

Abstract
Natural products chemistry is the discipline that lies at the heart of modern pharmacognosy. The field encompasses qualitative and quantitative analytical tools that range from spectroscopy and spectrometry to chromatography. Among other things, modern research on crude botanicals is engaged in the discovery of the phytochemical constituents necessary for therapeutic efficacy, including the synergistic effects of components of complex mixtures in the botanical matrix. In the phytomedicine field, these botanicals and their contained mixtures are considered the active pharmaceutical ingredient (API), and pharmacognosists are increasingly called upon to supplement their molecular discovery work by assisting in the development and utilization of analytical tools for assessing the quality and safety of these products. Unlike single-chemical entity APIs, botanical raw materials and their derived products are highly variable because their chemistry and morphology depend on the genotypic and phenotypic variation, geographical origin and weather exposure, harvesting practices, and processing conditions of the source material. Unless controlled, this inherent variability in the raw material stream can result in inconsistent finished products that are under-potent, over-potent, and/or contaminated. Over the decades, natural products chemists have routinely developed quantitative analytical methods for phytochemicals of interest. Quantitative methods for the determination of product quality bear the weight of regulatory scrutiny. These methods must be accurate, precise, and reproducible. Accordingly, this review discusses the principles of accuracy (relationship between experimental and true value), precision (distribution of data values), and reliability in the quantitation of phytochemicals in natural products.
Keywords: Accuracy, Precision, Validation, Analytical Methods, Natural Products, Herbals
1. INTRODUCTION
The word “pharmacognosy” was coined in the early 19th century to designate the discipline related to the study of medicinal plants [1]. The science of pharmacognosy became aligned with botany and plant chemistry, and until the early 20th century, dealt mostly with physical description and identification of whole and powdered plant drugs including their history, commerce, collection, preparation, and storage. Advances in organic chemistry added a new dimension to the description and quality control of these drugs, and the discipline has since expanded to include discovery of novel chemical therapeutic agents from the natural world.
While discovery of new chemical entities has become the modern focus of much natural products work, identification and quality control remain important for pharmacopoeial identification and quality control of goods traded as crude botanicals or extracts [2]. Books and courses on analytical chemistry often do not fully describe the overall process of analytical method design, development, optimization, and validation [3]. As a result, the chemical literature is rich in procedures that have been developed with variable rigor and conclusions that imply, rather than prove, correctness and validity of reported results. Peer-review of publications that report quantitative results but are not primarily analytical papers may not address method validity and the methods may not be useful for actual samples. The role of reliable measurements in regulatory settings has obvious public health implications; tight control over active ingredients, nutrients and other constituents of foods and supplements (including deleterious substances such as pesticides and toxic elements) are necessary for safety and efficacy.
While this review cannot capture the breadth of all existing rules surrounding measurements made on commercial goods, two excerpts from U.S. Good Manufacturing Practice (GMP) regulations for drugs and dietary supplements shall highlight the importance that the U.S. government places on the integrity of data. For drugs, 21 CFR Part 211.194 (a)(2) requires a “statement of each method used. … statement shall indicate the location of data that establish that the methods used in the testing… meet proper standards of accuracy and reliability…” [4]. For dietary supplements, 21 CFR Part 111.75 requires manufacturers to “ensure that the tests and examinations that you use to determine whether the specifications are met are appropriate, scientifically valid methods”, and notes that “a scientifically valid method is one that is accurate, precise, and specific for its intended purpose” [5]. The International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) [6] defines fitness for purpose as the “degree to which data produced by a measurement process enables a user to make technically and administratively correct decisions for a stated purpose.” This relates to scope and applicability. In order for a method to be of use, it needs to be tailored to specific analytes, matrices and expected concentration ranges.
However, method development and validation can be challenging when dealing with poorly defined analytes, such as antioxidants, flavonoids and phenolics, as well as the complex matrices of botanical raw materials and finished products. Defining analytes and matrices in the fitness for purpose statement is important for developing a successful method.
2. PARAMETERS OF VALIDATION
Various organizations are involved with analytical method validation: (a) the International Union of Pure and Applied Chemistry (IUPAC) publishes chemical data and standard methods for analytical, clinical, quality control and research laboratories, while ICH has developed validation guidelines [6]; (b) FDA s “Guidance for Industry: Analytical Procedures and Methods Validation” provides recommendations on submitting analytical procedures, validation data and samples to support the documentation of the identity, strength, quality, purity and potency of drug substances and drug products [7]. A more specific guidance document focuses on the “what” and “how” of chromatographic method validation [8]; (c) AOAC International (AOACI) produces rigorous, well recognized validation guidelines that range from single laboratory validation (SLV) guidelines [9] complete with acceptance criteria [10] and sample protocol [11] to guidelines for the conduct of interlaboratory collaborative studies [12].
