

Abstract
Alcoholism is a chronic relapsing disorder. Major characteristics observed in alcoholics during an initial period of alcohol abstinence are altered physiological functions and a negative emotional state. Evidence suggests that a persistent, cumulative adaptation involving a kindling/allostasis-like process occurs during the course of repeated chronic alcohol exposures that is critical for the negative symptoms observed during alcohol withdrawal. Basic studies have provided evidence for specific neurotransmitters within identified brain sites being responsible for the negative emotion induced by the persistent cumulative adaptation following intermittent-alcohol exposures. After an extended period of abstinence, the cumulative alcohol adaptation increases susceptibility to stress- and alcohol cue-induced negative symptoms and alcohol seeking, both of which can facilitate excessive ingestion of alcohol. In the alcoholic, stressful imagery and alcohol cues alter physiological responses, enhance negative emotion, and induce craving. Brain fMRI imaging following stress and alcohol cues has documented neural changes in specific brain regions of alcoholics not observed in social drinkers. Such altered activity in brain of abstinent alcoholics to stress and alcohol cues is consistent with a continuing ethanol adaptation being responsible. Therapies in alcoholics found to block responses to stress and alcohol cues would presumably be potential treatments by which susceptibility for continued alcohol abuse can be reduced. By continuing to define the neurobiological basis of the sustained alcohol adaptation critical for the increased susceptibility of alcoholics to stress and alcohol cues that facilitate craving, a new era is expected to evolve in which the high rate of relapse in alcoholism is minimized. 250
Keywords: alcoholism, alcohol, adaptation, stress, corticotropin releasing factor, cytokines, brain sites, substance P, relapse, brain imaging
1. Introduction
1.1. Alcoholism as a disease—relationship to alcohol responsiveness, genetics, and stress
Rhem et al. (2009) recently reviewed the extreme burden of alcohol use on society—a circumstance long recognized but inadequately addressed. Progress initiated in the middle of the 20th century began to provide views concerning the possible basis of alcoholism. One of the early conclusions was that offspring from a family with alcoholism were more likely to become alcoholic (Cadoret, 1980; Cotton, 1979; Devor and Cloninger, 1989; Schukit et al., 1985). One important biological clue linking this familial relationship to alcoholism was a lesser susceptibility for alcohol-induced ataxia and sway in sons of alcoholics compared to the response in individuals whose fathers were negative for alcoholism (Schukit, 1985, 1988,Schukit, 1994, 2009a,b. The logic gleaned from this work was that susceptible individuals become alcoholic because they can drink more alcohol before noting intoxication (Schukit, 1994). This view concerning the degree of alcohol intake received further support from knowledge that Asian individuals who became ill upon ingestion of small amounts of alcohol were less likely to become alcoholic (Peng et al., 2007). Thus, as a credible basis for the liability for alcoholism, this concept that sons of alcoholics could ingest more alcohol before observing symptoms of alcohol intoxication implicated a genetic susceptibility in alcoholism (Cloninger et al., 1981; Devor and Cloninger, 1989; Schuckit et al., 1985; Schweinsburg et al., 2004). A genetic basis is currently believed to be responsible for more than 50% of the individual risk for becoming alcoholic (Dick and Bierut, 2006). Ultimately such observations concerning a familial relationship to alcoholism have led to extensive investigations to identify specific gene changes that may relate to an increased susceptibility for this disease state (Bierut et al., 2010; Dick and Bierut, 2006; Edenberg and Foroud, 2006; Heilig, 2008; Ramchandani et al., 2010; Shuckit, 2009a).
In recent years, another population identified with a greater susceptibility for alcoholism includes individuals who drink alcohol excessively during adolescence (Barr et al., 1998; Behrendt et al., 2008; Brown and Tapert, 2004; Clapper et al., 1995; Dawson et al., 2007, 2008; Dewit et al., 2000; Grant and Dawson, 1997; Hingson et al., 2006; Tapert et al., 2003). In an effort to understand this association, studies found that the excess drinking during adolescence that contributed to the increased vulnerability of this population for alcoholism involved such factors as a shared environment and genetic susceptibility (Dawson et al., 2008; Hopfer et al., 2003; McGue and Iacono, 2008; Nelson et al., 2010; Prescott and Kendler, 1999; Rangaswamy et al., 2007).
Another generalization concerning susceptibility for alcoholism is that a stressful environment may contribute to excessive use of alcohol (Breese et al., 2004, 2005c; Brown et al, 1990; 1995; Kofoed et al., 1993; Koob, 2008; Sillaber and Henniger, 2004; Tate et al., 2005; Uhart and Wand, 2008). One of the earliest views to explain the role of stress in alcoholism was the tension-reduction hypothesis (Cappel and Herman, 1972; Conger, 1951; 1956). This concept proposed that individuals drank to reduce stress-induced tension and anxiety. While the pharmacology of alcohol can be viewed as demonstrating an “anti-tension” action (Gilman et al., 2008), the logic has long been questioned whether this property of alcohol to reduce “tension” alone relates to an excess of alcohol ingestion that ultimately produces an alcoholic state. In spite of the uncertainty of the tension-reduction hypothesis to alcoholism, the Marlatt laboratory examined the relationship of drinking to stress and consistently demonstrated that exposure to stress resulted in greater drinking in college students (Higgins and Marlatt, 1973; 1975; Marlatt and Gordon, 1980; Marlatt et al, 1975).
Cooper et al. (1992) suggested that some vulnerable individuals drink as a coping mechanism to overcome the consequence of stress. In this respect, individuals who drink alcohol for self-medication of symptoms of stress associated with existing psychiatric disorders such as depression, generalized anxiety disorder, social phobia or post-traumatic stress disorder have a higher degree of alcoholism (Collimore et al., 2010; Conway et al. 2006; Gilbertson et al., 2008; Grant et al., 2004; McLeod et al., 2001; Petrakis et al., 2002; Pirkola et al., 2005; Schuckit, 2006, 2009b; Shivani et al., 2002). Like alcoholics (Haddad, 2004; Wand and Dobs, 1991), these disorders have a dysregulation of the HPA axis (Nestler et al., 2002; Yehuda, 2001); however, it is unknown whether this dysregulation could influence susceptibility for alcoholism. Further not clear is whether a pre-existing genetic background for alcoholism may also be a contributing factor to excessive alcohol use in this psychiatric population. In these subjects, onset of alcohol use is typically later in life, and is predominantly driven by an ability of alcohol to transiently alleviate negative emotional states. Alcohol intakes in this population who drink for self-medication of symptoms use alcohol initially to dampen negative emotions—a coping mechanism. Subsequent drinking becomes excessive resulting in adaptive change that sensitizes stress-relevant brain systems—a change which enhances the incentive for further increases in alcohol to diminish escalating negative symptoms to stress that occur during alcohol abstinence (Heilig, 2008).
In a comprehensive model of stress as a factor in risk and relapse to alcoholism, Sinha (2001, 2008a, b, 2009) presents stress as a broader risk factor in the initiation and escalation of alcohol abuse that goes beyond self-medication of co-morbid psychiatric disorders, but rather discusses genetic risk and stress/adversity environment interactions that increase susceptibility for addiction. Such shared risk affects the reinforcing effects of alcohol and results in escalation of alcohol abuse. Furthermore, stress is proposed to reciprocally affect alcohol-related neuroadaptation. Such neuroadaptation in alcohol-dependent individuals is also manifested during abstinence as functional changes to stress and alcohol cues which induce craving that can increase vulnerability for relapse. Details concerning this alcoholic susceptibility to stress in relation to relapse are outlined in Sections 5 and 6. Thus, from the early tension-reduction hypothesis to the present views of stress in alcoholism being associated with genetic and co-morbitity with selected stress disorders and with stress facilitating negative symptoms during abstinence to sustain alcoholism, there has been continuing progress in defining a potential role of stress in alcohol abuse disorders.
Irrespective of the potential underlying basis for the susceptibility to drink, alcoholism is clearly related to excessive ingestion of alcohol over time—a circumstance which ultimately is responsible for the lack of control individuals have for minimizing alcohol intake. Alcoholics who attempt not to drink have a remarkable history of relapse ranging from 50 to 90% depending in part upon treatment (Hunt et al., 1971; Bradizza et al., 2006; Charney et al., 2010; Dawson et al., 2007; McKay et al., 2006). The phenomenon of relapse is so common that it can be considered a core characteristic of the alcoholic state.
1.2. Concept of chronic alcohol adaptation—the kindling/allostasis hypothesis
Since alcoholism is characterized by persistence of excessive alcohol intake (Beigleiter, 1975), concepts were proposed to understand how initiation of alcohol use might lead to alcoholism. While development of tolerance during chronic alcohol exposure had long been recognized, the possible relationship between adaptive change and maintenance of excessive alcohol use in the alcoholic was overlooked. A critical observation concerning the consequence of prolonged excessive alcohol use to adaptation came from a report to understand why only a small percentage of alcoholics exhibited seizure activity during abstinence from alcohol (i.e. during alcohol withdrawal). In this seminal report, Ballenger and Post (1978) hypothesized that a kindling-like cumulative adaptive process occurs in brain over time with excessive chronic alcohol exposure, an adaptation which ultimately results in seizure production (“epilepsy”) during cessation of alcohol intake (withdrawal). Subsequent assessments in alcoholics were consistent with this hypothesis (Booth and Blow, 1993; Lechtenberg and Worner, 1991). “Kindling” is a term defined as a change in brain physiology caused by repeated subthreshold levels of electrical stimulation until epileptiform activity results in seizure activity to a subthreshold stimulus. Importantly, this cumulative kindling process that increases the sensitivity to stimulus-induced seizure activity is relatively permanent. In critical support of repeated alcohol exposures and withdrawals being capable of producing this persistent change in CNS function, evidence in animals subsequently showed that withdrawals from repeated alcohol exposures could indeed facilitate kindling of seizure activity (McCown and Breese, 1990). In addition to proposing that a cumulative adaptive change was responsible for withdrawal-induced seizures in alcoholics, Ballenger and Post (1978) also noted that symptoms other than seizures worsened during withdrawal in alcoholic patients over the extended course of alcohol abuse. Based upon these findings, these investigators further contended that if a ‘kindling-like’ process occurs with excessive alcohol use, alcoholism would truly be a cumulative illness by which continued drinking adds to‘ previous drinking which overtime worsens the overall withdrawal syndrome.
The role of persisting adaptive consequences of chronic brain alcohol exposure being a contributor to alcoholism was not immediately embraced. However, some two decades later, the Koob laboratory (Koob, 2003; Koob and LeMoal, 1997; 2001; Roberts et al., 2000) proposed that prolonged excessive alcohol exposure produced an “allostatic change” in brain function to explain the characteristic components of alcoholism, particularly the symptoms of withdrawal. Allostasis, as a term, is stated to be an adaptive process of achieving functional stability by a physiological adjustment outside of normal homeostasis (McEwen, 2000; McEwen and Wingfield, 2003). Subsequently, Overstreet et al. (2002) found that intermittent exposures to chronic alcohol sensitized alcohol withdrawal anxiety, a change attributed to a “kindling-like” adaptive process (see Section 2). Regardless of whether one refers to the adaptive consequence of repeated exposures to excessive alcohol exposure as a “kindling”-like process (Ballenger and Post, 1978; Breese et al., 2005c; McCown and Breese, 1990) or to an increased “allostatic load” (Koob, 2003; Koob and LeMoal, 2001; Roberts et al. 2000), the hypothesized persistence of adaptation induced by alcohol is proposed to be responsible for the accentuated neurobiological sequalae observed in alcoholics upon cessation of alcohol drinking (Breese et al., 2005c; Koob, 2003). In fact, the worsening negative emotion that predominates during alcohol abstinence in the alcoholic is proposed to result in continued alcohol use to overcome the enhanced negative symptoms associated with withdrawal (Breese et al., 2005c; Heilig and Koob, 2007; Heilig et al., 2010a,b; Koob, 2008, 2009; Koob and LeMoal, 2005, 2008).
An additional point to be addressed is that repeated chronic alcohol exposures result in escalation of voluntary alcohol intake, measured either as simple two-bottle free-choice drinking or operant self-administration. This phenomenon is observed transiently following repeated periods of alcohol availability and deprivation [alcohol deprivation effect (ADE); Sinclair and Senter, 1968] or following repeated cycles of experimenter-imposed intoxication and withdrawal (O’Dell et al., 2004; Roberts et al, 2000; Sommer et al., 2008; Valdez et al., 2002, 2003, 2004). In respect to adaptation by chronic alcohol influencing alcohol intake, work showed that prolonged brain exposure to intermittent cycles of alcohol intoxication results in up-regulation of voluntary alcohol intake that persists long after acute withdrawal symptoms subside (Roberts et al., 2000; Valdez et al., 2002, 2004; See Section 3). This reflection of excessive drinking that occurs in animals may be comparable to the loss of control (i.e., excessive alcohol intake) observed in drinking alcoholics and in abstinent alcoholics during relapse (Keller, 1972).
After a period of time from excessive alcohol drinking, the alcoholic becomes free of the acute withdrawal symptoms. Nonetheless, an often quoted statement is “once an alcoholic always an alcoholic”. While the excessive relapse rate of abstinent alcohol dependent individuals would be consistent with this latter view (Fein and Landman, 2005), a neurobiological basis for this unfortunate consequence in the alcoholic remained elusive. In recent years, progress to understand the susceptibility of alcoholics for relapse provided an outcome that supports the brain of alcoholics being distinct from normal individuals (Fox et al., 2007; Gilman and Hommer, 2008; Sinha, 2001, 2008a; Sinha and O’Malley, 1999; Sinha et al., 2009, 2010) Consequently, this distinction in the alcoholic brain could be responsible for the dysfunctional behavioral and altered functional responses in alcoholics not observed in social drinkers (see details in Sections 5 & 6).
The purpose of this overview is to provide evidence that chronic alcohol induces a persistent cumulative adaptation that alters brain function by a kindling/allostatic process—a change that facilitates the many negative attributes associated with alcoholism. Section 2 provides evidence that repeated cycling of alcohol induces this kindling/allostatic process of cumulative adaptation more effectively than does continuous alcohol. Section 3 reviews material concerning models of alcohol intake. In Section 4, material is provided that stress after the cumulative adaptation induced by chronic intermittent alcohol exposure induces an increased negative behavioral responsiveness and enhanced alcohol self-administration (Breese et al., 2004; Valdez et al. 2003; Sommer et al. 2008). These latter observations are consistent with evidence presented in Section 5 that the abstinent alcoholic is more susceptible to induction of negative emotional state by “stresses” and alcohol cues (Gilman and Hommer, 2008; Sinha 2001, 2007, 2008a,b; Sinha et al., 2009, 2010). Another point emphasized in Section 5 is that the negative response in the alcoholic to stress and alcohol cues is accompanied by increased craving—a response which may be associated with the heightened likelihood of relapse (Sinha, 2001, 2008; Sinha and Li, 2007; Sinha et al., 2009, 2010). Section 6 will provide evidence from measurements of functional brain imaging that stress and alcohol-cues result in altered brain activity which differentiates the alcoholic from controls. This latter section will also emphasize how testing of drugs on brain imaging changes to stress and alcohol cues in alcoholics could provide a new strategy for identifying promising therapies for this disorder.
For each of these critical issues, available information which has evolved to understand the neurobiological basis of alcohol adaptation within specific brain sites will be provided. By understanding the biological and neuroanatomical factors that contribute to the cumulative adaptation associated with excessive drinking, avenues for future treatment may be possible that will minimize the vulnerability alcoholics have for relapse to excessive alcohol use. Identification of the progress made thus far provides optimism for a favorable future outlook for addressing alcoholism as a disease.
2. Neuroadaptation by Repeated Alcohol Cycling: Evidence for the Kindling/Allostasis Hypothesis
2.1. Repeated cycling of chronic alcohol induces a persistent adaptation
As noted earlier, Ballenger and Post (1978) implied that excessive alcohol ingestion over years induces an adaptation that accentuates detrimental consequences during repeated removals from alcohol. Similar in concept to the “alcohol kindling hypothesis” (Ballenger and Post, 1978), Koob and LeMoal (2001) later proposed that repeated alcohol exposures result in a cumulative adaptation that accentuates the negative consequences observed during alcohol withdrawal—an adaptation related to a change in “allostatic load” (McEwen, 2000). However, while an intermittent exposure to alcohol was a requirement for “kindling” of seizures (Becker and Hale, 1993; McCown and Breese, 1990; Ulrichsen et al., 1995, 1998a,b), critical investigations had not been performed to establish whether intermittent chronic cycles of alcohol were essential for enhancement of the anxiety and negative mood observed during alcohol removal.
A strategy of repeated cycling of alcohol exposure similar to that used to evaluate kindling of seizures was utilized to test if intermittent alcohol exposure was indeed required for induction of adaptation that resulted in negative emotional symptoms during withdrawal from alcohol. As illustrated in Figure 1, rats exposed to only 3 cycles of 5 days of 4.5% alcohol diet interrupted by two days of abstinence exhibited sensitization of anxiety-like behavior 5–6 hrs after removal of the final cycle of alcohol (Overstreet et al., 2002). This sensitization occurred equally in both male and female rats (Overstreet et al, 2004b). Importantly, removal of alcohol from rats that received the same amount of alcohol continuously did not induce negative symptoms upon alcohol removal. These findings indicate that cycling of alcohol exposure is a critical contributor to the adaptation that supports sensitization of anxiety during alcohol withdrawal, a process proposed to be comparable to that by which repeated alcohol cycling kindles seizures (McCown and Breese, 1990). Nonetheless, it is apparent that a considerably shorter period of alcohol cycling is required to sensitize an emotional response during withdrawal (Overstreet et al., 2002) than is needed to induce “kindling” of seizures (McCown and Breese, 1990). Similar conclusions concerning adaptation have been expressed using other means of brain alcohol exposure and readouts of negative emotionality (Gilpin et al., 2008a,b; O’Dell et al., 2004; Rimondini et al. 2002; Roberts et al. 2000; Sommer et al., 2008; Valdez et al., et al. 2002, 2004). These observations are consistent with the interpretation by Ballenger and Post (1978) that symptoms of alcohol withdrawal with alcohol abuse worsen over the course of time.
Figure 1. Comparison of three repeated cycles of alcohol (ETOH) with 1 ETOH cycle and continuous (Contin) ETOH diet on social interaction—a measure of anxiety-like behavior.

Social interaction was measured 5–6 hrs after ETOH removal. P < 0.01 compared to Control Diet (CD) and the other ETOH exposed groups. Modified from Overstreet et al. (2002) by permission of publisher.
Consistent with extended alcohol use resulting in a persistent adaptation, the Begleiter laboratory documented in rats as well as in male alcoholics that chronic alcohol dependence was associated with an extended increase in brain activity after withdrawal from alcohol (Begleiter and Porjesz, 1977; Porjesz and Begleiter, 1981). Later, Roberts et al. (2000) demonstrated that alcohol self-administration was elevated for 4–8 weeks after alcohol dependent rats were withdrawn from initial exposure. Based upon this finding, protracted withdrawal symptoms from chronic alcohol were proposed to be capable of triggering relapse. More recently, the repeated cycling of chronic alcohol in rats was also discovered to induce a persisting adaptation (Overstreet et al., 2002). Evidence for this persistence was demonstrated by observing sensitization of withdrawal-induced anxiety-like behavior following re-exposure to a single 5 days of alcohol diet 16 days following previous exposure to repeated cycles of alcohol (Figure 2). Removal from a single 5-day period of alcohol diet of animals not previously exposed to alcohol diet produced no anxiety-like behavior (Overstreet et al., 2002). Consistent with this report, intermittent alcohol exposures were later reported to produce an extension of the expression of alcohol withdrawal symptoms (Zhang et al., 2007). Likewise, Sommer et al. (2008) demonstrated fear suppression of behavior after an extended absence from intermittent alcohol exposure. Rimondini et al. (2008) found a long-lasting tolerance to alcohol following induction of dependence—an additional demonstration of a persistence of adaptation following chronic alcohol exposure. In Section 3, it will be demonstrated that previous extended intermittent exposure to alcohol produces an escalation of voluntary alcohol intake (Rimondini et al. 2002, 2003; O’Dell et al., 2004; Valdez et al., 2002, 2004). Collectively, these findings indicate that the outcome of continuing alcohol use results in neural alterations that persist beyond acute withdrawal. Such persistent functional changes induced by intermittent alcohol exposure are accompanied by equally persistent changes in brain gene expression patterns (McBride et al., 2009; Rimondini et al., 2002; Sommer et al., 2008). The importance of this extended adaptation in the susceptibility alcoholics have to stress is outlined in Sections 4 & 5.
Figure 2. Social interaction after withdrawal from alcohol (ET) re-exposure of rats previously exposed to repeated cycling of alcohol.

Animals were given three cycles of 5 days of 7% alcohol diet (ED-CY3) followed by an additional 5 days of alcohol diet 8 or 16 days after the initial repeated alcohol exposure (ED-CY3-R-8D & ED-CY3-R-16 D). CD rats received only control diet. The CY1 is withdrawal from a single cycle of alcohol diet to animals that previously received CD. Social interaction as a measure of anxiety-like behavior was measured 5–6 hrs after withdrawal from the re-exposure to alcohol diet. * P < 0.01 compared to CD and CY1. Modified from Overstreet et al. (2002) by permission of publisher.
Consequently, efforts over several decades (Ballenger and Post, 1978; Koob and LeMoal, 2001; McCown and Breese, 1990) have provided seminal support for the postulated concept that intermittent chronic alcohol exposures are capable of producing a persisting adaptation. Subsequently, to minimize the progressive increase in withdrawal symptoms to the intermittent alcohol exposures an escalation of alcohol drinking occurs—a circumstance that ultimately leads to addiction (Breese et al., 2005a,c; Heilig and Koob 2007; Heilig et al., 2010a,b; Koob, 2009). Importantly, documenting that intermittent alcohol exposures induce a persistent adaptation provided an excellent means by which to document whether a “cumulative adaptive process” is responsible for the intermittent alcohol exposures facilitating alcohol withdrawal symptoms.
2.2. Neuroadaptation induced by intermittent alcohol cycling facilitates alcohol withdrawal anxiety: Documentation of a cumulative adaptive process
In spite of the cumulative adaptation hypothesis being an attractive explanation for removal from intermittent alcohol treatments sensitizing withdrawal symptoms (Ballenger and Post, 1978; Koob, 2003; McCown and Breese, 1990), this interpretation had not been critically tested. Therefore, to confirm the hypothesis that the adaptation induced by the intermittent alcohol exposure was cumulative, it was reasoned that if drug treatments were given during withdrawal from the 1st and 2nd alcohol cycles of the repeated cycle protocol, sensitization of anxiety-like behavior would not be observed during withdrawal from the 3rd and final alcohol cycle. Since the benzodiazepine (BZD) diazepam was known to prevent withdrawal anxiety (Knapp et al, 2004), this BZD was administered during the initial two ethanol withdrawals. Consistent with a cumulative adaptation being responsible for the withdrawal-induced anxiety induced by the repeated alcohol exposures, the diazepam prevented the ethanol withdrawal anxiety (Knapp et al., 2005). Additional results implicating GABA function in the withdrawal sensitization was that the GABAB agonist, baclofen, administered during early withdrawals also prevented the adaptation that supports facilitation of withdrawal-induced anxiety-like behavior induced by the repeated alcohol cycling protocol (Knapp et al., 2007a). Paradoxically, flumazenil, a BZD receptor antagonist, administered during the initial withdrawals was also capable of blocking sensitization of withdrawal anxiety (Knapp et al., 2005). Likewise, buspirone, a 5-HT1A receptor agonist as well as a 5-HT2C receptor antagonist preventing sensitization of withdrawal-induced anxiety implicated altered serotonergic function in the sensitization (Overstreet et al., 2003, 2005). Importantly, a CRF1-receptor (CRF1R) antagonist also prevented the intermittent alcohol exposure adaptation responsible for sensitization of withdrawal-induced anxiety (Overstreet et al., 2004a, 2005). This action by the CRF1R antagonist was consistent with alcohol-induced neuroadaptation resulting in recruitment of CRF transmission. See Section 4.1 for changes in CRF measures by stress and chronic alcohol exposure.
Finding that selected drugs given during initial ethanol withdrawals prevent sensitization of alcohol withdrawal anxiety (Knapp et al., 2007a; Overstreet et al., 2003, 2004a, 2005) provided critical support for the Ballenger and Post (1978) and Koob (2003) postulate that the adaptation that occurs with the repeated alcohol exposures is indeed cumulative. Additionally elucidated was that the drug treatments given during the initial removals from alcohol cycling, which prevented the initial sensitization of withdrawal anxiety, also prevented sensitization of anxiety during withdrawal from a later re-exposure to a final cycle of alcohol diet—a reflection that the ethanol cumulative adaptation associated with the initial intermittent alcohol cycles persists (Breese et al., 2004; 2008; see Section 4). The fact that distinct drug classes block sensitization of alcohol-withdrawal anxiety-like behavior as well as the persistent adaptation by the multiple cycles of alcohol clearly implicates differing neural pathway involvement in the adaptive process induced by alcohol. However, while this observation is informative, yet to be resolved is the basis by which altering each of these several neural pathways individually prevents the adaptation responsible for sensitization of withdrawal symptoms and the persistence of adaptation by the repeated alcohol cycling.
