by X-ray crystallography as well as other techniques, progress is hampered by the difficulties in obtaining sufficient quantities of the purified membrane protein and by limitations due to method-specific sample preparation. As an alternative, modeling techniques can be employed to provide spatial information. Two such methods are homology modeling (comparative modeling) and fold recognition (threading). Homology modeling uses the experimentally determined structure of a homologous protein as a template to model a protein of unknown structure provided that the sequence similarity equals or exceeds 30% (Forrest et al., 2006). Fold recognition does not require a homologue with a known structure but relies on the premise that there is only a finite number of protein folds from which the most suitable fold(s) is selected to construct a model. While the technique of homology modeling is considered to produce the most reliable structures, especially as the sequence similarity increases between the template and the target protein, the method of fold recognition provides a viable alternative where appropriate templates do not exist (Liu and Hsu, 2005).
Many computationally generated structures have provided useful insights into the mechanisms of protein function. For instance, the model of the human ABC transporter MRP1 has been used to help elucidate the role of aromatic residues in substrate recognition (DeGorter et al., 2008). In other instances, the structural basis of the differences in substrate-specificity between the ABC transporters ABCB1 and ABCC5 was investigated through homology modeling (Ravna et al., 2007), and a comparison of the ABC transporter models of CFTR (the cystic fibrosis transmembrane conductance regulator) and ABCC4 facilitated the identification of conserved sites within the CFTR ion channel (Jordan et al., 2008). In this study, a homology model of Pdr5p with a pseudo two-fold symmetry was completed for the TMDs and NBDs based on available structural templates, incorporating prior knowledge of membrane protein structures. Fold recognition was employed for regions of Pdr5p where no suitable templates were available. The functional consequences of mutations reported in the literature are consistent with predictions based on our model, validating the approach we have taken. We believe that in the absence of an experimentally determined structure, the model represents a computational approach addressing the need for structural information to understand the interactions of the transporter with its ligands. This need is particularly pressing for the Pdr5p family of ABC transporters since all proteins of this group have unique features that make them significantly different from the non-fungal transporter models that have been developed to date.
MATERIALS AND METHODS
Compounds, media, and strains
All compounds were purchased from Sigma-Aldrich (St. Louis, MO). Cycloheximide (cyh) was dissolved in water whereas clotrimazole (clo) and tritylimidazole (trityl) were dissolved in dimethyl sulfoxide. Primers were obtained from Operon Biotechnologies (Huntsville, AL). All strains used in this study are isogenic and derived from the Δpdr5 strain R-1 that was created in another study (Sauna et al., 2008). Mutations were made using the QuikChange® site-directed mutagenesis kit (Stratagene, LaJolla, CA) and the plasmid pSS607, which bears the PDR5 gene (Sauna et al., 2008). All mutations were confirmed by sequencing the entire PDR5 gene (Retrogen, San Diego, CA). After confirmation, the mutated PDR5 from pSS607 was integrated at the pdr5 locus via the Gietz transformation kit (Medicorp, Montreal, QC, Canada).
Secondary structure prediction for transmembrane helices
The secondary structure prediction of TM segments of Pdr5p was made via a consensus approach (Albrecht et al., 2003) using the programs listed in Table I. From this methodology we predicted the locations and length of each transmembrane helix. The major differences in predictions from the various programs were the locations of the amino and carboxyl termini of each transmembrane helix. Therefore, the boundaries of the transmembrane helices were manually edited in light of the propensity of individual amino acids to participate in transmembrane helical structures (von Heijne, 1986; von Heijne, 1994).
Table I.
Boundary determination for TMHs by consensus method.
| Program | TM1 | TM2 | TM3 | TM4 | TM5 | TM6 | TM7 | TM8 | TM9 | TM10 | TM11 | TM12 | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SMART | 520–542 | 554–576 | 604–626 | 633–652 | 662–684 | N/A | 1206–1228 | 1238–1260 | 1281–1303 | 1326–1348 | 1355–1377 | 1477–1499 | (Schultz et al., 1998) |
| WinPep | 519–544 | 557–581 | 600–620 | 630–651 | 666–688 | 772–796 | N/A | 1238–1258 | 1279–1299 | 1325–1354 | 1355–1375 | 1477–1497 | (Hennig, 2001) |
| TopPred | 519–542 | 557–580 | 605–628 | 631–654 | 663–686 | 772–795 | 1206–1229 | 1237–1260 | 1290–1313 | 1325–1348 | 1357–1380 | 1477–1500 | (Claros and von Heijne, 1994) |
| TMpred | 522–542 | 559–579 | 607–626 | 634–653 | 662–686 | 774–793 | 1205–1225 | 1233–1253 | 1280–1306 | 1324–1343 | 1358–1386 | 1480–1499 | (Hofmann and Stoffel, 1993) |
| TMHMM | 520–542 | 554–576 | 604–626 | 633–652 | 662–684 | 774–793 | 1206–1228 | 1238–1260 | 1281–1303 | 1326–1348 | 1355–1377 | 1477–1499 | (Krogh et al., 2001) |
| PHD | 524–541 | 557–574 | 610–627 | 635–652 | 667–684 | 776–793 | 1208–1225 | 1237–1254 | 1286–1304 | 1327–1344 | 1355–1372 | 1480–1497 | (Rost, 1996) |
| Sousi | 519–542 | 554–580 | 604–628 | 631–654 | 662–686 | 772–795 | 1205–1229 | 1233–1260 | 1280–1313 | 1324–1348 | N/A | 1477–1500 | (Hirokawa et al., 1998) |
| Consensus | 521–542 | 557–576 | 607–626 | 634–652 | 662–684 | 776–793 | 1208–1225 | 1237–1254 | 1281–1304 | 1326–1344 | 1357–1377 | 1477–1499 | |
| Final TMHs | 519–545 | 553–581 | 602–626 | 632–653 | 663–686 | 772–795 | 1203–1228 | 1237–1260 | 1281–1303 | 1325–1348 | 1355–1379 | 1478–1502 |
Structure and sequence alignments
For a full-length multiple sequence alignment (MSA) with Pdr5p, 19 sequences having the same domain organization and pairwise sequence identities between 33% and 75% (supplementary materials, Fig. S1), were selected from the PFamA (Finn et al., 2006) database. The initial full-length MSA was made with M-Coffee (Moretti et al., 2007) and errors were corrected by the RASCAL program (Thompson et al., 2003) and by manual editing. The sequence of the Sav1866p (Dawson and Locher, 2006) protein (PDB entry: 2hyd) or P-gp (Aller et al., 2009) (3G5U) was aligned to the predicted transmembrane portion of the full-length MSA and refined according to its hydrophobicity profile using BioEdit (Hall, 1999). Independent of the full-length MSA, 19 structures of experimentally determined NBDs from the PDB (1B0U, 2D3W, 1G6H, 1GAJ, 1OXT, 1XEF, 1MT0, 1R0X, 1R0W, 1Z47, 2CBZ, 2GHI, 1VPL, 1JJ7, 1MV5, 1Q1B, 1L2T, 1V43, 1JI0, 2HYD, 3G5U) were aligned by structure superposition using the molecular modeling program O (Jones et al., 1991). The predicted NBD sequences of Pdr5p were then added to this structure-based alignment (supplementary materials, Fig. S2). Due to its overall good agreement in amino acid sequence, the NBD of the E. coli haemolysin B protein (Schmitt et al., 2003)(PDB entry: 1mt0) was chosen as the template to model both Pdr5p NBDs in the closed conformation with bound nucleotides.
Homology modeling
Homology modeling of TMDs and NBDs was done by making substitutions, insertions, and deletions in the respective template. For the closed conformation, the TMD of Sav18166p was used to model the TMDs of Pdr5p and the NBD of haemolysin B was used to model the NBDs. The open conformation, which lacks bound nucleotides, was modeled using the TMDs and NBDs of mouse P-gp as templates. Fold recognition, with the program LOOPP, (Meller and Elber, 2001) was used to model any remaining elements of the protein that were greater than 20 amino acids in length and for which no structural templates were available. These parts were then manually fitted into the homology model. The model was refined by energy minimization using CNS (Brunger et al., 1998) and by LIPS scoring (Adamian and Liang, 2006). Model quality was verified using Procheck (Laskowski et al., 1993). Molecular graphics images were produced using the UCSF Chimera package from the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIH P41 RR-01081) (Pettersen et al., 2004).