While there are numerous approaches to quantitative chemical analysis of natural products, space is limited and this review will focus on validation of chromatographic methods since they are the most widely used for determination of phytochemicals in raw materials and finished products. Analytical methods are not universal; characteristics, techniques, scope and applicability can differ substantially. Thus, it is impossible to have a single set of instructions that can be used to validate all methods. However they do share basic commonalities that can be addressed to ensure confidence in their use and the measurements obtained. Beyond the health implications of inaccurate measurements made on commercial products, practitioners should be aware that inaccurate quantitative measurements can cause significant bias when they are published.
2.1 ACCURACY AND PRECISION
A good starting point for basic definitions and descriptions of the key terms and concepts pertaining to the assurance of the quality of quantitative chemical measurements is the U.S. Food and Drug Administration s (FDA) Reviewer Guidance [8]. The two most important elements of a chromatographic test method are accuracy and precision. Accuracy is a measure of the closeness of the experimental value to the actual amount of the substance in the matrix. Precision measures of how close individual measurements are to each other.
2.1.1 Accuracy and Recovery
The purpose of analysis of botanicals and other natural products is quantitation of target compounds in the matrix in which the compounds occur. The most common technique for determining accuracy in natural product studies is the spike recovery method, in which the amount of a target compound is determined as a percentage of the theoretical amount present in the matrix. In a spike recovery experiment, a measured amount of the constituent of interest is added to a matrix (spiked) and then the analysis is performed on the spiked material, from the sample preparation through chromatographic determination. A comparison of the amount found versus the amount added provides the recovery of the method, which is an estimate of the accuracy of the method. In an ideal situation, such as the determination of a synthetic pesticide in food, the matrix will be devoid of the target analyte(s). However, this is seldom the case in phytochemical studies where the target analyte occurs naturally in the matrix. Therefore, analysts will frequently perform parallel analyses of spiked and un-spiked materials. The theoretical recovery of the target analyte from the spiked material is the sum of the amount of added analyte plus the amount of naturally occurring analyte (as determined in the parallel analysis of unspiked material). The difference between the theoretical amount and the amount analytically determined in the spiked matrix provides an estimate of accuracy. Other approaches to spike recovery studies include adding the target analyte to a similar matrix that does not contain the target and spiking the target analyte into natural matrix from which the target has been exhaustively extracted and then dried. Recovery is frequently concentration dependent; the FDA guidance for drugs [8] suggests that matrices be spiked at 80, 100, and 120% of the expected value, and that the experiment be performed in triplicate. For botanical materials and dietary supplements; where the analyte may be present over a large concentration range, recovery should be determined over the entire analytical range of interest for the method.
While analyte addition has both pros and cons, it is one commonly practiced in the natural products community. Other techniques such as exhaustive extraction can be used to help verify the accuracy of the method. In some cases a certified reference material may be available that contains the substance(s) of interest. These materials contain a known amount of the analyte with a given uncertainty and can be used in lieu of and/or in addition to analyte spiking. If available, certified reference materials can be obtained from national metrological laboratories such as the U.S. National Institute for Standards and Technology (NIST), the Environmental Protection Agency (EPA), or commercial suppliers.
Various factors affect the accuracy of an analytical method. These range from extraction efficiency to stability of the analyte to adequacy of the chromatographic separation and can generally be optimized during the method development and optimization phase of a study.
Important but frequently overlooked factors that affect accuracy are assumptions made in setting up and performing the assays. The first assumption involves the purity of the reference materials used to establish the identity of the analyte, create the calibration curve, and arrive at a quantitative analytical result. Available in milligram to gram quantities, these materials are usually accompanied by a label declaration of purity and/or a certificate of analysis that includes a purity declaration. Depending on their stability and the technique(s) used to determine their purity, the actual purity of these materials may differ from the claimed value, and investigators should take steps to assure identity and purity before using them.
The second assumption also involves calibration standards. There are many compounds that are not commercially available or that are prohibitively expensive. As a result, some analyses are designed to use a single compound that is nominally similar to all of the analytical targets, and quantitative results for the other compounds are expressed in terms of the one compound at hand (normalization). In UV detection, this may be appropriate if the specific extinction coefficients of the target compounds are similar; the less similar they are, the more inaccurate are the results.
2.1.2 Accuracy Case Study
An HPLC investigation [13] of cranberry (Vaccinium macrocarpon Aiton) was performed using two different means of constructing the calibration curve for the major cranberry anthocyanins. The first set of experiments was modeled after previous approaches [14] and compared results of the quantitation of individual anthocyanins in cranberry fruit using cyanidin-3-glucoside as calibrant for all compounds. The underlying assumption was that detector response at a wavelentgth of 520 nm would be the same for all of the anthocyanins. In the second experiment, the major anthocyanins were obtained and used to construct individual calibration curves for each. When individual calibration curves were used, the amounts of individual compounds were found to be different from those reported using normalization (Figure 1, Table 1).