2.3. Neuroanatomical sites support the cumulative adaptation Induced by intermittent alcohol cycling
Microinjection of drugs into specific brain sites were used previously to identify regions critical for their action to effect withdrawal seizures (Frye et al., 1983), alcohol sedation (Breese et al., 1984; Givens and Breese, 1990; McCown et al. 1985; 1986) as well as the anxiety that accompanies withdrawal from chronic alcohol exposure (Rassnick et al., 1993). With knowledge that specific pharmacological compounds prevent the cumulative adaptation induced by repeated cycling of chronic alcohol (Section 2.2), these drugs were microinjected into appropriate brain sites during the 1st and 2nd withdrawals, but not the final withdrawal of the protocol, to identify brain regions associated with the adaptation induced by repeated alcohol cycling. From this exploration to define specific sites of action of these drugs, it was hoped that some clarification would occur to resolved how differing neurotransmitter systems were capable of preventing sensitization of anxiety that follows withdrawal from the repeated alcohol cycling (Section 2.2).
Using the microinjection strategy outlined, Figure 3 provides examples of the site selectivity within the dorsal raphe and amygdala of drugs affecting differing 5-HT receptors as well as the site selectivity observed with flumazenil. When the 5-HT1A receptor (5-HT1AR) agonist was microinjected into the dorsal raphe (DRN), sensitization of withdrawal anxiety was prevented (Overstreet et al., 2006). However, microinjection of the 5-HT1AR agonist into the central amygdala (CeA) during the initial withdrawals was ineffective in preventing sensitization. In contrast to the 5-HT1AR agonist, microinjection of the 5-HT2CR antagonist into the CeA prevented the anxiety-like behavior induced during withdrawal from the repeated alcohol cycles, but was ineffective when administered into the DRN (Overstreet et al., 2006). Importantly, a 5-HT2CR agonist microinjected into the CeA prior to a single 5 days of alcohol diet induced sensitization of alcohol-withdrawal anxiety (Overstreet et al., 2006). Further shown in Figure 3 is that flumazenil microinjected into the CeA during early withdrawals blocked the sensitization of anxiety observed during the final alcohol withdrawal without producing this inhibition after administration into the DRN (Knapp et al., 2007b). These various drug exposures had no effect on sensitization of withdrawal symptoms when administered into the paraventricular nucleus (PVN) or the nucleus accumbens (NAC) (Knapp et al., 2007b). Importantly, the brain regions identified to influence withdrawal-induced anxiety have previously been found to be critical for support of anxiety-like behavior (Koob, 2008; Koob and LeMoal, 2005, 2008; Shin and Liberzon, 2009; Wand, 2005). In Section 4, the brain sites at which CRF1R antagonists influence sensitization of negative symptoms associated with alcohol withdrawal is discussed in relation to the role of CRF in stress-induced adaptive change (Breese et al., 2004).
Figure 3. Treatment of a 5-HT2C inverse agonist, a 5-HT1A agonist, or a BZD antagonist into either the dorsal raphe or the amygdala on sensitization of alcohol withdrawal-induced anxiety-like behavior.

Dorsal Raphe data are at the top and amygdala data are presented in the bottom illustration. The 5-HT2C inverse agonist (SB243213; SB), the 5-HT1A agonist (buspirone), and the BZD antagonist (Flumazenil; Flu) were administered into the sites during withdrawal from the initial two cycles. Social interaction as a measure of anxiety-like behavior was measured 5–6 hrs after withdrawal from the final cycle of alcohol. * P < 0.01 compared to control diet exposure. Modified from Overstreet et al (2006) and Knapp et al. (2007) by permission of the publishers.
An important conclusion from defining sites of action of drugs during intermittent alcohol exposure is that neurotransmitter systems in differing brain regions are capable of supporting the cumulative adaptation responsible for sensitization of emotional behavior during withdrawal from the repeated alcohol cycling. Notable from the drug microinjections into active sites was that the sensitized alcohol withdrawal response returned to the normality observed in control rats, just as seen after systemic administration of these agents (Section 2.2). While not resolving the basis of differing brain sites inducing a near complete blockade of the cumulative adaptive expression of withdrawal symptoms associated with the repeated cycling of alcohol, a logical conclusion could be that a neural system identified within a given brain site is in series with other sites involved in the negative symptoms observed during withdrawal from alcohol. Therefore, disruption of neurotransmitter signaling at any site associated with the proposed serial organization of differing neural pathways could prevent the exaggerated behavioral responses during withdrawal. Even though this hypothesis of an integrated circuit involving differing neural systems may support the adaptation induced by repeated alcohol exposures, no direct empirical support exists for this hypothesis, but it is certainly a view that should be tested.
3. Alcohol intake and relapse-like behavior
3.1. Reinstatement of operant responding as a model of alcohol seeking
An increasingly popular approach allows modeling of the motivational aspect of relapse-like behavior in the absence of drug. In this protocol, stable operant responding for alcohol is established followed by continued daily operant sessions in which the alcohol re-enforcement is absent. This absence will lead to extinction of responding on the previously alcohol-delivering lever over the course of 2–3 weeks (Mellow and Mendelson, 1964; Samson, 1986). However, following extinction from alcohol, categories of stimuli known to be potent relapse triggers in human alcoholics are able to produce a robust resumption or “reinstatement” of responding on the previously alcohol-associated lever in the absence of alcohol reinforcement. Reinstatement will occur with a priming dose of alcohol, an “alcohol cue”, exposure to inescapable shock, or to nicotine (see discussion in Section 4) (Lê et al., 1998, 2003; Lê and Shaham 2002). Using this model of alcohol seeking, preclinical studies have identified CRFR antagonists, α2-adrenergic agonists, α1-noradrenergic antagonist, dopamine receptor antagonists, and selected glutamatergic agents that reduce responding when given prior to initiation of a session (Besheer et al. 2006; Gehlert et al., 2007; Lê et al., 2005; Liu and Weiss, 2002a,b; Mantsch et al., 2008; Marinelli et al., 2007; Rasmussen et al., 2008; Shaham and Hope., 2005; Walker et al., 2008; Zhao et al., 2006). Investigations that explored drug treatments on reinstatement of alcohol seeking after extinction have been summarized in reviews (Heilig and Egli, 2006; Koob and LeMoal, 2008; Sanchis-Segura and Spanagel, 2006; Sinclair, 2001; Spanagel, 2009; Weiss, 2005). In respect to sites of action supporting responding, Lê et al. (2002) reported that a 5HT1A receptor agonist and CRF into the raphe nucleus reinstated alcohol seeking. Additionally, Chaudhri et al. (2009) reported differing roles for the core and shell of the NAC in context and cue-induced alcohol seeking.
3.2. Escalation of voluntary alcohol drinking and operant self-administration
Another question key to the concept of “kindling” of cumulative neuroadaptation by alcohol is whether such previous adaptation can increase the motivation to escalate either simple voluntary two-bottle free-choice drinking or operant self-administration, just as this adaptation supports emotional responses during withdrawal. Noteworthy is that the two approaches of assessing drinking may at times produce diverging results due to potential contributions of taste and appetite for calories in the simpler model. However, since neuroadaptation by these approaches consistently escalates alcohol drinking (see Heilig and Koob, 2007), they are presented together. The relationship between alcohol dependence, experience with withdrawal, and subsequent alcohol self-administration has been the subject of extensive research. Early studies generally yielded equivocal findings (Begleiter, 1975; Deutsch and Koopmans 1973; Hunter et al., 1974; Myers et al., 1972; Numan, 1981; Samson and Falk, 1974; Schulteis et al., 1996; Winger, 1988). This outcome was most likely because procedures did not sufficiently establish the reinforcing effects of alcohol prior to dependence induction. In these situations, minimal opportunity likely occurred to allow an association of alcohol drinking with its alleviation of withdrawal consequences (Meisch, 1983; Meisch and Stewart, 1994). Taking this into account, more recent studies in mice (Becker and Lopez 2004; Chu et al., 2007; Dhaher et al., 2008; Finn et al., 2007; Lopez and Becker, 2005) and rats (Funk et al., 2006b, 2007; Gilpin et al., 2008a,b; O’Dell et al., 2004; Rimondini et al. 2002; Roberts et al. 2000; Sommer et al. 2008; Valdez et al., 2002) have demonstrated “escalated” operant self-administration or increased free-choice drinking of alcohol in animals with a history of dependence compared to those without such a history. In agreement with a “kindling” process by alcohol exposure being responsible for the increase in drinking, repeated ethanol withdrawal (deprivation) experiences are more potent in inducing escalation than is continuous alcohol exposure (O’Dell et al., 2004; Rodd et al., 2003). Further, this consequence of alcohol dependence was shown to induce an extended peristence of self-administration (Gilpin et al., 2008a; Rimondini et al. 2002; Roberts et al. 2000; Sommer et al., 2008; Valdez et al., 2002).
Importantly, post-alcohol dependent self-administration can have a different pharmacology from that of baseline alcohol self-administration. For example, CRF1R antagonists blocked the increase in alcohol consumption in dependent animals without having an effect on intake in non-dependent rats (Funk et al., 2007; Gehlert et al., 2007; Gilpin et al., 2008b). Similar to CRFR1 antagonist action, a kappa-opioid antagonist, as well as acamprosate, selectively suppressed alcohol self-administration in dependent rats, but both had no consequence in non-dependent rats (Rimondini et al., 2002; Walker et al. 2008; Walker and Koob, 2008). Additionally, Rimondini et al. (2005) demonstrated that administration of the neuropeptide Y (NPY) type2 receptor antagonist to facilitate NPY release suppressed alcohol responding in dependent rats while being without effect on self-administration in non-dependent rats. The results with the CRF1 and NPY2 receptor antagonists are in accord with the belief that NPY and CRF exert a reciprocal regulation (Heilig et al., 1994; Sajdyk et al. 2006; Valdez and Koob, 2004). The involvement of CRF and NPY on alcohol drinking has been reviewed (Ciccocioppi et al., 2009).
Limited studies have been performed to identify sites at which drugs act to influence alcohol self-administration in animals. For example, in the absence of physical dependence, there is body of literature implicating GABA and nicotine receptors in the NAC in alcohol drinking (e.g., Nadal et al., 1998; Rewal et al., 2009). Additionally, Besheer et al. (2010) found that metabotropic-glutamate (mGLU)-5R activity in the NAC was required for maintenance of alcohol self-administration. Just as noted for the distinct effects of a CRFR antagonist in post-dependent and non-dependent animals on alcohol consumption, microinjection of a general CRF antagonist (D-Phe CRF12–41) into the CeA prevented alcohol self-administration in alcohol-dependent rats, while being without an effect on this measure in non-dependent rats (Funk et al., 2006b). Administration of this general CRFR antagonist into the lateral dBNST and the NAC did not alter responding for alcohol in either the alcohol-dependent or non-dependent rats. Collectively, these findings indicate that the elevation of alcohol self administration in dependent rats is associate only with specific regions of brain (Funk et al., 2006b). In contrast to CRF involvement in the support of alcohol self-administration in dependent rats, urocortin 3, an agonist for the CRF2R, injected intraventricularly (Valdez et al., 2004) or into the CeA (Funk and Koob, 2007) was capable of preventing alcohol self-administration in dependent rats, while increasing intake in non-dependent rats. This finding is consistent with CRF2Rs having an apposing action to CRF1Rs, which are clearly responsible for the elevated alcohol self administration in dependent rats. By defining neuroanatomical sites related to drugs that affect this excessive drinking in dependent rats, it will be possible to identify at a cellular level the neurobiological basis of the cumulative adaptation induced by the chronic alcohol exposure that facilitates alcohol drinking—a strategy that should be encouraged.
3.3. The alcohol deprivation effect (ADE) as a model of relapse-like drinking
Abstinent alcoholics are known to drink excessive amounts of alcohol upon relapse, a reflection of “loss of control”. An excess of alcohol drinking in animals after repeated intermittent chronic alcohol exposures was first described by Sinclair and Senter (1968) as an alcohol deprivation effect (ADE)—a phenomenon proposed as an animal model of the loss of control observed during relapse (Spanagel and Holter 2000). Wolffgram and Heyne (1995) demonstrated that the excessive drinking induced by the ADE protocol persisted for an extended period after the initial repeated alcohol drinking episodes. Other reports demonstrated that intermittent voluntary alcohol drinking produced a persisting consequence on ADE in a high alcohol drinking-1 (HAD1) rat line (Rodd-Henricks et al., 2000a) as well as in the alcohol preferring P-rat strain (Gilpin et al., 2008a,b; Rodd-Henricks et al., 2000b; Toalston et al., 2008). Repeated deprivations following an initial period of free access have recently also been shown to result in progressive escalation of alcohol drinking in C57BL/6 mice (Melendez et al., 2006). It is important to recognize that the excess drinking observed with the ADE protocol may not be equivalent to the excess drinking observed with the change observed with ethanol dependence discussed in Section 3.2.
Repeated (intermittent) cycling of alcohol drinking in the P-rat strain was recently shown to facilitate the ADE phenomenon within a short time period (Breese et al., 2004; Overstreet et al., 2007; see Figure 8). A reasonable assumption for repeated chronic alcohol exposures being required for the ADE is that the exposure to the repeated cycling of alcohol induces a cumulative adaptation which accentuates the loss of control to limit alcohol intake upon re-exposure to alcohol. Drugs have been tested during the final drinking cycle to determine what neural mechanisms would affect the excessive alcohol ingestion in this ADE model. Opiate antagonists minimize this excessive alcohol intake implicating a possible involvement of an opiate mechanism in this aspect of alcohol dependence (Altschuler et al., 1980; Hölter and Spanagel, 1999; Hubbel et al., 1986; Sinclair 1990). Subsequently, these early preclinical observations led to finding that this opiate antagonist was useful in minimizing the loss of control characteristic of the alcoholic during a relapse episode (O’Malley et al., 1992; Vollpicilli et al., 1992; See reviews by Bouza et al., 2004; Heilig and Egli, 2006; Sinclair, 2001; Soyka and Roosner, 2008). Likewise, the action of acamprosate to reduce excess alcohol drinking in the ADE protocol supported its clinical use in treatment of alcohol dependence (see review by Bouza et al., 2004; Heilig and Egli, 2006; Spanagel, 2009).
Figure 8. Stress during withdrawal from repeated voluntary alcohol drinking facilitates the Alcohol Deprivation effect (ADE).

P-rats were given either continuous voluntary access to water and alcohol solution (10%) or 3 cycles of voluntary access to water and alcohol solution with 2 days of abstinence between the 1st two cycles. Intake of alcohol was subtracted from intake on the final 5 days of fluid ingestion.
*Significantly different from Continuous. +Significantly different from Cycling Only. Modified from Breese et al. (2004) by permission of the publisher.
This induction of a rapid facilitation of ADE induced by repeated intermittent cycling of voluntary alcohol intake in P-rats (Breese et al., 2004) could provide a convenient approach to investigate whether an underlying cumulative adaptation associated with repeated deprivations indeed supports the progressive increase in alcohol drinking. In this respect, Roberto et al. (2010) showed that chronic repeated administration of a CRF-1 antagonist protocol blocks development of excess alcohol intake associated with an ADE in nondependent rats and prevents withdrawal-induced excessive drinking in dependent rats. Certainly, future studies are encouraged to fully understand the neural basis of the cumulative increase in the excessive voluntary alcohol drinking induced by the ADE. In Section 4.4, stress induction of a cumulative adaptation during repeated cycles of voluntary drinking will be shown to facilitate the ADE.
4. Stress Interacts with Chronic Alcohol Neuroadaptation
4.1. Repeated Stresses accentuate the maladaptation induced by chronic alcohol
Introductory remarks (Section 1.1) provided logic for stress involvement in alcoholism. Previous reports have supported CRF being a primary central mediator of stress (Bale and Vale, 2004; Dunn and Berridge, 1990; Imaki et al., 1993; Koob and Heinrichs, 1999; Vale et al., 1981). In confirmation of CRF involvement with chronic alcohol and stress exposure, Merlo-Pich et al. (1995) using dialysis found an increase in extracellular CRF levels in the amygdala of rats during restraint stress and after alcohol withdrawal. Likewise, Cook (2004) reported that sheep exposed to an acute predator (i.e., a stress) increased CRF release from the PVN and the amygdala.
Given the earlier observation that a CRF1R antagonist prevented the cumulative adaptation induced by repeated cycling of alcohol (Section 2.2; Overstreet et al., 2004a), there was an impetus to explore whether stress would substitute for initial withdrawals of the repeated alcohol protocol to sensitize withdrawal symptoms following a single alcohol cycle. Subsequently, Breese et al. (2004) found that restraint stress applied at weekly intervals prior to only 5-days of alcohol diet (i.e., the stress/alcohol withdrawal protocol) did indeed sensitize anxiety-like behavior during removal from the alcohol exposure (Figure 4). Likewise, sensitization of anxiety was not seen with just weekly stress application (i.e. no alcohol diet) or upon withdrawal from only 5-days of chronic alcohol exposure (i.e. no stress; Figure 4). Consequently, this investigation (Breese et al., 2004) demonstrates that repeated exposures to restraint stress is capable of inducing an adaptation that allows a lesser amount of alcohol to sensitize negative symptoms during withdrawal that would not otherwise be observed. At this time, the many types of stress noted by Nestler et al. (2002) have not been tested prior to ethanol to determine if all will facilitate ethanol adaptation.
Figure 4. Repeated Stresses Sensitization of alcohol (ET) withdrawal-induced anxiety.

The (Control Diet) group received only diet. The (No Stress-1 cycle ET) group received no stress and and only 1 cycle ET. The (Control Diet-3 Stress) group received 3 separate 60 min stresses at weekly intervals. The (2 Stresses—1 cycle ET) group was exposed to two 60 min restraint stresses at a weekly interval followed by exposure to 1 cycle of 5 days of 4.5% alcohol diet. Social interaction as a measure of anxiety was measured 5–6 hrs after removal of alcohol diet or the final stress. * P < 0.01 compared to all other groups. From Breese et al. (2004) by permission of publisher.
Additionally important was demonstrating that the systemic corticosterone increase induced by the repeated stress exposure was not responsible for the stress-induced sensitization of alcohol withdrawal-induced anxiety (Breese et al., 2004). This latter finding provided strong evidence that activation of neural pathways distinct from the hypothalamic-pituitary-adrenal (HPA) axis path is critical for the stress-induced behavioral sensitization during alcohol withdrawal (Breese et al., 2004). This latter conclusion is in agreement with other observations that extrahypothalamic neural systems linked to stress, rather than the hypothalamic-pituitary pathway, are involved in neuroadaptive mechanisms that influence ethanol actions (Gehlert et al. 2007; Lê et al. 2000; Lowery et al., 2010). A final critical observation concerning the adaptation induced by the repeated stress/alcohol withdrawal protocol was that re-exposure to an additional 5 days of alcohol diet 16 days after alcohol removal from the original protocol resulted in sensitization of anxiety during withdrawal from this later alcohol exposure (Breese et al., 2004). Animals re-exposed to a 5-day alcohol challenge that received only control diet did not display an alteration in behavior during withdrawal (Breese et al., 2004). This latter finding is compatible with the stress/alcohol withdrawal protocol supporting a persistent adaptation (Breese et al., 2004), just as observed with repeated cycles of chronic alcohol (Section 2.1). Collectively, stress-induced facilitation of alcohol action supports the view that stress induces a “kindling process” or an allostatic adaptive state (Korte et al., 2005) that facilitates a detrimental consequence during withdrawal from only 5 days of alcohol exposure. Documentation is presented in Section 4.2 that a cumulative adaptation is the means by which repeated stress facilitates alcohol adaptation.
As noted in our introductory remarks (Section 1.2), human studies have demonstrated that alcoholic patients exhibit an increased susceptibility to stress not observed in social drinking controls (Gilman and Hommer, 2008; Gilman et al., 2008; Sinha, 2001). In accord with these clinical studies, a preclinical investigation demonstrated that stress 3 days after exposure to cycling of chronic alcohol induced an anxiety-like response that was not observed when stress was applied to animals not previously exposed to alcohol (Breese et al., 2005b). Noteworthy is that others have also shown that chronic alcohol exposure enhances the consequences of stress (Liu and Weiss, 2003; Sommer et al. 2008; Valdez et al. 2003). Thus, such enhancement of stress symptoms in previously chronic alcohol-exposed animals is felt to be comparable to the increased susceptibility abstinent alcoholics have to stressful imagery (Sinha, 2001; See Section 5.1 for clinical details). In line with stress interacting with alcohol, Section 4.4b documents that stress during repeated cycles of alcohol escalates alcohol self-administration (Breese et al., 2004; Sommer et al. 2008).
Based upon both basic and clinical findings, a conclusion is that stress can produce a negative outcome associated with chronic alcohol exposure irrespective of whether the stress occurs before or after alcohol exposure. To account for the escalating negative behavioral pathology associated with stress during abstinence in the alcoholic, the “kindling”/stress hypothesis of alcoholism was proposed (Breese et al., 2005c). Therefore, this documentation that stress influences the pharmacology of alcohol provides a compelling rationale for continuing to clarify the basis of the neuroadaptive processes that accompany stress interactions with alcohol.
4.2. Documentation of a cumulative adaptive process in facilitation of alcohol adaptation by stress: Neural mechanisms involved
Based upon CRF involvement in the action of stress (Vale et al., 1981), it was reasoned that CRF was a plausible mediator in the stress facilitation of alcohol adaptation. In support of this view, Figure 5 shows that systemic administration of a CRF1R antagonist prior to the weekly stresses (i.e., the stress/alcohol withdrawal protocol) prevented sensitization of anxiety during alcohol withdrawal (Breese et al., 2004). As noted earlier for repeated cycling of alcohol (Section 2.2), the strategy of administering the CRF1R antagonist prior to the early repeated stresses, but not during the final withdrawal from alcohol, clearly indicates that the repeated stresses induce a cumulative adaptive process that is essential for facilitation of alcohol withdrawal symptoms (Figure 5). In addition, a CRF1R antagonist given during the initial cycles of the repeated withdrawal protocol prevented the later stress-induced anxiety (Breese et al., 2005b) indicative that the cumulative adaptation induced by the repeated alcohol exposures support this accentuated response to the stress challenge. In further confirmation of CRF importance to the cumulative adaptation responsible for sensitization of alcohol withdrawal anxiety by stress, CRF given repeatedly intracerebroventricularly (ICV) prior to a single 5-day alcohol exposure sensitized withdrawal-induced anxiety-like behavior that was prevented by a CRF1R antagonist (Overstreet et al., 2004a). Collectively, these findings support the conclusion that CRF mediation should be a critical focus of treatment for preventing negative symptoms and other consequences induced by withdrawal from chronic alcohol and by stress (Breese et al., 2005b; Gehlert et al., 2007; Heilig and Koob, 2007; Zorrilla and Koob, 2004).
Figure 5. A CRF1R antagonist, a BZD receptor antagonist and a 5-HT1A agonist prevent the repeated stress sensitization of alcohol (ETOH) withdrawal anxiety.

The CRF1R antagonist was CRA1000, the BZD antagonist was flumazenil (FLU) and the 5-HT1A agonist was buspirone. For each of the drug groups the drug was administered prior to each stress given prior to 5 days of 4.5% ethanol diet (ETOH) Social interaction as a measure of anxiety-like behavior was measured 5–6 hrs after ETOH Diet removal. * P < 0.01 compared to all other groups. From Breese et al. (2004) by permission of publisher.
In confirmation of CRF association with a history of alcohol dependence, a prolonged history of alcohol exposure induced a persistent up regulation of post-synaptic CRF1R expression and increased levels of CRF in brain for extended periods after alcohol removal (Roberto et al., 2010; Sommer et al., 2008; Zorrilla et al., 2001; Weiss et al., 2001). Conversely, the CRF2R, which in many circumstances has actions opposite to those of the CRF1 subtype (Valdez et al., 2004; Funk and Koob, 2007), was down-regulated within the amygdala complex following an extended period of ethanol withdrawal (Sommer et al. 2008). However, upon acute withdrawal from chronic intermittent exposure to alcohol vapor, Martin-Fardon et al. (2010) observed reduced CRF1R, but not CRF2R, binding in the CeA, medial amygdala, and the basolateral amygdala of Wistar rats. This latter reduction in CRF1R binding observed shortly after ethanol removal could be attributed to internalization of the CRF1R by the elevated CRF presence during ethanol exposure and withdrawal (Martin-Fardon et al., 2010; Olive et al., 2002; Weiss et al., 2001). However, upon extended ethanol withdrawal, this process must be reversed resulting in an increase in CRF1Rs, as noted by Roberto et al. (2010). See Section 4.4c for further modifications in CRF function by stress and alcohol.
Since drugs not directly associated with CRFR function prevented the ethanol adaptation induced by the repeated cycling of alcohol (See Section 2.3; Figure 3), studies of other drugs were undertaken to define if neural mechanisms other than CRF seen with adaptation by the repeated cycling of alcohol were involved in stress sensitization of alcohol withdrawal anxiety. As shown in Figure 5 both flumazenil and a 5-HT1A receptor agonist (buspirone) given prior to each of the stresses also prevented sensitization of ethanol withdrawal-induced anxiety (Breese et al., 2004). During this pharmacological testing, one distinct difference was noted between the stress/alcohol withdrawal protocol and the repeated alcohol withdrawal protocol. This difference was that a 5-HT2C-receptor antagonist did not block the cumulative adaptation induced by the repeated stress/withdrawal protocol (Breese et al., 2004), as it did for the repeated withdrawal protocol (Figure 3; Overstreet et al., 2003). Therefore, while results demonstrate considerable overlap in the neural mechanisms responsible for the adaptation that supports sensitization of withdrawal anxiety induced by the repeated alcohol withdrawal and stress/withdrawal protocols, it is equally apparent that a difference in the neural control of adaptation by these protocols exists.
