

Summary
Dengue virus (DENV) is a mosquito-borne flavivirus that has strained global healthcare systems throughout tropical and subtropical regions of the world. In addition to plaguing developing nations, it has re-emerged in several developed countries with recent outbreaks in the USA (CDC, 2010), Australia (Hanna et al., 2009), Taiwan (Kuan et al., 2010) and France (La Ruche et al., 2010). DENV infection can cause significant disease, including dengue fever, dengue hemorrhagic fever, dengue shock syndrome, and death. There are no approved vaccines or antiviral therapies to prevent or treat dengue-related illnesses. However, the viral NS2B-NS3 protease complex provides a strategic target for antiviral drug development since NS3 protease activity is required for virus replication. Recently, we reported two compounds with inhibitory activity against the DENV protease in vitro and antiviral activity against dengue 2 (DEN2V) in cell culture (Tomlinson et al., 2009a). Analogs of one of the lead compounds were purchased, tested in protease inhibition assays, and the data evaluated with detailed kinetic analyses. A structure activity relationship (SAR) identified key atomic determinants (i.e. functional groups) important for inhibitory activity. Four “second series” analogs were selected and tested to validate our SAR and structural models. Here, we report improvements to inhibitory activity ranging between ~2- and 60-fold, resulting in selective low micromolar dengue protease inhibitors.
Keywords: dengue virus, NS2B-NS3, protease, small-molecule inhibitor, flavivirus
1. Introduction
Dengue virus (DENV) is a member of the family Flaviviridae that includes other serious pathogens such as West Nile (WNV), yellow fever (YFV), and Japanese encephalitis (JEV) viruses. DENV exists as four distinct serotypes, each of which can cause dengue disease. DENV is estimated to infect 50–100 million individuals each year (CDC, 2008), and produce upwards of 1.5 million clinical cases of dengue disease annually. Approximately 500,000 of these cases develop into dengue hemorrhagic fever (DHF) and dengue shock syndrome (DSS), with ~25,000 fatalities every year. It has been suggested that subsequent infection by different serotypes increases the likelihood of developing the more serious forms of dengue disease such as DHF and DSS (Alvarez et al., 2006, Halstead, 2003).
DENV has primarily affected tropical and subtropical regions, and except for ~200 travel-associated cases each year, it was rarely isolated from the continental USA. However, in 2008, the US National Institutes of Health expressed concern that DENV was a potential threat to the USA (Morens et al., 2008). Those concerns were realized with recent dengue outbreaks in Florida, a southern USA state bordering the Caribbean (CDC, 2010). The continued emergence of DENV into previously temperate latitudes suggests the population at risk for DENV infection will increase in the future. Given the severity of dengue diseases, the increasing range of DENV, and the absence of approved antiviral drugs or vaccines to combat DENV infection, it is essential to develop potent anti-dengue drugs.
DENV is an enveloped virus with an ~11 kb positive-sense RNA genome that is transcribed as a single polyprotein (Figure 1A) and subsequently co- and post-translationally cleaved into three structural proteins at its 5’ end and seven nonstructural proteins at its 3’ end (Tomlinson et al., 2009b, Fields et al., 1996). Host and viral proteases, such as the N-terminal 180 residues of the NS3 protein, are responsible for post-translational processing (Bazan et al., 1989). It has been demonstrated that ~40 residues of the NS2B protein are required for full NS3 protease activity (Yusof et al., 2000). The NS2B-NS3 protease complex is required for viral replication (Falgout et al., 1991), and thus is one of the primary targets for development of anti-dengue drugs (Leyssen et al., 2000, Tomlinson et al., 2009b).
Figure 1.

(A) DENV genome including the 5’ and 3’ UTR (untranslated region), the 5’ cap, and the structural (C [capsid], PrM [premembrane] E [envelope]) and nonstructural (NS1, NS2A, NS2B, NS3, NS4A, NS4B, NS5) genes. (B) Schematic representation of the DEN2V protease expression construct (DEN2V NS2B-NS3pro) which includes the central hydrophilic domain of NS2B (cofactor) and the N-terminal protease domain of NS3.
Recent studies have used various strategies, including high throughput screening and peptide substrate analogs (Bodenreider et al., 2009, Chanprapaph et al., 2005, Ganesh et al., 2005, Kiat et al., 2006, Leung et al., 2001, Yang et al., 2011, Yin et al., 2006a, Yin et al., 2006b) to identify dengue protease inhibitors. Our previous study employed a virtual screen to identify compounds that inhibited dengue 2 virus (DEN2V) protease in vitro and prevented DEN2V replication in cell culture (Tomlinson et al., 2009a).
In this study, we present a detailed biochemical characterization of anthracene-based compounds that strongly inhibited the DEN2V NS2B-NS3 protease (NS2B-NS3pro). Inhibition constants and mechanisms of action were determined from non-linear fitting of reaction models to kinetic data. These results allowed development of a structure activity relationship (SAR) for anthracene-based small molecule inhibitors of DEN2V NS2B-NS3pro that identified key functional groups for effective binding and inhibition. The anthracene fused ring system provided a robust “scaffold” to support pharmacophores that were predicted to interact with the protease catalytic triad (His51, Asp75, Ser135) and P1 binding site residues (Gly151, Gly153, Tyr150). Furthermore, modeling analysis suggested that ligands that interacted with Tyr161 had decreased binding affinity to the protease, likely resulting from disrupted interactions with the catalytic and P1 binding site residues.
2. Materials and Methods
2.1 Analogs
Pubchem and Sigma Aldrich structure similarity search programs were employed to identify commercially-available analogs of the ARDP0006 lead (Tomlinson et al., 2009a). The OSIRIS property explorer (www.organic-chemistry.org) was used to determine clogP and predicted solubility, and compounds with predicted solubility significantly less than ARDP0006 were not selected for testing. Analogs were purchased from Sigma Aldrich (St. Louis, MO.) and Chembridge Corporation (San Diego, CA).
2.2 Solubility assays
Compounds were tested for solubility in DMSO and aqueous buffer according to the previously described protocol (Tomlinson et al., 2009a). Briefly, compounds were dissolved in DMSO at 10 mM and 1 mM. Compounds that appeared soluble by visual inspection were centrifuged at 11,000 × g (Spectrafuge 16M, LabNet International, Edison, NJ) for 30 min and inspected for insoluble pellet formation. Compounds soluble in DMSO were diluted 100-fold into aqueous assay buffer (200 mM Tris [pH 9.0], 20% glycerol) and vortexed. Compounds that appeared soluble in aqueous buffer by visual inspection were centrifuged as described above and inspected for pellet formation. Compounds that precipitated at concentrations of 1 mM in DMSO or 10 µM in aqueous assay buffer were removed from further study.