Figure 1.
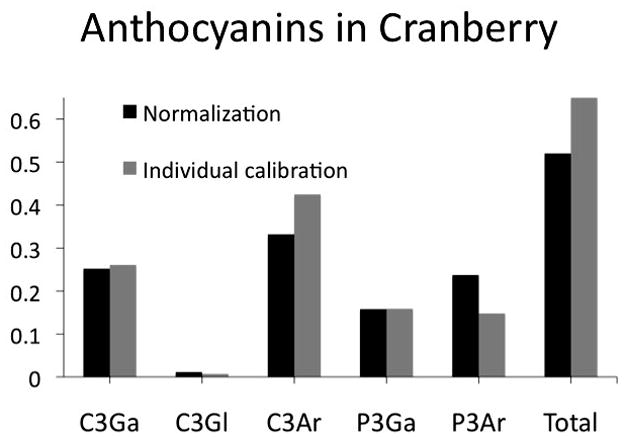
Graphical comparison of anthocyanin content in cranberry fruit when determined by normalization using cyanidin 3-O-glucoside as the external calibrant as compared to quantitation using calibration curves generated for each individual anthocyanin [7]. C3Ga=Cyanidin-3-O-galactoside, C3Gl=Cyanidin-3-O-glucoside, C3Ar= Cyanidin-3-O-Arabinoside, P3Ga=Peonidin-3-O-galactoside, P3Ar=Peonidin-3-O-galactoside.
Table 1.
Anthocyanin content of cranberries determined by HPLC using normalization to cyanidin 3-O-glucoside [13,14] vs. anthocyanin content determined using individual anthocyanins as calibrants [13]
| Anthocyanin | Calibration by normalization against C3GI [14] | Calibration with individual anthocyanins [13] |
|---|---|---|
| Cyanidin-3-O-galactoside (C3Ga) | 25.2 | 26.1 |
| Cyanidin-3-O-glucoside (C3GI) | 1.1 | 0.7 |
| Cyanidin-3-O-arabinoside (C3Ar) | 33.2 | 42.5 |
| Peonidin-3-O-galactoside (P3Ga) | 15.8 | 15.9 |
| Peonidin-3-O-arabinoside (P3Ar) | 23.7 | 14.8 |
Purity of reference materials can also affect accuracy. An illustration of the importance of verifying the purity of chemicals used as calibrants is provided in Table 2. In the HPLC investigation of cranberry anthocyanins described above [13], calibration standards for the five major cranberry anthocyanins were purchased from a commercial supplier. In preparation for the analysis, the investigator determined the purity of the purchased standards using a standard approach [15]. While the manufacturer’s certificates of analysis declared that all five compounds were > 97% pure (as determined by HPLC), the investigators found that their actual purity ranged from 66–97%. Calculation of individual anthocyanin content of cranberry using the declared purity of the calibration standards would have resulted in inaccurate results for several of the compounds. In addition, actual purities were different for different lots of the same material.
Table 2.
Claimed and actual purity of commercial cranberry anthocyanins [13]
| Anthocyanin | Supplier Purity Claim (%) | Determined Purity [15] (%) | |
|---|---|---|---|
| Lot1 | Lot 2 | ||
| Cyanidin-3-O-galactoside | >97 | 95.8 | 95.6 |
| Cyanidin-3-O-glucoside | >97 | 96.7 | 97.7 |
| Cyanidin-3-O-arabinoside | >97 | 66.1 | 87.1 |
| Peonidin-3-O-galactoside | >97 | 94.2 | 83.4 |
| Peonidin-3-O-arabinoside | >97 | 69.1 | 78.3 |
2.1.3 Precision
The FDA guidance document on validation of chromatographic methods [8] breaks the overall concept of precision into three components: repeatability, intermediate precision, and reproducibility. Repeatability is a measure of the within-laboratory uncertainty. It takes into account the reproducibility of injections and other aspects of the analysis such as weighing, fluid dispensing and handling, serial dilution, and adequacy of extraction. Among other factors, calibration of balances and glassware can increase repeatability. The guidance recommends that a validation package include data from a minimum of 10 injections that show a relative standard deviation of less than one percent. Intermediate precision is a measure of the ruggedness of the method, i.e., reliability when performed in different environments. Demonstration of intermediate precision requires that the method be run on multiple days by different analysts and on different instruments. At a minimum, such studies should be run on at least two separate occasions. Reproducibility is an indication of the precision that can be achieved between different laboratories and is evaluated using multi-laboratory collaborative studies.