4.2. a. Consideration of cytokine mediation of stress facilitation of alcohol adaptation
In addition to convincing evidence that CRF is involved in the action of stress (Vale et al., 1981), literature has also shown that stress increases cytokines in brain (Deak et al., 2005; Minami et al., 1991; Nguyen et al. 1998; O’Connor et al., 2003; Shintani et al., 1995a; Shizuya et al., 1997; Suzuki et al., 1997). Others have expressed the view that cytokines act as neuro-modulators (Adler et al., 2006; Bauer et al., 2007; Rostène et al., 2007; Shintani et al., 1995b) and may contribute to depressive, anxiety and PTSD symptoms (Anisman and Merali, 2003; Dunn et al., 2005; Hayley et al., 2005; Pucak and Kaplin, 2005; Raison et al., 2006; Uddin et al., 2010.
To explore the possible involvement of cytokines in the action of restraint stress used for the stress/withdrawal protocol, a single stress exposure nearly doubled the cytokine TNFα in the cortex (unpublished data). Based upon this observation, the possibility was considered that cytokines could substitute for stress to facilitate the alcohol adaptation responsible for sensitization of alcohol withdrawal-induced anxiety. Upon substituting several cytokines for the weekly stresses applied prior to 5 days of alcohol diet, Breese et al. (2008) found that these selected cytokines (i.e., TNFα, IL1β, and CCL2) sensitized alcohol withdrawal-induced anxiety (Figure 6). During this investigation, flumazenil administered prior to the cytokine exposures prevented this sensitization of alcohol withdrawal anxiety—evidence that the repeated cytokine exposure induced a cumulative adaptation, just as observed with the repeated stress/withdrawal protocol (Breese et al., 2004). Additionally, anxiety was observed during withdrawal from re-exposure to an additional 5 days of alcohol 16 days after termination of the original protocol—evidence that the cytokine/alcohol withdrawal protocol induced a persistent adaptation. Flumazenil administration prior to each of the repeated cytokine exposures prevented this sensitization to re-exposure to alcohol—evidence that this persistence depends upon the cumulative adaptation initiated by the repeated cytokine/alcohol protocol (Breese et al., 2008). Given these observations, the possibility is likely that cytokines released by stress contribute to the mediation of stress sensitization of alcohol withdrawal-induced symptoms (Breese et al., 2008).
Figure 6. Repeated cytokines sensitize alcohol-withdrawal anxiety (decrease social interaction).

Rats were injected with individual cytokines [CCL2 = chemokine (C-C motif) ligand 2; IL1β = interleukin-1β; TNFα = tumor necrosis factor-α] at a weekly interval prior to exposure to 5 days of 4.5% alcohol diet (ED). CD VEH = vehicle during control diet; ED-Veh = vehicle prior to alcohol diet. CD-CCL2 = CCL2 given weekly during control diet exposure. Social interaction as a measure of anxiety-like behavior was determined 5–6 hrs after alcohol removal. * P< 0.001 compared to CD-Veh and ED-Veh groups. From Breese et al., (2008).
An impetus for continuing to define the role of cytokines in stress influences on alcohol adaptation comes from a recent report that a genetic polymorphism occurs in the cytokine gene for IL1β that is associated with alcohol dependence (Liu et al., 2009). Furthermore, cytokines reportedly are elevated in plasma of alcoholics (Achur et al., 2010; Kiefer et al., 1991). Most importantly, He and Crews (2008) found elevated levels of the cytokine MCP1 (CCL2) in alcoholic brain, evidence that cytokines could be contributing to ethanol adaptation. Additionally, just as implied that alterations in CRF function are implicated in anxiety and depression (Hauger et al., 2009; Zobel et al., 2000; Zorrilla and Koob 2004), literature has also proposed cytokine involvement in these central disorders (Anisman and Merali, 2003; Dunn et al., 2005; Hayley et al., 2005; Pucak and Kaplin, 2005; Raison et al., 2006). Section 5 notes that such symptoms are elicited to stress in the abstinent alcoholic. Therefore, an intriguing consideration would be that the susceptibility of alcoholics to stress induction of depressive and anxiety symptoms may be linked not only to CRF, but also to cytokines (Fox et al., 2010).
In spite of the background consistent with cytokines contributing to the action of stress (Breese et al., 2008; Anisman, 2009), a CRF1R antagonist prevents stress-induced facilitation of ethanol adaptation (Breese et al., 2004). Integration of several reports may provide a means by which CRF and cytokines could interact for involvement in the action of stress to influence alcohol adaptation that would be prevented by a CRF1R antagonist. For example, Del-Cerro and Borrell (1990) reported that the cytokine interleukin-1 facilitated forced swim immobility, a “behavioral despair response”, which was minimized by neutralization of CRF with anti-serum. Additionally, Wang et al. (2002, 2003) demonstrated that CRF activation of CRF1Rs on microglia can facilitate release of cytokines. Thus, it is hypothesized that stress release of CRF can cause a subsequent release of cytokines (Wang et al., 2002, 2003), which in turn facilitate further CRF release (Del-Cerro and Borrell, 1990). In spite of background and knowledge that cytokines contribute to CRF control of HPA axis activation (See Turnbill and Rivier, 1999), it remains unknown whether sensitization of alcohol withdrawal anxiety that follows repeated cytokine exposures depends upon a CRF mechanism. Given the importance of this postulate concerning cytokine and CRF involvement in extra-hypothalamic function, future research can be expected to resolve whether indeed cytokines acting as central neuromodulators (Adler et al., 2006; Bauer et al., 2007; Rostène et al., 2007) depend upon CRF to contribute to the stress-induced facilitation of alcohol adaptation (Breese et al., 2004, 2008).
4.3. Neuroanatomical sites involved in the repeated stress-induced facilitation of alcohol withdrawal anxiety
Based upon CRF being released within the amygdala during withdrawal from alcohol (Merlo-Pich et al., 1995) and finding that CRF mediates alcohol-withdrawal anxiety (Baldwin et al., 1991) through actions within this structure (Rassnick et al., 1993), a strategy was undertaken to identify brain sites at which CRF mediated the action of stress to sensitize alcohol withdrawal anxiety. As before, the focus was on brain sites believed to be involved in anxiety-like behavior that had a relationship to stress (Koob, 2003; 2008; 2009; Shin and Liberzon, 2009). The approach taken was to microinject a CRF1R antagonist into selected brain sites prior to each stress applied before the 5-day exposure to chronic alcohol (Huang et al., 2010). As summarized in Figure 7, microinjection of a CRF1R antagonist into the CeA, DRN, or dBNST prior to each weekly stress exposure prevented sensitization of anxiety observed during alcohol withdrawal.
Figure 7. Microinjection of a CRF1-R antagonist (SSR) into the CeA, DRN, or dBNST prior to restraint stress prevents sensitization of alcohol withdrawal-induced anxiety-like behavior (reduction in social interaction).

The CRF-1 receptor antagonist SSR125543 (SSR; 10 μg/0.5 μl) was microinjected into the CeA, DRN, or the dorsal BNST (BNST) 15 min prior to the 2 weekly 60-min restraint stresses applied before exposure to 5 days of 4.5% alcohol diet (ED). For the CD-Vehicle and ED-Vehicle groups, vehicle was administered into each of the brain sites (N = 3–6 for each site) and these data combined. Social interaction, as a measure of anxiety-like behavior, was determined 5–6 hrs after alcohol removal. The CD-Vehicle group was not significantly different from the ED-vehicle group. *P < 0.001 when compared to the CD-Vehicle, ED-Vehicle, and CD Stress groups as well as the groups that received the CRF1R antagonist (SSR) into the three brain sites prior to the repeated stresses. From Huang et al. (2009) by permission of the publisher.
Following the results obtained with the CRF1R antagonist on stress facilitation of withdrawal anxiety (Figure 7), CRF, as a substitute for stress, was microinjected into various brain sites at a weekly interval prior to the single cycle of alcohol. This repeated site administration of CRF into the CeA, the DRN, or the dBNST was found to sensitize alcohol withdrawal-induced anxiety (Huang et al. 2010), providing further evidence for CRF involvement in the action of stress to sensitize anxiety during alcohol withdrawal. In contrast to the identification of brain sites positive for CRF action (Huang et al., 2010), repeated microinjection of CRF into the hypothalamic PVN, the NAC, and the CA1 region of the hippocampus prior to alcohol exposure did not support sensitization of alcohol withdrawal anxiety (Huang et al., 2010). The CRF microinjection into the PVN being without an effect provided further evidence that the HPA axis is not a component of the negative emotional state that accompanies withdrawal from chronic alcohol (Breese et al., 2004). In another study, flumazenil administered into the amygdala prior to each stress prevented the stress-induced cumulative adaptive process responsible for this sensitization of alcohol withdrawal anxiety (Knapp et al., 2007). Presently unknown is whether buspirone, which also prevents stress sensitization of anxiety (Breese et al., 2004) will do so at a site where this drug prevented sensitization of withdrawal anxiety induced by the repeated cycling of alcohol (See Figure 4).
Importantly, results obtained with CRF and the CRF1R antagonist administration into the CeA are consistent with earlier work that supported an involvement of CRF in withdrawal anxiety at this brain site (Baldwin et al., 1991; Rassnick et al., 1993); however, the other CRF-positive brain sites identified had not previously been implicated in the anxiety following alcohol withdrawal. When a CRF1R antagonist was administered systemically prior to CRF microinjection into the CeA, the d-raphe (DRN) and the d-BNST, the antagonist prevented the CRF-induced sensitization, just as this antagonist prevented stress sensitization of withdrawal anxiety (Figure 7; Huang et al., 2010). Collectively, this work not only supports the importance of the CRF1R being critical to the action of CRF and stress, but also demonstrates that CRF action on this receptor subtype induces the cumulative adaptation that supports sensitization of alcohol withdrawal anxiety. This latter conclusion concerning the importance of the CRF1R in the action of CRF at these sites was confirmed by finding that microinjection of urocortin3 into selected CRF-positive sites prior to alcohol produced no sensitization of withdrawal anxiety (Huang et al., 2010).
CRF being without effect when microinjected into some brain sites, while having selectivity to induce sensitization in several brain regions, supports brain region selectivity for expression of anxiety—a conclusion noted with the microinjection studies performed for the anxiety associated with withdrawal from the repeated cycling protocol (Section 2.3). Even though this effort to identify brain sites involved in the sensitization related to CRF and stress is important and informative, the results do not provide a definitive explanation for the means by which CRF action in differing brain regions supports an adaptation responsible for sensitization of alcohol withdrawal-induced anxiety. Consequently, just as proposed earlier for repeated ethanol withdrawal adaptation, a future focus should be on how these various brain sites interact within a neural circuit to allow stress to induce sensitization of anxiety during ethanol withdrawal. Additionally, given a potential relationship between CRF and cytokines mediating stress facilitation of ethanol adaptation, a future investigation should define whether cytokine substitution for stress sensitization of alcohol withdrawal-induced anxiety (Breese et al., 2008) depends upon those brain sites that support CRF sensitization or whether cytokine action occurs at sites distinct from those sensitive to CRF (Huang et al., 2010).
4.4. Influences of stress on alcohol consumption, the ADE, and reinstatement of alcohol seeking: Drug actions and neuroanatomical substrates
For sometime, investigators have attempted to initiate greater drinking with stress as a means to address the role of stress in alcohol ingestion predicted by the tension-reduction hypothesis (Conger 1956). However, considerable controversy has existed concerning whether stress can increase alcohol drinking. Pohorecky (1990) evaluated results obtained on alcohol drinking by stress and concluded that stress applied when animals were drinking alcohol resulted in minimal changes or a reduction in drinking. Nonetheless, more recently, stress has been demonstrated to influence alcohol self-administration, the ADE, and reinstatement of alcohol seeking. Because of the distinct characteristics of stress on these models, stress influences on each are discussed separately.
4.4. a. Effect of stress on alcohol self-administration
As mentioned above, in acute paradigms commonly used in alcohol research, stress exposure leaves alcohol drinking unaffected or even reduces it when applied while drinking. However, Lynch et al. (1999) demonstrated that restraint stress increased alcohol intake in Wistar rats. While Chester et al. (2004) reported that stress did not increase excessive drinking when applied during drinking, excessive drinking was facilitated at a later time by the stress. Furthermore, when restraint stress was applied during periods of abstinence from repeated cycling of voluntary alcohol drinking of P-rats, a consistent accentuation of the ADE was observed (Breese et al., 2004; Overstreet et al., 2007). An example of this facilitation of ADE is presented in Figure 8. Importantly, during the final 5 days of drinking after stress application the level of intake remains elevated rather than rapidly returning to baseline as is normally the case when only cycling of voluntary alcohol drinking is performed (Overstreet et al., 2007). Following the stress application during multiple cycles of voluntary alcohol drinking (ADE) in P-rats, the stressed animals displayed sensitization of anxiety-like behavior during withdrawal from the final drinking bout (Overstreet et al., 2007).
Other work has demonstrated that a prolonged history of voluntary access to alcohol alters stress influences on alcohol drinking. For example, following 4 months of free-choice drinking, which also included a cycle of forced deprivation, both forced swim stress and footshock stress resulted in approximately a doubling of voluntary alcohol intake (Vengeliene et al., 2003). Further, by evaluating the differing effects of stress in two rat lines genetically selected for high alcohol preference, Vengeliene et al. (2003) effectively demonstrated the importance of genetic background for stress influence on alcohol intake. More recently, the effects of forced swim stress on subsequent alcohol consumption was evaluated in Wistar rats with and without chronic intermittent alcohol vapor exposure that induced dependence (Sommer et al., 2008). This effort demonstrated that alcohol intake was unaffected by stress in animals without a history of dependence, whereas animals with a history of dependence not only started out at a higher–escalated level of consumption, but the stress induced a further increased in the voluntary alcohol intake. An intriguing additional finding was that the stress-induced increase in drinking remained for an extended period after the stress exposure was terminated (Sommer et al. 2008)—an outcome similar to that previously reported (Valdez et al. 2003) (see also Section 3.1). Based upon this observation by Sommer et al. (2008), an aspect needing clarification is whether producing dependence to alcohol prior to stress exposure in the ADE protocol in P-rats will induce an even greater facilitation in voluntary alcohol drinking than seen with stress alone (Overstreet et al., 2007).
4.4. b. Drug actions and neuroanatomical support of stress on alcohol self-administration
Even though the pharmacology and neurocircuitry of stressed-potentiated drinking has generally been neglected, a link between stress escalation of alcohol self-administration or consumption in dependent animals and increased behavioral responses to stress in the post-dependent state are both selectively sensitive to inhibition by CRF1R antagonists (Chu et al., 2007; Finn et al., 2007; Funk et al., 2007; Gehlert et al., 2007; Gilpin et al., 2008b; Sommer et al., 2008). The importance of CRF in stress-induced alcohol drinking was further emphasized by finding that a genetic variant of CRF in primates increased alcohol consumption by stress (Barr et al, 2009). Surprisingly, CRF-deficient mice consumed twice as much alcohol as wild type mice (Olive et al., 2003). Likewise, mice lacking CRF1Rs exhibited enhanced drinking to stress that persisted (Sillaber et al., 2002), another unexpected finding. An elevation of the NR2B receptor subunit was provided as an explanation for the CRF1R deletion enhancing drinking (Sillaber et al., 2002).
Overstreet et al. (2007) examined the effect of several drugs on the stress-induced increase in ADE in P-rats as well as on accompanying sensitization of alcohol withdrawal-induced anxiety. In this latter case, buspirone, flumazenil, and a CRF1R antagonist given prior to each stress applied during deprivation from repeated cycling of voluntary alcohol drinking successfully prevented the stress-induced facilitation of ADE as well as the sensitization of anxiety observed during the final alcohol withdrawal (Overstreet et al., 2007). Of further interest to the mediation of stress on drinking is the finding that a D2-dopamine receptor antagonist and naloxone blocked the stress-induced increase in ADE without preventing the stress-induced sensitization of withdrawal-induced anxiety during the final withdrawal from this repeated voluntary alcohol ingestion (Overstreet et al., 2007)—an outcome that contrasts with blockade of the repeated alcohol sensitization of withdrawal-induced anxiety as well as the ADE by buspirone, flumazenil and a CRF1R antagonist (Breese et al., 2004; Knapp et al., 2004, 2005; Overstreet et al., 2003, 2004a, 2007). Thus, distinct neural mechanisms must contribute to the stress-induced cumulative adaptation responsible for facilitation of the ADE and sensitization of alcohol withdrawal anxiety in the P-rats. Further, genetic deletion of the neurokinin-1 receptors (NK1Rs) or blockade of NK1Rs to minimize substance P (SP) action was found to markedly reduce voluntary alcohol consumption in C57BL/6 mice (George et al., 2008; Thorsell et al., 2010). This observation is consistent with reports that SP acting at NK1Rs within the amygdala complex mediates behavioral responses to stress both in rodents (Ebner et al. 2004) and in humans (Furmark et al. 2005; Michelgard et al. 2007). In Section 6, information will indicate that a NK1R antagonist prevented challenge-induced symptoms in human abstinent alcoholics.
Just as investigations sought to define brain regions involved in the negative emotional state observed during withdrawal from chronic alcohol (See Section 2.3), subsequent studies sought to elucidate brain sites at which stress facilitated the ADE. Because CRF is a major contributor to the actions of stress, initial studies utilized microinjection of CRF into various brain sites during deprivation to identify sites supporting stress facilitation of the ADE and the accompanying sensitization of alcohol withdrawal anxiety. Since CRF1R expression was up-regulated within the amygdala as well as in other components of the extended amygdala with alcohol dependence (Sommer et al., 2008), the CeA appeared to be a plausible site for stress to facilitate alcohol drinking by the ADE. However, CRF microinjection into the CeA did not support CRF-induced facilitation of the ADE, but did support sensitization of alcohol withdrawal-induced anxiety (unpublished data). Likewise, CRF microinjection into the raphe sensitized alcohol withdrawal anxiety without facilitation of the ADE. Given the relationship of the ventral tegmental area (VTA) (Gatto et al., 1994; Rodd et al., 2004) and the NAC to alcohol self-administration and responding for alcohol (Besheer et al., 2010; Chaudhri et al., 2009), CRF was microinjected into these sites during deprivation from the voluntary alcohol drinking to determine if the ADE would be facilitated. The repeated CRF treatment into the NAC enhanced the ADE without altering anxiety during alcohol withdrawal. Injection of CRF into the VTA affected neither measure. Subsequently, it was determined that a CRF1R antagonist microinjected into the NAC prior to stress prevented the stress-induced increase in the ADE, while being without an effect on stress sensitization of alcohol withdrawal anxiety (Breese et al., 2010). This evidence that an action of stress in the NAC increased drinking without inducing sensitization of alcohol withdrawal anxiety negated a previous view that the degree of alcohol intake induced by stress during intermittent voluntary alcohol drinking was responsible for the sensitization of alcohol withdrawal-induce anxiety (Overstreet et al., 2007).
Additionally noteworthy is finding that CRF in the CeA is involved in facilitation of alcohol self-administration in rats following alcohol physical dependence (Funk et al., 2006b). However, as noted for non-dependent P-rats, the CeA is not involved in stress facilitation of the ADE (Breese et al., 2010). Therefore, future efforts should attempt to clarify the neurobiological basis for the dependence associated adaptation involving a brain site for CRF facilitation of alcohol self administration distinct from that observed for the non-dependent P-rats. One aspect that could be explored would be whether stress facilitation of alcohol self administration in physically dependent P-rats would involve both the CeA and the NAC.
Demonstration of the site selectivity of CRF-related drinking and induction of sensitization of withdrawal anxiety provides strong evidence that the neural mechanisms associated with these central changes induced by stress can involve distinct brain regions. Yet to be determined is whether buspirone and flumazenil, which also prevent stress facilitation of the ADE, will act within the NAC to prevent the stress facilitation of drinking, just as observed with the CRF1R antagonist, or whether other brain sites will be involved. Regardless of the site of action of these drugs on the stress facilitation of the ADE, a presumption would be that buspirone and flumazenil will prevent sensitization of withdrawal anxiety induced by the stress facilitation of the ADE at sites where they minimized sensitization of anxiety induced by intermittent chronic alcohol exposures (Figure 3; Knapp et al., 2007b; Overstreet et al., 2006).
4.4. c. Influence of stress on reinstatement of alcohol seeking: Drug actions and neuroanatomical substrates
As mentioned above (Section 3.3), stress can reinstate alcohol-seeking behavior in rodents following extinction of operant self-administration (Lê et al., 1998, 1999; 2000; Lê and Shaham, 2002; Liu and Weiss, 2002a, 2003; Martin-Fardon et al., 2000; Weiss et al., 2001), just as stress increased ADE. Lê et al. (1998) reported that stress was a more powerful stimulus for reinstating alcohol seeking than was an alcohol-priming dose. Further, Liu and Weiss (2002a, 2003) also reported that post-alcohol dependent rats exhibit increased alcohol seeking behavior and a greater facilitation of responding to stress—a finding consistent with other observations which demonstrated that previous chronic alcohol exposure facilitates the consequences of stress on operant reinstatement responding (Rimondini et al 2002; Sommer et al. 2008; Valdez et al 2003). This latter outcome supports the concept (Breese et al., 2005a,c) that a persisting adaptation that follows previous intermittent chronic alcohol exposures induces an allostatic state that enhances responsiveness to a subsequently stress.
Liu and Weiss (2002a) demonstrated that a general CRFR antagonist inhibited stress-induced reinstatement of alcohol seeking. However, this CRFR antagonist had no effect on the increase in cue-induced responding in these previously dependent rats (Liu and Weiss, 2002a). Lê et al. (2000) was first to demonstrate that blockade of CRF1 receptor function would prevent stress-induced reinstatement of alcohol seeking. More recently, Gehlert et al. (2007) documented that CRF1R blockade prevented stress-induced alcohol seeking in post-dependent rats. Consistent with CRF involvement in stress, CRF itself reinstated alcohol seeking (Lê et al., 2000; Lê and Shaham, 2002). In further agreement with a link of stress to CRF, the α2-adrenoceptor antagonist yohimbine, which potently induces anxiety responses in alcoholics (Krystal et al. 1996), also reinstates alcohol seeking and induces excessive alcohol self-administration in a CRF1R-dependent manner (Marinelli et al. 2007). Consistent with CRF involvement in stress influences on alcohol, Funk et al. (2006a) found that stress-induced alcohol reinstatement was associated with an increase in CRF mRNA in the CeA and basolateral amygdala.
Additionally, Hansson et al. (2006) reported that Marchigian-Sardinian-prefering (msP) rats, which have high alcohol preference, had an upregulation of the Crhr1 transcript in the CeA, medial amygdala, and the basolateral amygdala that was accompanied by elevated CRF1R density. These changes in CRF function in the msP rats were accompanied by an increase in stress-induced increase in alcohol seeking that was blocked by a CRF1R antagonist (Hansson et al., 2006), as was the increased alcohol self-administration in these msP rats. However, the CRF1R antagonist was without effect in unselected Wistar rats on alcohol self-administration and did not suppress the stress-induce increase in alcohol seeking in these rats (Hansson et al., 2006). Of interest is the down regulation of elevated Crhr1 mRNA in various brain sites of msP rats after about 2 weeks of ad lib access to 10% ethanol (Hansson et al., 2007). Collectively, these observations suggest that a genetic predisposition for excess CRF function in the msP rats can mimic the neuroadaptation induced by a chronic alcohol state (Sommer et al., 2008)—a circumstance that enhances the action of stress in Wistar rats (Hansson et al., 2006).
Comparison of various other peptides or their antagonists on cue versus stress reinstatement responding for alcohol has provided mixed results. Martin-Fardon et al. (2000, 2010) reported that nociceptin was capable of reducing stress influences on reinstatement of alcohol seeking as well as cue-induced reinstatement. A cannabinoid-1 receptor (CB1R) antagonist reduced cue induced, but not stress reinstatement responding for alcohol ingestion (Economidou et al., 2006b). Cippitelli et al. (2010) found that neuropeptide Y (NPY) blocked alcohol reinstatement responding induced by yohimbine-induced stress, but did not prevent cue-induced alcohol reinstatement responding. In previously dependent Wistar rats, a mu-opioid antagonist effectively inhibited cue-induced reinstatement to an alcohol paired lever, but was ineffective in blocking the stress-induced alcohol seeking (Liu and Weiss, 2002a)—findings which contrast to the action of a CRFR antagonist on these challenges (Liu and Weiss, 2002a). Such findings clearly denote that the mechanisms driving cue and stress reinstatement responding for alcohol may have a differing neural basis particularly after alcohol dependence. Ciccocioppo et al. (2009) have reviewed the potential involvement of several peptides that may relate to stress and the consequences of chronic alcohol.
5. Chronic Alcohol Neuroadaptation in Alcoholism: Relationship to Stress-induced Negative Emotion and Craving in Alcoholism
In addition to neuroadaptations that underlie tolerance and alcohol withdrawal symptoms associated with chronic alcohol, an additional clinical construct relevant to the desire to use drugs of abuse was first described by Wikler (1948). In this respect, Wikler (1948) noted that abstinent opioid addicted patients in residential treatment described an intense desire and physiological need for opiates—a longing referred to as “craving”. Later studies in abstinent alcoholics also documented a strong desire and intent to use alcohol (Ludwig et al., 1974). This strong emotional and physiological need in the abstinent alcoholic that results in this intense desire for alcohol ingestion was likewise defined as craving (Sayette et al., 2000)—a hallmark feature of alcohol dependence. In the following sections, laboratory challenges are described that reliably induce craving and alter physiological and neural measures in alcoholics.