2.3 Expression and purification of DEN2V NS2B-NS3pro
The expression and purification of DEN2V (strain TSV01; Genbank accession number AY037116) NS2B cofactor linked to the protease domain of NS3 (NS2B-NS3pro (Figure 1); plasmid a generous gift from Dr. Lim Siew Pheng of the Novartis Institute for Tropical Diseases, Singapore) was modified from previously described protocols (Li et al., 2005). Briefly, expression was identical to that previously described with the exception that cultures were grown at 25°C for 8 hrs after IPTG induction. For purification, pelleted cells were first resuspended in chilled lysis buffer (50 mM HEPES [pH 7.5], 300 mM NaCl, 5% glycerol). Cell lysis was facilitated by the addition of DNase (30 µg/ml), MgCl2 (10 mM), lysozyme (300 µg/ml), and Triton X-100 (final concentration 0.5% v/v). The lysis mixture was incubated on ice, rocked gently for 1 hr, and centrifuged at 4°C and ~12,500 × g for 30 min. The soluble fraction was applied to a nickel affinity column formed from nickel sephadex beads (Amersham Biosciences) pre-equilibrated with lysis buffer. The beads were washed with lysis buffer and increasing concentrations of imidazole (5 mM, 10 mM, and 20 mM, in lysis buffer) to remove contaminating proteins. Bound NS2B-NS3pro was eluted from the column with lysis buffer and 150 mM imidazole in 1 ml aliquots, dialyzed into storage buffer (50 mM Tris [pH 7.5], 300 mM NaCl), portioned into 1 ml aliquots with 25% glycerol, flash-frozen in liquid nitrogen, and stored at −80°C. Protein concentration was determined by UV spectroscopy.
2.4 Preliminary inhibition assays
Protease activity experiments were performed in vitro using purified DEN2V NS2B-NS3pro and the 7-amino-4-methylcoumarin (AMC) fluorophore-linked peptide substrate Boc-GRR-AMC (Bachem, USA). Preliminary activity experiments were performed by incubating each soluble compound with 100 nM DEN2V NS2B-NS3pro and 100 µM Boc-GRR-AMC (Bachem, USA) in cleavage buffer (200 mM Tris [pH 9.5], 20% glycerol) for 30 min at 25°C. Release of free AMC was monitored using a Fluorolog FL3-22 spectrofluorometer (Horiba Jobin Yvon) to record fluorescence emitted at 465 nm following excitation at 380 nm. Assays were performed in duplicate. Protease reactions performed with 100 µM aprotinin, a known broad-spectrum serine protease inhibitor, showed fluorescence levels that were similar to that of the “substrate alone” background control (data not shown).
2.5 Steady-state kinetics of inhibitors of DEN2V NS2B-NS3pro
Detailed kinetic studies were performed under similar reaction conditions as described above using a broad range of substrate concentrations. Reaction progress was monitored by release of free AMC every five minutes for at least 30 minutes. All assays were performed at least two times in duplicate.
To correct for potential variations in instrument response, fluorescence from an AMC dilution series was recorded in conjunction with each protease reaction. These measurements defined the linear range and response of the spectrofluorometer, and established an AMC standard curve to correct for concentration-dependent absorption changes due to colored compounds. Briefly, each concentration of tested analog and a “no inhibitor” control were incubated with a two-fold dilution series of AMC. Relative fluorescence unit data were converted to absolute AMC product concentrations using EXCEL (Microsoft, Redmond, WA), where the data were transformed using the slope from the linear regression of the AMC dilution series. Linear regression analysis was performed using GraphPad Prism (GraphPad Software San Diego, CA).
For each tested analog, the mechanism of inhibition and inhibition constant(s) were determined from rigorous kinetic assays. Three concentrations of each inhibitor were separately mixed with cleavage buffer and DEN2V NS2B-NS3pro (100 nM final concentration). Kinetic assays were performed in duplicate in 96-well black plates (100 ul final volume/well). Serial dilutions of substrate were added to the wells for final substrate concentrations of 37.5 µM, 75 µM, 150 µM, 300 µM, 600 µM, and 1200 µM. Fluorescence of released AMC was monitored every 5 min for 30 min by emission at 465 nm (excitation 380 nm). To convert relative fluorescence units to absolute AMC concentrations, an AMC dilution series was performed as described above. Linear regression analysis was performed using GraphPad Prism (GraphPad Software San Diego, CA) to determine initial velocities for each reaction from AMC product concentrations and reaction times. Errors associated with each initial velocity measurement were consistently < 2%.
2.6 Trypsin inhibition assays
Bovine pancreatic trypsin (Sigma Aldrich, St. Louis) and N-α-Benzoyl-DL-arginine 4-nitroanilide hydrochloride (BAPNA) substrate (Sigma Aldrich, St. Louis) were used for trypsin inhibition assays. Trypsin stock solutions were prepared in sodium phosphate buffer (67 mM [pH 7.6]). Trypsin (60 µM) was incubated with chromogenic BAPNA (500 µM) and release of the para-nitroanilide product monitored using a DU640 spectrophotometer (Beckman Coulter, USA) to measure absorption at 415 nm. The slope of the progress curve was determined using linear regression and used as a baseline for comparison with inhibitor reactions. Each analog was tested at 100 µM final concentration for inhibition of trypsin. Benzamidine (Sigma Aldrich, St. Louis), a well-documented inhibitor of trypsin (Markwardt et al., 1968), was used as a trypsin inhibitor control.
2.7 Steady-state kinetics of inhibitors of trypsin
Detailed kinetic experiments were completed for analogs that inhibited trypsin in assays with the chromogenic BAPNA substrate. Experiments used the BOC-GRR-AMC substrate and were performed as described above with a 50 nM final concentration of trypsin. The fluorescence of AMC released in the trypsin assays was monitored every three minutes for twenty-four minutes.
2.8 Kinetic analysis
Initial reaction velocity versus substrate concentration data were analyzed with Dynafit (Biokin, Watertown, MA) (Kuzmic, 1996) to determine the kinetic parameters, reaction mechanism, and inhibition model. Models tested included competitive, uncompetitive, and mixed noncompetitive inhibition, with and without substrate inhibition. Inhibition data were analyzed using global non-linear least square fitting.