As with accuracy, precision can be affected by a number of factors. Use of inappropriate or uncalibrated equipment such as pipets or analytical balances, failure to control light or moisture when required, or inadequately trained analysts can all reduce precision. Inadequate chromatographic resolution, tailing peaks, and attempts to measure different analytes across an excessive dynamic range can also decrease precision as data handling systems struggle to perform integrations against unstable baselines. The problem is especially acute when simultaneously determining low and high levels of analytes in complex natural products. Finally, the lack of homogeneity between test portions in multi-laboratory studies can result in apparent imprecision.
2.1.4 Precision-Case Study
Decoctions of Má Huáng or ephedra (Ephedra sinica Stapf., E. equisetina Bunge, E. intermedia var. tibetica Stapf., or E. distachya L.) are used in Traditional Chinese Medicine to expel cold wind. In western allopathic medicine, ephedrine and pseudoephedrine, first isolated from Ephedra spp. [16], are used for treatment of asthma and as a decongestant. Until banned from use as a dietary supplement ingredient by FDA in 2004 [17], ephedra plants and their extracts were used as ingredients in dietary supplements intended for weight loss and to “increase energy” [16]. Early FDA attempts to analyze ephedra-containing products for alkaloid content met with mixed success as the available published analytical methods were designed primarily for ephedrine and/or pseudoephedrine in finished pharmaceutical dosage forms or for a single plant species. Ephedra products marketed in the US as dietary supplements were almost always sold as mixtures of several plant species and often included caffeine and other alkaloids. Figure 2A is typical a HPLC chromatogram [18] of a multi-botanical ephedra product using a published method for separation of ephedrine alkaloids in ephedra herb [19]. The sample was run as part of an FDA investigation [18], and sample preparation involved a solvent extraction without additional cleanup. Note the complexity of the chromatogram and the incomplete resolution of the pseudoephedrine (P) and N-methylephedrine (N-ME) peaks from non-ephedra botanical constituents. The separation was sufficient to allow identification of the major alkaloids, but repeat injections of the same sample yielded different area under the curve values due to difficulties in integration.
Figure 2.

Comparison LC-UV chromatograms of dietary supplement products containing ephedra, caffeine, and other botanical ingredients using three different analytical methods. A: Extraction with no cleanup [18,19]. B: Solid-phase extraction [20,21]. C: AOAC Official Method of Analysis [22–24]. E=Ephedrine, P=Pseudoephedrine, N-ME=N-methylephedrine, NE=Norephedrine, NPE=Norpseudoephedrine, N-MPE=N-methylpseudoephedrine, Ph=Phentermine.
Figure 2B shows a chromatogram of a multi-herb ephedra product obtained [20] using a method [21] that included a solid-phase extraction cleanup step and phentermine (Ph) as internal standard. It provides for near-baseline separation of the six ephedra alkaloids in the complex multi-botanical product because the sample cleanup has removed most of the interfering substances. This method gave good precision for ephedrine (E) and pseudoephedrine (P) measurements, but norpseudoephedrine (NPE) was present in small quantities relative to E and was not well resolved from a small inflection in the baseline at about the same retention time. Thus, unreliable integration of the peak reduced precision for NPE. In addition, column performance and mobile-phase composition had to be carefully monitored for this separation. The peak eluting at 11.219 minutes in Figure 2B (just after pseudoephedrine) was identified by LC/MS as a phthalate that was leached from the solid-phase extraction (SPE) column used for cleanup. Consequently, small deviations in the organic content of the mobile-phase or column aging caused loss of resolution and imprecise integration of the pseudoephedrine peak.
Finally, Figure 2C shows a typical HPLC chromatogram of a multi-botanical ephedra product [22] obtained using the AOAC Official Method of Analysis [23]. This method yields much improved resolution and lack of interference for NPE, E, PE, and N-ME. A small interference with an unknown constituent remains with the NE peak. In the validation study that led to the approval of the official method, overall precision was deemed adequate only for E and PE [24]. Quantitative determination of the other four compounds was not sufficiently precise due to a lack of homogeneity in the blind duplicate test articles sent to the individual investigators in the collaborative study rather than to any fault of the method itself [24].
3. ADDITIONAL VALIDATION PARAMETERS
Additional parameters to be evaluated when demonstrating accuracy and precision are part of the method development and optimization process, or are performed during the validation process when demonstrating acceptable method performance. These parameters include limits of detection and quantification, linearity of the method, range, recovery, robustness and selectivity.