5.1. Stress increases susceptibility for craving and alters physiological measures in alcoholics: A reflection of stress dysregulation
Basic studies reviewed in the previous sections clearly delineate that withdrawal related physiological and affective changes occur that can persist for an extended period after removal from alcohol exposure. In abstinent alcoholics, the “craving” to consume alcohol is a common symptom precipitated by negative emotional states that follow stress (Breese et al., 2005a; Brown et al. 1990; Cooney et al., 1997; Fox et al., 2005, 2007; Sinha, 2007; Sinha & O’Malley, 1999) and alcohol cues (Cooney et al., 1987; Monti et al., 1987; Payne et al., 1992). Furthermore, any craving induced by “stress” during acute alcohol withdrawal is not only accompanied by a negative emotional state but as well by aspects of physiological dysregulation, such as elevated levels of blood pressure, heart rate, and selected behavioral symptoms. This susceptibility for craving to stress and alcohol cues persists in the abstinent alcoholic. Importantly, upon alcohol removal other alterations are also known to persist for an extended period in the alcoholic, including residual anxiety, negative affect, sleep disturbances, and cognitive and behavioral changes (Brower, 2001; Drummond et al., 1998; Sinha et al., 2009, 2010). The time required for normalization (recovery) of the neural dysregulation in the alcoholic, which underlies the continuing susceptibility for craving to stress or negative symptoms, remains unclear. Nonetheless, the sizable progress made from these investigations resolved a lack of previous background on measures by which a persisting adaptation induced by alcohol abuse and dependence could be evaluated.
In early studies, findings indicated that stress imagery elicited alcohol craving as well as multiple emotions of fear, sadness and anger in alcohol addicted individuals, a change which contrasted with the stress of public speaking that elicited increases in fear, but no anger and sadness, and most importantly no increase in alcohol craving (Sinha and O’Malley, 1999). In a comprehensive assessment of subjective, neuroendocrine and physiological responses to exposure to stress and alcohol cues and neutral circumstances, the Sinha laboratory demonstrated that abstinent alcoholics exposed to stress and alcohol cues reported alcohol craving that was accompanied by anxiety, negative emotions and increases in physiological measures and an elevation of cortisol and adreno-corticotrophic hormone (ACTH) caused by activation of the hypothalamic pituitary adrenal (HPA) axis (Fox et al., 2007, 2009b; Sinha and O’Malley, 1999). In this respect, alcoholics with a greater severity of alcohol abuse show significantly higher stress and cue-induced alcohol craving, greater levels of stress and cue-induced anxiety, and higher levels of physiological and neuroendocrine activation to these challenges (Fox et al., 2009b). These data support the idea that a greater degree of chronic alcohol abuse is associated with concomitantly increasing levels of anxiety and craving as well as elevated alterations in physiological and biochemical indices of stress arousal.
The Sinha Laboratory has also pursued the possibility that the persistence of neuroadaptation associated with alcoholism contributes to compulsive alcohol seeking and high relapse rates in alcoholism (Sinha, 2007; Sinha et al., 2009, 2010; Fox et al., 2009a). Recovering alcoholics after 4-weeks of abstinence exhibit greater basal heart rate and salivary cortisol levels compared to control drinkers. Upon stress and alcohol cue exposure, alcoholics show persistently greater subjective distress, alcohol craving and blood pressure responses, but a suppressed heart rate and cortisol response to stress compared to controls (Sinha et al., 2009). In controls, the stress exposure did not increase alcohol craving, even though they showed robust physiological and endocrine responses. On the other hand, alcohol cue exposure in the controls produced a lower magnitude desire for alcohol than observed in alcoholics without exhibiting a concomitant negative emotional distress or an increased neuroendocrine response (Sinha et al., 2009, 2010). This assessment agrees with previous reports that recently abstinent alcoholics or smokers not only display increased craving (Yoon et al., 2006), but show an altered basal HPA axis level and a suppressed HPA cortisol response to stress compared to their non-addicted counterparts (Al-absi et al., 2005; Badrick et al., 2007; Junghanns et al., 2003; Lovallo et al., 2000; Munro et al., 2005). Furthermore, continued monitoring of physiological and subjective responses across multiple time points demonstrated that alcoholic patients show persistent stress-induced increases in alcohol craving, subjective distress, and blood pressure changes compared to stress in social drinkers—a finding suggestive of an inability of alcoholics to regulate their state of emotional distress and physiological responses linked with elevated craving (Sinha et al., 2009). These data support the view that stress-related alcohol craving and the enhanced emotional distress observed during alcohol cue exposure are associated with chronic alcohol use, but not with light to moderate alcohol exposure.
In summary, the studies reviewed indicate that the enhanced susceptibility to stress and cue-induced drug seeking observed in the alcoholic is not seen in healthy non-addicted individuals. Furthermore, there are basal alterations in peripheral markers of stress in the alcoholic which are indicative of stress-related dysregulation of CRF-HPA axis and autonomic function. High morning basal salivary cortisol and heart rate levels were observed in recovering alcohol-dependent patients that were associated with lower or blunted stress-related arousal. Such a pattern of HPA and autonomic dysregulation is similar to what has been documented in other chronic high distress states (Li et al., 2007; Steptoe and Ussher, 2006). It is important to note that these alterations were not accounted for by smoking status or lifetime history of anxiety or mood disorders; therefore, these changes observed to stress can be attributed to chronic alcoholism. Additionally, the persistence of emotional distress and alcohol craving induced by stress and alcohol cue exposure in the dependent alcoholic suggests a dysfunction in mechanisms regulating emotion. Since stress-induced HPA axis and autonomic responses are associated with regaining homeostasis, dysfunction of these responses in the alcoholic can serve as markers of the dysregulation of central stress mechanisms involved in perpetuating alcohol craving and relapse susceptibility. Certainly, the dysregulation of stress responses resulting in high levels of craving or compulsive alcohol seeking for an extended period in the abstinent, recovering alcoholic is consistent with the concept that adaptation induced by chronic alcohol use is dependent upon a sustained increase in allostatic load induced by a kindling-like process.
5.2. Gender differences in alcoholics to stress and alcohol cues
Sex differences are well documented in responses to stress as well as to changes induced by abusive drugs (Becker et al., 2007; Kudielka et al., 2005). Therefore, Chaplin et al. (2008) examined whether there were significant sex differences in the emotional stress responses and alcohol craving in healthy social drinkers exposed to stressful, alcohol related and neutral imagery. With this strategy, sex differences in emotional and physiological responses to stress and alcohol cues were observed. Interestingly, in contrast to women who reported greater sadness and anxiety in the stress condition, men showed a positive correlation between emotional distress and alcohol craving (Chaplin et al., 2008). Diastolic, but not systolic, blood pressure responses to stress were greater in men than women. Although men reported somewhat higher levels of alcohol consumption than women, the level of alcohol consumption did not account for this sex-specific association between negative affect and alcohol craving (Chaplin et al., 2008). This greater vulnerability to alcohol craving in men is consistent with other findings indicating a tendency of men having an inability to cope with stress (Gentry et al., 2007; Lindquist et al., 1997; Tamares et al., 2002). In contrast to men, women show rumination and negative thinking in stressful situations (Nolen-Hoeksema, 1987). These distinct tendencies seen with gender may contribute to the well known sex differences observed in the prevalence of alcohol use disorders (Kessler et al., 1993; 1994).
When men and women who drink socially are compared to their abstinent alcohol dependent counterparts on biological stress levels, Fox et al. (2009b) found clear sex differences in the pattern of stress dysregulation associated with chronic alcoholism. Alcoholic males showed increased ACTH and epinephrine basal tone compared with control males and alcoholic females. However, the alcoholic males demonstrated no increase in ACTH and cortisol levels following stress and alcohol cue imagery exposure compared to the neutral condition. Control males and alcoholic females demonstrated a typical stress response in both measures. In addition, alcoholic males showed no increase in cardiovascular responses to either stress or cue, and no increase in catecholamine response to cue compared with their response to neutral imagery. Again, this flattening of the stress response was not observed in control males who produced significantly higher levels of cue-related heart rate, epinephrine, and significantly higher stress-related diastolic blood pressure. In contrast, alcoholic females showed an enhanced ACTH and cortisol response to stress and an alcohol cue compared with neutral imagery—a change that was not observed in the healthy control females. Alcoholic females also showed blunted norepinephrine and epinephrine levels and reduced heart rate to stress compared with healthy females. Therefore, while alcoholic males show the previously discussed increased basal stress levels and a generalized suppression of HPA cortisol, sympatho-adrenal response as well as markers of cardiovascular function following both stress and alcohol cues, females showed a selective sympatho-adrenal suppression to stress and an enhanced HPA response to both stress and alcohol cues (Fox et al., 2009b). Interestingly, these differences in physiological and neuroendocrine patterns were not seen in alcohol craving levels which were comparable across men and women. These differential patterns of physiological and neuroendocrine findings with sex may reflect involvement of distinct neurobiological processes in the compulsive alcohol craving state in alcoholic men and women. Therefore, such manifested differences between men and women should be an important consideration in therapy development for alcoholics of differing sex.
5.3. Investigations of the relationship of craving and physiological measures to relapse in alcoholism
The potential role of stress in increasing risk of alcohol relapse was initially support by work showing that heavier drinkers, and not light drinkers, were more susceptible to stress-induced alcohol consumption in laboratory situations (Marlat et al., 1975; Marlatt & Gordon, 1980). In addition, alcohol-related stimuli or “cues” or alcohol itself are potent factors to increase the risk of relapse in the alcoholic. Thus, it is not surprising that stress and alcohol cues are associated with significant increases in alcohol craving and compulsive alcohol seeking in the abstinent alcoholic—responses which in turn are predictive of alcohol relapse (Cooney et al., 1997; Litt et al., 2000; McKay et al., 2005; Miller et al., 1996; Sinha, 2007; Tate et al., 2005; Zywiak et al., 2003). In alcoholics with varying levels of abstinence, the degree of negative mood and stress-induced alcohol craving (Cooney et al., 1997, Breese et al., 2005a; Sinha, 2007) as well as blunted stress-induced cortisol responses were associated with alcohol relapse (Adinoff et al., 2005a,b,c; Brady et al., 2006a,b; Junghanns et al., 2003).
In a previous study with cocaine-dependent individuals, Sinha et al. (2006) examined whether stress-induced cocaine craving in the laboratory significantly predicted time to cocaine relapse. The findings indicated that stress-induced cocaine craving was indeed predictive of a shorter time to relapse. In this study, stress-induced ACTH and cortisol responses were not associated with time to relapse, but these responses were predictive of amounts of cocaine consumed during follow-up. More recently, these studies were extended to abstinent recovering alcoholics to understand whether the increased subjective distress and alcohol craving is predictive of alcohol relapse. Sinha (2008b) prospectively followed abstinent, alcohol dependent individuals who completed a 5-week inpatient treatment for a 90-day period post-discharge to assess whether laboratory craving and subjective and biological responses to stress and alcohol cues were predictive of a later relapse to drinking. Subjects returned for repeated assessments 14, 30 and 90 days after discharge with follow-up rates of compliance of 96%, 89% and 92%, respectively. Findings indicated that both stress-induced and alcohol cue-induced craving were predictive of time to alcohol relapse (Sinha, 2008b). Furthermore, higher cortisol levels and cortisol/ACTH ratio (a measure of adrenal sensitivity) across all conditions also significantly predicted a decreased time to initial alcohol lapse (time to first drink) and time to relapse to the first heavy drinking day of 5 drinks or more in men and 4 or more in women. While a high basal HPA axis tone occurs in alcoholics that is accompanied by a blunted HPA responsivity to stress (Sinha, 2008b), the higher morning basal HPA tone is a better predictor of time to alcohol relapse than is the acute stress-related HPA response.
6. Imaging of Brain Regions Activated in Alcoholics by Alcohol Cues and Stressful Imagery
Modell et al. (1990) proposed that alcoholics suffer from dysfunction of basal ganglia/limbic striatal and thalamocortical neural circuits. Based upon this view that specific regions of the corticostriatal limbic circuits would be altered during exposure to emotional stress and alcohol cues in alcoholics, compared to social drinkers, brain imaging of alcoholic subjects was initiated to determine if alcohol cues that resulted in craving could be associated with specific regions of brain (Braus et al., 2001; George et al., 2001; Modell and Mountz, 1995; Park et al., 2007; Schneider et al., 2001). As outlined below, these early investigations of brain imaging in the alcoholic were the impetus to explore not only alcohol cues, but also the consequences of stress on brain activity. Recently, drug testing has also been performed on brain activity changes induced by alcohol cues in the alcoholic.
6.1. Brain regions activated by stress in alcoholics that initiate negative emotion and craving: Evidence for neuroadaptation and alcohol relapse
An early study to measure blood flow with SPECT imaging found a change in the caudate nucleus during induction of craving in alcoholics (Modell and Mountz, 1995). Subsequently, George et al. (2001) found a greater increase in brain activity to alcohol cues in the anterior thalamus and left dorsal lateral prefrontal cortex using functional magnetic resonance imaging (fMRI). Using a memory task during fMRI to evaluate brain, Tapert et al. (2001) found dysfunctional changes in the cortex of alcoholics distinct from that of controls. Subsequently, other imaging studies with alcoholic patients showed an association between increased activity of dorsal striatum regions and alcohol craving in response to presentation of alcohol-related stimuli (Grusser et al., 2004; Wrase et al., 2002). Myrick et al. (2004) reported that alcohol cues produced changes in the left orbital frontal cortex, anterior cingulate cortex and NAC in alcoholics, which were not seen in control subjects. Figure 9 provides an illustration of brain imaging demonstrating the distinct fMRI response of an alcoholic to an alcohol cue compared to the response in a corresponding control. The brain regions exhibiting the greatest activation in the alcoholic were the right and left insula, the right inferior frontal cortex, the left lingual cortex, and the right medial temporal cortex (Gilman and Hommer, 2008). Additionally, PET imaging of alcohol-dependent individuals demonstrated a significant association of D2-dopamine receptor binding in the ventral striatum with alcohol craving (Heinz et al., 2004, 2005) and a motivation for self-administration of alcohol (Martinez et al., 2005; 2007). To emphasize the importance of this approach to investigating drugs of abuse, recent PET studies of D2-receptor binding have shown significant positive correlations between selected dorsal striatum brain regions and cue-induced cocaine craving (Wong et al., 2006; Volkow et al., 2006).
Figure 9. Sensitized brain responses to aversive visual stimuli healthy controls and in alcohol dependent patients approximately 3 weeks into abstinence.
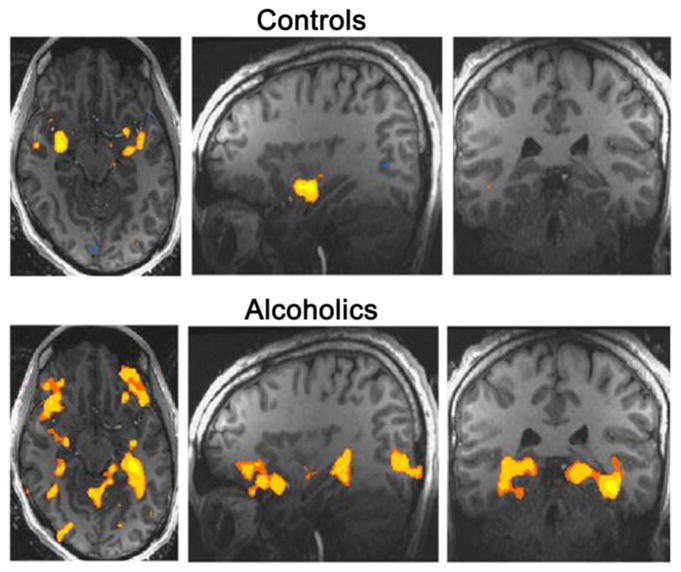
Among 10 distinct areas altered (Gilman and Hommer, 2008), the brain regions exhibiting the greatest activation (P< 0.001) in the alcoholic were the right and left insula, the right inferior frontal cortex, the left lingual cortex, and the right medial temporal cortex. Reproduced with permission from Gilman and Hommer, 2008).
In investigations by the Sinha laboratory (Sinha, 2007; Sinha and Li, 2007), dependent alcoholics abstinent from alcohol for 4-weeks were compared to social drinkers who were exposed a single fMRI session. Using structured guided imagery, these participants received two trials each of personalized stress, alcohol cues and neutral situations (Sinha et al., 2007). Significant group main effects were observed in the ventromedial prefrontal cortex, insula, the ventral striatum, and specific regions of the thalamus and cerebellum, with alcoholic patients showing greater activity in limbic-striatal regions across the imagery conditions (Sinha, 2007; Sinha and Li, 2007) and lower activity in the medial frontal and cingulated regions. Specific condition effects were observed in the caudate and posterior cingulate regions (Sinha, 2007; Sinha and Li, 2007). Consistent with this description of change in alcoholics (Sinha et al., 2007; Sinha and Li, 2007), Figure 10 provides visual examples of the fMRI changes in neural activity observed in selected brain regions of alcoholics (AD) in relation to changes in social drinkers (SD). Note the increased activity in the NAC/ventral striatum region and in the insular cortex in the alcoholic in both the neutral and stress conditions. In contrast, hypo-responsivity was observed to stress and alcohol cues in the orbito-frontal cortex and the medial-prefrontal cortex (mPFC) in alcoholic brain compared to the change in social drinkers (Figure 10).
Figure 10. Functional magnetic resonance imaging (fMRI) responses during exposure to stress (S), alcohol cue (C; Cues), and neutral relaxing (N; Neutral) imagery trials.

Twenty-eight recently abstinent alcohol dependent (AD) and 24 healthy social drinkers (SD) are included in the trials. Whole brain voxel-based mixed effects model analyses for significant Group 2 (AD, SD) main effects and Group X Condition 3 (S, C, N) interaction effects are shown for the selected brain regions at specified thresholds of significance (p<.05, p<.01, p<.005 as noted). Talaraich coordinates (x,y,z) designate the center of activation in each brain region. Whole brain voxel based maps for main and interaction effects are followed by group difference maps (AD minus SD) depicted in the right three images for each test condition (Neutral, Stress, Alcohol Cue).
For (a), the nucleus accumbens/ventral striatum (NAc/VS) region, a significant group main effect is shown at two separate thresholds ([i] p<.05; [ii] p<.01 on the left in red/yellow). Simple effect contrasts show greater activation in AD subjects than SD controls in the neutral and stress conditions (p<.01; depicted in red/yellow), but not in the alcohol cue condition (i.e., the three images shown on the right). For (b), the orbitofrontal cortex (OFC), a significant group main effect was seen at two separate thresholds ([i] p<.05; [ii] p<.01 shown on the left). Simple effects to explain this significant group effect in the OFC indicated lower activity in AD patients relative to SD controls as shown in pink for the OFC region across all three conditions (p<.01; AD less than SD).
For (c), the left insula region, the Group X Condition significant interaction effect is shown on the left in red/yellow at [i] p<.01 and [ii] p<.005 thresholds. Simple effect contrasts to explain the significant interaction effect indicated greater activity in AD patients relative to SD controls in the neutral and stress condition shown on the right (significant contrast area shown in red/yellow, p<.01), but not in the alcohol cue condition.
For (d), the medial prefrontal cortex (mPFC) region, group by condition significant effect is shown on the left in red/yellow at [i] p<.005 and [ii] p<.01 thresholds. Simple effect contrasts to explain this significant interaction effect indicated that it was a result of lower activity in mPFC in AD patients relative to SD controls in the stress and alcohol cue conditions shown in pink on the right (p<.01) and not due to a activity change in the neutral condition.
Recently, Seo et al. (2010), using structured guided imagery and fMRI (Sinha et al., 2007) found robust activity changes to stress and alcohol cues relative to the neutral condition in the ventro-medial-PFC, dorsal-lateral PFC, cingulated cortex, amygdala, as well as several other brain sites in the alcoholics. Hyper-responsivity within the ventro-medial-PFC, anterior cingulated cortex and ventral striatum of the alcoholics during neutral relaxed states was associated with a blunted response to stress and alcohol-cue exposure. Such hyper-responsivity during relaxation within brain sites being accompanied by blunted responses during stress predicted stress- and alcohol cue-induced craving and concomitant anxiety. This difference in brain responses between the neutral imagery and stressful cues shown in alcohol-dependent patients is suggestive of a poor ability to differentiate between challenging and relaxed states (Sinha et al., 2007; Sinha and Li, 2007). Importantly, both the hyper-responsivity of the ventral-medial-PFC and anterior-cingulated cortex during neutral relaxed states and the blunted response during stress were also predictive of a greater propensity for a shorter time to relapse (Seo et al., 2010).
The fMRI findings in abstinent alcoholics, which show an overall hyper-responsivity of the striatal-limbic regions associated with emotion processing, but hypo-responsivity in medial frontal regions associated with emotional regulation, self control, and executive functioning, likely accounts for the central dysregulation that accounts for the increased susceptibility for relapse (Sinha, 2007; Sinha and Li, 2007; Seo et al., 2010; Figure 10). Such central activity changes in these brain sites of the alcoholics are consistent with an overall kindling process increasing an allostatic load that results in this dysfunction in neural processing. Therefore, it can be presumed that normalization of these alcohol dysregulated brain regions is critical for restoring integration of emotional and motivational functions. Consequently, normalizing this dysfunction within these brain sites in the alcoholic would be an important strategy for improving recovery and minimizing relapse to alcohol abuse. In 6.2 to follow, a particularly promising approach to identify potential new pharmaco-therapies is to employ testing drug inhibition of craving and brain imaging changes induced by stress and alcohol cues in alcoholics.
6.2. Drug treatments on brain imaging in alcoholics to stress and alcohol cues: Approach for human drug testing
While several efficacious behavioral and pharmacological therapies in the treatment of alcoholism exist, it is well known that relapse rates in alcoholism remain high (Sinha, 2001; O’Brien, 2005; Vocci et al., 2005). Exposure to stress, alcohol-related stimuli and alcohol itself can each reinstate drug-seeking behavior in animals and increase relapse susceptibility in alcohol dependent individuals (Sinha, 2007; Shaham and Hope, 2005; Shaham et al., 2003; Weiss, 2005). Such data underscore the need for a specific understanding of the phenomena of relapse susceptibility in order to target development of pharmacological therapeutics that can minimize this liability alcoholics have for relapse. Findings from basic science laboratories have identified several pharmacological treatment targets to address stress-induced anxiety and reinstatement of drug seeking. As noted in Sections 2 & 3, basic science data suggest that CRF1R antagonists, α2-adrenergic agonists, buspirone, baclofen, a NK1R antagonist and selected glutamatergic agents could be promising treatments to address stress-related relapse in alcoholism. Therefore, human laboratory investigations are needed that will screen these agents in alcoholics to assess their potential efficacy on surrogate markers that predict suppression of stress-related relapse.
A particularly promising strategy for identification of new pharmaco-therapies would be to determine if drugs, which have been found to have efficacy in preclinical investigations against models of alcohol action, would prevent stress- and alcohol cue-induced craving, craving related anxiety, HPA measures, heart rate variability as well as the brain imaging changes in specific brain regions induced by these stressful challenges (Sinha, 2007; Sinha and Li, 2007). Thus, future human laboratory studies are needed that will screen new as well as established agents in alcoholics to assess their effectiveness against the stress and cue-induced changes. The next course would be to determine whether actions on these measures associated with the alcoholic would be predictive of minimizing or preventing relapse. Given that human laboratory investigations have documented the neural and physiological changes induced in alcoholics by stress and alcohol cues, this strategy outlined could be a potentially powerful approach for identifying drugs for further clinical testing against the susceptibility for relapse observed in alcoholics.
From preclinical background, an obvious candidate mechanism to target alcohol-induced neuroadaptation in alcoholism would be antagonism of CRF1Rs. Based upon preclinical evidence that CRF1Rs are involved in the increased stress responsiveness in rodents (Breese et al., 2005b; Sommer et al. 2008; Valdez et al., 2002), it can be predicted that testing of a CRF1R antagonist would reduce craving and exaggerated brain responses to stress in alcohol-dependent patients. However, clinical testing of CRF1R antagonists to reduce relapse in alcoholism is only now being considered. Nonetheless, as noted in Section 4.4b, substance P (SP), like CRF, has been found to have an involvement in stress responsiveness (Ebner et al. 2004; Furmark et al. 2005; Michelgard et al. 2007) and in ethanol drinking (George et al. 2008). Consequently, given the similarities of CRF and SP to stress, the assumption was that some insight might be gained by investigating whether SP mediation influences behavioral and functional neural responses of alcoholics to stress and alcohol cues.
Based upon this background, the NIAAA intramural clinical program tested a whether a NKIR antagonist administered to human alcoholics would affect alcohol craving and brain imaging responses to negative affective stimuli (George et al. 2008). This investigation demonstrated that pharmacological blockade of NK1Rs in alcohol-dependent patients suppressed subjective cravings for alcohol and cortisol responses induced by exposure to stress and alcohol associated stimuli (George et al. 2008). Figure 11 documents that the NK1R antagonist normalized the exaggerated brain responses to the negative affective stimuli in key brain regions in this alcoholic population, including most importantly the metabolic changes in the anterior insula induced by the stimuli (George et al. 2008; Figure 10). This latter structure is of particular interest because it is reciprocally interconnected with the amygdala, encodes aversive interoceptive states, and has been implicated in mechanisms of craving and relapse (see review by Naqvi and Bechara, 2009).
Figure 11. NK1R antagonist (LY686017) blockade of fMRI BOLD responses to visual affective stimuli in hospitalized alcoholics.