2.9 Modeling and structure activity relationship (SAR)
The AutoDock Vina program (Trott et al., 2010) was used to computationally bind each small molecule inhibitor to DEN2V NS2B-NS3pro (PDB identifier 2FOM; (Erbel et al., 2006). The inhibitor conformation with the lowest docking score was assumed to represent the inhibitor/protease structure and the intermolecular interactions analyzed in detail using Swiss-PDBViewer (Guex et al., 1997). Protease residues that interacted with inhibitor functional groups were tabulated if the interacting atoms were within 4 Å of each other. These interactions were used to develop a preliminary SAR (Patani et al., 1996) for this system.
3. Results
3.1 Protein expression and purification
The DEN2V NS2B-NS3pro plasmid construct (Figure 1B) included an ~40 residue central hydrophilic domain from DEN2V NS2B joined to the N-terminal protease domain of the DEN2V NS3 protein by a protease-resistant linker (Gly4-Ser-Gly4) (Li et al., 2005). The nucleotide sequence of the plasmid construct was verified by direct sequencing (S. Smith, UTMB). DEN2V NS2B-NS3pro was expressed and purified to homogeneity as visualized by Coomassie blue staining of proteins separated by SDS polyacrylamide gel electrophoresis (data not shown).
3.2 Solubility and preliminary inhibition assays
Twenty-three analogs of the previously identified DEN2V protease inhibitor ARDP0006 were purchased for testing against DEN2V NS2B-NS3pro (Table 1). Compound selection was based on commercial availability, computer-predicted aqueous solubility, occurrence of an “anthracene-like” scaffold similar to lead ARDP0006, and distribution of unique functional groups on the scaffold. Only 10 analogs were soluble to at least 10 µM in aqueous assay buffer and 1% DMSO, highlighting the need for more effective computational algorithms to predict compound solubility. The insoluble compounds were removed from further testing. The 10 soluble compounds were tested in a preliminary protease inhibition screen. Four of the 10 compounds demonstrated inhibition significantly better than the parent compound ARDP0006 (Figure 2), while an additional 4 compounds demonstrated inhibition similar to ARDP0006.
Table 1.
Analogs of lead compound ARDP0006 that were purchased for testing against DEN2V NS2B-NS3 protease. Compounds soluble in aqueous buffer were indicated with “S” and insoluble compounds indicated with “NS.” Relative solubilities (clogS, calculated with the OSIRIS program http://www.organic-chemistry.org/prog/peo) were listed in parenthesis.
| ID | Structure | Solubility (clogS*) |
ID | Structure | Solubility (clogS) |
ID | Structure | Solubility (clogS) |
|---|---|---|---|---|---|---|---|---|
| ARDP0006 | 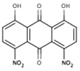 |
S (−5.1) | 6A31 | NS (−1.8) | 6A46 | 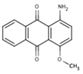 |
S (−4.8) | |
| 6A24 | NS (−4.8) | 6A33 | 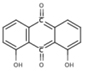 |
NS (−4.1) | 6A47 | 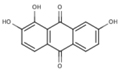 |
S (−3.8) | |
| 6A25 |  |
NS (−4.9) | 6A35 |  |
NS (−5.3) | 6A48 |  |
S (−4.5) |
| 6A26 |  |
NS (−4.9) | 6A40 | 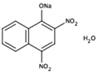 |
NS (−3.9) | 6A49 | 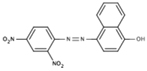 |
S (−6.4) |
| 6A27 | 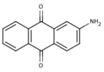 |
NS (−4.8) | 6A41 | 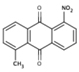 |
NS(−5.5) | 6A50 | 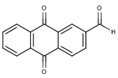 |
S (−5.1) |
| 6A28 | 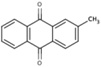 |
NS (−5.1) | 6A42 | 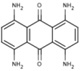 |
S(−5.0) | 6A51 | 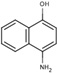 |
S (−3.0 |
| 6A29 | NS (−2.8) | 6A44 | 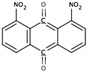 |
S(−5.7) | 6A52 | 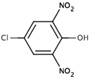 |
S (−3.0) | |
| 6A30 | 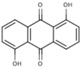 |
NS (−4.1) | 6A45 | 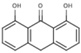 |
S(−3.8) | 6A53 | 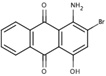 |
NS (−5.3) |
calculated log of the compound’s aqueous solubility (S), with units for S being mol l−1
Figure 2.

In vitro NS2B-NS3 protease inhibition assay for soluble analogs of lead inhibitor ARDP0006. Compounds were assayed for in vitro protease inhibition along with “no inhibitor” and no protease controls. Protease activities of each reaction were normalized to the “no inhibitor” controls.
3.3 Kinetics of inhibitors of DEN2V NS2B-NS3pro
Detailed kinetic analyses were completed on analogs 6A42, 6A45, 6A47, and 6A49 to determine their kinetic parameters, inhibition constants, and mechanism of inhibition. These compounds showed significant inhibition of DEN2V NS2B-NS3pro in the preliminary inhibition assay. Several inhibition mechanisms, including competitive, uncompetitive, and mixed noncompetitive, were examined for each compound. Analyses were performed using Dynafit and kinetic parameters for each model were optimized to provide the best global fit to the experimental data. Final model selection was based on fitting accuracy, parameter errors, and model discrimination analysis within the Dynafit program (Kuzmic, 2009). Analysis of kinetic data clearly showed that ARDP0006 and analogs 6A42, 6A45, 6A47, and 6A49 inhibited DEN2V NS2B-NS3pro in this assay (Table 2). The selected inhibition models with computed model parameters (Table 2) had excellent fits to the experimental data as shown by representative curves for the parent compound ARDP0006 (Figure 3A) and analog 6A42 (Figure 3B). A competitive inhibition model best described the ARDP0006 kinetic inhibition data. In contrast, mixed noncompetitive inhibition models (Scheme 1) best described the kinetic inhibition data for analogs 6A42, 6A45, 6A47, and 6A49.
Table 2.
Inhibition constants for analogs with in vitro activity against DEN2V NS2B-NS3pro.
| Compound | Ki1 (µM) | Ki2 (µM) | Mechanism |
|---|---|---|---|
| ARDP0006 | 432 ± 46 | - | Competitive |
| 6A42 | 158 ± 32 | 43 ± 3 | Mixed |
| 6A45 | 47 ± 15 | 77 ± 35 | Mixed |
| 6A47 | 215 ± 119 | 20 ± 2 | Mixed |
| 6A49 | 15 ± 3 | 10 ± 1 | Mixed |
Figure 3.