Drug sensitivity testing
Minimal inhibitory concentrations (MICs) of drugs for mutant cultures were determined by first preparing overnight cultures in 5 ml YPD broth (1% yeast extract, 2% peptone, 2% dextrose) at 30°C in a shaking water bath. The next day, the medium was removed by centrifugation; the cells were washed with 10 ml of sterile water, and resuspended in sterile water. Cell concentrations were determined spectrophotometrically (absorbance at λ = 600 nm). An aliquot of 0.5 × 105 cells was added to 2 ml of YPD broth containing tritylimidazole (0–40 μM), clotrimazole (0–5 μM), or cycloheximide (0–40 μM) and incubated at 30°C in a shaking water bath for 48 h. Relative inhibition was determined spectrophotometrically.
Purification of plasma membrane and detection of Pdr5p expression by Western blot
Plasma membranes were prepared as described previously (Sauna et al., 2008) Briefly, frozen cells were thawed on ice, suspended in homogenization buffer (HB, 50 mM Tris pH 7.5, 2 mM PMSF, 1 mM EDTA) at a ratio of 1 gram of cell to 2 ml of HB. The glass beads were then added in a 1:1 ratio (v/v) and vortexed 8 times in a 50 ml Falcon tube with 1 minute vortexing following by 1 minute incubation on an ice bath. After removing the beads (3500 × g for 15 minutes), the sample was further centrifuged at 120,000 × g for 30 minutes and the pellet (crude membrane) was resuspened in a buffer (RB, 10 mM tris pH 7.5, 10% glycerol (v/v), 1 mM EDTA, 1 mM AEBFS). After adjusting the protein concentration to 10 mg/ml with RB, 3 ml of the crude membrane was overlaid onto the top of a preset sucrose gradient centrifuge tube (53.5% sucrose on bottom and 43.5% sucrose on top, w/v), which was centrifuged at 25,000 rpm with the Beckman SW28 rotor for 5 hours. The plasma membrane was collected at the interface of the 43.5% and 53.5% sucrose layers, diluted with RB to at least 15-fold and centrifuge again at 120,000 × g for 30 minutes to pellet the plasma membrane. The pellet was resuspended in RB and store at −80°C. For Western blots, protein (20 μg each lane) was separated by SDS-PAGE with 10% gel, transferred onto a nitrocellulose membrane and blocked in TBST containing 5% skimmed milk. The blot was incubated with an anti-Pdr5p polyclonal antibody (Santa Cruz Biotechnology, Santa Cruz, CA) with 300-fold dilution (recommended 100–1000 fold) for 1 hour at room temperature followed by several washes, and further incubated with donkey anti-goat IgG-HRP for 45 min at room temperature. After several washes, the blot was developed following the instruction of the chemiluminescence kit to detect Pdr5p.
RESULTS AND DISCUSSION
Overview of the modeling and the model structure
The yeast protein Pdr5p has 1511 amino acid residues. Sequence analysis shows that the protein has two of each of the major ABC transporter domains (Fig. 1A), arranged in the reverse orientation (NBD1-TMD1-NBD2-TMD2). As predicted, each TMD consists of 6 TM helices. Pdr5p has two large extracellular loops, ECL3 (86 amino acid residues) and ECL6 (97 amino acid residues), that connect the terminal two helices of each TMD, and eight short loops that connect the extracellular or intracellular (ECL1-2, ECL4-5, and ICL1-4) termini of the other helices. An amino terminal extension of 150 amino acids (NTE:1–150) precedes the first NBD, and long intracellular linkers (L1, L2 and L3) connect the NBDs (NBD1:151–420 and NBD2:861–1110) to the TMDs. Neither the NTE nor the linkers L1–L3 are considered to be part of either the NBDs or the TMDs.
Figure 1. Structural model of the yeast ABC transporter Pdr5p.

(A) Predicted topology of Pdr5p. Pdr5p is predicted to have 12 TMHs and 2 NBDs, organized in reverse order. The two NBDs are shown as blue and red ovals, respectively. Helices in TMD1 (519–795) are in cyan and those in TMD2 in magenta. Extracellular loops (ECLs), intracellular loops (ICLs), linkers (L1–L3), and the amino terminal extension (NTE) are indicated. (B) Stereoscopic view of the Pdr5p model based on the Sav1866 and haemolysin B templates. The two modeled TMDs, colored magenta and cyan, are embedded in a membrane bilayer, which is indicated by the two horizontal lines. Underneath the TMDs are the NBDs (NBD1 in blue, NBD2 in red). The NBDs are surrounded by connecting loops L1–L3 in yellow and orange. The amino terminal extension (NTE) is located below the NBDs (black) and the extracellular loops (ECL3 and ECL6) sit above the TMDs (yellow). (C) Stereoscopic view of the Pdr5p model based on the P-gp template. The colors are the same as above.
Sequence comparisons failed to identify a homolog of known structure with significant similarity to the entire Pdr5p sequence; however, templates with adequate sequence similarity were available for the NBDs and TMDs. Thus, homology modeling could not be used for the NTE, linkers (L1–L3), or the two large extracellular inserts between TMH5 and TMH6 (ECL3) and between TMH11 and TMH12 (ECL6). Therefore, we considered it necessary to apply different approaches for modeling different parts of the protein. The strategy employed for modeling Pdr5p (Fig. 2) consists of multiple steps: high sequence similarity in the NBDs allowed homology modeling for this domain. The relatively low sequence similarity in the TMDs required that the homology modeling be aided with additional information, whereas the lack of suitable templates for the various insertions required ab initio techniques. In the last step, a complete Pdr5p model was constructed by combining all of the separate parts. Pdr5p is significantly larger in molecular mass than its modeling templates; therefore, the placements of the NTE and insertions in the model may contain errors and require future experimental verification. Although this approach resulted in Pdr5p modules of different degrees of reliability, listed in descending order of NBD, TMD, and those built ab initio, the quality and reliability of the NBDs and TMDs were validated by mutagenesis data.
Figure 2.
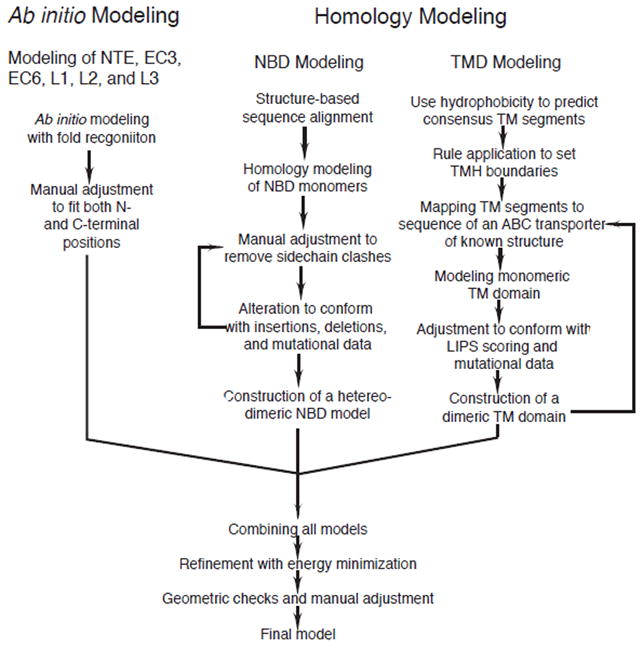
Flowchart of the strategy for modeling the yeast ABC transporter Pdr5p.
We have modeled Pdr5p in both an open and closed conformation. The closed conformation model (Fig. 1B) resembles a transporter with the TMDs open to the extracellular side and the NBDs, located on the cytoplasmic side, close together with bound nucleotides. Unique to ABC transporters with a reverse topology, the ICLs are considerably shorter, which is also true for Pdr5p. The mean Kyte-Doolittle hydrophobicity of the NTE is below 0.5 (data not shown), indicating that this portion of Pdr5p is probably located outside the membrane and exposed to the cytosol of the cell. We positioned the NTE beneath both NBDs (Fig. 1B), similar to the location of regulatory domains of some ABC metal-chelate transporters (Gerber et al., 2008). The three linkers, L1–L3, were placed on the side of the NBDs, consistent with their roles in connecting NBDs with TMDs. In the open conformation model (Fig. 1C), which lacks bound nucleotides, the separation between the NBDs increases by as much as 18Å (supplementary materials, Fig. S5). The TMDs are closed to the extracellular environment (Fig. 1C), but the distance between helices 3 and 4 of TMD1 increases relative to that in the closed conformation (supplementary materials, Fig. S6), giving intracellular substrates access to the binding pocket of the transporter. The same widening occurs between helices 9 and 10 of TMD2.