In the placebo group of alcoholics, there were robust significant activations to the application of the negative stimuli in the inferior frontal gyrus, insula, and middle temporal gyrus. The NK1R antagonist-treated group (LY686017) had significantly less activation in these areas. Likewise, the placebo control group had little activation in response to positive emotional stimuli. The NK1R antagonist group of controls exhibited activation in the thalamus, caudate (including the ventral putamen), lingual gyrus and several temporal areas (see George et al., 2008 for details). Modified from George et al. (2008) by permission of the publisher.
This pioneering approach taken by George et al. (2008) to evaluate a drug action on stimulus induced craving and changes in brain imaging of alcoholics demonstrates the potential of this proposed clinical model to identify potential compounds for extended clinical testing in alcoholism. Obviously, the next step would be to test the degree to which this strategy predicts whether a drug effective in this proposed model of drug evaluation is capable of minimizing the high susceptibility of relapse observed in alcoholics. In this respect, the results obtained by George et al., (2008) would warrant a future investigative clinical trial to assess if this NK1R antagonist would minimize relapse in alcoholic patients, particularly those who demonstrate anxiety and depressive symptoms during abstinence. Certainly, future inclusion of this proposed strategy of clinical testing of drugs identified with an action against the craving and the brain change in fMRI in alcoholics to stress and alcohol cues is worthy of pursuit to determine if these drugs would indeed affect the degree of susceptibility for relapse.
7. Implications that Understanding “Kindling/Allostasis-Like” Neuroadaptation Induced by Alcoholism Provides Optimism for Future Treatment
The background material provided clearly outlined several aspects that contribute to the alcoholic state (Section 1.1). Contributions by genetic susceptibility, early environmental adversity, drinking during adolescence, or co-morbidity with other stress- related psychiatric disorders are factors that have been linked to susceptibility for alcoholism. Based upon this diversity of factors associated with alcoholism, there have been several efforts to classify alcoholics (Cloninger et al., 1981; Heilig, 2008; Penick et al., 1990; and see Report from NIAAA). Regardless of the classification or basis of the alcoholism, excess ingestion of alcohol over time facilitates negative emotions during abstinence, resulting in an escalation of future alcohol use. Heilig et al. (2010a) have reviewed aspects of acute withdrawal, negative emotion and protracted symptoms observed during abstinence in the alcoholic. Preclinical data support the view that worsening of withdrawal symptoms during periods of abstinence in the alcoholic results from a cumulative adaptation by intermittent alcohol exposures. This adaptive process by alcohol has been shown to facilitate alcohol self administration. A critical conclusion from such preclinical studies is that consequence of chronic alcohol depends upon adaptation by a kindling-like process that increases an allostatic load (Koob, 2003; Breese et al., 2004, 2005c). Importantly, stress alone substitutes for initial alcohol withdrawals to produce a cumulative adaptation that sensitizes anxiety-like symptoms during withdrawal from alcohol. To illustrate that mediators of stress induce a cumulative adaptation that increases alcohol adaptation, literature demonstrated that repeated CRF and cytokines, like stress, sensitize withdrawal symptoms by facilitating adaptation of a single chronic alcohol exposure that alone was without effect. Consequently, stress facilitation of ethanol adaptation may provide a basis for the elevated susceptibility individuals with various “stress-related disorders”, such as depression, anxiety disorders or post-traumatic stress disorder, have for alcoholism.
A critical component of clinical findings in alcoholics under treatment is their extraordinary relapse rate to abusive drinking. An important clue for this susceptibility for relapse of alcoholics was the demonstrated craving in alcoholics challenged with stress or alcohol-cues (George et al. 2008; Seo et al., 2010; Sinha, 2001; Sinha et al., 2009, 2010). Subsequently, specific brain regions in the alcoholic were shown to be activated by these challenges, a change which is not observed in social drinkers. These data have provided convincing clinical evidence that the brains of alcoholics are altered by persistent alcohol abuse. Further, documentation of accompanying dysfunction of the HPA axis and of blood pressure reaction to stress in the alcoholic, with these distinct alterations in specific brain sites, clearly provided further support for the view that a persisting adaptation is present after the extended use of alcohol (Sinha et al., 2009). Importantly, these demonstrations of altered functions being persistently present in alcoholics are consistent with the proposed allostasis/kindling hypothesis of alcoholism (Breese et al., 2005c).
As treatment strategies for alcoholism are investigated, the extent of the persisting adaptation in brain by alcohol abuse must be considered. An argument is made that there is a strong relationship between the degree of craving induced by the alcoholic and relapse. Base upon finding that administration of a drug prior to brain imaging can prevent changes in brain of alcoholics to stress, utilization of drug action on brain imaging changes induced by alcohol cues in the alcoholic may be a useful approach for identifying new agents that can disrupt both the craving as well as the altered brain response to stress and alcohol cues. In this regard, George et al. (2008) demonstrated the feasibility of this approach. Hopefully, drugs found effective in minimizing the susceptibility shown by alcoholics to stress and alcohol cues will be found to reduce the degree of relapse. Nonetheless, future investigations remain to determine whether identified drugs, which minimize craving and the changes in brain responses in the alcoholic, will indeed minimize relapse and loss of control.
In closing, the neurobiology that provided a theoretical basis for the increased susceptibility alcoholics have to stress has primarily evolved in the past decade. In particular, identification of brain regions being associated with specific symptoms of withdrawal and to stress challenges can be expected to facilitate identification of treatments that will minimize the vulnerability of the alcoholic for relapse. Although addictions to other drugs would be expected to have a differing neurobiological basis from that of alcohol, many of the same drugs that interrupt stress-induced responding to alcohol in animals also minimize responding induced by other addicting drugs (see Aujla et al., 2008; Shaham et al., 2003). Therefore, defining the basis of the similarities of treatments effective in both circumstances of drug and alcohol addiction may be critical to isolating effective therapies to minimize relapse in alcoholism. Convincing basic evidence for ethanol inducing a kindling/allostasis-like adaptation has provided a biological basis for the persistent pathology associated with chronic alcohol abuse. Based upon this background, the documentation that stress and alcohol cue-induced craving in alcoholics is associated with an altered neurobiology of brain provides optimism for future research establishing a means by which the disease of alcoholism can be treated and controlled.
Acknowledgments
The preparation of this review was supported by funds from NIAAA and from the Bowles Center for Alcohol Studies.
Abbreviations
- ACTH
Adrenocorticotropic hormone
- ADE
Alcohol deprivation effect
- CeA
Central Amygdala
- CB1R
Cannabinoid-1 Receptor
- CRF
Corticotropin releasing factor
- CRF1R
Corticotropin releasing factor-1 receptor
- CRF2R
Corticotropin releasing factor-2 receptor
- HPA
Hypothalamic-pituitary-adrenal
- dBNST
Dorsal bed nucleus of the stria terminalis
- DNR
Dorsal nucleus of the raphe
- fMRI
Functional magnetic resonance imaging
- mPFC
Medial prefrontal cortex
- mGLUR
Metabotropic glutamate receptor
- NK1R
Neurokinin-1 receptor
- NAC
Nucleus accumbens
- R
Receptor
- 5-HT
Serotonin
- SP
Substance P
- VTA
Ventral tegmental area
Footnotes
The authors have no conflicts.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Achur RN, Freeman WM, Vrana KE. Circulating cytokines as biomarkers of alcohol abuse and alcoholism. J Neuroimmune Pharmacol. 2010;5:83–91. doi: 10.1007/s11481-009-9185-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adinoff B, Krebaum SR, Chandler PA, Ye W, Brown MB, Williams MJ. Dissection of hypothalamic-pituitary-adrenal axis pathology in 1-month-abstinent alcohol-dependent men, part 1: adrenocortical and pituitary glucocorticoid responsiveness. Alcohol Clin Exp Res. 2005a;29:517–527. doi: 10.1097/01.ALC.0000158940.05529.0A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adinoff B, Krebaum SR, Chandler PA, Ye W, Brown MB, Williams MJ. Dissection of hypothalamic-pituitary-adrenal axis pathology in 1-month-abstinent alcohol-dependent men, part 2: response to ovine corticotropin-releasing factor and naloxone. Alcohol Clin Exp Res. 2005b;29:528–537. doi: 10.1097/01.ALC.0000158939.25531.EE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adinoff B, Junghanns K, Kiefer F, Krishnan-Sarin S. Suppression of the HPA axis stress-response: implications for relapse. Alcohol Clin Exp Res. 2005c;29:1351–1355. doi: 10.1097/01.ALC.0000176356.97620.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adler M, Geller E, Chen X, Rogers T. Viewing chemokines as a third major system of communication in the brain. AAPS J. 2006;7:E865–870. doi: 10.1208/aapsj070484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- al’Absi M, Hatsukami D, Davis GL. Attenuated adrenocorticotropic responses to psychological stress are associated with early smoking relapse. Psychopharmacology. 2005;181:107–117. doi: 10.1007/s00213-005-2225-3. [DOI] [PubMed] [Google Scholar]
- Altschuler HL, Phillips PE, Feinhandler DA. Alteration of alcohol self-administration by naltrexone. Life Sci. 1980;26:679–688. doi: 10.1016/0024-3205(80)90257-x. [DOI] [PubMed] [Google Scholar]
- Anisman H. Cascading effects of stressors and inflammatory immune system activation: implications for major depressive disorder. Rev Psychiatr Neurosci. 2009;34:4–20. [PMC free article] [PubMed] [Google Scholar]
- Anisman H, Merali Z. Cytokines, stress and depressive illness: brain-immune interactions. Ann Med. 2003;35:2–11. doi: 10.1080/07853890310004075. [DOI] [PubMed] [Google Scholar]
- Aujla H, Martin-Fardon R, Weiss F. Rats with extended access to cocaine exhibit increased stress reactivity and sensitivity to the anxiolytic-like effects of the mGluR 2/3 agonist LY379268 during abstinence. Neuropsychopharm. 2008;33:1818–1826. doi: 10.1038/sj.npp.1301588. [DOI] [PubMed] [Google Scholar]
- Badrick E, Bobak M, Britton A, Kirschbaum C, Marmot M, Kumari M. The relationship between smoking status and cortisol secretion. J Clin Endocrinol Metab. 2007;92:819–824. doi: 10.1210/jc.2006-2155. [DOI] [PubMed] [Google Scholar]
- Baldwin HA, Rassnick S, Rivier J, Koob GF, Britton TK. CRF antagonist reverses the “anxiogenic” response to alcohol withdrawal in the rat. Psychopharmacology. 1991;103:227–232. doi: 10.1007/BF02244208. [DOI] [PubMed] [Google Scholar]
- Bale TL, Vale WW. CRF and CRF receptors: Role in stress responsivity and other behaviors. Ann Rev Pharmacol Toxicol. 2004;44: 525–557. doi: 10.1146/annurev.pharmtox.44.101802.121410. [DOI] [PubMed] [Google Scholar]
- Ballenger JC, Post RM. Kindling as a model for alcohol withdrawal syndromes. Br J Psychiatry. 1978;133:1–14, 1978. doi: 10.1192/bjp.133.1.1. [DOI] [PubMed] [Google Scholar]
- Barr CS, Dvoskin RL, Gupte M, Grant GF. The impact of a family history of alcoholism on the relationship between age of onset of alcohol use and DSM-IV alcohol dependence: results of the National Longitudinal Alcohol Epidemiological Survey. Alcohol Health & Research World. 1998;22:144–147. [PMC free article] [PubMed] [Google Scholar]
- Barr CS, Dvoskin RL, Gupte M, Sommer W, Sun H, Schwandt ML, Lindell SG, Kasckow JW, Suomi SJ, Goldman D, Higley JD, Heilig M, et al. Functional CRH variation increases stress-induced alcohol consumption in primates. Proc Natl Acad Sci. 2009;25(106):14593–14598. doi: 10.1073/pnas.0902863106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer S, Kerr BJ, Patterson PH. The neuropoietic cytokine family in development, plasticity, disease and injury. Nature Neurosci Rev. 2007;8:221–232. doi: 10.1038/nrn2054. [DOI] [PubMed] [Google Scholar]
- Becker HC, Hale RL. Repeated episodes of alcohol withdrawal potentiate the severity of subsequent withdrawal seizures: an animal model of alcohol withdrawal “kindling”. Alcohol Clin Exp Res. 1993;17:94–98. doi: 10.1111/j.1530-0277.1993.tb00731.x. [DOI] [PubMed] [Google Scholar]
- Becker HC, Lopez MF. Increased alcohol drinking after repeated chronic alcohol exposure and withdrawal experience in C57BL/6 mice. Alcohol Clin Exp Res. 2004;28:1829–1838. doi: 10.1097/01.alc.0000149977.95306.3a. [DOI] [PubMed] [Google Scholar]
- Becker JB, Monteggia LM, Perrot-Sinal TS, Romeo RS, Taylor JR, Yehuda R, et al. Stress and disease: is being female a predisposing factor? J Neurosci. 2007;27:11851–11855. doi: 10.1523/JNEUROSCI.3565-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begleiter H. Alcohol consumption subsequent to physical dependence. Adv Exp Med Biol. 1975;59:373–378. doi: 10.1007/978-1-4757-0632-1_26. [DOI] [PubMed] [Google Scholar]
- Begleiter H, Porjesz B. Persistence of brain hyperexcitability following chronic alcohol exposure in rats. Adv exp Med Biol. 1977;85B:209–222. doi: 10.1007/978-1-4615-9038-5_14. [DOI] [PubMed] [Google Scholar]
- Behrendt S, Wittchen HU, Höfler M, Lieb R, Low NC, Rehm J, Beesdo K. Risk and speed of transitions to first alcohol dependence symptoms in adolescents: a 10-year longitudinal community study in Germany. Addiction. 2008;103:1638–1647. doi: 10.1111/j.1360-0443.2008.02324.x. [DOI] [PubMed] [Google Scholar]
- Besheer J, Grondin JJ, Cannady R, Sharko AC, Faccidomo S, Hodge CW. Metabotropic glutamate receptor 5 activity in the nucleus accumbens is required for the maintenance of alcohol self-administration in a rat genetic model of high alcohol intake. Biol Psychiatry. 2010 doi: 10.1016/j.biopsych.2009.09.016. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besheer J, Stevenson RA, Hodge CW. mGlu5 receptors are involved in the discriminative stimulus effects of self-administered alcohol in rats. Eur J Pharmacol. 2006;551:71–75. doi: 10.1016/j.ejphar.2006.08.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierut LJ, Agrawal A, Bucholz KK, Doheny KF, Laurie C, Pugh E, et al. A genome-wide association study of alcohol dependence. Proc Nat Acad Sci. 2010;107:5082–5087. doi: 10.1073/pnas.0911109107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouza C, Angeles M, Munoz A, Amate JM. Efficacy and safety of naltrexone and acamprosate in the treatment of alcohol dependence: a systematic review. Addiction. 2004;99:811–828. doi: 10.1111/j.1360-0443.2004.00763.x. [DOI] [PubMed] [Google Scholar]
- Booth BM, Blow FC. The kindling hypothesis: further evidence from a U.S. national study of alcohol men. Alcohol Alcohol. 1993;28:593–598. [PubMed] [Google Scholar]
- Bradizza CM, Stasiewicz PR, Paas ND. Relapse to alcohol and drug use among individuals diagnosed with co-occurring mental health and substance use disorders: a review. 2006;26:162–178. doi: 10.1016/j.cpr.2005.11.005. [DOI] [PubMed] [Google Scholar]
- Brady KT, Waldrop AE, McRae AL, Back SE, Saladin ME, Upadhyaya HP, et al. The impact of alcohol dependence and posttraumatic stress disorder on cold pressor task response. J Stud Alcohol. 2006a;67:700–706. doi: 10.15288/jsa.2006.67.700. [DOI] [PubMed] [Google Scholar]
- Brady KT, Back SE, Waldrop AE, McRae AL, Anton RF, Upadhyaya HP, et al. Cold pressor task reactivity: predictors of alcohol use among alcohol-dependent individuals with and without comorbid posttraumatic stress disorder. Alcohol Clin Exp Res. 2006b;30:938–946. doi: 10.1111/j.1530-0277.2006.00097.x. [DOI] [PubMed] [Google Scholar]
- Braus DF, Wrase J, Grüsser S, Hermann D, Ruf M, Flor H, et al. Alcohol-associated stimuli activate the ventral striatum in abstinent alcoholics. J Neural Transm. 2001;108:887–894. doi: 10.1007/s007020170038. [DOI] [PubMed] [Google Scholar]
- Breese G, Chu K, Dayas C, Funk D, Knapp D, Koob G, et al. Stress enhancement of craving during sobriety: risk of relapse. Alcohol Clin Exp Res. 2005a;29:185–195. doi: 10.1097/01.alc.0000153544.83656.3c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breese GR, Coyle S, Towle AC, Mueller RA, McCown TJ, Frye GD. Alcohol-induced locomotor stimulation in rats following thyrotropin releasing hormone. J Pharmacol Exp Ther. 1984;229:731–737. [PubMed] [Google Scholar]
- Breese GR, Knapp DJ, Huang M, Wills TA, Angel R, Sinnett SE, Whitman BA. Stress between cycles of access to voluntary ethanol increases both ethanol intake and sensitizes anxiety-like behavior in alcoholpreferring p rats. Alcohol Clin Exp Res. 2010;34:200A. [Google Scholar]
- Breese GR, Knapp DJ, Overstreet DH, Navarro M, Wills TA, Angel RA. Repeated lipopolysaccharide (LPS) or cytokine treatments sensitize alcohol withdrawal-induced anxiety-like behavior. Neuropsychopharm. 2008;33:867–876. doi: 10.1038/sj.npp.1301468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breese GR, Overstreet H, Knapp D, Navarro M. Prior multiple alcohol withdrawals enhance stress-induced anxiety-like behavior during extended abstinence: Inhibition by CRF-1 & BZD receptor antagonists & by a 5-HT1A-receptor agonist. Neuropsychopharm. 2005b;30:1662–1669. doi: 10.1038/sj.npp.1300706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breese GR, Overstreet H, Knapp D. Stress sensitization of the alcohol withdrawal-induced reduction in social interaction: inhibition by CRF-1 and benzodiazepine receptor antagonists and a 5-HT1A agonist. Neuropsychopharm. 2004;29:470–482. doi: 10.1038/sj.npp.1300282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breese GR, Overstreet DH, Knapp DJ. Conceptual framework for the etiology of alcoholism—a “kindling”/stress hypothesis. Psychopharmacology. 2005c;178:367–380. doi: 10.1007/s00213-004-2016-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brower KJ. Alcohol’s effects on sleep in alcoholics. Alcohol Res Health. 2001;25:110–125. [PMC free article] [PubMed] [Google Scholar]
- Brown SA, Tapert SF. Adolescence and the trajectory of alcohol use: basic to clinical studies. Ann NY Acad Sci. 2004;1021:234–244. doi: 10.1196/annals.1308.028. [DOI] [PubMed] [Google Scholar]
- Brown SA, Vik P, McQuaid JR, Patterson T, Irwin MR, Grant I. Severity of psychosocial stress and outcome of alcoholism treatment. J Abnorm Psychol. 1990;99:344–348. doi: 10.1037//0021-843x.99.4.344. [DOI] [PubMed] [Google Scholar]
- Brown S, Vik P, Patterson TL, Grant I, Schuckit MA. Stress vulnerability and adult alcohol relapse. J Studies Alcohol. 1995;56:538–545. doi: 10.15288/jsa.1995.56.538. [DOI] [PubMed] [Google Scholar]
- Cadoret RJ, Cain CA, Grove WM. Development of alcoholism in adoptees raised apart from alcoholic biologic relatives. Arch Gen Psychiatry. 1980;37:561–563. doi: 10.1001/archpsyc.1980.01780180075008. [DOI] [PubMed] [Google Scholar]
- Cappel H, Herman CP. Alcohol and tension reduction: a review. Q J Stud Alcohol. 1972;33:33–64. [PubMed] [Google Scholar]
- Chaplin TM, Hong K, Bergquist K, Sinha R. Gender differences in response to emotional stress: an assessment across subjective, behavioral, and physiological domains and relations to alcohol craving. Alcohol Clin Exp Res. 2008;32:1242–1250. doi: 10.1111/j.1530-0277.2008.00679.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhri N, Sahuque LL, Schairer WW, Janak PH. Separable roles of the nucleus accumbens core and shell in context- and cue-induced alcohol-seeking. Neuropsychopharm. 2010;35:783–789. doi: 10.1038/npp.2009.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charney DA, Zikos E, Gill KJ. Early recovery from alcohol dependence: Factors that promote or impede abstinence. J Subst Abuse Treat. 2010;38:42–50. doi: 10.1016/j.jsat.2009.06.002. [DOI] [PubMed] [Google Scholar]
- Chester JA, Blose AM, Zweifel M, Froehlich JM. Effects of stress on alcohol consumption in rats selectively bred for high or low alcohol drinking. Alcohol Clin Exp Res. 2004;28:385–393. doi: 10.1097/01.alc.0000117830.54371.7a. [DOI] [PubMed] [Google Scholar]
- Chu K, Koob GF, Cole M, Zorrilla EP, Roberts AJ. Dependence-induced increases in alcohol self-administration in mice are blocked by the CRF1 receptor antagonist antalarmin and by CRF1 receptor knockout. Pharmacol Biochem Behav. 2007;86:813–821. doi: 10.1016/j.pbb.2007.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccocioppo R, Gehlert DR, Ryabinin A, Kaur S, Cippitelli A, Thorsell A, et al. Stress-related neuropeptides and alcoholism: CRH, NPY, and beyond. Alcohol. 2009;43:491–498. doi: 10.1016/j.alcohol.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cippitelli A, Damadzic R, Hansson AC, Singley E, Sommer WH, Eskay R, Thorsell A, Heilig M. Neuropeptide Y (NPY) suppresses yohimbine-induced reinstatement of alcohol seeking. Psychopharmacology. 2010;208:417–426. doi: 10.1007/s00213-009-1741-y. [DOI] [PubMed] [Google Scholar]
- Clapper RL, Buka SL, Goldfield EC, Lipsitt LP, Tsuang MT. Adolescent problem behaviors as predictors of adult alcohol diagnoses. Int J Addict. 1995;30:507–523. doi: 10.3109/10826089509048741. [DOI] [PubMed] [Google Scholar]
- Cloninger CR, Bohman M, Sigvardsson S. Inheritance of alcohol abuse. Cross-fostering analysis of adopted men. Arch Gen Psychiatry. 1981;38:861–868. doi: 10.1001/archpsyc.1981.01780330019001. [DOI] [PubMed] [Google Scholar]
- Collimore KC, Carleton RN, Hofmann SG, Asmundson GJ. Posttraumatic stress and social anxiety: the interaction of traumatic events and interpersonal fears. Depress Anxiety. 2010 doi: 10.1002/da.20728. In press. [DOI] [PubMed] [Google Scholar]
- Conger JJ. The effects of alcohol on conflict behavior in the albino rat. Q J Stud Alcohol. 1951;12:1–29. [PubMed] [Google Scholar]
- Conger JJ. Alcoholism: theory, problem, and challenge. II. Reinforcement theory and the dynamics of alcoholism. Q J Stud Alcohol. 1956;17:296–305. [PubMed] [Google Scholar]