DEN2V NS2B-NS3 protease inhibition curves for lead inhibitor ARDP0006 (A) and analog 6A42 (B). Concentrations of inhibitor tested were 0 (circles), 50 (squares), and 100 (diamonds) µM. Data were analyzed with the program Dynafit according to Scheme 1.
Scheme 1.

Control experiments were performed with substrate concentrations ranging from 0 to 5 mM to allow unequivocal determination of Vmax and thus obtain highly accurate kinetic parameters. It should be noted that similar to findings for WNV NS2B-NS3pro (Tomlinson et al., 2008), DEN2V NS2B-NS3pro demonstrated substrate inhibition though at high ([S] = 5 mM) substrate concentrations. Kinetic analysis performed with different ranges of substrate concentrations produced similar kinetic parameters, although the calculated error estimates were slightly smaller for experiments performed with the largest range of substrate concentrations ([S] varied from 0 to 5 mM). For this reason, Km and kcat were constrained to the values obtained from the higher substrate experiments for all analyses. The kinetic parameters were essentially the same for all kinetic assays with Km = 673 ± 49 µM and kcat = 0.02 ± 0.002 s−1
3.4 Trypsin inhibition
To determine if the inhibitors were selective for DEN2V NS2B-NS3pro, each compound was tested for its ability to inhibit trypsin cleavage of the chromogenic substrate N-α-benzoyl-DL-arginine 4-nitroanilide hydrochloride (BAPNA) and the fluorogenic substrate BOC-GRR-AMC. Detailed analysis of trypsin cleavage of the fluorogenic substrate with DEN2V NS2B-NS3pro inhibitors showed that 6A49 did not inhibit trypsin, 6A42 and 6A47 were mixed noncompetitive trypsin inhibitors, ARDP0006 was an uncompetitive trypsin inhibitor, and 6A45 and benzamidine were competitive trypsin inhibitors (Table 3). Analysis of the trypsin reactions were highly reproducible, with kinetic parameters calculated as Kd = 34 ± 2 µM and kcat = 0.06 ± 0.005 s−1. Compounds that had a Ki1 value for the fluorogenic substrate (either competitive or mixed noncompetitive inhibition mechanisms) also inhibited trypsin cleavage of the chromogenic substrate, with the two competitive inhibitors showing the strongest degree of inhibition (data not shown). This was noteworthy, as we have observed compounds (e.g., ARDP0006 and inhibitors from unrelated high-throughput screening studies) that inhibited trypsin cleavage of the fluorogenic substrate but not the chromogenic substrate. In these cases, kinetic studies with the fluorogenic substrate revealed an uncompetitive inhibition mechanism (Ki2 only), which implied interactions between the inhibitor (I) and enzyme-substrate (ES) complex, but not the apo-enzyme (see Scheme 1).
Table 3.
Trypsin inhibition constants from kinetic studies.
| Compound | Ki1 trypsin (µM) | Ki2 trypsin (µM) | Mechanism |
|---|---|---|---|
| ARDP0006 | - | 13 ± 0.1 | Uncompetitive |
| 6A42 | 60 ± 13 | 167 ± 20 | Mixed |
| 6A45 | 0.6 ± 0.07 | - | Competitive |
| 6A47 | 4 ± 0.7 | 25 ± 3 | Mixed |
| 6A49 | - | - | No inhibition |
| Benzamidine | 21.6 ± 1 | - | Competitive |
Compound 6A49 inhibited the DEN2V NS2B-NS3 protease but not trypsin. In contrast, compounds 6A42, 6A45, and 6A47 inhibited both the DEN2V NS2B-NS3 protease and trypsin, and thus may not be good candidates for further optimization. However, these compounds provide useful information to understand determinants of binding affinity.
3.5 Combined modeling and SAR of the inhibitor-protease complex
The program AutoDock Vina (Trott et al., 2010) was used to initially position the DEN2V NS2B-NS3pro inhibitors ARDP0006, 6A42, 6A45, 6A47, and 6A49 into the DEN2V apo-enzyme (PDB identifier 2FOM; (Erbel et al., 2006) active site to predict likely intermolecular interactions. For each inhibitor, the AutoDock Vina program predicted several similar bound conformations that had energy differences of <1 kcal mol−1. Upon examination and comparison of the top scored conformations for all ligands, it was apparent that there was a prevailing low energy conformation that was similar for all inhibitors. In this conformation, interactions were between catalytic or P1 pocket residues of the active site and functional groups on only one edge of the inhibitors. Significant interactions occurred between hydroxyl and nitro groups of the inhibitors and conserved residues that constituted the catalytic triad (His51, Asp75, Ser135) and P1 pocket (Gly151, Gly153, Tyr150) of the protease. Inhibitors 6A42 and 6A47 shared the anthraquinone scaffold of the parent compound ARDP0006; these three compounds all “docked” such that the central ring’s carbonyl oxygen on the inhibitor’s “interacting edge” contacted the hydroxyl group of Ser135 (Figure 4). The hydroxyl groups attached to the flanking rings of the anthraquinones were predicted to interact with the imidazole ring of His51 and P1 pocket residues Tyr150, Gly151, and Gly153. Compound 6A45 had a similar scaffold but with functional groups along only one edge (termed the “interacting edge”), and demonstrated better inhibitory activity than ARDP0006, 6A42, and 6A47. Key interactions were between the hydroxyl groups of 6A45 and the imidazole ring of His51 and the hydroxyl group of Tyr150. The 6A49 inhibitor did not have the anthracene triplet ring structure, but had a comparable extended planar structure formed from the two aromatic rings connected by an azo linkage. This inhibitor had the lowest Ki1 (i.e., tightest binding) of the initial set of analogs tested, and was the only inhibitor that formed an additional contact between a nitro group of the inhibitor and the carboxyl group of Asp75. Finally, the docked structures of ARDP0006, 6A42, and 6A47 had functional groups on the “exposed edge” of the anthracene scaffold that interacted with Tyr161 (Figure 4). Since these compounds exhibited relatively high Ki1 values (i.e., low activity), these interactions likely contributed to reduced inhibitor binding.
Figure 4.

SAR suggested favorable (A) and unfavorable (B) arrangements of functional groups around the anthracene-based scaffold. Boxes represent protease residues predicted to interact with inhibitor pharmacophores based on the described computational docking studies. Grey shaded boxes represent residues that were invariant among dengue, West Nile, and Japanese encephalitis viruses.