Modeling the transmembrane domains
The ABC transporter Sav1866p from Staphylococcus aureus was used as a template to model the transmembrane domains of Pdr5p in the closed conformation and the recently solved mouse P-glycoprotein transporter (Aller et al., 2009) was used as the template for the open conformation. In order to obtain a clear delineation for the transmembrane segments of Pdr5p, a multiple sequence alignment of Pdr5p and 19 homologues was made and manually edited before adding Sav1866p and P-gp to the alignment. Since Sav1866p is a half transporter and P-gp has the forward topology, their transmembrane domains were aligned separately to each of the TMDs of Pdr5p. We sought to maximize both sequence similarity and matching hydrophobicity profiles. The hydrophobicity profile analysis of the alignment with Sav1866p (supplementary materials, Fig. S3, S10) shows that all of the transmembrane segments align well with each other except the last two transmembrane helices of Sav1866p, which completely fail to align with those of Pdr5p. This is due to the fact that Sav1866p has longer ICLs than Pdr5p and that Sav1866p has no domain analogous to ECL3 or ECL6. As a result, the alignment had to be manually corrected to have matching hydrophobicity profiles by introducing large gaps to accommodate the longer ICLs of Sav1866p and the larger ECLs of Pdr5p. The same mismatch occurred in the P-gp alignment (supplementary materials, Fig. S4, S9) and was corrected manually as described above. In the final alignments, the sequence identities were 9.7% and 9.6%, respectively, and similarities of 36% and 28.9%, respectively, for the Sav1866p based TMD1 and TMD2 (Table II). For the P-gp based TMD1 and TMD2, the identities were 20% and 12%, respectively, the similarities were 36% and 34%, respectively, and the matching of the two hydrophobicity profiles improve significantly.
Table II.
Sequence alignment of TMDs with Sav1866p or P-gp and NBDs with haemolysin B (1MT0)
| TMD1 | TMD2 | NBD1 | NBD2 | |||||
|---|---|---|---|---|---|---|---|---|
| Sav1866 | P-gp | Sav1866 | P-gp | 1MT0 | P-gp | 1MT0 | P-gp | |
| % Identity | 9.7 | 20 | 9.6 | 12 | 19.4 | 19.4 | 23.7 | 20.2 |
| % Similarity | 36 | 36 | 28.9 | 34 | 45.0 | 41.3 | 50.9 | 45.7 |
Since Pdr5p has such low sequence identity and similarity to both Sav1866p and P-gp, additional steps were taken to improve the modeling of the TMDs with respect to the length or boundaries of each transmembrane helix and the orientation or lipid-facing surface of each helix. A Survey of known membrane protein structures at high resolution shows that lipid-facing patches on a TM helix consist of residues that are very hydrophobic and often non-conserved (Adamian and Liang, 2006), whereas residues forming part of the protein interior or exposed to the ligand translocation channel are less hydrophobic and more conserved. The hydrophobicity and Shannon entropy for sequence conservation at each residue position of each helix were used to determine which face of the helix most likely contacts the lipid environment. This was done by calculating the LIPS score, a product of the lipophilicity and sequence entropy for each position in each helix face (Adamian and Liang, 2006). Residues with a high LIPS score will have both high sequence entropy and high lipophilicity and are expected to face a lipid environment. Residues that contact other amino acids will have lower LIPS score because they are expected to have lower sequence entropy and lower hydrophobicity. By simultaneously considering both entropy and lipophilicity along a particular helix face, a more accurate prediction of the helix surface in contact with lipids (the LIPS surface) is expected. The LIPS surface of each transmembrane helix was used as a guide to rotate each helix of the model so that the most probable lipid-facing residues were positioned to face the lipid environment. For example, in Fig. 3A, residues in helices 7–11 are depicted by helical wheels, which are arranged in the same relative positions as these helices in the closed conformation model. The LIPS surfaces (red curves) were rotated as needed to contact lipid, which has the effect of positioning the opposite side of the helix to face the substrate-binding pocket of the transporter. This was done for both the open and closed conformations of the model.
Figure 3. Structure of the TMD of Pdr5p.

(A) LIPS surfaces. The segments of transmembrane helices 7–11 that intersect the hydrophobic core of the plasma membrane are shown as helical wheels. The red curve on each wheel marks the location of the LIPS surface. Residues in red (T1213 and Q1253) are those tested in this paper, and those in blue (E1289, Y1311, S1360 and T1364) were tested by other authors. Inset 1 shows the relationship between LIPS surfaces and the tested residues from this study, where the LIPS surface is pictured as a minimal backbone with the atoms represented by green spheres. Clearly, T1213 and Q1253 are non-LIPS residues and face the substrate-binding cavity. Inset 2 shows the position of Tyr1311 in the loop (blue) above TMH 9 (red pipe). (B) Bottom view of TMDs based on the open conformation model which has P-gp as the template. The 12 TMHs are colored in a way that the two-fold pseudo molecular symmetry is represented. The TMHs 1 and 7 are colored red, 2 and 8 yellow, 3 and 9 green, 4 and 10 cyan, 5 and 11 blue, and 6 and 12 magenta. Amino acid positions where mutations are either generated in this work or documented in literature were shown in stick models and labeled. (C) Bottom view of the TMDs based on the closed conformation model in which Sav1866 was used the template. Color codes and labels are identical to (B). From these views, the 12 TMHs are associated in two groups, helices 1, 2, and 9–11 form one group and those of 3–6, 7 and 8 form the other.
Structure and validation of the transmembrane domains
Our models for the Pdr5p TMDs start at the residues Asn519 and Pro1203 and end at residues Tyr795 and Pro1502, respectively, for TMD1 and TMD2 (Fig. 1A). Two prominent extracellular gaps in sequence alignment between TMHs 5 and 6 and between TMHs 11 and 12 are filled by ECL3 and ECL6. Viewed from the extracellular side, the 12 TMHs can be roughly divided into two groups: TM1-2, TM9-12 form one group; TM3-6 and TM7-8 form the other (Fig. 3B, C). The pseudo-dimer encloses a large, central cavity, representing the most prominent feature of the TMDs. In both the open and closed conformations, residues from TMHs 1, 3 and 6 of TMD1 and those from TMHs 7, 9 and 12 of TMD2 contribute most to the cavity wall, with a few residues from TMHs 2, 5, 8, and 11 but none from TM4 and 10.
Predicted N-glycosylation of external loops indicates that the protein is correctly oriented with respect to the membrane bilayer. The program PredictProtein (Rost et al., 2004) identified several potential Asn-glycosylation sites, but only Asn734 and Asn1447 (Fig. 3B, C, supplementary materials, Fig. S8A, B) were considered to be genuine possibilities. Only these sites meet the requirements of N-glycosylation in that they are located in extracellular loops of at least 30 residues (ECL3 and ECL6 respectively) and are more than 14 residues away from upstream or downstream transmembrane helices (van Geest and Lolkema, 2000). Another indicator of correct orientation with respect to the membrane is the positive inside charge rule. The positive charge bias (Juretic et al., 2002) is greater for the cytoplasmic loops (84) than for the extracellular loops (24), which is expected if the protein model is properly oriented.