3.1 LIMITS OF DETECTION AND QUANTIFICATION
The Limit of Detection (LOD) is defined as the smallest amount or concentration of an analyte that can be reliably detected in a given type of sample or medium by a specific measurement process [25]. The United States Pharmacopeia defines the LOD as 2 or 3 times the baseline noise [26]. This is derived from the assumption that 3 times the noise will contain approximately 100% of the data from a normal distribution. Alternatively, the AOAC [9] and IUPAC [27] calculate limits from the variability of a blank matrix. With this methodology, the LOD is based on a minimum of 6 independent determinations of a matrix blank, where the LOD will equal the sum of the mean of blank measures and the product of the standard deviation of the blank measures and a numerical factor chosen according to the confidence level desired. The confidence level should be the Student t statistic with α = 0.05 [28], Alternatively, a value of 3 can also be used according to AOAC [9] and IUPAC [27].
The FDA chromatography guidance document notes that simply using instrument noise to estimate the limits is not adequate [8]. According to FDA, the value obtained from the chromatogram can be considered as an instrument detection limit rather than a method detection limit because the baseline noise technique does not take into consideration errors that occur during sample preparation. Although a blank that has gone through the entire sample preparation procedure may account for some of these errors, it is important to consider analyte specific effects, such as the UV extinction coefficient, which may contribute to the detection limit. Therefore, it is recommended that the LODs be calculated from the analysis of samples containing the analyte of interest [8,27,28]. The U.S. Environment Protection Agency (EPA) defines the Method Detection Limit (MDL) to be product of the standard deviation and Student t value calculated from the analysis of at least seven samples containing a low level of analyte that is near the actual detection limit [29]. All of the described methods are statistical estimates of the limit of detection and the levels should be verified under actual conditions of use.
Another limit to consider for an analytical method is the Limit of Quantification (LOQ). The LOQ is the amount of substance that can reliably be assigned a quantitative value. This limit is usually defined as 10% RSD [27] or as a fixed multiple (typically 10) of the noise [26] or standard deviation [29] used to calculate the detection limit.
3.2 LINEARITY & RANGE
In a validated method, the detector response should be linear over the anticipated range of analyte concentrations. Linearity is determined by creating a minimum 5 level calibration curve using the analyte(s) of interest. The resulting plot of detector response versus analyte concentration should have a regression coefficient of at least 0.999, and should be visually inspected for areas of non-linearity. Figures 3A and 3B [8] show plots of area under the curve versus concentration for two different analytes. Figure 3A shows an acceptable linearity over the entire range of concentrations evaluated, while Figure 3B does not. Figure 3C is a gas chromatogram of an extract of an ephedra product [16] obtained using a nitrogen/phosphorous detector. The chromatogram is enlarged to allow visualization of the minor alkaloid peaks (N-MPE, PE, N-ME, NE), and the ephedrine peak was truncated in this view. Truncation can result in integration errors, and in fact the calibration curve across the entire range of analytes was not linear. In this case, the sample had to be analyzed twice: the first analysis was performed on an undiluted sample, and the second on a diluted sample in order to bring the detector response for the ephedrine peak into the linear portion of the calibration curve. Both analyses were necessary, because the dilution step dropped the minor alkaloid concentrations below their limits of detection. Knowing the working range, (i.e., the interval between the high and low levels of analytes to be determined) of a method prevents erroneous interpretation of results.
Figure 3.

Linearity and dynamic range. A, B: Calibration curves plotting generic detector response versus concentration [8]. C: Gas chromatogram of an ephedra product extract using a Nitrogen/Phosphorous Detector [16]. E=Ephedrine, P=Pseudoephedrine, N-ME=N-methylephedrine, NPE=Norephedrine, N-MPE=N-methylpseudoephedrine, Ph=Phentermine, ?=Unknown.
3.3 ROBUSTNESS
Robustness is typically evaluated during method development/optimization, but can have a pronounced effect on the validation of a method. Robustness experiments measure a method s ability to remain unaffected by small but deliberate variations in method parameters. Examples of potentially sensitive processes include extraction time, extraction temperature, and extraction process (soxhlet, wrist shaker, orbital shaker). Column oven temperature, the percent organic phase, pH, or buffer concentration of mobile phase may also be important for chromatographic separations. Figure 4 provides a graphic comparison between chromatography outcomes in LC/MS analyses of ephedrine alkaloids and shows the differences in baseline noise, chromatographic resolution, peak shape, and analysis time achieved when HPLC columns with different carbon loading (4A) or ion-pairing reagents (4B) were used [30]. Impact of ion-pairing reagents and other factors on detector response is not addressed, but may be important to overall method performance.
Figure 4.

Representative total ion chromatogram of mixtures of six ephedra alkaloid standards plus ephedrine-d5. A: Separation using 3 different ion-pairing reagents. B: Separation on LC columns with different amounts of carbon loading. 1=norephedrine, 2=norpseudoephedrine, 3=ephedrine-d5, 4=ephedrine, 5=pseudoephedrine, 6=N-methylephedrine, and 7=methylpseudoephedrine [23].