This structure-based analysis suggested an improved inhibitor design (at least within the constraints of the compounds analyzed) for DEN2V NS2B-NS3pro (Table 4). The inhibitors’ core was an anthracene (or extended planar) scaffold and stabilizing H-bonding interactions occurred between functional groups located on the interacting edge of the inhibitor and catalytic (His51, Asp75, Ser135) and P1 pocket (Gly151, Gly153, Tyr150) residues of the protease. Since these protease residues were highly conserved among flaviviruses, interactions between them and an inhibitor would be preferred to minimize drug resistance. An additional interaction occurred with Pro132 that was located adjacent to Ser135. Interaction with Pro132 may interfere with its interactions with Ser135. Hydroxyl or carbonyl groups on the interacting edge of the central ring were predicted to interact with similar protease residues, although the latter group correlated with decreased activity perhaps because of increased bond distances. Finally, functional groups on the exposed edge of the scaffold that interacted with Tyr161 should be removed since they correlated with decreased inhibitor binding.
Table 4.
“Second series” analogs used for SAR validation. Inhibition parameters were determined from kinetic studies with DEN2V NS2B-NS3 and trypsin proteases. Compounds soluble in aqueous buffer were indicated with “S” and insoluble compounds indicated with “NS.” Relative solubilities (clogS, calculated with the OSIRIS program) were listed in parenthesis.
| Compound | Structure | Solubility (clogS*) |
DEN2V NS2B-NS3 | Trypsin | ||||
|---|---|---|---|---|---|---|---|---|
| Ki1 (µM) |
Ki2 (µM) |
Mechanism | Ki1 (µM) |
Ki2 (µM) |
Mechanism | |||
| 6A60 | 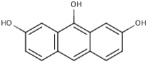 |
S (−3.9) | 7 ± 5 | 3 ± 1 | Mixed | - | - | No inhibition |
| 6A61 | 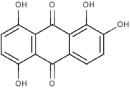 |
S (−3.6) | 72 ± 15 | 10 ± 2 | Mixed | - | - | No inhibition |
| 6A62 | 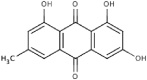 |
S (−4.2) | 508 ± 47 | - | Competitive | - | 5 ± 0.4 | Uncompetitive |
| 6A63 |  |
NS (−4.4) | ||||||
calculated log of the compound’s aqueous solubility (S), with units for S being mol l−1
To test the qualitative predictive power of the SAR, 4 “second series” anthracene-based analogs (6A60, 6A61, 6A62, 6A63) were purchased for testing (Table 4). Compound 6A60 was predicted to show better activity relative to the initial set of analogs as it contained functional groups in locations that correlated with improved activity. Compound 6A61 was predicted to have intermediate activity since it had key functional groups on the interacting edge, but additional functional groups on the exposed edge of rings 1 and 2. Compound 6A62 was predicted to be a relatively poor inhibitor due to the presence of functional groups on the exposed edge of each ring of the anthracene scaffold. Similar to 6A62, compound 6A63 was also predicted to be a relatively poor binder. Unfortunately, 6A63 was not soluble in the kinetic reaction buffer and was not tested further. Kinetic assays to determine inhibition constants for the three soluble analogs validated our predictions (Table 4), with 6A60 found to have the lowest Ki1 of all anthracene-based analogs. Moreover, this small molecule was a specific protease inhibitor since it did not inhibit trypsin cleavage activity (Table 4).
Compound 6A60 was predicted to bind to the DEN2V NS2B-NS3 active site such that functional groups on the interacting edge of the anthracene made contacts with residues that were conserved among flavivirus proteases (Figure 5). Favorable contacts were predicted between the hydroxyl of ring 1 and the NH of the side-chain of catalytic residue His51 (2.3 Å) and the carboxyl group of the catalytic residue Asp75 (2.8 Å), between the central hydroxyl of 6A60 and the hydroxyl groups of catalytic residue Ser135 (2.4 Å) and the P1 pocket residue Tyr150 (3.3 Å), and between the hydroxyl of ring 3 and the hydroxyl groups of catalytic residue Ser135 (2.4 Å) and P1 pocket residue Tyr150 (3.3 Å). Compound 6A61 demonstrated the next lowest Ki1 value and was predicted to interact with Ser135, Tyr150, and Tyr160. This compound also did not inhibit trypsin.
Figure 5.

Predicted interaction of compound 6A60 with DEN2V NS2B-NS3. Compound 6A60 was docked into the active site of the dengue protease using Vina docking software. Conserved residues were colored green, the conserved catalytic residues were colored red, and other (nonconserved) protease residues were colored blue.
A combined SAR and docking analysis using all tested anthracene-based analogs suggested that the inhibitors (with the exception of ARDP0006) formed contacts with the catalytic residue His51 of the protease. In addition, predicted interactions with Tyr150 correlated with improved (i.e., lower) Ki1 values. Inhibitors that were predicted to favorably interact with the side-chain of catalytic residue Asp75 (i.e., 6A49 and 6A60) were observed to have the lowest Ki1 values. The weak activity of 6A62 provided additional support that functional groups on the exposed edge of ring 3 (positioned to interact with Tyr161) compromised inhibitor binding. Finally, interactions with the side-chain of catalytic residue Asp75 (as predicted for 6A60 and 6A49) correlated with improved activity.
4. Discussion
Dengue virus is an important insect-borne pathogen with significant impact on global health, and thus ranks as an important target for developing small molecule drug candidates. A number of strategies have been suggested for the development of dengue antivirals (Tomlinson et al., 2009b) including targeting dengue structural proteins (Hrobowski et al., 2005, Marks et al., 2001, Modis et al., 2003, Yang et al., 2007), and nonstructural proteins such as the NS5 polymerase (Latour et al., 2010) and the NS2B-NS3 protease (Chanprapaph et al., 2005, Ganesh et al., 2005, Leung et al., 2001, Mueller et al., 2008, Tomlinson et al., 2009a, Yin et al., 2006b, Yusof et al., 2000). Viral proteases are a particularly attractive drug targets, in particular since HIV protease inhibitors have been licensed (Hsu et al., 2006, Wlodawer et al., 1998) and inhibitors of hepatitis C virus (Lamarre et al., 2003) and human rhinovirus (Hayden et al., 2003) proteases have entered clinical trials.