The structure of Sav1866p and the recent structure of mouse P-gp (Aller et al., 2009) show a large substrate-binding pocket formed by the transmembrane domains. Amino acid residues lining the pocket have been shown to be polyspecific with some residues that are specific to one group of compounds and others to a different group. We expect that mutations in residues lining this substrate-binding pocket in our model should have a detectable effect on drug sensitivity. To test our model, two residues, Thr1213 and Gln1253, that face the substrate-binding pocket were substituted individually with alanine. These residues were chosen specifically because of their predicted locations (Fig. 3, supplementary materials, Fig. S8, and Table III) and because of their ability to form hydrogen bonds, which is a possible requirement for interacting with several Pdr5p substrates (Hanson et al., 2005). They are situated in the same area of the substrate-binding pocket on TMHs 7 and 8, and face away from their respective LIPS surfaces (Fig. 3A, Inset 1). Residues Thr1213 and Gln1253 are non-LIPS residues and are pointing toward the predicted substrate-binding pocket. When either residue Thr1213 or Gln1253 is replaced by alanine, the strain bearing the mutation becomes more sensitive to clotrimazole (Fig. 4A). On cycloheximide (Fig. 4B), however, the Thr1213Ala strain shows nearly wild-type levels of resistance, but the Gln1253Ala mutation renders the strain more sensitive than the wild-type. Plasma membranes were isolated from these strains and the Western blot shows no decrease in the amount of Pdr5p compared to wild-type (Fig. 4C). Taken together, these data provide strong support for our model in that Thr1213 and Gln1253 face the substrate-binding pocket and help to determine the substrate specificity of Pdr5p. The Thr1213Ala and Gln1253Ala mutants were also tested on tritylimidazole (supplementary materials, Fig. S11) and they show increased sensitivity (especially Thr1213Ala) when compared with the wild-type. The altered sensitivities of the Thr1213Ala and Gln1253Ala mutants reflect a change in drug specificity by the transporter.
Table III.
Predicted location of Pdr5p mutations and their functional correlations
| Location | Substitution | Phenotype | Ref | |
|---|---|---|---|---|
| NTE | Gly138Asp | wild-type | (Egner et al., 1998) | |
| Ser140Asn | wild-type | (Egner et al., 1998) | ||
| Val149Met | wild-type | (Egner et al., 1998) | ||
| Val150Leu | wild-type | (Egner et al., 1998) | ||
| NBD1 | Near switch region | Leu183Pro | unstable Pdr5p | (de Thozee et al., 2007) |
| 363: Switch region | Leu183Pro/Thr363Ile | Leu183Pro suppressor | (de Thozee et al., 2007) | |
| Walker A | Cys199Tyr | wild-type | (Egner et al., 1998) | |
| Near Q-loop | Asn242Lys | altered specificity | (Sauna et al., 2008) | |
| Near Q-loop | Thr257Ile | altered specificity | (Tutulan-Cunita et al., 2005) | |
| Near C-loop | Gly302Asp | altered specificity | (Egner et al., 1998) | |
| Near Walker B and switch | Thr360Ile | wild-type | (Egner et al., 1998) | |
| TMD1 | TMH2: LIPS residue, outside | Ser558Tyr | sensitive to all drugs | (Sauna et al., 2008) |
| TMH2/NBD1 | Ser558Tyr/Asn242Lys | Ser558Tyr suppressor | (Sauna et al., 2008) | |
| TMH4: LIPS residue, outside | Ser648Phe | altered specificity | (Tutulan-Cunita et al., 2005) | |
| TMH5: non-LIPS, inside | Ala676Val | wild-type | (Egner et al., 1998) | |
| TMH6: LIPS, inside | Val782Ile | wild-type | (Egner et al., 1998) | |
| TMH6: non-LIPS, inside | Val783Ile | wild-type | (Egner et al., 1998) | |
| NBD2 | Walker A | Gly905Ser/Gly908Ser | sensitive to all drugs | (Egner et al., 1998) |
| Between ATP and C-loop | Gly1009Cys | altered specificity | (Egner et al., 1998) | |
| D-loop | Gly1040Asp | altered specificity | (Tutulan-Cunita et al., 2005) | |
| 1311: ECL5 | Gly1040Asp/Tyr1311Ser | altered specificity | (Tutulan-Cunita et al., 2005) | |
| Near C-loop | Ser1048Val | altered specificity | (Tutulan-Cunita et al., 2005) | |
| Switch region | His1068Ala | altered specificity | (Ernst et al., 2008) | |
| TMD2 | TMH7: non-LIPS, inside | Thr1213Ala | altered specificity | This study |
| TMH8: non-LIPS, inside | Gln1253Ala | altered specificity | This study | |
| TMH9: non-LIPS, inside | Glu1289Lys | altered specificity | (Tutulan-Cunita et al., 2005) | |
| TMH9/ECL5 | Glu1289Lys/Tyr1311Ser | altered specificity | (Tutulan-Cunita et al., 2005) | |
| ECL5, inside | Tyr1311Ser | altered specificity | (Tutulan-Cunita et al., 2005) | |
| TMH11: non-LIPS, inside | Ser1360Phe | altered specificity | (Egner et al., 2000; Egner et al., 1998) | |
| TMH11: non-LIPS, inside | Ser1360Ala | altered specificity | (Egner et al., 2000) | |
| TMH11: non-LIPS, inside | Ser1360Thr | near wild-type | (Egner et al., 2000) | |
| TMH11: non-LIPS, inside | Thr1364Phe | altered specificity | (Egner et al., 2000) | |
| TMH11: non-LIPS, inside | Thr1364Ala | altered specificity | (Egner et al., 2000) | |
| TMH11: non-LIPS, inside | Thr1364Ser | near wild-type | (Egner et al., 2000) | |
| ECL6, outside | Thr1393Ile | altered specificity | (Tutulan-Cunita et al., 2005) | |
| ECL6, inside | Cys1427Tyr | mislocalization | (Egner et al., 1998) | |
| ECL6, outside/inside | Thr1460Ile/Val1467Ile | altered specificity | (Egner et al., 1998) | |
Figure 4. Altered drug sensitivity of Pdr5p TMD mutants.

(A) Yeast strains were tested for sensitivity to clotrimazole. Strains with a wildtype PDR5 gene are indicated by the symbol WT. The symbol Δpdr5 denotes a strain in which the pdr5 gene has been deleted. The other strains are single point mutations in the PDR5 gene as labeled. (B) Yeast strains were tested for sensitivity to cycloheximide. (C) Colloidal blue-stained SDS-PAGE gel (left) and western blot (right) of the same strains. Both crude membrane and plasma membrane preparations are shown.
Numerous other residues, located primarily within TMD2, have been reported as altering the drug specificity when mutated (Table III). By altered drug specificity, we are referring to mutations that are reported as causing a change in sensitivity to only some of the Pdr5p substrates tested. In our model, all these mutations are located either inside the substrate-binding pocket or at the entrance to the pocket. Three such residues (Glu1289, Ser1360, and Thr1364) are non-LIPS residues and face the substrate-binding pocket (Figs. 3A, 3B, 3C, and supplementary materials, Fig. S8). A fourth residue, Tyr1311, is apparently outside the TM portion of TMH9 (Fig. 3B, C, and supplementary materials, Fig. S8). Adding this residue to the helical wheel (Fig. 3A) places it on the LIPS surface of the helix; however, because Pro1307 breaks the helix before this point (Fig. 3A inset 2), Tyr1311 faces the opening to the substrate-binding pocket on the extra-cellular side of the membrane and does not align with the LIPS face in our model. Pro1307 has the same effect on the helix in both the Sav1866 and P-gp versions of the model. The Glu1289Lys/Tyr1311Ser double mutant exhibits a more severe phenotype than either single substitution, producing increased sensitivity to all drugs tested except to staurosporine, for which it shows wild-type resistance. Three different substitutions of the Ser1360 residue have been reported; the phenylalanine and alanine mutations altered specificity, whereas the threonine substitution displayed an almost wild type phenotype with only a slightly increased sensitivity. This suggests that a polar residue at this position is needed to recognize a particular set of substrates. Similar results were found for Thr1364. These four residues appear to interact in a substrate-specific manner suggesting that they are facing the substrate-binding pocket, which is in agreement with their positions in the current model of the TMDs.
In TMD1, the LIPS surface residue Ser648, located near the cytoplasmic terminus of TMH4 (Table III, Figs. 3B, 3C, and supplementary materials, Fig. S8), is reported as altering the drug specificity of Pdr5p when mutated to phenylalanine. Based on our models, it is expected that during the catalytic cycle of Pdr5p, TMH3 and TMH4 transiently separate from each other and may serve as a portion of the portal for substrate uptake (Fig. S6). Conceivably, the Ser648Phe mutation would alter the way substrates gain access to the substrate-binding pocket through the portal, therefore altering substrate specificity. The Ser558Tyr mutation affects all drugs tested (Table III, (Sauna et al., 2008)) and was shown to be defective in crosstalk between TMDs and NBDs. Consistently, in our model, this mutation does not contribute to the substrate-binding pocket. Three other mutations have been reported for hydrophobic residues in TMD1: Ala676Val, Val782Ile, and Val783Ile (Table III, Fig. 3B, C). Each of these exhibits a wild-type phenotype. The model places the three mutations in the substrate-binding pocket. Since they do not produce drug-sensitive phenotypes this may simply reflect the subtle nature of the mutations and suggests that a different substitution, such as a polar residue, may be required to produce a phenotypic change or that the drugs tested do not interact with these residues. In summary, these mutants of Pdr5p produced phenotypes that were consistent with the proposed model and demonstrated its predictive power.