Although the parameters affecting the method can be explored using an approach that tests one variable at a time, the use of factorial studies can be much more efficient when facing a large number of factors [9, 31]. For instance, the AOAC International recommends the use of a Youden Ruggedness Trial that permits the examination of up to 7 factors in a single experiment requiring only 8 determinations [9].
3.4 SPECIFICITY (SELECTIVITY)
It is vital to ensure the identity of the chromatographic peak that will be measured. When evaluating the previously mentioned HPLC method for determination of ephedrine-alkaloids in botanical supplements [21], a matrix blank was run using Ephedra nevadensis as the test article. This North American species was once thought to contain pseudoephedrine [32], but this claim has been controversial. Analysis using the method shown in Figure 2B produced a chromatogram (not shown) that had a flat baseline except for a small, unexpected peak that the HPLC/UV data system erroneously identified as pseudoephedrine. As noted previously, LC/MS analysis found this peak to be a phthalate from the solid-phase extraction column. Instead of an confirming the presence of pseudoephedrine in E. nevadensis, this only showed that certain solvents are incompatible with certain brands of SPE columns. The claim that E. nevadensis contains ephedrine-types alkaloids was subsequently dismissed [33]. A classical technique for verifying, but not proving, analyte identity is standard addition to a natural matrix that contains the compound of interest. Other techniques for analyte verification include the use of a photodiode array detector or a mass spectrometer. An earlier technique collects the eluted peak and performs subsequent mass spectrometry or another identity analysis.
3.5 REFERENCE MATERIALS
Finally, identity, purity, and stability of reference compounds must be confirmed. While the case for reference material purity was already made above, the authors have experienced instances in which commercial chemicals intended for use as reference materials have been incorrectly identified. In one case, proton NMR was used to confirm the identity of purchased hydrastine when received from the supplier. The experiment demonstrated that the alkaloid dimer (hydrastine) had decomposed into hydrastinine, its constituent monomers. In a second case, the detergent, 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS), had been shipped labeled as caffeine. These incidents typically do not make it into the peer-reviewed literature, but do occur.
In the age of reliable autosamplers, it is also important to assure the stability of analytical standards and target analytes in solution for the duration of the test-run. In the gas chromatogram seen in Figure 3C, the small peak eluting a few minutes before the N-MPE peak was not present when the extract was first made. As the solution aged, it turned from clear and colorless to yellow. As the color of the solution increased, so did the size of the unidentified peak. Solutions of the pure compound NE also turned yellow with time, even at refrigerator temperatures, and the size of the unknown peak increased as the intensity of the yellow color increased. More important, the size of the NE peak decreased as the size of the unknown peak increased.
In practice, it is often difficult or impossible to confirm the purity of reference materials due to their limited availability and cost. In these situations, certificates of analyses should be examined for accuracy and completeness. Determination of moisture, residual solvents, residue on ignition (inorganics), and chromatographic purity (preferably by two independent methods) are all needed to obtain an accurate assessment of material suitability. Moisture in particular can be problematic, and it is important to equilibrate the standards before use under the same conditions used prior to the moisture determination.
3.6 CHROMATOGRAPHIC PERFORMANCE
While extraction efficiency, analyte stability and purity, linearity, recovery, and selectivity are important to the final result, they must all lead to a viable separation. This is evaluated by determining system suitability. A typical approach involves development of an optimized method with adequate system suitability, prior to performing validation studies. The FDA reviewer guidance [8] suggests that the peak of interest should have a capacity factor (k′) greater than or equal to 2 and a resolution (RS) greater than 2. Additional desirable characteristics are provided in detail in the FDA guidance [6] and in numerous other sources [3, 9, 12, 26, 27, 34–37].
4. CONCLUSIONS
Systematic evaluation of analytical method performance is critical to the utility of analytical methods and to the integrity of scientific research. While accuracy, precision, and fitness for purpose are often assumed in published methods, this assumption does not bear close scrutiny in many cases. Accurate measurements are as important in clinical- and pre-clinical studies as they are in regulatory or manufacturing environments. While demonstration of performance should be a pre-requisite for any quantitative method used in a laboratory, the burden of proving that any measurements made are correct and reproducible depends on the intended use and pedigree of the method being evaluated.
There are a number of validation study designs available, and each is intended to accomplish certain pre-defined goals. In-house or single laboratory validation (SLV) studies can demonstrate applicability of the method to the analysis at hand, evaluate intra-laboratory performance, ruggedness, accuracy, and repeatability while identifying interferences and critical control points [9]. Inter-laboratory collaborative studies, including but not limited to studies for the purpose of creating AOAC Official Methods of Analysis, provide information on inter-laboratory reproducibility [12].