- Conway KP, Compton W, Stinson FS, Grant BF. Lifetime comorbidity of DSM-IV mood and anxiety disorders and specific drug use disorders: results from the National Epidemiologic Survey on Alcohol and Related Conditions. J Clin Psychiatry. 2006;67:247–257. doi: 10.4088/jcp.v67n0211. [DOI] [PubMed] [Google Scholar]
- Cooney NL, Litt MD, Morse PA, Bauer LO, Gaupp L. Alcohol cue reactivity, negative-mood reactivity, and relapse in treated alcoholic men. J Abnorm Psychol. 1997;106:243–250. doi: 10.1037//0021-843x.106.2.243. [DOI] [PubMed] [Google Scholar]
- Cook CJ. Stress induces CRF release in the paraventricular nucleus, and both CRF and GABA release in the amygdala. Physiol Behav. 2004;82:751–762. doi: 10.1016/j.physbeh.2004.06.013. [DOI] [PubMed] [Google Scholar]
- Cooper ML, Russell M, Skinner JB, Frone MR, Mudar P. Stress and alcohol use: moderating effects of gender, coping and alcohol expectancies. J Abnorm Psychol. 1992;101:139–152. doi: 10.1037//0021-843x.101.1.139. [DOI] [PubMed] [Google Scholar]
- Cotton NS. The familial incidence of alcoholism: a review. J Stud Alcohol. 1979;40:89–116. doi: 10.15288/jsa.1979.40.89. [DOI] [PubMed] [Google Scholar]
- Dawson DA, Goldstein RB, Chou SP, Ruan WJ, Grant BF. Age at first drink and the first incidence of adult-onset DSM-IV alcohol use disorders. Alcohol Clin Exp Res. 2008;32: 2149–2160. doi: 10.1111/j.1530-0277.2008.00806.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson DA, Goldstein RB, Grant BF. Rates and correlates of relapse among individuals in remission from DSM-IV alcohol dependence: a 3-year follow-up. Alcohol Clin Exp Res. 2007;31:2036–2045. doi: 10.1111/j.1530-0277.2007.00536.x. [DOI] [PubMed] [Google Scholar]
- Deak T, Bordner K, McElderry N, Barnum C, Blandino P, Jr, Deak M, Tammariello S. Stress-induced increases in hypothalamic IL-1: systematic analysis of multiple stressor paradigms. Br Res Bull. 2005;64:541–556. doi: 10.1016/j.brainresbull.2004.11.003. [DOI] [PubMed] [Google Scholar]
- Del Cerro S, Borrrell J. Interleukin-1 affects the behavioral despair response in rats by an indirect mechanism which requires endogenous CRF. Brain Res. 1990;528:162–164. doi: 10.1016/0006-8993(90)90212-t. [DOI] [PubMed] [Google Scholar]
- Deutsch JA, Koopmans HS. Preference enhancement for alcohol by passive exposure. Science. 1973;179:1242–1243. doi: 10.1126/science.179.4079.1242. [DOI] [PubMed] [Google Scholar]
- Devor EJ, Cloninger CR. Genetics of alcoholism. Annu Rev Genet. 1989;23:19–36. doi: 10.1146/annurev.ge.23.120189.000315. [DOI] [PubMed] [Google Scholar]
- DeWit DJ, Adlaf EM, Offord DR, Ogborne AC. Age at first alcohol use: a risk factor for the development of alcohol disorders. Am J Psychiatry. 2000;157:745–750. doi: 10.1176/appi.ajp.157.5.745. [DOI] [PubMed] [Google Scholar]
- Dhaher R, Finn D, Snelling C, Hitzemann R. Lesions of the extended amygdala in C57BL/6J mice do not block the intermittent alcohol vapor-induced increase in alcohol consumption. Alcohol Clin Exp Res. 2008;32:197–208. doi: 10.1111/j.1530-0277.2007.00566.x. [DOI] [PubMed] [Google Scholar]
- Dick DM, Bierut LJ. The genetics of alcohol dependence. Curr Psychiatry Rep. 2006;8:151–157. doi: 10.1007/s11920-006-0015-1. [DOI] [PubMed] [Google Scholar]
- Drummond SP, Gillin JC, Smith TL, Demodena A. The sleep of abstinent pure primary alcoholic patients: natural course and relationship to relapse. Alcohol Clin Exp Res. 1998;22:1796–1802. [PubMed] [Google Scholar]
- Dunn AJ, Berridge CW. Physiological and behavioral response to corticotropin-releasing factor administration: Is CRF a mediator of anxiety-like behavior or stress responses? Brain Res Rev. 1990;15:71–100. doi: 10.1016/0165-0173(90)90012-d. [DOI] [PubMed] [Google Scholar]
- Dunn AJ, Swiergiel AH, de Beaurepaire R. Cytokines as mediators of depression: what can we learn from animal studies? Neurosci Biobehav Rev. 2005;29:891–909. doi: 10.1016/j.neubiorev.2005.03.023. [DOI] [PubMed] [Google Scholar]
- Ebner K, Rupniak NM, Saria A, Singewald N. Substance P in the medial amygdala: emotional stress-sensitive release and modulation of anxiety-related behavior in rats. Proc Natl Acad Sci. 2004;101:4280–4285. doi: 10.1073/pnas.0400794101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Economidou D, Mattioli L, Cifani C, Perfumi M, Massi M, Cuomo V, et al. Effect of the cannabinoid CB1 receptor antagonist SR-141716A on alcohol self-administration and alcohol-seeking behaviour in rats. Psychopharmacology. 2006;183:394–403. doi: 10.1007/s00213-005-0199-9. [DOI] [PubMed] [Google Scholar]
- Edenberg HJ, Foroud T. The genetics of alcoholism: identifying specific genes through family studies. Addiction Biol. 2006;11:386–396. doi: 10.1111/j.1369-1600.2006.00035.x. [DOI] [PubMed] [Google Scholar]
- Fein G, Landman B. Treated and treatment-naive alcoholics come from different populations. Alcohol. 2005;36:19–26. [PMC free article] [PubMed] [Google Scholar]
- Finn DA, Snelling C, Fretwell AM, Tanchuck MA, Underwood L, Cole M, Crabbe JC, Roberts AJ. Increased drinking during withdrawal from intermittent alcohol exposure is blocked by the CRF receptor antagonist D-Phe-CRF(12–41) Alcohol Clin Exp Res. 2007;31:939–949. doi: 10.1111/j.1530-0277.2007.00379.x. [DOI] [PubMed] [Google Scholar]
- Fox HC, D’SA C, Sinha R. Altered cytokine responses are associated with enhanced stress-induced craving in alcohol dependent individuals. Alcohol Clin Exp Res. 2010;34(Supplement):46A. [Google Scholar]
- Fox HC, Jackson ED, Sinha R. Difficulties in emotion regulation and impulse control in recently abstinent alcoholics compared with social drinkers. Psychoneuroendocrinology. 2009a;34:1198–1207. doi: 10.1016/j.addbeh.2007.10.002. [DOI] [PubMed] [Google Scholar]
- Fox HC, Bergquist KL, Hong KI, Sinha R. Stress-induced and alcohol cue-induced craving in recently abstinent alcohol-dependent individuals. Alcohol Clin Exp Res. 2007;31:395–403. doi: 10.1111/j.1530-0277.2006.00320.x. [DOI] [PubMed] [Google Scholar]
- Fox HC, Hong KI, Siedlarz K, Sinha R, Bergquist K, Anderson G, Kreek MJ, Sinha R. Sex-specific dissociations in autonomic and HPA responses to stress and cues in alcohol-dependent patients with cocaine abuse. Alcohol Alcohol. 2009b;44:575–585. doi: 10.1093/alcalc/agp060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox H, Talih M, Malison R, Anderson G, Kreek M, Sinha R. Frequency of recent cocaine and alcohol use affects drug craving and associated responses to stress and drug-related cues. Psychoneuro-endocrinology. 2005;30:880–891. doi: 10.1016/j.psyneuen.2005.05.002. [DOI] [PubMed] [Google Scholar]
- Frye GD, McCown TJ, Breese GR. Characterization of susceptibility to audiogenic seizures in alcohol-dependent rats after microinjection of gamma-aminobutyric acid (GABA) agonists into the inferior colliculus, substantia nigra or medial septum. J Pharmacol Exp Ther. 1983;227:663–670. [PMC free article] [PubMed] [Google Scholar]
- Funk CK, Koob GF. A CRF2 agonist administered into the central nucleus of the amygdala decreases alcohol self-administration in alcohol-dependent rats. Brain Res. 2007;1155:172–178. doi: 10.1016/j.brainres.2007.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funk D, Li Z, Lê AD. Effects of environmental and pharmacological stressors on c-fos and corticotropin-releasing factor mRNA in rat brain: Relationship to the reinstatement of alcohol seeking. Neuroscience. 2006a;138:235–243. doi: 10.1016/j.neuroscience.2005.10.062. [DOI] [PubMed] [Google Scholar]
- Funk CK, O‘Dell LE, Crawford EF, Koob GF. Corticotropin-releasing factor within the central nucleus of the amygdala mediates enhanced alcohol self-administration in withdrawn, alcohol-dependent rats. J Neurosci. 2006b;26:11324–11332. doi: 10.1523/JNEUROSCI.3096-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funk CK, Zorrilla EP, Lee MJ, Rice KC, Koob GF. Corticotropin-releasing factor 1 antagonists selectively reduce alcohol self-administration in alcohol-dependent rats. Biol Psychiat. 2007;61:78–86. doi: 10.1016/j.biopsych.2006.03.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furmark T, Appel L, Michelgard A, Wahlstedt K, Ahs F, Zancan S, Jacobsson E, et al. Cerebral blood flow changes after treatment of social phobia with the neurokinin-1 antagonist GR205171, citalopram, or placebo. Biol Psychiatry. 2005;58:132–142. doi: 10.1016/j.biopsych.2005.03.029. [DOI] [PubMed] [Google Scholar]
- Gatto GJ, McBride WJ, Murphy JM, Lumeng L, Li TK. Alcohol self-infusion into the ventral tegmental area by alcohol-preferring rats. Alcohol. 1994;11:557–564. doi: 10.1016/0741-8329(94)90083-3. [DOI] [PubMed] [Google Scholar]
- Gehlert DR, Cippitelli A, Thorsell A, Lê AD, Hipskind PA, Hamdouchi C, et al. 3-(4-Chloro-2-morpholin-4-yl-thiazol-5-yl)-8-(1-ethylpropyl)-2,6-dimethyl-imidazo[1,2-b]pyridazine: a novel brain-penetrant, orally available corticotropin-releasing factor receptor 1 antagonist with efficacy in animal models of alcoholism. J Neurosci. 2007;27:2718–2726. doi: 10.1523/JNEUROSCI.4985-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentry L, Chung J, Aung N, Keller S, Heinrich K, Maddock J. Gender Differences in Stress and Coping among Adults living in Hawaii. Cal J Health Promotion. 2007;5:89–102. [Google Scholar]
- George MS, Anton RF, Bloomer C, Teneback C, Drobes DJ, Lorberbaum JP, Nahas Z, Vincent DJ. Activation of prefrontal cortex and anterior thalamus in alcoholic subjects on exposure to alcohol-specific cues. Arch Gen Psychiatry. 2001;58:345–352. doi: 10.1001/archpsyc.58.4.345. [DOI] [PubMed] [Google Scholar]
- George DT, Gilman J, Hersh J, Thorsell A, Herion D, Geyer C, et al. Neurokinin 1 receptor antagonism as a possible therapy for alcoholism. Science. 2008;319:1536–1539. doi: 10.1126/science.1153813. [DOI] [PubMed] [Google Scholar]
- Gilbertson R, Prather R, Nixon SJ. The role of selected factors in the development and consequence of alcohol dependence. Alcohol Res Health. 2008;31:389–399. [PMC free article] [PubMed] [Google Scholar]
- Gilman JM, Hommer DW. Modulation of brain response to emotional images by alcohol cues in alcohol-dependent patients. Addict Biol. 2008;13:423–434. doi: 10.1111/j.1369-1600.2008.00111.x. [DOI] [PubMed] [Google Scholar]
- Gilman JM, Ramchandani VA, Davis MB, Bjork JM, Hommer DW. Why we like to drink: a functional magnetic resonance imaging study of the rewarding and anxiolytic effects of alcohol. J Neurosci. 2008;28:4583–4591. doi: 10.1523/JNEUROSCI.0086-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilpin NW, Richardson HN, Lumeng L, Koob GF. Dependence-induced alcohol drinking by alcohol-preferring (P) rats and outbred Wistar rats. Alcohol Clin Exp Res. 2008a;32:1688–1696. doi: 10.1111/j.1530-0277.2008.00678.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilpin NW, Richardson HN, Koob GF. Effects of CRF1-receptor and opioid-receptor antagonist on dependence-induced increases in alcohol drinking by alcohol-preferring (P) rats. Alcohol Clin Exp Res. 2008b;32:1535–1542. doi: 10.1111/j.1530-0277.2008.00745.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Givens BS, Breese GR. Site-specific enhancement of gamma-aminobutyric acid-mediated inhibition of neural activity by alcohol in the rat medial septal area. J Pharmacol Exp Ther. 1990;254:528–538. [PMC free article] [PubMed] [Google Scholar]
- Grant BF, Dawson DA. Age at onset of alcohol use and its association with DSM-IV alcohol abuse and dependence: results from the National Longitudinal Alcohol Epidemiologic Survey. J Subst Abuse. 1997;9:103–110. doi: 10.1016/s0899-3289(97)90009-2. [DOI] [PubMed] [Google Scholar]
- Grant BR, Stinson FS, Dawson DA, Chou SP, Dufour MC, Compton W, et al. Prevalence and co-occurrence of substance use disorders and independent mood and anxiety disorders results from the national epidemiologic survey on alcohol and related conditions. Arch Gen Psychiatry. 2004;61:807–816. doi: 10.1001/archpsyc.61.8.807. [DOI] [PubMed] [Google Scholar]
- Grüsser SM, Wrase J, Klein S, Hermann D, Smolka MN, Ruf M, Weber-Fahr W, Flor H, Mann K, Braus DF, Heinz A. Cue-induced activation of the striatum and medial prefrontal cortex is associated with subsequent relapse in abstinent alcoholics. Psychopharmacology. 2004;175:296–302. doi: 10.1007/s00213-004-1828-4. [DOI] [PubMed] [Google Scholar]
- Haddad JJ. Alcoholism and neuro-immune-endocrine interactions: physiochemical aspects. Biochem Biophys Res Commun. 2004;323:361–371. doi: 10.1016/j.bbrc.2004.08.119. [DOI] [PubMed] [Google Scholar]
- Hansson AC, Cippitelli A, Sommer WH, Fedeli A, Björk K, Soverchia L, Terasmaa A, Massi M, Heilig M, Ciccocioppo R. Variation at the rat Crhr1 locus and sensitivity to relapse into alcohol seeking induced by environmental stress. Proc Natl Acad Sci. 2006;103:15236–15241. doi: 10.1073/pnas.0604419103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansson AC, Cippitelli A, Sommer WH, Ciccocioppo R, Heilig M. Region-specific down-regulation of Crhr1 gene expression in alcohol-preferring msP rats following ad lib access to alcohol. Addict Biol. 2007;12:30–34. doi: 10.1111/j.1369-1600.2007.00050.x. [DOI] [PubMed] [Google Scholar]
- Hauger RL, Risbrough V, Oakley RH, Olivares-Reyes JA, Dautzenberg FM. Role of CRF receptor signaling in stress vulnerability, anxiety, and depression. Ann NY Acad Sci. 2009;1179:120–143. doi: 10.1111/j.1749-6632.2009.05011.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayley S, Poulter MO, Merali Z, Anisman H. The pathogenesis of clinical depression: stressor- and cytokine-induced alterations of neuroplasticity. Neuroscience. 2005;135:659–678. doi: 10.1016/j.neuroscience.2005.03.051. [DOI] [PubMed] [Google Scholar]
- He J, Crews FT. Increased MCP-1 and microglia in various regions of the human alcoholic brain. Exp Neurol. 2008;210:349–358. doi: 10.1016/j.expneurol.2007.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heilig M. Molecular biology teases out two distinct forms of alcoholism. The Scientist. 2008;22:31–35. [Google Scholar]
- Heilig M, Egli M. Pharmacological treatment of alcohol dependence: target symptoms and target mechanisms. Pharmacol Ther. 2006;111:855–876. doi: 10.1016/j.pharmthera.2006.02.001. [DOI] [PubMed] [Google Scholar]
- Heilig M, Koob GF. A key role for corticotropin-releasing factor in alcohol dependence. Trends Neurosci. 2007;30:399–406. doi: 10.1016/j.tins.2007.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heilig M, Egli M, Crabbe JC, Becker HC. Acute withdrawal, protracted abstinence and negative affect in alcoholism: are they linked? Addict Biol. 2010a;15:169–184. doi: 10.1111/j.1369-1600.2009.00194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heilig M, Koob GF, Ekman R, Britton KT. Corticotropin-releasing factor and neuropeptide Y: role in emotional integration. Trends Neurosci. 1994;17:80–85. doi: 10.1016/0166-2236(94)90079-5. [DOI] [PubMed] [Google Scholar]
- Heilig M, Thorsell A, Sommer WH, Hansson AC, Ramchandani VA, George DT, et al. Translating the neuroscience of alcoholism into clinical treatments: From blocking the buzz to curing the blues. Neurosci Biobehav Rev. 2010b doi: 10.1016/j.neubiorev.2009.11.018. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz A, Siessmeier T, Wrase J, Hermann D, Klein S, Grüsser SM, et al. Correlation between dopamine D2 receptors in the ventral striatum and central processing of alcohol cues and craving. Am J Psychiatry. 2004;161:1783–1789. doi: 10.1176/appi.ajp.161.10.1783. [DOI] [PubMed] [Google Scholar]
- Heinz A, Siessmeier T, Wrase J, Buchholz HG, Gründer G, Kumakura Y, et al. Correlation of alcohol craving with striatal dopamine synthesis capacity and D2/3 receptor availability: a combined [18F]DOPA and [18F]DMFP PET study in detoxified alcoholic patients. Am J Psychiatr. 2005;162:1515–1520. doi: 10.1176/appi.ajp.162.8.1515. [DOI] [PubMed] [Google Scholar]
- Higgins RL, Marlatt GA. Effects of anxiety arousal on the consumption of alcohol by alcoholics and social drinkers. J Consult Clin Psychol. 1973;41:426–433. doi: 10.1037/h0035366. [DOI] [PubMed] [Google Scholar]
- Higgins RL, Marlatt GA. Fear of interpersonal evaluation as a determinant of alcohol consumption in male social drinkers. J Abnorm Psychol. 1975;84:644–645. doi: 10.1037//0021-843x.84.6.644. [DOI] [PubMed] [Google Scholar]
- Hingson RW, Heeren T, Winter MR. Age at drinking onset and alcohol dependence: age at onset, duration, and severity. Arch Peiatr Adolesc Med. 2006;160:739–746. doi: 10.1001/archpedi.160.7.739. [DOI] [PubMed] [Google Scholar]
- Hölter SM, Spanagel R. Effects of opiate antagonist treatment on the alcohol deprivation effect in long-term alcohol-experienced rats. Psychopharmacology. 1999;145:360–369. doi: 10.1007/s002130051069. [DOI] [PubMed] [Google Scholar]
- Hopfer CJ, Crowley TJ, Hewitt JK. Review of twin and adoption studies of adolescent substance use. J Am Acad Child Adolesc Psychiatry. 2003;42:710–719. doi: 10.1097/01.CHI.0000046848.56865.54. [DOI] [PubMed] [Google Scholar]
- Huang M, Overstreet DH, Knapp DJ, Angel R, Wills TA, Navarro M, et al. Corticotropin releasing factor (CRF) sensitization of alcohol withdrawal-induced anxiety-like behavior is brain site specific and mediated by CRF-1 receptors: relation to stress-induced sensitization. J Pharmacol Exp Ther. 2010;332:298–307. doi: 10.1124/jpet.109.159186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbell CL, Czirr SA, Hunter GA, Beaman CM, LeCann NC, Reid LD. Consumption of alcohol solution is potentiated by morphine and attenuated by naloxone persistently across repeated daily administrations. Alcohol. 1986;2:1–16. doi: 10.1016/0741-8329(86)90070-4. [DOI] [PubMed] [Google Scholar]
- Hunt WA, Barnett LW, Branch LG. Relapse rates in addiction programs. J Clin Psychol. 1971;27:455–456. doi: 10.1002/1097-4679(197110)27:4<455::aid-jclp2270270412>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- Hunter BE, Walker DW, Riley JN. Dissociation between physical dependence and volitional alcohol consumption: role of multiple withdrawal episodes. Pharmacol Biochem Behav. 1974;2:523–529. doi: 10.1016/0091-3057(74)90013-6. [DOI] [PubMed] [Google Scholar]
- Imaki T, Shibasaki T, Hotta M, Demura H. Intracerebroventricular administration of corticotropin releasing factor induces c-fos mRNA expression in brain regions related to stress responses: comparison with pattern of c-fos mRNA induction by stress. Brain Res. 1993;616:114–125. doi: 10.1016/0006-8993(93)90199-w. [DOI] [PubMed] [Google Scholar]
- Junghanns K, Backhaus J, Tietz U, Lange W, Bernzen J, Wetterling T, et al. Impaired serum cortisol stress response is a predictor of early relapse. Alcohol Alcohol. 2003;38:189–193. doi: 10.1093/alcalc/agg052. [DOI] [PubMed] [Google Scholar]
- Keller M. On the loss-of-control phenomenon in alcoholism. Br J Addict Alcohol Other Drugs. 1972;67:153–166. doi: 10.1111/j.1360-0443.1972.tb01188.x. [DOI] [PubMed] [Google Scholar]
- Kessler RC, McGonagle KA, Swartz M, Blazer DG, Nelson CB. Sex and depression in the National Comorbidity Survey. I: Lifetime prevalence, chronicity and recurrence. J Affect Disord. 1993;29:85–96. doi: 10.1016/0165-0327(93)90026-g. [DOI] [PubMed] [Google Scholar]
- Kessler RC, McGonagle KA, Zhao S, Nelson CB, Hughes M, Eshleman S, et al. Lifetime and 12-month prevalence of DSM-III-R psychiatric disorders in the United States. Results from the National Comorbidity Survey. Arch Gen Psychiatry. 1994;51:8–19. doi: 10.1001/archpsyc.1994.03950010008002. [DOI] [PubMed] [Google Scholar]
- Kiefer F, Jahn H, Schick M, Wiedemann K. Alcohol intake, tumor necrosis factor-α, leptin and craving: Factors of possible vicious circle? Alcohol Alcoholism. 2002;37:401–404. doi: 10.1093/alcalc/37.4.401. [DOI] [PubMed] [Google Scholar]
- Knapp D, Overstreet D, Moy S, Breese G. SB242084, flumazenil, and CRA1000 block alcohol withdrawal-induced anxiety. Alcohol. 2004;32:101–111. doi: 10.1016/j.alcohol.2003.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knapp D, Overstreet D, Breese G. Modulation of alcohol withdrawal-induced anxiety-like behavior during later withdrawals by treatment of early withdrawals with benzodiazepine/GABA ligands. Alcohol Clin Exp Res. 2005;29:553–563. doi: 10.1097/01.alc.0000158840.07475.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knapp D, Overstreet D, Breese G. Baclofen blocks expression and sensitization of anxiety-like behavior in an animal model of repeated stress and alcohol withdrawal. Alcohol Clin Exp Res. 2007a;31:582–595. doi: 10.1111/j.1530-0277.2007.00342.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knapp DJ, Overstreet DH, Angel RA, Navarro M, Breese GR. The amygdala regulates the antianxiety sensitization effect of flumazenil during repeated chronic alcohol or repeated stress. Alcohol Clin Exp Res. 2007b;31:1872–1882. doi: 10.1111/j.1530-0277.2007.00514.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kofoed L, Friedman MJ, Peck R. Alcoholism and drug abuse in patients with PTSD. Psychiat Q. 1993;64:151–171. doi: 10.1007/BF01065867. [DOI] [PubMed] [Google Scholar]
- Koob GF. Alcoholism: Allostasis and beyond. Alcohol Clin Exp Res. 2003;27:232–243. doi: 10.1097/01.ALC.0000057122.36127.C2. [DOI] [PubMed] [Google Scholar]
- Koob GF. A role of brain stress systems in addiction. Neuron. 2008;59:11–34. doi: 10.1016/j.neuron.2008.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF. Neurobiological substrates for the dark side of compulsivity in addiction. Neuropharmacology. 2009;56(Suppl 1):18–31. doi: 10.1016/j.neuropharm.2008.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF, Heinrichs SC. A role of corticotropin releasing factor and urocortin in behavioral responses to stressors. Brain Res. 1999;848:141–152. doi: 10.1016/s0006-8993(99)01991-5. [DOI] [PubMed] [Google Scholar]
- Koob GF, Le Moal M. Drug abuse: hedonic homeostatic dysregulation. Science. 1997;278:52–58. doi: 10.1126/science.278.5335.52. [DOI] [PubMed] [Google Scholar]
- Koob GF, Le Moal M. Drug addiction, dysregulation of reward, and allostasis. Neuropsychopharm. 2001;24:97–129. doi: 10.1016/S0893-133X(00)00195-0. [DOI] [PubMed] [Google Scholar]
- Koob GF, Le Moal M. Plasticity of reward neurocircuitry and the ‘dark side’ of drug addiction. Nat Neurosci. 2005;8:1442–1444. doi: 10.1038/nn1105-1442. [DOI] [PubMed] [Google Scholar]
- Koob GF, Le Moal M. Addiction and the brain antireward system. Annu Rev Psychol. 2008;59:29–53. doi: 10.1146/annurev.psych.59.103006.093548. [DOI] [PubMed] [Google Scholar]
- Korte SM, Koolhaas JM, Wingfield JC, McEwen BS. The Darwinian concept of stress: benefits of allostasis and costs of allostatic load and the trade-offs in health and disease. Neurosci Biobehav Rev. 2005;29:3–38. doi: 10.1016/j.neubiorev.2004.08.009. [DOI] [PubMed] [Google Scholar]