Modeling of the nucleotide-binding domains
The template for the NBDs for the ATP bound, closed conformation of Pdr5p was selected by aligning the predicted sequences of the Pdr5p NBDs to a structure-based alignment of twenty proteins from the Protein Data Bank. The NBD of the haemolysin B (1MT0) ABC transporter from E. coli was the closest homologue with 19.4% identity and 45% similarity to the first NBD and with 23.7% identity and 50.9% similarity to the second (Table II). For the open conformation, the P-gp NBDs were used as the template, with 19.4% identity and 41.3% similarity to the first NBD and with 20.2% identity and 45.7% similarity to the second (Table II). Both models of the Pdr5p NBDs are hetero-dimeric and possess all of the highly conserved sequence motifs expected in an NBD (Fig. 5B, C); however, several of these motifs are unusual, a fact which has also been observed for other proteins such as MRP1p (Qin et al., 2008) and Cdr1p (Jha et al., 2004). The most notable differences are found in the Walker A motif of NBD1 and the C-loop of NBD2.
Figure 5. Structure of NBDs of Pdr5p.

(A) A portion of the structure-based multiple sequence alignment of NBDs with known structures aligned with Pdr5p; non-fungal (left) and fungal (right). The Pdr5p sequences are boxed in blue. The Walker A motif is boxed in red. Only NBD1 is shown for the fungal sequences since NBD2 has the typical lysine in the Walker A motif. The sequence pattern of typical and atypical Walker A motifs is shown above the alignments. The names of the sequences from the left alignment are the PDB codes from the Protein Data Bank for non-fungal NBDs. The names of the sequences from the right alignment are abbreviations of the PFamA code. (B) Top view of NBD1 and NBD2 heterodimer from the closed conformation model based on haemolysin B. Color codes are listed on the figure. Magnesium ions appear as green spheres near the γ-phosphate of ATP. Each ATP interacts with both NBDs. (C) Top view of NBD1 and NBD2 heterodimer from the open conformation model based on P-gp. (D) Close-up view of Nucleotide-binding site 1. (E) Close-up view of Nucleotide-binding site 2.
A remarkable change occurs in NBD1 of Pdr5p: the position of the typical lysine residue of the generic Walker A motif (Gly-X4-Gly-Lys-[Thr/Ser]) (Fig. 5A) is now occupied by cysteine, while NBD2 still carries the classical lysine residue. Conceivably, the consequences for the structure and function of NBD1 could be dramatic because of the different properties of Lys and Cys: the ability of lysine to reach out and H-bond to the β- and γ-phosphates of ATP and to facilitate the hydrolysis of the nucleotide by virtue of its positive charge, are not only unmatched but practically opposed by the short and possibly negatively charged cysteine residue (Fig. 5D, E). This cysteine is perfectly conserved in NBD1 of Pdr5p and its fungal homologues (Fig. 5A, supplementary materials, Fig. S1, S2). Previously it has been demonstrated that the Cys residue is required for ATP hydrolysis in Cdr1 (Jha et al., 2004) - a close homologue of Pdr5p. However, a Cys199Tyr mutation in Pdr5p (Table III), has no observable effect on drug resistance which suggests that the polar hydroxyl group of tyrosine is sufficient to maintain the interaction with ATP that were contributed by cysteine in the wild-type protein.
As the Walker A motif from one NBD cooperates with the C-loop of the other NBD to form a major portion of the respective tri-phosphate-binding site (Fig. 5B–E), the unusual character of NBD1 is reflected in an equally unusual C-loop of NBD2. Normally, the C-loop interacts with the γ-phosphate and ribose moieties of ATP. In NBD1, the C-loop sequence Val-Ser-Gly-Gly-Glu matches the generic sequence Leu-Ser-Gly-Gly-[Gln/Glu] well. In contrast, the C-loop of NBD2 features the sequence Leu-Asn-Val-Glu-Gln in which the central residues Asn1011, Val1012, and Glu1013 are much larger than their equivalents (Ser310, Gly311, Gly312) from NBD1. Among this set of residues the most dramatic difference in terms of size and charge is Gly312 and its corresponding residue Glu1013. The acidic residue Glu1013 adds potentially a negative charge near the γ-phosphate of the ATP from the first ATP site, whereas the corresponding residue from the other NBD is glycine. This might lead to charge repulsion which appears to be neutralized by Arg337 from the D-loop by forming a salt bridge with Glu1013 (Fig. 5D). This type of interaction should stabilize the two NBDs in the ATP-bound state, perhaps making dissociation of ADP more difficult after hydrolysis. Paralleling the conservation of the Lys-to-Cys change in one of the Walker A motifs in Pdr5p and its homologues, the unusual “Asn-Val-Glu” sequence of the C-loop and the unusual Arg of the D-loop are correspondingly well conserved (supplementary materials, Fig. S1). Taken together, this suggests that changes in the C-loop of NBD2, D-loop of NBD1 and Walker A of NBD1 form a novel interacting unit that is evolutionarily independent of the one proposed for canonical ABC transporters and which maintains a functional role. Consequently, complete assessment of the function of Cys199 will not be as simple as mutating a single residue but will require that concomitant changes from the C-loop of NBD2 and D-loop of NBD1 be considered.
Other unusual residues from NBD1 are found in Walker B and the switch region. Again, the anomalies are centered around the first ATP site and are highly conserved among Pdr5p fungal homologues (supplementary materials, Fig. S1). The Walker B motif of Pdr5p NBD1 possesses the expected catalytic aspartate (Asp333), but the following residue is asparagine (Asn334) rather than the usual glutamate. Additionally, a polar residue (Gln330 in Pdr5p) is found at the beginning of Walker B from NBD1. Normally, this is the strictly hydrophobic site of the motif. The NBD2 of Pdr5p and its homologues have neither of these deviations. In the switch region, NBD1 lacks the conserved histidine that forms a hydrogen bond with the γ-phosphate of ATP. The second NBD has the expected histidine (His1068). In NBD1 it is replaced by tyrosine (Tyr367) (Fig. 5D) that is perfectly conserved among the Pdr5p fungal homologues for the first NBD only. The residues Tyr367 and His1068 are positioned in line with their respective ATP molecules capable of forming an H-bond to the γ-phosphate. The need for the larger Tyr as opposed to a shorter His residue may be a consequence of the sterically demanding Asn1011-Val-Glu motif (C-loop of NBD2 compared to Ser310-Gly-Gly of NBD1) which laterally displaces the tri-phosphate moiety of NBD1. A tyrosine residue unlike histidine may be able to maintain the proper H-bond distance to the γ-phosphate and with it to also maintain this site’s function. Thus, in our NBD model all deviant motifs (Walker A, Walker B, the C-loop, the D-loop, and the switch region) are found surrounding the same ATP molecule. This situation may represent a complicated series of coordinated substitutions that act in concert to maintain an active site for ATP hydrolysis in NBD1.
The models of Pdr5p that we have created depict it as a monomer, which is consistent with other ABC transporters such as CFTR (Chen et al., 2002; Riordan, 2005) and P-gp (Loo and Clarke, 1996). A dimeric form of Pdr5 with rotational movements of NBDs during a catalytic cycle was suggested by an electron microscopic study (Ferreira-Pereira et al., 2003). Our model is not in conflict with a dimeric association, even though it favors a dimer of monomers (Fig. S7A). A true dimer would require pairing of N- and C-terminal domains of one monomer with opposite domains from another monomer, forming a full transporter with two TMDs and two NBDs (Figs. S7B, C). However, this arrangement is already achieved and more easily so in a dimer of monomers. It would be a far stretch for our Pdr5p model to support the rotational movement mechanism for the following reasons: (1) the requirement for pairing-up the markedly dissimilar NBD1 and NBD2 or asymmetric pairing will not be met (Figs. S7A) and (2) the perpendicular arrangement among NBDs would make the interactions observed in all crystal structures between TMD and NBD impossible to preserve, as the two NBDs and two TMDs in a transporter are intertwined.