Finally, performing validation experiments is often viewed as “technician s work”. However, designing an appropriate validation protocol that will demonstrate the functional qualities required of the method, performing the appropriate statistics on the results, and drawing the correct conclusions from those statistics requires considerable knowledge and intellectual input. Knowledgeable senior scientists should be involved in assuring integrity of published quantitative chemical data of natural product analysis.
Supplementary Material
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ganzinger K. Zur Geschichte der Termini Pharmakognosie und Pharmakodynamik. Sci Pharm. 1982;50:351–354. [Google Scholar]
- 2.Evans WC. Trease and Evans Pharmacognosy. 15. W.B. Saunders; Edinburgh: 2002. [Google Scholar]
- 3.Swartz ME, Krull IS. Analytical Method Development and Validation. Marcel Dekker, Inc; New York: 1997. [Google Scholar]
- 4.Food and Drug Administration. Current Good Manufacturing Practice for Finished Pharmaceuticals (Title 21 Code of Federal Regulations Part 211) Department of Health and Human Services; Washington, DC: 2010. [accessed 5/11/10]. http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?CFRPart=211. [Google Scholar]
- 5.Food and Drug Administration. Current Good Manufacturing Practice in Manufacturing, Packaging, Labeling, or Holding Operations for Dietary Supplements (Title 21 Code of Federal Regulations Part 111) Department of Health and Human Services; Washington, DC: 2010. [accessed 5/11/10]. http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?CFRPart=111. [Google Scholar]
- 6.International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use, Validation of Analytical Methods: Definitions and Terminology; Geneva, Switzerland: ICH Secretariat; 2010. [accessed 5/11/10]. http://www.ich.org/cache/compo/276-254-1.html. [Google Scholar]
- 7.Center for Drug Evaluation and Research, Center for Biologicals Evaluation and Research, Food and Drug Administration. Guidance for Industry- Analytical Procedures and Methods Validation- Chemistry, Manufacturing, and Controls Documentation Draft Guidance. Department of Health and Human Services; Rockville, MD: 2000. [accessed 5/11/10]. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm122858.pdf. [Google Scholar]
- 8.Center for Drug Evaluation and Research, Food and Drug Administration. Reviewer Guidance: Validation of Chromatographic Methods. Department of Health and Human Services; Washington, DC: 1994. [accessed 4/09/10]. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM134409.pdf. [Google Scholar]
- 9.AOAC International. AOAC Guidelines for Single Laboratory Validation of Chemical Methods for Dietary Supplements and Botanicals. AOAC International; Gaithersburg, MD: 2002. [accessed 5/11/10]. http://www.aoac.org/dietsupp6/Dietary-Supplement-web-site/slv_guidelines.pdf. [Google Scholar]
- 10.AOAC International. Single Laboratory Validation Acceptance Criteria (Chemical Methods) AOAC International; Gaithersburg, MD: 2002. [accessed 5/11/10]. http://www.aoac.org/dietsupp6/Dietary-Supplement-web-site/SLV_criteria.pdf. [Google Scholar]
- 11.AOAC International. Single Laboratory Validation- Generic Protocol. AOAC International; Gaithersburg, MD: 2002. [accessed 5/11/10]. http://www.aoac.org/dietsupp6/Dietary-Supplement-web-site/SLVGeneric.pdf. [Google Scholar]
- 12.AOAC International. Method Validation. AOAC International; Gaithersbrg, MD: 2010. [accessed 5/11/10]. http://www.aoac.org/vmeth/omamanual/omamanual.htm. [Google Scholar]
- 13.Brown PN. unpublished data. [Google Scholar]
- 14.Prior RL, Lazarus SA, Cao G, Muccutelli H, Hammerstone JF. Identification of Procyanidins and Anthocyanins in Blueberries and Cranberries (Vaccinium spp.) Using High-Performance Liquid Chromatography/Mass Spectrometry. J Agric Food Chem. 2001;49:1270–6. doi: 10.1021/jf001211q. [DOI] [PubMed] [Google Scholar]
- 15.May W, Parris R, Beck C, II, Fassett J, Greenberg R, Guenther F, Kramer G, Wise S, Gills T, Colbert J, Gettings R, MacDonald B. Definition of Terms and Modes Used at NIST for Value-Assignment of Reference Materials for Chemical Measurements. [accessed Sep 2010];NIST Special Publication 260–136. 2000 http://ts.nist.gov/MeasurementServices/ReferenceMaterials/PUBLICATIONS.cfm.