This study analyzed the activity of several analogs of a previously identified anthracene-based DEN2V NS2B-NS3 protease inhibitor termed ARDP0006 (Tomlinson et al., 2009a) to better understand the molecular determinants that were associated with inhibition activity. Rigorous kinetic analyses provided both accurate inhibition constants and mechanisms of inhibition. Interestingly, 6 of 8 analogs exhibited a mixed noncompetitive mechanism of inhibition with both Ki1 and Ki2 values, which suggested these inhibitors bound the apoenzyme (E) and the enzyme-substrate (ES) complex (see scheme 1). The observation of a non-competitive mode of inhibition implied that there was a substrate-dependent binding event that could inhibit the protease even at high substrate concentrations; this could have significant in vivo ramifications since the protease would be inhibited during high levels of replication (high levels of polyprotein substrate). Alternatively, the observed uncompetitive component of inhibition could be an artifact of the AMC-coupled substrate. Evidence for this latter interpretation was observed in trypsin inhibition kinetic studies using substrates with either chromogenic or fluorogenic leaving groups. In these experiments, some inhibitors prevented cleavage of the fluorogenic substrate (via an uncompetitive mechanism) but not the chromogenic substrate (data not shown). Thus, this dengue protease inhibition study utilized Ki1 values (competitive and mixed noncompetitive mechanisms) to develop a SAR, since the Ki2 values may have included uncompetitive interactions with the ES complex and/or substrate-dependent artifacts due to interactions between inhibitors and the AMC moiety. Rigorous kinetic analyses, as opposed to simple determination of IC50 values, were necessary to decide which compounds were incorporated into SAR and developed as antiviral leads.
SAR and docking studies of anthracene-based compounds suggested potential interactions between the NS2B-NS3 protease and functional groups flanking the inhibitor scaffold. Interactions involved conserved catalytic and active site residues. Highest inhibitory activity was associated with hydroxyl groups situated on a common edge of all three anthracene rings. Functional groups on the opposite (i.e., exposed) edge of the anthracene scaffold correlated with reduced activity. Screening of a limited number of commercially-available anthracene-based “second series” analogs resulted in a sixty-fold decrease in the Ki1 value relative to lead compound ARDP0006 and an increase in selectivity relative to trypsin inhibition (Table 1, 4). The above SAR and structural models can guide rational modification of the anthracene scaffold to develop selective sub-micromolar binding inhibitors.
RNA virus such as DENV were estimated to have a mutation rate between 10−3 and 10−5 per base per generation (Drake, 1993). The emergence of drug resistant strains are well documented in HIV (Trono et al., 2010) and influenza (Holmes, 2010), and have resulted in the eventual ineffectiveness of some antiviral therapeutics. The emergence of drug-resistant strains is an issue that will need to be addressed before DENV antiviral drugs are approved for use in humans. One promising approach to this issue will be to develop dengue antivirals that interact with conserved residues of the protease, thereby increasing the likelihood that drug-resistant mutations will be detrimental to the fitness and survival of the mutated virus. Since the above anthracene-based inhibitors were predicted to interact with conserved residues of the catalytic triad and active site, drugs developed from these compounds may delay the emergence of drug-resistant dengue viruses. Moreover, since these residues were invariant in all dengue virus serotypes and in distant flaviviruses such as West Nile, Japanese encephalitis, and yellow fever viruses, these inhibitors may serve as the basis for developing broad-spectrum antivirals.
5. Conclusions
This study reported the biochemical analysis and SAR of anthracene-based analogs of a lead DEN2V NS2B-NS3pro inhibitor. From SAR and structural models, we developed a design strategy from which to proceed with inhibitor improvement. Comprehensive kinetic studies identified selective low molecular weight analogs with an ~60-fold increase in inhibition (as evidenced by decreased Ki1) over the parent compound. Future studies will include cell culture and small animal studies.
Acknowledgements
We would like to thank Dr. Siew Pheng (Novartis Institute for Tropical Diseases) for the plasmid encoding DEN2V NS2B-NS3pro. In addition, we thank Drs. A. Barrett, S. Gilbertson, J. Halpert, J. Lee, and J. Stevens for helpful discussions.
Abbreviations
- DENV
dengue virus
- DEN2V
dengue 2 virus
- NS
nonstructural
- NS2B-NS3pro
NS3 protease domain with NS2B cofactor
- DHF
dengue hemorrhagic fever
- DSS
dengue shock syndrome
- GKR
glycine-lysine-arginine
- AMC
7-amino-4-methylcoumarin
- SAR
structure activity relationship
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
This research was supported in part by NIH/NIAID AI066160 (SJW), the Welch Foundation H-1642 (SJW), a training fellowship to SMT from the Computational and Structural Biology in Biodefense Training Program of the W. M. Keck Center for Interdisciplinary Bioscience Training of the Gulf Coast Consortia (NIAID Grant No. 1 T32 AI065396-01), and a National Library of Medicine training fellowship in Computational Biology and Biomedical Informatics to the Keck Center for Interdisciplinary Bioscience Training of the Gulf Coast Consortia (NLM Grant No. T15 LM007093).