Support of the NBD model from reported functional studies
Several mutations have been reported in the NBDs that render the yeast cell sensitive to drugs (Table III). One such case is the double mutation Gly905Ser/Gly908Ser that is located in the Walker A motif of NBD2 (Fig. 6A). Apparently the double mutation disrupts the ATP catalytic cycle since the cells bearing it are sensitive to all Pdr5p drugs. Other NBD mutations result in a differential sensitivity (de Thozee et al., 2007). Those mutations include Asn242Lys, Thr257Ile, Gly302Asp, Gly1009Cys, Gly1040Asp, Ser1048Val, and His1068Ala. Each of these is close to one of the major structural elements involved in the ATP catalytic cycle of the NBD. Conceivably, these NBD mutations should induce an altered interaction with the TMDs, which bind drugs. Indeed, the Asn242Lys and Thr257Ile mutations are situated near the Q-loop (Fig. 6B) and it seems likely that they could affect the cross-talk between the TMDs and the NBDs by interrupting existing interactions with neighboring residues. This, in turn, would influence the effectiveness of drug efflux. Unlike Thr257Ile and Asn242Lys, the Gly302Asp substitution is located near the C-loop (Fig. 6B), and the Gly1009Cys, Gly1040Asp, Ser1048Val (Fig. 6C), and His1068Ala (Fig. 6A) substitutions are all located at the interface between the two NBDs, which is the location of the ATP binding sites. Gly1009Cys is between Walker A and the C-loop. Gly1040Asp is in the D-loop, and in close proximity to the C-loop. Ser1048Val is near the D-loop and the C-loop, and His1068 is near the γ-phosphate of ATP, between the C-loop and Walker A. It is easy to see that disrupting the ATP catalytic cycle would result in drug sensitivity, but it is difficult to understand how altering this cycle results in differential sensitivity. Among many hypotheses, an intriguing possibility (Ernst et al., 2008) suggested that the kinetics of non-rate-limiting steps of the ATP catalytic cycle are altered by certain mutations in the NBD and that a change in kinetics leads to changes in the duration of equilibrium between the drug and its substrate-binding site in the TMDs. Since the on and off rates of binding vary for each drug, this decreases the efficiency with which some substrates are effluxed, producing an increased sensitivity for those drugs. In the current model, the mutations around the ATP-binding sites at the interface of the two NBDs are well-situated to disturb the kinetics of the ATP cycle.
Figure 6. Locations of NBD mutations in the model.

Color codes are listed at the bottom of the figure. (A) Glycine residues 905 and 908 of the Walker A motif and His1068 of switch region are proximal to ATP and the interface between NBD1 (orange backbone) and NBD2 (green backbone). (B) Asn242 and Thr257 are near the Q-loop while Gly302 is close to the C-loop. (C) Residues Gly1009, Gly1040, Ser1048, and C199 are located near the interface between NBD1 (orange backbone) and NBD2 (green backbone). (D) Thr 363 and I183 are proximal to each other and Thr 360 is positioned at the edge of NBD1, pointing toward the cytosol.
Several other mutants in the NBDs also provide valuable feedback about the model. The mutation Leu183Pro (Fig. 6D and Table III) results in an unstable protein that is degraded. The double mutation Leu183Pro/Thr363Ile is a partial suppressor and has the consequence that some of the Pdr5p protein is able to make it to the plasma membrane. The authors of that study (de Thozee et al., 2007) speculated that these two residues must be near each other, which is indeed the case in the model (Fig. 6D). A second interesting mutation is Thr360Ile (Fig. 6D). While this residue is located near the Walker B motif and switch region, it is positioned on the edge of the NBD, pointing to the cytosol. Therefore, it is not surprising that the Thr360Ile substitution does not produce a drug sensitive phenotype. Five other mutations fail to produce drug sensitive phenotypes. They are Gly138Asp, Ser140Asn, Val149Met, Val150Leu, and Cys199Tyr (Table III). The first four are located in the predicted NTE and therefore display expectedly wild-type phenotype.
Extracellular domains and linkers
The Pdr5p model is a hybrid created through homology modeling for TMDs and NBDs and fold recognition for the extracellular domains and linkers, which lack suitable templates. The extracellular domains and linkers are big in size, attached to the periphery of the core of the NBD-TMD (Fig. 1A, B), and assumed to fold independently. For example, L3 was generated without restraints from the nearby NBD residues. As a result, the tertiary structure of the LOOPP-generated domains had to be changed to fit into the rest of the model, leaving secondary structural elements intact. Consequently, L1–L3, NTE, ECL3, and ECL6 are less reliable than the homology modeled domains because they were modeled out of context and then manually fitted into place. However, having these elements in the model helps to delineate some TMD-NBD transition points, serves to demonstrate just how many residues are part of neither the NBDs nor TMDs, and provides visualization of possible locations where these elements may belong.
Integration of various fragments into a complete Pdr5p model and evaluation of model quality
Integration of various modules of Pdr5p became necessary because of the different approaches that were taken to model different parts of the transporter: the TMDs of the closed conformation were modeled using Sav1866p as a template; the NBDs of the closed conformation were based on the haemolysin B transporter, while the linkers and extracellular domains were modeled ab initio. The most important integration is the assembly of the TMDs and the NBDs. Even in the open conformation, which was based solely on the structure of mouse P-gp to model both the NBDs and TMDs, the NBDs had to be repositioned due to the longer intracellular linkers of P-gp compared with Pdr5p. In the closed conformation model, we aligned the pseudo two-fold axes of both domains. Since the ICLs of Sav1866p are much longer than those of Pdr5p, it was necessary to shorten the gap between the TMDs and the NBDs when combining the two domains. This was done based on the following observations. (1) By comparing structures of various bacterial ABC transporters, we found that the sites of interaction between the two domains are conserved and can be used as a guide to bring TMD and NBD into contact. (2) Sauna et al. identified a key residue Asn242, an NBD1 residue (Fig. 6B), that is essential in the crosstalk between NBD1 and TMD1 of Pdr5p (Sauna et al., 2008). This residue is indeed located at the TMD-NBD interaction site in our model, providing essential experimental support for the integration. Residue Asn242 sits directly below ICL1, which connects TMH2 and TMH3, and is positioned near the Q-loop and the Walker B motif. Moving the NBDs closer to the TMDs to reflect their interaction also necessitated placing L1, L2, and L3 (Fig. 1B) around the outside of the NBDs rather than between the NBDs and TMDs. The same rationale was followed when repositioning the NBDs closer to the TMDs in the open conformation model. Essentially, the NBDs were moved closer to the TMDs and then the NBDs were moved closer to each other until they occupied the same position relative to ICL1 and ICL3 of the P-gp template (Fig. S12).
ECL3 and ECL6 sit like a cap on top of both transmembrane domains (Fig. 1B). Rather than modeling these domains as extra helices inside of the lipid bilayer, we chose to place them near the membrane interfacial zone due to their mean hydrophobicity, which is below 0.5 on the Kyte-Doolittle scale (data not shown). We further adjusted the orientation of these two domains so that their glycosylation sites were exposed to the extracellular surface. The position of ECL3 and ECL6 is analogous to that of the binding proteins of ABC transporters from some gram-negative bacteria (Davidson and Maloney, 2007), but it is not known whether ECL3 and ECL6 have a comparable function. Reported mutations of four different ECL6 residues (Table III) suggest that these extracellular domains may have more than one function.
As mentioned at the beginning of this report, the purpose of this work is to fill a knowledge vacuum created by the lack of structural data for the fungal family of ABC transporter proteins; it is by no means a substitute for a real experimental structure at atomic resolution, nor is it expected to be a final model. Since it is only a model, a realistic evaluation of its quality, reliability and applicability should be provided. Based primarily on sequence similarities, our confidence on the quality and reliability of NBD is relatively high. The quality and reliability for the TMDs were improved significantly by incorporating prior structural knowledge despite the lower sequence similarity between Pdr5p and Sav1866p. As it is, our Pdr5p model fits all experimental data available to us, including structural, genetic, and biochemical studies on the NBDs and TMDs. We are quite confident in the general features of our model in terms of overall folding, the nucleotide binding, the TMD pocket and various interfaces. However, we are less certain about the amino acid side chain positions and their exact neighbors. The same confidence cannot be placed on the quality and reliability of NTE, ECL3, ECL6, and L1–L3 as they are not built based on homology modeling. Our current model satisfies standard stereochemical parameters derived from atomic resolution structures as verified by the Ramachandran plot. The majority of the residues (90.4% and 88% for the closed and open conformations, respectively) fall within the most favored regions. Currently, we are making a number of mutations based on the model to further improve its quality and accuracy.