- 16.Betz JM, Gay ML, Portz BS, Mossoba MM, Adams S. Chiral Gas Chromatographic Determination of ephedrine-type alkaloids in Ma-Huang Containing Dietary Supplements. J AOAC Int. 1997;80:303–315. [PubMed] [Google Scholar]
- 17.Department of Health and Human Services. 21 CFR Part 119 Final Rule Declaring Dietary Supplement Containing Ephedrine Alkaloids Adulterated Because They Present an Unreasonable Risk. Federal Register. 2004;69:6787–6854. [PubMed] [Google Scholar]
- 18.Food and Drug Administration. Unpublished data. [Google Scholar]
- 19.Zhang JS, Tian Z, Lou ZC. Simultaneous Determination of Six Alkaloids in Ephedrae Herba by High Performance Liquid Chromatography. Planta Medica. 1987;54:69–70. doi: 10.1055/s-2006-962342. [DOI] [PubMed] [Google Scholar]
- 20.Betz JM. Unpublished data. [Google Scholar]
- 21.Hurlbut JA, Carr JR, Singleton ER, Faul KC, Madson MR, Storey JM, Thomas TL. Solid-phase extraction cleanup and liquid chromatography with ultraviolet detection of ephedrine alkaloids in herbal products. J AOAC Int. 1998;81:1121–1127. [PubMed] [Google Scholar]
- 22.Roman MC. Unpublished data. [Google Scholar]
- 23.AOAC International. Official Methods of Analysis of AOAC International. 18. Gaithersburg, MD: 2008. Official Method 2003.13 Ephedrine and Pseudoephedrine in Botanicals and Dietary Supplements. [Google Scholar]
- 24.Roman M. Determination of Ephedrine Alkaloids in Botanicals and Dietary Supplements by HPLC-UV: Collaborative Study. J AOAC Int. 2004;87:1–14. [PMC free article] [PubMed] [Google Scholar]
- 25.Currie LA. Detection: International update, and some emerging dilemmas involving calibration, the blank and multiple detection decisions. Chemometr Intell Lab. 1997;37:151–181. [Google Scholar]
- 26.United States Pharmacopeia. The Official Compendia of Standards USP 32/NF29. United States Pharmacopeial Convention; Rockville, MD: 2009. Validation of Compendial Methods <1225>. [Google Scholar]
- 27.Thompson M, Ellison SLR, Wood R. Harmonized guidelines for single laboratory validation of methods of analysis (IUPAC Technical Report) Pure Appl Chem. 2002;74:835–855. [Google Scholar]
- 28.Currie LA. IUPAC nomenclature in evaluation of analytical methods including detection and quantification capabilities. Pure Appl Chem. 1995;67:1699–1723. [Google Scholar]
- 29.Environmental Protection Agency. Appendix B rev 1.11. 2003. 40 CFR Part 136 Guidelines Establishing Test Procedures for the Analysis of Pollutants; Procedures for Detection and Quantification. [Google Scholar]
- 30.Gay ML, White KD, Obermeyer WR, Betz JM, Musser SM. Determination of Ephedrine-Type Alkaloids in Dietary Supplements by LC/MS Using a Stable-Isotope Labeled Internal Standard. J AOAC Int. 2001;84:761–769. [PubMed] [Google Scholar]
- 31.Plackett RL, Burman JP. The Design of Optimum Multifactorial Experiments. Biometrika. 1946;33:305–325. [Google Scholar]
- 32.Willaman JJ, Scheubert BG. Technical Bulletin 1234. Agricultural Research Service, United States Department of Agriculture; Washington, DC, USA: 1961. Alkaloid-bearing plants and their contained alkaloids. [Google Scholar]
- 33.Caveney S, Charlet DA, Freitag H, Maier-Stolte M, Starratt AN. New Observations on the Secondary Chemistry of World Ephedra (Ephedraceae) Am J Botany. 2001;88:1199–1208. [PubMed] [Google Scholar]
- 34.United States Pharmacopeia. The Official Compendia of Standards USP 32/NF29. United States Pharmacopeial Convention; Rockville, MD: 2009. Verification of Compendial Methods <1226>. [Google Scholar]
- 35.ICH Harmonised Tripartate Guideline- Validation of Analytical Procedures: Text and Methodology Q2(R1); International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use; 2001. [accessed 4/29/2010]. http://www.ich.org/LOB/media/MEDIA417.pdf. [Google Scholar]
- 36.AOAC International. [accessed 4/29/2010];Horwitz on Validation. 2003 http://www.aoac.org/dietsupp6/Dietary-Supplement-web-site/HorwitzValid.pdf.
- 37.AOAC International. Dietary Supplements Training Materials. 2010 http://www.aoac.org/dietsupp6/Dietary-Supplement-web-site/DSHomePage2.html.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.