- Krystal JH, Webb E, Cooney NL, Kranzler HR, Southwick SW, Heninger GR, Charney DS. Serotonergic and noradrenergic dysregulation in alcoholism: m-chlorophenylpiperazine and yohimbine effects in recently detoxified alcoholics and healthy comparison subjects. Am J Psychiatry. 1996;153:83–92. doi: 10.1176/ajp.153.1.83. [DOI] [PubMed] [Google Scholar]
- Kudielka BM, Kirschbaum C. Sex differences in HPA axis responses to stress: a review. Biol Psychiat. 2005;69:113–132. doi: 10.1016/j.biopsycho.2004.11.009. [DOI] [PubMed] [Google Scholar]
- Lê AD, Shaham Y. Neurobiology of relapse to alcohol in rats. Pharmacol Ther. 2002;94:137–156. doi: 10.1016/s0163-7258(02)00200-0. [DOI] [PubMed] [Google Scholar]
- Lê AD, Harding S, Juzytsch W, Fletcher PJ, Shaham Y. The role of corticotropin-releasing factor in the median raphe nucleus in relapse to alcohol. J Neurosci. 2002;22:7844–7849. doi: 10.1523/JNEUROSCI.22-18-07844.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lê AD, Harding S, Juzytsch W, Funk D, Shaham Y. Role of alpha-2 adrenoceptors in stress-induced reinstatement of alcohol seeking and alcohol self-administration in rats. Psychopharmacology. 2005;179:366–373. doi: 10.1007/s00213-004-2036-y. [DOI] [PubMed] [Google Scholar]
- Lê AD, Harding S, Juzytsch W, Watchus J, Shalev U, Shaham Y. The role of corticotrophin-releasing factor in stress-induced relapse to alcohol-seeking behavior in rats. Psychopharmacology. 2000;150:317–324. doi: 10.1007/s002130000411. [DOI] [PubMed] [Google Scholar]
- Lê AD, Poulos CX, Harding S, Watchus J, Juzytsch W, Shaham Y. Effects of naltrexone and fluoxetine on alcohol self-administration and reinstatement of alcohol seeking induced by priming injections of alcohol and exposure to stress. Neuropsychopharm. 1999;21:435–444. doi: 10.1016/S0893-133X(99)00024-X. [DOI] [PubMed] [Google Scholar]
- Lê AD, Quan B, Juzytsch W, Fletcher PJ, Joharchi N, Shaham Y. Reinstatement of alcohol-seeking by priming injections of alcohol and exposure to stress in rats. Psychopharmacology. 1998;135:169–174. doi: 10.1007/s002130050498. [DOI] [PubMed] [Google Scholar]
- Lê AD, Wang A, Harding S, Juzytsch W, Shaham Y. Nicotine increases alcohol self-administration and reinstates alcohol seeking behavior in rats. Psychopharmacology. 2003;168:216–221. doi: 10.1007/s00213-002-1330-9. [DOI] [PubMed] [Google Scholar]
- Lechtenberg R, Worner TM. Relative kindling effect of detoxification and non-detoxification admissions in alcoholics. Alcohol Alcohol. 1991;26:221–225. doi: 10.1093/oxfordjournals.alcalc.a045104. [DOI] [PubMed] [Google Scholar]
- Li L, Power C, Kelly S, Kirschbaum C, Hertzman C. Lifetime socioeconomic position and cortisol patterns in mid-life. Psychoneuroendocrinology. 2007;32:824–833. doi: 10.1016/j.psyneuen.2007.05.014. [DOI] [PubMed] [Google Scholar]
- Lindquist TL, Beilin LJ, Knuiman MW. Influence of lifestyle, coping, and job stress on blood pressure in men and women. Hypertension. 1997;29:1–7. doi: 10.1161/01.hyp.29.1.1. [DOI] [PubMed] [Google Scholar]
- Litt MD, Cooney NL, Morse P. Reactivity to alcohol-related stimuli in the laboratory and in the field: predictors of craving in treated alcoholics. Addiction. 2000;95:889–900. doi: 10.1046/j.1360-0443.2000.9568896.x. [DOI] [PubMed] [Google Scholar]
- Liu L, Hutchinson MR, White JM, Somogyi AA, Coller JK. Association of IL-1B genetic polymorphisms with an increased risk of opioid and alcohol dependence. Pharmacogenetics and Genomics. 2009;19:869–876. doi: 10.1097/FPC.0b013e328331e68f. [DOI] [PubMed] [Google Scholar]
- Liu X, Weiss F. Additive effect of stress and drug cues on reinstatement of alcohol seeking: Exacerbation by history of dependence and role of concurrent activation of corticotropin-releasing factor and opioid mechanisms. J Neurosci. 2002a;22:7856–786. doi: 10.1523/JNEUROSCI.22-18-07856.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Weiss F. Reversal of alcohol-seeking behavior by D1 and D2 antagonists in an animal model of relapse: differences in antagonist potency in previously alcohol-dependent versus nondependent rats. J Pharmaco Exp Ther. 2002b;300:882–889. doi: 10.1124/jpet.300.3.882. [DOI] [PubMed] [Google Scholar]
- Liu X, Weiss F. Stimulus conditioned to foot-shock stress reinstates alcohol-seeking behavior in an animal model of relapse. Psychopharmacology. 2003;168:184–191. doi: 10.1007/s00213-002-1267-z. [DOI] [PubMed] [Google Scholar]
- Lopez MF, Becker HC. Effect of pattern and number of chronic alcohol exposures on subsequent voluntary alcohol intake in C57BL/6J mice. Psychopharmacology. 2005;181:688–696. doi: 10.1007/s00213-005-0026-3. [DOI] [PubMed] [Google Scholar]
- Lovallo WR, Dickensheets SL, Myers DA, Thomas TL, Nixon SJ. Blunted stress cortisol response in abstinent alcoholic and polysubstance-abusing men. Alcohol Clin Exp Res. 2000;24:651–658. [PubMed] [Google Scholar]
- Lowery EG, Spanos M, Navarro M, Lyons AM, Hodge CW, Thiele TE. CRF-1 antagonist and CRF-2 agonist decrease binge-like alcohol drinking in C57BL/6J mice independent of the HPA axis. Neuropsychopharm. 2010;35:1241–1252. doi: 10.1038/npp.2009.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig AM, Wikler A, Stark LH. The first drink: Psychobiological aspects of craving. Arch Gen Psychiat. 1974;30:539–547. doi: 10.1001/archpsyc.1974.01760100093015. [DOI] [PubMed] [Google Scholar]
- Lynch WJ, Kushner MG, Rawleigh JM, Fiszdon J, Carroll ME. The effects of restraint stress in voluntary ethanol consumption in rats. Exp Clin Psychopharmacol. 1999;7:318–323. doi: 10.1037//1064-1297.7.4.318. [DOI] [PubMed] [Google Scholar]
- Mantsch JR, Baker DA, Francis DM, Katz ES, Hoks MA, Serge JP. Stressor- and corticotropin releasing factor-induced reinstatement and active stress-related behavioral responses are augmented following long-access cocaine self-administration by rats. Psychopharmacology. 2008;195:591–603. doi: 10.1007/s00213-007-0950-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinelli PW, Funk D, Juzytsch W, Harding S, Rice KC, Shaham Y, Lê AD. The CRF1 receptor antagonist antalarmin attenuates yohimbine-induced increases in operant alcohol self-administration and reinstatement of alcohol seeking in rats. Psychopharmacology. 2007;195:345–355. doi: 10.1007/s00213-007-0905-x. [DOI] [PubMed] [Google Scholar]
- Marlatt G, Gordon J. Determinants of relapse: Implications for the maintenance of behavioral change. In: Davidson P, Davidson S, editors. Behavioral Medicine: Changing health lifestyles. New York: Brunner/Mazel; 1980. pp. 410–452. [Google Scholar]
- Marlatt GA, Kosturn CF, Lang AR. Provocation to anger and opportunity for retaliation as determinants of alcohol consumption in social drinkers. J Abnorm Psychol. 1975;84:652–659. [PubMed] [Google Scholar]
- Martin-Fardon R, Ciccocioppo R, Massi M, Weiss F. Nociceptin prevents stress-induced alcohol- but not cocaine-seeking behavior in rats. Neuroreport. 2000;11:1939–1943. doi: 10.1097/00001756-200006260-00026. [DOI] [PubMed] [Google Scholar]
- Martin-Fardon R, Zorrilla EP, Ciccocioppo R, Weiss F. Role of innate and drug-induced dysregulation of brain stress and arousal systems in addiction: Focus on corticotropin-releasing factor, nociceptin/orphanin FQ, and orexin/hypocretin. Brain Res. 2010;1314:145–146. doi: 10.1016/j.brainres.2009.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez D, Gil R, Slifstein M, Hwang DR, Huang Y, Perez A, et al. Alcohol dependence is associated with blunted dopamine transmission in the ventral striatum. Biol Psychiatry. 2005;58:779–786. doi: 10.1016/j.biopsych.2005.04.044. [DOI] [PubMed] [Google Scholar]
- Martinez D, Kim JH, Krystal J, Abi-Dargham A. Imaging the neurochemistry of alcohol and substance abuse. Neuroimaging Clin N Am. 2007;17:539–555. doi: 10.1016/j.nic.2007.07.004. [DOI] [PubMed] [Google Scholar]
- McBride WJ, Schultz JA, Kimpel MW, McClintick JN, Wang M, You J, Rodd ZA. Differential effects of alcohol in the nucleus accumbens shell of alcohol-preferring (P), alcohol-non-preferring (NP) and Wistar rats: A proteomics study. Pharmacol Biochem Behav. 2009;92:304–313. doi: 10.1016/j.pbb.2008.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCown TJ, Breese GR. Multiple withdrawals from chronic alcohol “kindles” inferior collicular seizure activity: evidence for kindling of seizures associated with alcoholism. Alcohol Clin Exp Res. 1990;14:394–399. doi: 10.1111/j.1530-0277.1990.tb00492.x. [DOI] [PubMed] [Google Scholar]
- McCown TJ, Moray LJ, Kizer JS, Breese GR. Interactions between TRH and alcohol in the medial septum. Pharmacol Biochem Behav. 1986;24:1269–1274. doi: 10.1016/0091-3057(86)90183-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCown TJ, Frye GD, Breese GR. Evidence for site specific alcohol actions in the CNS. Alcohol Drug Res. 1985;6:423–429. [PubMed] [Google Scholar]
- McEwen BS. Allostasis and allostatic load: implications for neuropsychopharmacology. Neuropsychopharm. 2000;22:108–124. doi: 10.1016/S0893-133X(99)00129-3. [DOI] [PubMed] [Google Scholar]
- McEwen BS, Wingfield JC. The concept of allostasis in biology and biomedicine. Hormone Behav. 2003;43:2–15. doi: 10.1016/s0018-506x(02)00024-7. [DOI] [PubMed] [Google Scholar]
- McGue M, Iacono WG. The adolescent origins of substance use disorders. Int J Methods Psychiatr Res. 2008;17(1 suppl):S30–S38. doi: 10.1002/mpr.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKay JR, Foltz C, Stephens RC, Leahy PJ, Crowley EM, Kissin W. Predictors of alcohol and crack cocaine use outcomes over a 3-year follow-up in treatment seekers. J Subst Abuse Treat. 2005;28(Suppl 1):S73–S82. doi: 10.1016/j.jsat.2004.10.010. [DOI] [PubMed] [Google Scholar]
- McLeod DS, Koenen KC, Meyer JM, Lyons MJ, Eisen S, True W, Goldberg J. Genetic and environmental influences on the relationship among combat exposure, posttraumatic stress disorder symptoms, and alcohol use. J Trauma Stress. 2001;14:259–275. doi: 10.1023/A:1011157800050. [DOI] [PubMed] [Google Scholar]
- Meisch RA. Relationship between physical dependence on alcohol and reinforcing properties of alcohol in animals. NIAAA Res Monogr. 1983;13:27–32. [Google Scholar]
- Meisch RA, Stewart RB. Alcohol as a reinforcer: a review of laboratory studies of non-human primates. Behav Pharmacol. 1994;5:425–440. [PubMed] [Google Scholar]
- Melendez RI, Middaugh LD, Kalivas PW. Development of an alcohol deprivation and escalation effect in C57BL/6J mice. Alcohol ClinExpRes. 2006;30:2017–2025. doi: 10.1111/j.1530-0277.2006.00248.x. [DOI] [PubMed] [Google Scholar]
- Mello NK, Mendelson JH. Ethanol and whisky drinking patterns in rats under free-choice and forced-choice conditions. Q J Stud Alcohol. 1964;25:1–25. [PubMed] [Google Scholar]
- Merlo-Pich E, Lorang M, Yeganeh M, deFonseca FR, Raber J, Koob GF, Weiss F. Increase of extracellular corticotropin-releasing factor-like immunoreactivity levels in the amygdala of awake rats during restraint stress and alcohol withdrawal as measured by microdialysis. J Neurosci. 1995;15:5439–5447. doi: 10.1523/JNEUROSCI.15-08-05439.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelgard A, Appel L, Pissiota A, Frans O, Langstrom B, Bergstrom M, Fredrikson M. Symptom provocation in specific phobia affects the substance P neurokinin-1 receptor system. Biol Psychiatry. 2007;61:1002–1006. doi: 10.1016/j.biopsych.2006.07.003. [DOI] [PubMed] [Google Scholar]
- Miller WR, Westerberg VS, Harris RJ, Tonigan JS. What predicts relapse? Prospective testing of antecedent models. Addiction. 1996;91(Suppl):155–171. [PubMed] [Google Scholar]
- Minami M, Kuraishi Y, Yamaguchi T, Nakai S, Hirai Y, Satoh M. Immobilization stress induces interleukin-1 beta mRNA in the rat hypothalamus. Neurosci Lett. 1991;123:254–256. doi: 10.1016/0304-3940(91)90944-o. [DOI] [PubMed] [Google Scholar]
- Modell JG, Mountz JM. Focal cerebral blood flow change during craving for alcohol measured by SPECT. J Neurophysiol. 1995;7:15–22. doi: 10.1176/jnp.7.1.15. [DOI] [PubMed] [Google Scholar]
- Modell JG, Mountz JM, Beresford TP. Basal ganglia/limbic striatal and thalamocortical involvement in craving and loss of control in alcoholism. J Neuropsychiat Clin Neurosci. 1990;2:123–144. doi: 10.1176/jnp.2.2.123. [DOI] [PubMed] [Google Scholar]
- Monti PM, Binkoff JA, Abrams DB, Zwick WR, Nirenberg TD, Liepman MR. Reactivity of alcoholics and nonalcoholics to drinking cues. J Abnorm Psychol. 1987;96:122–126. doi: 10.1037//0021-843x.96.2.122. [DOI] [PubMed] [Google Scholar]
- Munro CA, Oswald LM, Weerts EM, McCaul ME, Wand GS. Hormone responses to social stress in abstinent alcohol-dependent subjects and social drinkers with no history of alcohol dependence. Alcohol Clin Exp Res. 2005;29:1133–1138. doi: 10.1097/01.alc.0000172459.71517.05. [DOI] [PubMed] [Google Scholar]
- Myers RD, Stoltman WP, Martin GE. Effects of alcohol dependence induced artificially in the rhesus monkey on the subsequent preference for ethyl alcohol. Physiol Behav. 1972;9:43–48. doi: 10.1016/0031-9384(72)90262-4. [DOI] [PubMed] [Google Scholar]
- Myrick H, Anton RF, Li X, Henderson S, Drobes D, Voronin K, George MS. Differential brain activity in alcoholics and social drinkers to alcohol cues: relationship to craving. Neuropsychopharm. 2004;29:393–402. doi: 10.1038/sj.npp.1300295. [DOI] [PubMed] [Google Scholar]
- Nadal R, Chappell AM, Samson HH. Effects of nicotine and mecamylamine microinjections into the nucleus accumbens on ethanol and sucrose self-administration. Alcohol Clin Exp Res. 1998;22:1190–1198. [PubMed] [Google Scholar]
- Naqvi NH, Bechara A. The hidden island of addiction: the insula. Trends Neurosci. 2009;32:56–67. doi: 10.1016/j.tins.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson EC, Agrawal A, Pergadia ML, Wang JC, Whitfield JB, Saccone FS, et al. H2 haplotype at chromosome 17q21.31 protects against childhood sexual abuse-associated risk for alcohol consumption and dependence. Addict Biol. 2010;15:1–11. doi: 10.1111/j.1369-1600.2009.00181.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestler EJ, Barrot M, DiLeone RJ, Eisch AJ, Gold SJ, Monteggia LM. Neurobiology of depression. Neuron. 2002;34:13–25. doi: 10.1016/s0896-6273(02)00653-0. [DOI] [PubMed] [Google Scholar]
- Nguyen KT, Deak T, Owens SM, Kohno T, Fleshner M, Watkins LR, Maier SF. Exposure to acute stress induces brain IL-1β protein in the rat. J Neurosci. 1998;18:2239–2246. doi: 10.1523/JNEUROSCI.18-06-02239.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolen-Hoeksema S. Sex differences in unipolar depression: Evidence and theory. Psych Bulln. 1987;101:259–282. [PubMed] [Google Scholar]
- Numan R. Multiple exposures to alcohol facilitate intravenous self-administration of alcohol by rats. Pharmacol Biochem Behav. 1981;15:101–108. doi: 10.1016/0091-3057(81)90346-4. [DOI] [PubMed] [Google Scholar]
- O’Brien CP. Anticraving medications for relapse prevention: a possible new class of psychoactive medications. Am J Psychiatry. 2005;162:1423–1431. doi: 10.1176/appi.ajp.162.8.1423. [DOI] [PubMed] [Google Scholar]
- O’Connor KA, Johnson JD, Hansen MK, Wieseler Frank JL, Maksimova E, Watkins LR, et al. Peripheral and central proinflammatory cytokine response to a severe acute stressor. Brain Res. 2003;991:123–132. doi: 10.1016/j.brainres.2003.08.006. [DOI] [PubMed] [Google Scholar]
- O’Dell LE, Roberts AJ, Smith RT, Koob GF. Enhanced alcohol self-administration after intermittent versus continuous alcohol vapor exposure. Alcohol Clin Exp Res. 2004;28:1676–1682. doi: 10.1097/01.alc.0000145781.11923.4e. [DOI] [PubMed] [Google Scholar]
- Olive MF, Koenig HN, Nannini MA, Hodge CW. Elevated extracellular CRF levels in the bed nucleus of the stria terminalis during ethanol withdrawal and reduction by subsequent ethanol intake. Pharmacol Biochem Behav. 2002;72:213–220. doi: 10.1016/s0091-3057(01)00748-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olive MF, Mehmert KK, Camarini R, Kim JA, Ou CJ, Hodge CW. A role for corticotropin releasing factor (CRF) in alcohol consumption, sensitivity, and reward as revealed by CRF-deficient mice. Psychopharmacology. 2003;165:181–187. doi: 10.1007/s00213-002-1248-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Malley SS, Jaffe AJ, Chang G, Schottenfeld RS, Meyer RE, Rounsaville B. Naltrexone and coping skills therapy for alcohol dependence: a controlled study. Arch Gen Psychiat. 1992;49:881–887. doi: 10.1001/archpsyc.1992.01820110045007. [DOI] [PubMed] [Google Scholar]
- Overstreet D, Knapp D, Breese G. Accentuated decrease in social interaction in rats subjected to repeated multiple alcohol withdrawals. Alcohol Clin Exp Res. 2002;26:1259–1268. doi: 10.1097/01.ALC.0000023983.10615.D7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overstreet D, Knapp D, Breese GR. Modulation of multiple alcohol withdrawal-induced anxiety-like behavior by CRF and CRF1 receptors. Pharmacol Biochem Behav. 2004a;77:405–413. doi: 10.1016/j.pbb.2003.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overstreet D, Knapp D, Breese GR. Similar anxiety-like responses in male and female rats exposed to repeated withdrawals from alcohol. Pharmacol Biochem Behav. 2004b;78:459–464. doi: 10.1016/j.pbb.2004.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overstreet DH, Knapp DJ, Breese GR. Pharmacological modulation of repeated alcohol withdrawal-induced anxiety-like behavior differs in alcohol-preferring P and Sprague-Dawley rats. Pharmacol Biochem Behav. 2005;81:122–130. doi: 10.1016/j.pbb.2005.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overstreet DH, Knapp DJ, Breese GR. Drug challenges reveal differences in mediation of stress facilitation of voluntary alcohol drinking and withdrawal-induced anxiety in alcohol-preferring P rats. Alcohol Clin Exp Res. 2007;31:1473–1481. doi: 10.1111/j.1530-0277.2007.00445.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overstreet DH, Knapp DJ, Angel RA, Breese GR. Reduction in repeated alcohol withdrawal-induced anxiety-like behavior by site-selective injections of 5-HT1A and 5-HT2c ligands. Psychopharmacology. 2006;187:1–12. doi: 10.1007/s00213-006-0389-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overstreet D, Knapp D, Moy S, Breese GR. A 5-HT1A agonist and 5-HT2C antagonist reduce social interaction deficit induced by multiple withdrawals in rats. Psychopharmacology. 2003;167:344–352. doi: 10.1007/s00213-003-1425-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park MS, Sohn JH, Suk JA, Kim SH, Sohn S, Sparacio R. Brain substrates of craving to alcohol cues in subjects with alcohol use disorder. Alcohol Alcohol. 2007;42:417–22. doi: 10.1093/alcalc/agl117. [DOI] [PubMed] [Google Scholar]
- Payne TJ, Rychtarik RG, Rappaport NB, Smith PO, Etscheidt M, Brown TA, Johnson CA. Reactivity to alcohol-relevant beverage and imaginal cues in alcoholics. Addict Behav. 1992;17:209–217. doi: 10.1016/0306-4603(92)90026-r. [DOI] [PubMed] [Google Scholar]
- Peng GS, Chen YC, Tsao TP, Wang MF, Yin SJ. Pharmacokinetic and pharmacodynamic basis for partial protection against alcoholism in Asians, heterozygous for the variant ALDH2*2 gene allele. Pharmacogenet Genomics. 2007;17:845–855. doi: 10.1097/FPC.0b013e3282609e67. [DOI] [PubMed] [Google Scholar]
- Penick EC, Powell BJ, Nickel EJ, Read MR, Gabrielli WF, Liskow BI. Examination of Cloninger’s type I and type II alcoholism with a sample of men alcoholics in treatment. Alcohol Clin Exp Res. 1990;14:623–629. doi: 10.1111/j.1530-0277.1990.tb01213.x. [DOI] [PubMed] [Google Scholar]
- Petrakis IL, Gonzalez G, Rosenheck R, Krystal JH. Comorbity of alcoholism and psychiatric disorders. Alcohol Res & Health. 2002;26:81–89. [Google Scholar]
- Pirkola SP, Pirkola SP, Isometsä E, Suvisaari J, Aro H, Joukamaa M, et al. DSM-IV mood-, anxiety- and alcohol use disorders and their comorbidity in the Finnish general population--results from the Health 2000 Study. Soc Psychiatry Psychiatr Epidemiol. 2005;40:1–10. doi: 10.1007/s00127-005-0848-7. [DOI] [PubMed] [Google Scholar]
- Pohorecky LA. Interaction of alcohol and stress: research with experimental animals—an update. Alcohol Alcohol. 1990;25:263–276. doi: 10.1093/oxfordjournals.alcalc.a045000. [DOI] [PubMed] [Google Scholar]
- Porjesz B, Begleiter H. Human evoked brain potentials and alcohol. Alcohol Clin Exp Res. 1981;5:304–317. doi: 10.1111/j.1530-0277.1981.tb04904.x. [DOI] [PubMed] [Google Scholar]
- Prescott CA, Kendler KS. Age at first drink and risk for alcoholism: a noncausal association. Alcohol Clin Exp Res. 1999;23:101–107. [PubMed] [Google Scholar]
- Pucak ML, Kaplin Al. Unkind cytokines: current evidence for the potential role of cytokines in immune-mediated depression. Int Rev Psychiatry. 2005;17:477–483. doi: 10.1080/02646830500381757. [DOI] [PubMed] [Google Scholar]
- Raison CL, Capuron L, Miller AH. Cytokines sing the blues: inflammation and the pathogenesis of depression. Trends Immunol. 2006;27:24–31. doi: 10.1016/j.it.2005.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramchandani VA, Umhau J, Pavon FJ, Ruiz-Velasco V, Margas W, Sun H, et al. A genetic determinant of the striatal dopamine response to alcohol in men. Mol Psychiatry. 2010 doi: 10.1038/mp.2010.56. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangaswamy M, Jones KA, Porjesz B, Chorlian DB, Padmanabhapillai A, Kamarajan C, Kuperman S, Rohrbaugh J, O’Connor SJ, Bauer LO, Schuckit MA, Begleiter H. Delta and theta oscillations as risk markers in adolescent offspring of alcoholics. Int J Psychophysiol. 2007;63:3–15. doi: 10.1016/j.ijpsycho.2006.10.00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen DD, Raskind MA, Koob GF. alpha1-noradrenergic receptor antagonism blocks dependence-induced increases in responding for alcohol. Alcohol. 2008;42:91–97. doi: 10.1016/j.alcohol.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rassnick S, Heinrichs SC, Britton KT, Koob GF. Microinjection of a corticotropin-releasing factor antagonist into the central nucleus of the amygdala reverses anxiogenic-like effects of alcohol withdrawal. Brain Res. 1993;605:25–32. doi: 10.1016/0006-8993(93)91352-s. [DOI] [PubMed] [Google Scholar]
- Rehm J, Mathers C, Popova S, Thavorncharoensap M, Teerawattananon Y, Patra J. Global burden of disease and injury and economic cost attributable to alcohol use and alcohol-use disorders. Lancet. 2009;373:2223–2233. doi: 10.1016/S0140-6736(09)60746-7. [DOI] [PubMed] [Google Scholar]