References
- Alvarez M, Rodriguez-Roche R, Bernardo L, Vazquez S, Morier L, Gonzalez D, Castro O, Kouri G, Halstead SB, Guzman MG. Dengue hemorrhagic Fever caused by sequential dengue 1–3 virus infections over a long time interval: Havana epidemic, 2001–2002. Am J Trop Med Hyg. 2006;75:1113–1117. [PubMed] [Google Scholar]
- Bazan JF, Fletterick RJ. Detection of a trypsin-like serine protease domain in flaviviruses and pestiviruses. Virology. 1989;171:637–639. doi: 10.1016/0042-6822(89)90639-9. [DOI] [PubMed] [Google Scholar]
- Bodenreider C, Beer D, Keller TH, Sonntag S, Wen D, Yap L, Yau YH, Shochat SG, Huang D, Zhou T, Caflisch A, Su XC, Ozawa K, Otting G, Vasudevan SG, Lescar J, Lim SP. A fluorescence quenching assay to discriminate between specific and nonspecific inhibitors of dengue virus protease. Anal Biochem. 2009;395:195–204. doi: 10.1016/j.ab.2009.08.013. [DOI] [PubMed] [Google Scholar]
- CDC. Dengue Fever. 2008 [Google Scholar]
- CDC. Locally acquired Dengue--Key West, Florida, 2009–2010. MMWR Morb Mortal Wkly Rep. 2010;59:577–581. [PubMed] [Google Scholar]
- Chanprapaph S, Saparpakorn P, Sangma C, Niyomrattanakit P, Hannongbua S, Angsuthanasombat C, Katzenmeier G. Competitive inhibition of the dengue virus NS3 serine protease by synthetic peptides representing polyprotein cleavage sites. Biochem Biophys Res Commun. 2005;330:1237–1246. doi: 10.1016/j.bbrc.2005.03.107. [DOI] [PubMed] [Google Scholar]
- Drake JW. Rates of spontaneous mutation among RNA viruses. Proc Natl Acad Sci U S A. 1993;90:4171–4175. doi: 10.1073/pnas.90.9.4171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erbel P, Schiering N, D'Arcy A, Renatus M, Kroemer M, Lim SP, Yin Z, Keller TH, Vasudevan SG, Hommel U. Structural basis for the activation of flaviviral NS3 proteases from dengue and West Nile virus. Nat Struct Mol Biol. 2006;13:372–373. doi: 10.1038/nsmb1073. [DOI] [PubMed] [Google Scholar]
- Falgout B, Pethel M, Zhang Y, Lai C. Both nonstructural proteins NS2B and NS3 are required for the proteolytic processing of dengue virus nonstructural proteins. J Virol. 1991;65:2467–2475. doi: 10.1128/jvi.65.5.2467-2475.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields B, Knipe D, Howley P, Chanock R, Melnick J, Monath T, Roizman B, Strauss S. Field's Virology. Third Edition. Philadelphia: Lippincott Williams & Wilkins; 1996. [Google Scholar]
- Ganesh VK, Muller N, Judge K, Luan CH, Padmanabhan R, Murthy KH. Identification and characterization of nonsubstrate based inhibitors of the essential dengue and West Nile virus proteases. Bioorg Med Chem. 2005;13:257–264. doi: 10.1016/j.bmc.2004.09.036. [DOI] [PubMed] [Google Scholar]
- Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- Halstead SB. Neutralization and antibody-dependent enhancement of dengue viruses. Adv Virus Res. 2003;60:421–467. doi: 10.1016/s0065-3527(03)60011-4. [DOI] [PubMed] [Google Scholar]
- Hanna JN, Ritchie SA, Richards AR, Humphreys JL, Montgomery BL, Ehlers GJ, Pyke AT, Taylor CT. Dengue in north Queensland, 2005–2008. Commun Dis Intell. 2009;33:198–203. doi: 10.33321/cdi.2009.33.19. [DOI] [PubMed] [Google Scholar]
- Hayden FG, Turner RB, Gwaltney JM, Chi-Burris K, Gersten M, Hsyu P, Patick AK, Smith GJ, 3rd, Zalman LS. Phase II, randomized, double-blind, placebo-controlled studies of ruprintrivir nasal spray 2-percent suspension for prevention and treatment of experimentally induced rhinovirus colds in healthy volunteers. Antimicrob Agents Chemother. 2003;47:3907–3916. doi: 10.1128/AAC.47.12.3907-3916.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes EC. Virology. Helping the resistance. Science. 2010;328:1243–1244. doi: 10.1126/science.1190994. [DOI] [PubMed] [Google Scholar]
- Hrobowski YM, Garry RF, Michael SF. Peptide inhibitors of dengue virus and West Nile virus infectivity. Virol J. 2005;2:49. doi: 10.1186/1743-422X-2-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu JT, Wang HC, Chen GW, Shih SR. Antiviral drug discovery targeting to viral proteases. Curr Pharm Des. 2006;12:1301–1314. doi: 10.2174/138161206776361110. [DOI] [PubMed] [Google Scholar]
- Kiat TS, Pippen R, Yusof R, Ibrahim H, Khalid N, Rahman NA. Inhibitory activity of cyclohexenyl chalcone derivatives and flavonoids of fingerroot, Boesenbergia rotunda (L.), towards dengue-2 virus NS3 protease. Bioorganic & Medicinal Chemistry Letters. 2006;16:3337–3340. doi: 10.1016/j.bmcl.2005.12.075. [DOI] [PubMed] [Google Scholar]
- Kuan MM, Chang FY, Lin T, Chuang JH, Wu HS. Epidemiological trends and the effect of airport fever screening on prevention of domestic dengue fever outbreaks in Taiwan, 1998–2007. Int J Infect Dis. 2010 doi: 10.1016/j.ijid.2009.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzmic P. Program DYNAFIT for the analysis of enzyme kinetic data: application to HIV proteinase. Anal Biochem. 1996;237:260–273. doi: 10.1006/abio.1996.0238. [DOI] [PubMed] [Google Scholar]
- Kuzmic P. DynaFit--a software package for enzymology. Methods Enzymol. 2009;467:247–280. doi: 10.1016/S0076-6879(09)67010-5. [DOI] [PubMed] [Google Scholar]
- La Ruche G, Souares Y, Armengaud A, Peloux-Petiot F, Delaunay P, Despres P, Lenglet A, Jourdain F, Leparc-Goffart I, Charlet F, Ollier L, Mantey K, Mollet T, Fournier J, Torrents R, Leitmeyer K, Hilairet P, Zeller H, Van Bortel W, Dejour-Salamanca D, Grandadam M, Gastellu-Etchegorry M. First two autochthonous dengue virus infections in metropolitan France, September 2010. Euro Surveill. 2010;15 [PubMed] [Google Scholar]
- Lamarre D, Anderson PC, Bailey M, Beaulieu P, Bolger G, Bonneau P, Bos M, Cameron DR, Cartier M, Cordingley MG, Faucher AM, Goudreau N, Kawai SH, Kukolj G, Lagace L, LaPlante SR, Narjes H, Poupart MA, Rancourt J, Sentjens RE, St George R, Simoneau B, Steinmann G, Thibeault D, Tsantrizos YS, Weldon SM, Yong CL, Llinas-Brunet M. An NS3 protease inhibitor with antiviral effects in humans infected with hepatitis C virus. Nature. 2003;426:186–189. doi: 10.1038/nature02099. [DOI] [PubMed] [Google Scholar]