Supplementary Material
Acknowledgments
This research was supported by the intramural research program of NIH, National Cancer Institute, Center for Cancer Research and by an NIH grant GM07721 to John Golin. We thank Prof. Dr. John Golin for confirming the results initially obtained by Jakub Janecka and for reading several drafts of this paper.
Abbreviations
- ABC
ATP-binding cassette
- ECL
extracellular loop
- ICL
intracellular loop
- LIPS
lipid-facing surface
- MDR
multidrug resistance
- MSA
multiple sequence alignment
- NBD
nucleotide-binding domain
- NTE
amino terminal extension
- PDB
Protein Data Bank
- P-gp
P-glycoprotein
- S. cerevisiae
Saccharomyces cerevisiae
- TM
transmembrane
- TMD
transmembrane domain
- TMH
transmembrane helix
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adamian L, Liang J. Prediction of transmembrane helix orientation in polytopic membrane proteins. BMC Struct Biol. 2006;6:13. doi: 10.1186/1472-6807-6-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albrecht M, Tosatto SC, Lengauer T, Valle G. Simple consensus procedures are effective and sufficient in secondary structure prediction. Protein Eng. 2003;16:459–62. doi: 10.1093/protein/gzg063. [DOI] [PubMed] [Google Scholar]
- Aller SG, Yu J, Ward A, Weng Y, Chittaboina S, Zhuo R, Harrell PM, Trinh YT, Zhang Q, Urbatsch IL, Chang G. Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science. 2009;323:1718–22. doi: 10.1126/science.1168750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–21. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- Chen JH, Chang XB, Aleksandrov AA, Riordan JR. CFTR is a monomer: biochemical and functional evidence. J Membr Biol. 2002;188:55–71. doi: 10.1007/s00232-001-0174-2. [DOI] [PubMed] [Google Scholar]
- Claros MG, von Heijne G. TopPred II: an improved software for membrane protein structure predictions. Comput Appl Biosci. 1994;10:685–6. doi: 10.1093/bioinformatics/10.6.685. [DOI] [PubMed] [Google Scholar]
- Davidson AL, Chen J. ATP-binding cassette transporters in bacteria. Annu Rev Biochem. 2004;73:241–68. doi: 10.1146/annurev.biochem.73.011303.073626. [DOI] [PubMed] [Google Scholar]
- Davidson AL, Maloney PC. ABC transporters: how small machines do a big job. Trends Microbiol. 2007;15:448–55. doi: 10.1016/j.tim.2007.09.005. [DOI] [PubMed] [Google Scholar]
- Dawson RJ, Locher KP. Structure of a bacterial multidrug ABC transporter. Nature. 2006;443:180–5. doi: 10.1038/nature05155. [DOI] [PubMed] [Google Scholar]
- de Thozee CP, Cronin S, Goj A, Golin J, Ghislain M. Subcellular trafficking of the yeast plasma membrane ABC transporter, Pdr5, is impaired by a mutation in the N-terminal nucleotide-binding fold. Mol Microbiol. 2007;63:811–25. doi: 10.1111/j.1365-2958.2006.05562.x. [DOI] [PubMed] [Google Scholar]
- DeGorter MK, Conseil G, Deeley RG, Campbell RL, Cole SP. Molecular modeling of the human multidrug resistance protein 1 (MRP1/ABCC1) Biochem Biophys Res Commun. 2008;365:29–34. doi: 10.1016/j.bbrc.2007.10.141. [DOI] [PubMed] [Google Scholar]
- Egner R, Bauer BE, Kuchler K. The transmembrane domain 10 of the yeast Pdr5p ABC antifungal efflux pump determines both substrate specificity and inhibitor susceptibility. Mol Microbiol. 2000;35:1255–63. doi: 10.1046/j.1365-2958.2000.01798.x. [DOI] [PubMed] [Google Scholar]
- Egner R, Rosenthal FE, Kralli A, Sanglard D, Kuchler K. Genetic separation of FK506 susceptibility and drug transport in the yeast Pdr5 ATP-binding cassette multidrug resistance transporter. Mol Biol Cell. 1998;9:523–43. doi: 10.1091/mbc.9.2.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst R, Kueppers P, Klein CM, Schwarzmueller T, Kuchler K, Schmitt L. A mutation of the H-loop selectively affects rhodamine transport by the yeast multidrug ABC transporter Pdr5. Proc Natl Acad Sci U S A. 2008;105:5069–74. doi: 10.1073/pnas.0800191105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira-Pereira A, Marco S, Decottignies A, Nader J, Goffeau A, Rigaud JL. Three-dimensional reconstruction of the Saccharomyces cerevisiae multidrug resistance protein Pdr5p. J Biol Chem. 2003;278:11995–9. doi: 10.1074/jbc.M212198200. [DOI] [PubMed] [Google Scholar]
- Finn RD, Mistry J, Schuster-Bockler B, Griffiths-Jones S, Hollich V, Lassmann T, Moxon S, Marshall M, Khanna A, Durbin R, Eddy SR, Sonnhammer EL, Bateman A. Pfam: clans, web tools and services. Nucleic Acids Res. 2006;34:D247–51. doi: 10.1093/nar/gkj149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forrest LR, Tang CL, Honig B. On the accuracy of homology modeling and sequence alignment methods applied to membrane proteins. Biophys J. 2006;91:508–17. doi: 10.1529/biophysj.106.082313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galperin MY. The Molecular Biology Database Collection: 2008 update. Nucleic Acids Res. 2008;36:D2–4. doi: 10.1093/nar/gkm1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber S, Comellas-Bigler M, Goetz BA, Locher KP. Structural basis of trans-inhibition in a molybdate/tungstate ABC transporter. Science. 2008;321:246–50. doi: 10.1126/science.1156213. [DOI] [PubMed] [Google Scholar]
- Golin J, Ambudkar SV, Gottesman MM, Habib AD, Sczepanski J, Ziccardi W, May L. Studies with novel Pdr5p substrates demonstrate a strong size dependence for xenobiotic efflux. J Biol Chem. 2003;278:5963–9. doi: 10.1074/jbc.M210908200. [DOI] [PubMed] [Google Scholar]
- Golin J, Kon ZN, Wu CP, Martello J, Hanson L, Supernavage S, Ambudkar SV, Sauna ZE. Complete inhibition of the Pdr5p multidrug efflux pump ATPase activity by its transport substrate clotrimazole suggests that GTP as well as ATP may be used as an energy source. Biochemistry. 2007;46:13109–19. doi: 10.1021/bi701414f. [DOI] [PubMed] [Google Scholar]
- Gottesman MM, Ambudkar SV. Overview: ABC transporters and human disease. J Bioenerg Biomembr. 2001;33:453–8. doi: 10.1023/a:1012866803188. [DOI] [PubMed] [Google Scholar]
- Hall TA. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symposium Series. 1999;41:95–98. [Google Scholar]
- Hanson L, May L, Tuma P, Keeven J, Mehl P, Ferenz M, Ambudkar SV, Golin J. The role of hydrogen bond acceptor groups in the interaction of substrates with Pdr5p, a major yeast drug transporter. Biochemistry. 2005;44:9703–13. doi: 10.1021/bi0502994. [DOI] [PubMed] [Google Scholar]
- Hennig L. WinPep 2.11: novel software for PC-based analyses of amino acid sequences. Prep Biochem Biotechnol. 2001;31:201–7. [PubMed] [Google Scholar]
- Higgins CF. ABC transporters: physiology, structure and mechanism--an overview. Res Microbiol. 2001;152:205–10. doi: 10.1016/s0923-2508(01)01193-7. [DOI] [PubMed] [Google Scholar]
- Hirokawa T, Boon-Chieng S, Mitaku S. SOSUI: classification and secondary structure prediction system for membrane proteins. Bioinformatics. 1998;14:378–9. doi: 10.1093/bioinformatics/14.4.378. [DOI] [PubMed] [Google Scholar]
- Hofmann K, Stoffel W. TMbase - A database of membrane spanning proteins segments. Biol Chem Hoppe-Seyler. 1993;347:166. [Google Scholar]
- Jha S, Dabas N, Karnani N, Saini P, Prasad R. ABC multidrug transporter Cdr1p of Candida albicans has divergent nucleotide-binding domains which display functional asymmetry. FEMS Yeast Res. 2004;5:63–72. doi: 10.1016/j.femsyr.2004.07.002. [DOI] [PubMed] [Google Scholar]
- Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A. 1991;47(Pt 2):110–9. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- Jordan IK, Kota KC, Cui G, Thompson CH, McCarty NA. Evolutionary and functional divergence between the cystic fibrosis transmembrane conductance regulator and related ATP-binding cassette transporters. Proc Natl Acad Sci U S A. 2008;105:18865–70. doi: 10.1073/pnas.0806306105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juretic D, Zoranic L, Zucic D. Basic charge clusters and predictions of membrane protein topology. J Chem Inf Comput Sci. 2002;42:620–32. doi: 10.1021/ci010263s. [DOI] [PubMed] [Google Scholar]
- Kolaczkowski M, van der Rest M, Cybularz-Kolaczkowska A, Soumillion JP, Konings WN, Goffeau A. Anticancer drugs, ionophoric peptides, and steroids as substrates of the yeast multidrug transporter Pdr5p. J Biol Chem. 1996;271:31543–8. doi: 10.1074/jbc.271.49.31543. [DOI] [PubMed] [Google Scholar]
- Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305:567–80. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Cryst. 1993;26:283–291. [Google Scholar]
- Leppert G, McDevitt R, Falco SC, Van Dyk TK, Ficke MB, Golin J. Cloning by gene amplification of two loci conferring multiple drug resistance in Saccharomyces. Genetics. 1990;125:13–20. doi: 10.1093/genetics/125.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu HL, Hsu JP. Recent developments in structural proteomics for protein structure determination. Proteomics. 2005;5:2056–68. doi: 10.1002/pmic.200401104. [DOI] [PubMed] [Google Scholar]
- Loo TW, Clarke DM. The minimum functional unit of human P-glycoprotein appears to be a monomer. J Biol Chem. 1996;271:27488–92. doi: 10.1074/jbc.271.44.27488. [DOI] [PubMed] [Google Scholar]
- Meller J, Elber R. Linear programming optimization and a double statistical filter for protein threading protocols. Proteins. 2001;45:241–61. doi: 10.1002/prot.1145. [DOI] [PubMed] [Google Scholar]
- Meyers S, Schauer W, Balzi E, Wagner M, Goffeau A, Golin J. Interaction of the yeast pleiotropic drug resistance genes PDR1 and PDR5. Curr Genet. 1992;21:431–6. doi: 10.1007/BF00351651. [DOI] [PubMed] [Google Scholar]
- Mishra NN, Prasad T, Sharma N, Payasi A, Prasad R, Gupta DK, Singh R. Pathogenicity and drug resistance in Candida albicans and other yeast species. A review. Acta Microbiol Immunol Hung. 2007;54:201–35. doi: 10.1556/AMicr.54.2007.3.1. [DOI] [PubMed] [Google Scholar]
- Moretti S, Armougom F, Wallace IM, Higgins DG, Jongeneel CV, Notredame C. The M-Coffee web server: a meta-method for computing multiple sequence alignments by combining alternative alignment methods. Nucleic Acids Res. 2007;35:W645–8. doi: 10.1093/nar/gkm333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–12. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Qin L, Zheng J, Grant CE, Jia Z, Cole SP, Deeley RG. Residues responsible for the asymmetric function of the nucleotide binding domains of multidrug resistance protein 1. Biochemistry. 2008;47:13952–65. doi: 10.1021/bi801532g. [DOI] [PubMed] [Google Scholar]
- Ravna AW, Sylte I, Sager G. Molecular model of the outward facing state of the human P-glycoprotein (ABCB1), and comparison to a model of the human MRP5 (ABCC5) Theor Biol Med Model. 2007;4:33. doi: 10.1186/1742-4682-4-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rea PA. MRP subfamily ABC transporters from plants and yeast. Journal of Experimental Biology. 1999;50:895–913. [Google Scholar]
- Riordan JR. Assembly of functional CFTR chloride channels. Annu Rev Physiol. 2005;67:701–18. doi: 10.1146/annurev.physiol.67.032003.154107. [DOI] [PubMed] [Google Scholar]
- Rogers B, Decottignies A, Kolaczkowski M, Carvajal E, Balzi E, Goffeau A. The pleitropic drug ABC transporters from Saccharomyces cerevisiae. J Mol Microbiol Biotechnol. 2001;3:207–14. [PubMed] [Google Scholar]
- Rost B. PHD: predicting one-dimensional protein structure by profile-based neural networks. Methods Enzymol. 1996;266:525–39. doi: 10.1016/s0076-6879(96)66033-9. [DOI] [PubMed] [Google Scholar]
- Rost B, Yachdav G, Liu J. The PredictProtein server. Nucleic Acids Res. 2004;32:W321–6. doi: 10.1093/nar/gkh377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauna ZE, Bohn SS, Rutledge R, Dougherty MP, Cronin S, May L, Xia D, Ambudkar SV, Golin J. Mutations define cross-talk between the N-terminal nucleotide-binding domain and transmembrane helix-2 of the yeast multidrug transporter Pdr5: possible conservation of a signaling interface for coupling ATP hydrolysis to drug transport. J Biol Chem. 2008;283:35010–22. doi: 10.1074/jbc.M806446200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt L, Tampe R. Structure and mechanism of ABC transporters. Curr Opin Struct Biol. 2002;12:754–60. doi: 10.1016/s0959-440x(02)00399-8. [DOI] [PubMed] [Google Scholar]
- Schmitt L, Benabdelhak H, Blight MA, Holland IB, Stubbs MT. Crystal structure of the nucleotide-binding domain of the ABC-transporter haemolysin B: identification of a variable region within ABC helical domains. J Mol Biol. 2003;330:333–42. doi: 10.1016/s0022-2836(03)00592-8. [DOI] [PubMed] [Google Scholar]
- Schultz J, Milpetz F, Bork P, Ponting CP. SMART, a simple modular architecture research tool: identification of signaling domains. Proc Natl Acad Sci U S A. 1998;95:5857–64. doi: 10.1073/pnas.95.11.5857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharom FJ. ABC multidrug transporters: structure, function and role in chemoresistance. Pharmacogenomics. 2008;9:105–27. doi: 10.2217/14622416.9.1.105. [DOI] [PubMed] [Google Scholar]
- Sipos G, Kuchler K. Fungal ATP-binding cassette (ABC) transporters in drug resistance & detoxification. Curr Drug Targets. 2006;7:471–81. doi: 10.2174/138945006776359403. [DOI] [PubMed] [Google Scholar]
- Thompson JD, Thierry JC, Poch O. RASCAL: rapid scanning and correction of multiple sequence alignments. Bioinformatics. 2003;19:1155–61. doi: 10.1093/bioinformatics/btg133. [DOI] [PubMed] [Google Scholar]
- Tutulan-Cunita AC, Mikoshi M, Mizunuma M, Hirata D, Miyakawa T. Mutational analysis of the yeast multidrug resistance ABC transporter Pdr5p with altered drug specificity. Genes Cells. 2005;10:409–20. doi: 10.1111/j.1365-2443.2005.00847.x. [DOI] [PubMed] [Google Scholar]
- van Geest M, Lolkema JS. Membrane topology and insertion of membrane proteins: search for topogenic signals. Microbiol Mol Biol Rev. 2000;64:13–33. doi: 10.1128/mmbr.64.1.13-33.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Heijne G. Mitochondrial targeting sequences may form amphiphilic helices. EMBO J. 1986;5:1335–42. doi: 10.1002/j.1460-2075.1986.tb04364.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Heijne G. Membrane proteins: from sequence to structure. Annu Rev Biophys Biomol Struct. 1994;23:167–92. doi: 10.1146/annurev.bb.23.060194.001123. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.