- Latour DR, Jekle A, Javanbakht H, Henningsen R, Gee P, Lee I, Tran P, Ren S, Kutach AK, Harris SF, Wang SM, Lok SJ, Shaw D, Li J, Heilek G, Klumpp K, Swinney DC, Deval J. Biochemical characterization of the inhibition of the dengue virus RNA polymerase by beta-d-2'-ethynyl-7-deaza-adenosine triphosphate. Antiviral Res. 2010;87:213–222. doi: 10.1016/j.antiviral.2010.05.003. [DOI] [PubMed] [Google Scholar]
- Leung D, Schroder K, White H, Fang NX, Stoermer MJ, Abbenante G, Martin JL, Young PR, Fairlie DP. Activity of recombinant dengue 2 virus NS3 protease in the presence of a truncated NS2B co-factor, small peptide substrates, and inhibitors. J Biol Chem. 2001;276:45762–45771. doi: 10.1074/jbc.M107360200. [DOI] [PubMed] [Google Scholar]
- Leyssen P, De Clercq E, Neyts J. Perspectives for the treatment of infections with Flaviviridae. Clin Microbiol Rev. 2000;13:67–82. doi: 10.1128/cmr.13.1.67-82.2000. table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Lim SP, Beer D, Patel V, Wen D, Tumanut C, Tully DC, Williams JA, Jiricek J, Priestle JP, Harris JL, Vasudevan SG. Functional profiling of recombinant NS3 proteases from all four serotypes of dengue virus using tetrapeptide and octapeptide substrate libraries. J Biol Chem. 2005;280:28766–28774. doi: 10.1074/jbc.M500588200. [DOI] [PubMed] [Google Scholar]
- Marks RM, Lu H, Sundaresan R, Toida T, Suzuki A, Imanari T, Hernaiz MJ, Linhardt RJ. Probing the interaction of dengue virus envelope protein with heparin: assessment of glycosaminoglycan-derived inhibitors. J Med Chem. 2001;44:2178–2187. doi: 10.1021/jm000412i. [DOI] [PubMed] [Google Scholar]
- Markwardt F, Landmann H, Walsmann P. Comparative studies on the inhibition of trypsin, plasmin, and thrombin by derivatives of benzylamine and benzamidine. Eur J Biochem. 1968;6:502–506. doi: 10.1111/j.1432-1033.1968.tb00473.x. [DOI] [PubMed] [Google Scholar]
- Modis Y, Ogata S, Clements D, Harrison SC. A ligand-binding pocket in the dengue virus envelope glycoprotein. Proc Natl Acad Sci U S A. 2003;100:6986–6991. doi: 10.1073/pnas.0832193100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morens DM, Fauci AS. Dengue and hemorrhagic fever: a potential threat to public health in the United States. JAMA. 2008;299:214–216. doi: 10.1001/jama.2007.31-a. [DOI] [PubMed] [Google Scholar]
- Mueller NH, Pattabiraman N, Ansarah-Sobrinho C, Viswanathan P, Pierson TC, Padmanabhan R. Identification and biochemical characterization of smallmolecule inhibitors of west nile virus serine protease by a high-throughput screen. Antimicrob Agents Chemother. 2008;52:3385–3393. doi: 10.1128/AAC.01508-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patani GA, LaVoie EJ. Bioisosterism: A Rational Approach in Drug Design. Chem Rev. 1996;96:3147–3176. doi: 10.1021/cr950066q. [DOI] [PubMed] [Google Scholar]
- Tomlinson SM, Malmstrom RD, Russo A, Mueller N, Pang YP, Watowich SJ. Structure-based discovery of dengue virus protease inhibitors. Antiviral Res. 2009a;82:110–114. doi: 10.1016/j.antiviral.2009.02.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomlinson SM, Malmstrom RD, Watowich SJ. New approaches to structurebased discovery of dengue protease inhibitors. Infect Disord Drug Targets. 2009b;9:327–343. doi: 10.2174/1871526510909030327. [DOI] [PubMed] [Google Scholar]
- Tomlinson SM, Watowich SJ. Substrate inhibition kinetic model for West Nile virus NS2B-NS3 protease. Biochemistry. 2008;47:11763–11770. doi: 10.1021/bi801034f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trono D, Van Lint C, Rouzioux C, Verdin E, Barre-Sinoussi F, Chun TW, Chomont N. HIV persistence and the prospect of long-term drug-free remissions for HIV-infected individuals. Science. 2010;329:174–180. doi: 10.1126/science.1191047. [DOI] [PubMed] [Google Scholar]
- Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wlodawer A, Vondrasek J. Inhibitors of HIV-1 protease: a major success of structure-assisted drug design. Annu Rev Biophys Biomol Struct. 1998;27:249–284. doi: 10.1146/annurev.biophys.27.1.249. [DOI] [PubMed] [Google Scholar]
- Yang CC, Hsieh Y-C, Lee S-J, Wu S-H, Liao C-L, Tsao C-H, Chao Y-S, Chern J-H, Wu C-P, Yueh A. Novel Dengue Virus-Specific NS2B/NS3 Protease Inhibitor, B 1 P2109, Discovered by a High-Throughput Screening Assay. Antimicrob. Agents Chemother. 2011 doi: 10.1128/AAC.00855-10. doi:10.1128/AAC.00855-10 (published online ahead of print on 11 October 2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JM, Chen YF, Tu YY, Yen KR, Yang YL. Combinatorial computational approaches to identify tetracycline derivatives as flavivirus inhibitors. PLos One. 2007;2:e428. doi: 10.1371/journal.pone.0000428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Z, Patel SJ, Wang WL, Chan WL, Ranga Rao KR, Wang G, Ngew X, Patel V, Beer D, Knox JE, Ma NL, Ehrhardt C, Lim SP, Vasudevan SG, Keller TH. Peptide inhibitors of dengue virus NS3 protease. Part 2: SAR study of tetrapeptide aldehyde inhibitors. Bioorg Med Chem Lett. 2006a;16:40–43. doi: 10.1016/j.bmcl.2005.09.049. [DOI] [PubMed] [Google Scholar]
- Yin Z, Patel SJ, Wang WL, Wang G, Chan WL, Rao KR, Alam J, Jeyaraj DA, Ngew X, Patel V, Beer D, Lim SP, Vasudevan SG, Keller TH. Peptide inhibitors of Dengue virus NS3 protease. Part 1: Warhead. Bioorg Med Chem Lett. 2006b;16:36–39. doi: 10.1016/j.bmcl.2005.09.062. [DOI] [PubMed] [Google Scholar]
- Yusof R, Clum S, Wetzel M, Murthy HM, Padmanabhan R. Purified NS2B/NS3 serine protease of dengue virus type 2 exhibits cofactor NS2B dependence for cleavage of substrates with dibasic amino acids in vitro. J Biol Chem. 2000;275:9963–9969. doi: 10.1074/jbc.275.14.9963. [DOI] [PubMed] [Google Scholar]