- Rewal M, Jurd R, Gill TM, He DY, Ron D, Janak PH. Alpha4-containing GABAA receptors in the nucleus accumbens mediate moderate intake of alcohol. J Neurosci. 2009;29:543–549. doi: 10.1523/JNEUROSCI.3199-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rimondini R, Arlinde C, Sommer W, Heilig M. Long-lasting increase in voluntary alcohol consumption and transcriptional regulation in the rat brain after intermittent exposure to alcohol. FASEB J. 2002;16:27–35. doi: 10.1096/fj.01-0593com. [DOI] [PubMed] [Google Scholar]
- Rimondini R, Sommer W, Heilig M. A temporal threshold for induction of persistent alcohol preference: behavioral evidence in a rat model of intermittent intoxication. J Stud Alcohol. 2003;64:445–449. doi: 10.15288/jsa.2003.64.445. [DOI] [PubMed] [Google Scholar]
- Rimondini R, Sommer W, Dall’Olio R, Heilig M. Long-lasting tolerance to alcohol following a history of dependence. Addict Biol. 2008;13:26–30. doi: 10.1111/j.1369-1600.2007.00079.x. [DOI] [PubMed] [Google Scholar]
- Rimondini R, Thoirsell A, Heilig MJ. Suppression of alcohol self-administration by the neuropeptide Y (NPY) Y2 receptor antagonist BIIE0246: evidence for sensitization in rats with a history of dependence. Neurosci Lett. 2005;375:129–133. doi: 10.1016/j.neulet.2004.10.084. [DOI] [PubMed] [Google Scholar]
- Roberto M, Cruz MT, Gilpin NW, Sabino V, Schweitzer P, Bajo M, et al. Corticotropin releasing factor-induced amygdala gamma-aminobutyric acid release plays a key role in alcohol dependence. Biol Psychiatry. 2010;67:831–839. doi: 10.1016/j.biopsych.2009.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts AJ, Heyser CJ, Cole M, Griffin P, Koob GF. Excessive alcohol drinking following a history of dependence: animal model of allostasis. Neuropsychopharm. 2000;22:581–594. doi: 10.1016/S0893-133X(99)00167-0. [DOI] [PubMed] [Google Scholar]
- Rodd ZA, Bell RL, Kuc KA, Murphy JM, Lumeng L, Li TK, McBride WJ. Effects of repeated alcohol deprivations on operant alcohol self-administration by alcohol-preferring (P) rats. Neuropsychopharm. 2003;28:1614–1621. doi: 10.1038/sj.npp.1300214. [DOI] [PubMed] [Google Scholar]
- Rodd ZA, Melendez RI, Bell RL, Kuc KA, Zhang Y, Murphy JM, McBride WJ. Intracranial self-administration of alcohol within the ventral tegmental area of male Wistar rats: evidence for involvement of dopamine neurons. J Neurosci. 2004;24:1050–1057. doi: 10.1523/JNEUROSCI.1319-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodd-Henricks ZA, McKinzie DL, Murphy JM, McBride WJ, Lumeng L, Li TK. The expression of an alcohol deprivation effect in the high-alcohol-drinking replicate rat lines is dependent on repeated deprivations. Alcohol Clin Exp Res. 2000a;24:747–753. [PubMed] [Google Scholar]
- Rodd-Henricks ZA, McKinzie DL, Shaikh SR, Murphy JM, McBride WJ, Lumeng L, Li TK. Alcohol deprivation effect is prolonged in the alcohol preferring (P) rat after repeated deprivations. Alcohol Clin Exp Res. 2000b;24:8–16. [PubMed] [Google Scholar]
- Rostène W, Kitabgi P, Parsadaniantz SM. Chemokines: a new class of neuromodulator? Nat Rev Neurosci. 2007;8:895–903. doi: 10.1038/nrn2255. [DOI] [PubMed] [Google Scholar]
- Sajdyk TJ, Fitz SD, Shekhar A. The role of neuropeptide Y in the amygdala on corticotropin-releasing factor receptor mediated behavioral stress response in the rat. Stress. 2006;9:21–28. doi: 10.1080/10253890600557315. [DOI] [PubMed] [Google Scholar]
- Samson HH. Initiation of alcohol reinforcement using a sucrose-substitution procedure in food and water sated rats. Alcohol Clin exp Res. 1986;10:436–442. doi: 10.1111/j.1530-0277.1986.tb05120.x. [DOI] [PubMed] [Google Scholar]
- Samson HH, Falk JL. Alteration of fluid preference in alcohol-dependent animals. J Pharmacol Exp Ther. 1974;190:365–376. [PubMed] [Google Scholar]
- Sanchis-Segura C, Spanagel R. Behavioral assessment of drug reinforcement and addictive features in rodents: an overview. Addiction Biol. 2006;11:2–38. doi: 10.1111/j.1369-1600.2006.00012.x. [DOI] [PubMed] [Google Scholar]
- Sayette MA, Shiffman S, Tiffany ST, Niaura RS, Martin CS, Shadel WG. The measurement of drug craving. Addiction. 2000;95 (Supplement 2):S189–210. doi: 10.1080/09652140050111762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider F, Habel U, Wagner M, Franke P, Sallaoum JB, et al. Subcortical correlates of craving in recently abstinent alcoholic patients. Am J Psychiatr. 2001;158:1075–1083. doi: 10.1176/appi.ajp.158.7.1075. [DOI] [PubMed] [Google Scholar]
- Schukit MA. Alcohol-induced changes in body sway in men at high alcoholism risk. Arch Gen Psychiatry. 1985;42:375–379. doi: 10.1001/archpsyc.1985.01790270065007. [DOI] [PubMed] [Google Scholar]
- Schuckit MA. Reactions to alcohol in sons of alcoholics and controls. Alcohol Clin Exp Res. 1988;12:465–470. doi: 10.1111/j.1530-0277.1988.tb00228.x. [DOI] [PubMed] [Google Scholar]
- Schuckit MA. The 1994 Isaacson Award Lecture: a prospective study of sons of alcoholics. Alcohol Alcohol. 1994;(Suppl 2):1–6. [PubMed] [Google Scholar]
- Schuckit MA. Comorbidity between substance use disorders and psychiatric conditions. Addiction. 2006;101:76–88. doi: 10.1111/j.1360-0443.2006.01592.x. [DOI] [PubMed] [Google Scholar]
- Schuckit MA. An overview of genetic influences in alcoholism. J Subst Abuse Treat. 2009a;36:S5–S14. [PubMed] [Google Scholar]
- Schuckit MA. Alcohol use disorders. Lancet. 2009b;373:492–501. doi: 10.1016/S0140-6736(09)60009-X. [DOI] [PubMed] [Google Scholar]
- Schuckit MA, Li TK, Cloninger CR, Deitrich RA. Genetics of alcoholism. Alcohol Clin Exp Res. 1985;9:475–492. doi: 10.1111/j.1530-0277.1985.tb05588.x. [DOI] [PubMed] [Google Scholar]
- Schulteis G, Hyytiä P, Heinrichs SC, Koob GF. Effects of chronic alcohol exposure on oral self-administration of alcohol or saccharin by Wistar rats. Alcohol Clin Exp Res. 1996;20:164–171. doi: 10.1111/j.1530-0277.1996.tb01060.x. [DOI] [PubMed] [Google Scholar]
- Schweinsburg AD, Paulus MP, Barlett VC, Killeen LA, Caldwell LC, Pulido, et al. An FMRI study of response inhibition in youths with a family history of alcoholism. Ann N Y Acad Sci. 2004;1021:391–394. doi: 10.1196/annals.1308.050. [DOI] [PubMed] [Google Scholar]
- Seo D, Lacadie C, Constable TC, Bergquist KT, Sinha R. Disrupted ventromedial prefrontal cortex function during stress and neutral relaxed states is predictive of alcohol relapse outcomes. Alcohol Clin Exp Res. 2010;34(Supplement):161A. [Google Scholar]
- Shaham Y, Hope BT. The role of neuroadaptations in relapse to drug seeking. Nat Neurosci. 2005;8:1437–1439. doi: 10.1038/nn1105-1437. [DOI] [PubMed] [Google Scholar]
- Shaham Y, Shalev U, Lu L, De Wit H, Stewart J. The reinstatement model of drug relapse: history, methodology and major findings. Psychopharmacology. 2003;168:3–20. doi: 10.1007/s00213-002-1224-x. [DOI] [PubMed] [Google Scholar]
- Shin LM, Liberzon I. The neurocircuitry of fear, stress, and anxiety disorders. Neuropsychopharm Rev. 2010;35:169–191. doi: 10.1038/npp.2009.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shintani F, Nakaki T, Kanba S, Sato K, Yagi G, Shiozawa M, Aiso S, Kato R, Asai M. Involvement of interleukin-1 in immobilization stress-induced increase in plasma adrenocorticotropic hormone and in release of hypothalamic monoamines in the rat. J Neurosci. 1995a;15:1961–1970. doi: 10.1523/JNEUROSCI.15-03-01961.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shintani F, Nakaki T, Kanba S, Kato R, Asai M. Role of interleukin-1 in stress responses. A putative neurotransmitter. Mol Neurobiol. 1995b;10:47–71. doi: 10.1007/BF02740837. [DOI] [PubMed] [Google Scholar]
- Shivani R, Goldsmith RJ, Anthenelli RM. Alcoholism and psychiatric disorder. Alcohol Res Health. 2002;26:90–98. [Google Scholar]
- Shizuya K, Komori T, Fujiware R, Miyahara S, Ohmori M, Nomura J. The influence of restraint stress on the expression of mRNAs for IL-6 and the IL-6 receptor in the hypothalamus and midbrain of the rat. Life Sci. 1997;61:PL 135–140. doi: 10.1016/s0024-3205(97)00608-5. [DOI] [PubMed] [Google Scholar]
- Sillaber I, Henniger MS. Stress and alcohol drinking. Ann Med. 2004;36:596–605. doi: 10.1080/07853890410018862. [DOI] [PubMed] [Google Scholar]
- Sillaber I, Rammes G, Zimmermann S, Mahal B, Zieglgänsberger W, Wurst W, et al. Enhanced and delayed stress-induced alcohol drinking in mice lacking functional CRH1 receptors. Science. 2002;296:931–933. doi: 10.1126/science.1069836. [DOI] [PubMed] [Google Scholar]
- Sinclair JD. Drugs to decrease alcohol drinking. Ann Med. 1990;22:357–362. doi: 10.3109/07853899009147920. [DOI] [PubMed] [Google Scholar]
- Sinclair JD. Evidence about the use of naltrexone and for different ways of using it in the treatment of alcoholism. Alcohol Alcohol. 2001;36:2–10. doi: 10.1093/alcalc/36.1.2. [DOI] [PubMed] [Google Scholar]
- Sinclair JD, Senter RJ. Development of an alcohol-deprivation effect in rats. Q J Stud Alcohol. 1968;29:863–867. [PubMed] [Google Scholar]
- Sinha R. How does stress increase risk of drug abuse and relapse. Psychopharmacology. 2001;158:343–359. doi: 10.1007/s002130100917. [DOI] [PubMed] [Google Scholar]
- Sinha R. The role of stress in addiction relapse. Curr Psychiatry Rep. 2007;9:388–395. doi: 10.1007/s11920-007-0050-6. [DOI] [PubMed] [Google Scholar]
- Sinha R. Chronic stress, drug use, and vulnerability to addiction. Ann NY Acad Sci. 2008a;1141:105–130. doi: 10.1196/annals.1441.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha R. Chronic alcohol related neuroadaptations in stress responses: Effects on compulsive alcohol seeking and relapse outcomes. Alcohol Clin Exp Res. 2008b;32:341a. [Google Scholar]
- Sinha R. Modeling stress and drug craving in the laboratory: implications for addiction treatment development. Addict Biol. 2009;14:84–98. doi: 10.1111/j.1369-1600.2008.00134.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha R, Li CS. Imaging stress and cue-induced drug and alcohol craving: Association with relapse and clinical implications. Drug Alcohol Rev. 2007;26:25–31. doi: 10.1080/09595230601036960. [DOI] [PubMed] [Google Scholar]
- Sinha R, O’Malley SS. Craving for alcohol: Findings from the clinic and the laboratory. Alcohol Alcohol. 1999;34:223–230. doi: 10.1093/alcalc/34.2.223. [DOI] [PubMed] [Google Scholar]
- Sinha R, Fox HC, Hong KA, Bergquist K, Bhagwagar Z, Siedlarz KM. Enhanced negative emotion and alcohol craving, and altered physiological responses following stress and cue exposure in alcohol dependent individuals. Neuropsychopharm. 2009;34:1198–1208. doi: 10.1038/npp.2008.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha R, Garcia M, Paliwal P, Kreek MJ, Rounsaville BJ. Stress-induced cocaine craving and hypothalamic-pituitary-adrenal responses are predictive of cocaine relapse outcomes. Arch Gen Psychiatry. 2006;63:324–331. doi: 10.1001/archpsyc.63.3.324. [DOI] [PubMed] [Google Scholar]
- Sinha R, Hong KA, Seo D, FoxHCBerguist K. Neural and endocrine predictions of alcohol relapse risk. Clin Exp Res. 34(Supplement):290A. [Google Scholar]
- Sinha R, Lacadie C, Li CSR, Milivojevic V, Bergquist KT. Altered corticostriatal-limbic responses to stress, alcohol-related and neutral relaxing cues in abstinent alcoholics compared to controls. Alcohol Clin Exp Res. 2007;31(Supplement):453A. [Google Scholar]
- Sommer WH, Rimondini R, Hansson AC, Hipskind PA, Gehlert DR, Barr CS, Heilig MA. Upregulation of voluntary alcohol intake, behavioral sensitivity to stress, and amygdala crhr1 expression following a history of dependence. Biol Psychiatry. 2008;63:139–145. doi: 10.1016/j.biopsych.2007.01.010. [DOI] [PubMed] [Google Scholar]
- Soyka M, Rösner S. Opioid antagonists for pharmacological treatment of alcohol dependence - a critical review. Curr Drug Abuse Rev. 2008;1:280–291. doi: 10.2174/1874473710801030280. [DOI] [PubMed] [Google Scholar]
- Spanagel R. Alcoholism: A systems approach from molecular physiology to addictive behavior. Physiol Rev. 2009;80:649–705. doi: 10.1152/physrev.00013.2008. [DOI] [PubMed] [Google Scholar]
- Spanagel R, Hölter SM. Pharmacological validation of a new animal model of alcoholism. J Neural Transm. 2000;107:669–680. doi: 10.1007/s007020070068. [DOI] [PubMed] [Google Scholar]
- Steptoe A, Ussher M. Smoking, cortisol and nicotine. Int J Psychophysiol. 2006;59:228–235. doi: 10.1016/j.ijpsycho.2005.10.011. [DOI] [PubMed] [Google Scholar]
- Suzuki E, Shintani F, Kanba S, Asai M, Nakaki T. Immobilization stress increases mRNA levels of interleukin-1 receptor antagonist in various rat brain regions. Cell Mol Neurobiol. 1997;17:557–562. doi: 10.1023/A:1026319107528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamres LK, Janicki D, Helgeson VS. Sex differences in coping behavior: A meta-analytic review and an examination of relative coping. Pers Soc Psychol Rev. 2002;6:2–30. [Google Scholar]
- Tapert SF, Brown GG, Kindermann SS, Cheung EH, Frank LR, Brown SA. fMRI measurement of brain dysfunction in alcohol-dependent young women. Alcohol Clin Exp Res. 2001;25:236–245. [PubMed] [Google Scholar]
- Tapert SF, Cheung EH, Brown GG, Frank LR, Paulus MP, Schweinsburg AD, et al. Neural response to alcohol stimuli in adolescents with alcohol use disorder. Arch Gen Psychiatry. 2003;60:727–735. doi: 10.1001/archpsyc.60.7.727. [DOI] [PubMed] [Google Scholar]
- Tate S, McQuaid J, Brown S. Characteristics of life stressors predictive of substance treatment outcomes. J Subst Abuse Treat. 2005;29:107–115. doi: 10.1016/j.jsat.2005.05.003. [DOI] [PubMed] [Google Scholar]
- Thorsell A, Schank JR, Singley E, Hunt SP, Heilig M. Neurokinin-1 receptors (NK1R:s), alcohol consumption, and alcohol reward in mice. Psychopharmacology. 2010;209:103–111. doi: 10.1007/s00213-010-1775-1. [DOI] [PubMed] [Google Scholar]
- Toalston JE, Oster SM, Kuc KA, Pommer TJ, Murphy JM, Lumeng L. Effects of alcohol and saccharin deprivations on concurrent alcohol and saccharin operant self-administration by alcohol-preferring (P) rats. Alcohol. 2008;42:277–284. doi: 10.1016/j.alcohol.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbull AV, Rivier CL. Regulation of the hypothalamic-pituitary-adrenal axis by cytokines: actions and mechanisms of action. Physiol Rev. 1999;79:1–71. doi: 10.1152/physrev.1999.79.1.1. [DOI] [PubMed] [Google Scholar]
- Uddin M, Aiello AE, Wildman DE, Koenen KC, Pawelec G, de Los Santos R, Goldmann E, Galea S. Epigenetic and immune function profiles associated with posttraumatic stress disorder. Proc Natl Acad Sci. 2010;107:9470–9475. doi: 10.1073/pnas.0910794107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhart M, Wand GS. Stress, alcohol, and drug interaction: an update of human research. Addiction Biol. 2008;14:43–64. doi: 10.1111/j.1369-1600.2008.00131.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulrichsen J, Bech B, Allerup P, Hemmingsen R. Diazepam prevents progression of kindled alcohol withdrawal behaviour. Psychopharmacology. 1995;121:451–460. doi: 10.1007/BF02246493. [DOI] [PubMed] [Google Scholar]
- Ulrichsen J, Haugbøl S, Brandt CF, Allerup P, Hemmingsen R. Irreversibility of kindled alcohol-withdrawal behavior in rats. Alcohol Alcohol. 1998a;33:230–243. doi: 10.1093/oxfordjournals.alcalc.a008387. [DOI] [PubMed] [Google Scholar]
- Ulrichsen J, Woldbye DP, Madsen TM, Clemmesen L, Haugbøl S, Olsen CH, Laursen H, Bolwig TG, Hemmingsen R. Electrical amygdala kindling in alcohol-withdrawal kindled rats. Alcohol Alcohol. 1998b;33:244–254. doi: 10.1093/oxfordjournals.alcalc.a008388. [DOI] [PubMed] [Google Scholar]
- Valdez GR, Koob GF. Allostasis and dysregulation of corticotropin-releasing factor and neuropeptide Y systems: implications for the development of alcoholism. Pharmacol Biochem Behav. 2004;79:671–679. doi: 10.1016/j.pbb.2004.09.020. [DOI] [PubMed] [Google Scholar]
- Valdez GR, Roberts AJ, Han K, Davis H, Brennan M, Zorrilla EP, Koob GF. Increased alcohol self-administration and anxiety-like behavior during acute alcohol withdrawal and protracted abstinence: regulation by corticotropin-releasing factor. Alcohol Clin Exp Res. 2002;26:1494–1501. doi: 10.1097/01.ALC.0000033120.51856.F0. [DOI] [PubMed] [Google Scholar]
- Valdez GR, Zorrilla EP, Roberts AJ, Koob GF. Antagonism of corticotropin-releasing factor attenuates the enhanced responsiveness to stress observed during protracted alcohol abstinence. Alcohol. 2003;29:55–60. doi: 10.1016/s0741-8329(03)00020-x. [DOI] [PubMed] [Google Scholar]
- Valdez GR, Sabino V, Koob GF. Increased anxiety-like behavior and alcohol self-administration in dependent rats: reversal via corticotropin-releasing factor-2 receptor activation. Alcohol Clin Exp Res. 2004;28:865–872. doi: 10.1097/01.alc.0000128222.29875.40. [DOI] [PubMed] [Google Scholar]
- Vale W, Spiess J, Rivier C, Rivier J. Characterization of a 41-residue ovine hypothalamic peptide that stimulates secretion of corticotrophin and beta-endorphin. Science. 1981;213:1394–1397. doi: 10.1126/science.6267699. [DOI] [PubMed] [Google Scholar]
- Vengeliene V, Siegmund S, Singer MV, Sinclair JD, Li TK, Spanagel R. A comparative study on alcohol-preferring rat lines: effects of deprivation and stress phases on voluntary alcohol intake. Alcohol Clin Exp Res. 2003;27:1048–1054. doi: 10.1097/01.ALC.0000075829.81211.0C. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Wang GJ, Telang F, Fowler JS, Logan J, Childress AR, et al. Cocaine cues and dopamine in dorsal striatum: mechanism of craving in cocaine addiction. J Neurosci. 2006;26:6583–6588. doi: 10.1523/JNEUROSCI.1544-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpicelli J, Alterman AI, Hayashida M, O’brien CP. Naltrexone in the treatment of alcohol dependence. Arch Gen Psychiat. 1992;49:876–880. doi: 10.1001/archpsyc.1992.01820110040006. [DOI] [PubMed] [Google Scholar]
- Vocci FJ, Acri J, Elkashef A. Medication development for addictive disorders: the state of the science. Am J Psychiatry. 2005;162(8):1432–1440. doi: 10.1176/appi.ajp.162.8.1432. [DOI] [PubMed] [Google Scholar]
- Walker BM, Koob GF. Pharmacological evidence for a motivational role of K-opioid systems in alcohol dependence. Neuropsychopharm. 2008;33:643–652. doi: 10.1038/sj.npp.1301438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker BM, Rasmussen DD, Raskind MA, Koob GF. alpha1-noradrenergic receptor antagonism blocks dependence-induced increases in responding for alcohol. Alcohol. 2008;42:91–97. doi: 10.1016/j.alcohol.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wand G. The anxious amygdala: CREB signaling and predisposition to anxiety and alcoholism. J Clin Investig. 2005;115:2762–2773. doi: 10.1172/JCI26436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wand GS, Dobs AS. Alterations in the hypothalamic-pituitary-adrenal axis in actively drinking alcoholics. J Clin Endocrin Metab. 1991;72:1290–1295. doi: 10.1210/jcem-72-6-1290. [DOI] [PubMed] [Google Scholar]
- Wang W, Ji P, Dow KE. CRF induces proliferation and TNFα release in cultured rat microglia via MAP-kinase signaling pathways. J Neurochem. 2003;84:189–195. doi: 10.1046/j.1471-4159.2003.01544.x. [DOI] [PubMed] [Google Scholar]
- Wang W, Ji P, Riopelle RJ, Dow KE. Functional expression of CRH receptor 1 in cultured rat microglia. J Neurochem. 2002;80:287–294. doi: 10.1046/j.0022-3042.2001.00687.x. [DOI] [PubMed] [Google Scholar]
- Weiss F. Neurobiology of craving, conditioned reward and relapse. Curr Opin Pharmacol. 2005;5:9–19. doi: 10.1016/j.coph.2004.11.001. [DOI] [PubMed] [Google Scholar]
- Weiss F, Ciccocioppo R, Parsons LH, Katner S, Liu X, Zorrilla EP, et al. Compulsive drug-seeking behavior and relapse. Neuroadaptation, stress, and conditioning factors. Ann N Y Acad Sci. 2001;937:1–26. doi: 10.1111/j.1749-6632.2001.tb03556.x. [DOI] [PubMed] [Google Scholar]
- Wikler A. Recent progress in research on the neurophysiological basis of morphine addiction. Am J Psychiat. 1948;105:329–338. doi: 10.1176/ajp.105.5.329. [DOI] [PubMed] [Google Scholar]
- Winger G. Effects of alcohol withdrawal on alcohol-reinforced responding in rhesus monkeys. Drug Alcohol Depend. 1988;22:235–240. doi: 10.1016/0376-8716(88)90023-3. [DOI] [PubMed] [Google Scholar]
- Wolffgramm J, Heyne A. From controlled drug intake to loss of control: the irreversible development of drug addiction in the rat. Behav Br Res. 1995;70:77–94. doi: 10.1016/0166-4328(95)00131-c. [DOI] [PubMed] [Google Scholar]
- Wong DF, Kuwabara H, Schretlen DJ, Bonson KR, Zhou Y, Nandi A, et al. Increased occupancy of dopamine receptors in human striatum during cue-elicited cocaine craving. Neuropsychopharm. 2006;31: 2716–2727. doi: 10.1038/sj.npp.1301194. [DOI] [PubMed] [Google Scholar]
- Wrase J, Grüsser SM, Klein S, Diene C, Hermann D, Flor H, et al. Development of alcohol-associated cues and cue-induced brain activation in alcoholics. Eur Psychiatry. 2002;17:287–291. doi: 10.1016/s0924-9338(02)00676-4. [DOI] [PubMed] [Google Scholar]
- Yehuda R. Biology of post-traumatic stress disorder. J Clin Psychiatry. 2002;62(Suppl 17):41–46. [PubMed] [Google Scholar]
- Yoon GK, Kim SW, Thuras P, Grant JE. Alcohol craving in outpatients with alcohol dependence: rate and clinical correlates. J Stud Alcohol. 2006;67:770–777. doi: 10.15288/jsa.2006.67.770. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Morse AC, Koob GF, Schulteis G. Dose- and time-dependent expression of anxiety-like behavior in the elevated plus-maze during withdrawal from acute and repeated intermittent alcohol intoxication in rats. Alcohol Clin Exp Res. 2007;31:1811–1819. doi: 10.1111/j.1530-0277.2007.00483.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Dayas CV, Aujla H, Baptista MA, Martin-Fardon R, Weiss F. Activation of Group II metabotropic glutamate receptors attenuates both stress and cue-induced alcohol-seeking and modulates c-fos expression in the hippocampus and amygdala. J Neurosci. 2006;26:9967–9967. doi: 10.1523/JNEUROSCI.2384-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zobel AW, Nickel T, Künzel HE, Ackl N, Sonntag A, Ising M, Holsboer F. Effects of the high-affinity corticotropin-releasing hormone receptor 1 antagonist R121919 in major depression: the first 20 patients treated. J Psychiatr Res. 2000;34:171–118. doi: 10.1016/s0022-3956(00)00016-9. [DOI] [PubMed] [Google Scholar]
- Zorrilla EP, Koob GF. The therapeutic potential of CRF1 antagonists for anxiety. Expert Opin Investig Drugs. 2004;13:799–828. doi: 10.1517/13543784.13.7.799. [DOI] [PubMed] [Google Scholar]
- Zorrilla EP, Valdez GR, Weiss F. Changes in levels of regional CRF-like-immunoreactivity and plasma corticosterone during protracted drug withdrawal in dependent rats. Psychopharmacology. 2001;158:374–381. doi: 10.1007/s002130100773. [DOI] [PubMed] [Google Scholar]
- Zywiak WH, Westerberg VS, Connors GJ, Maisto SA. Exploratory findings from the Reasons for Drinking Questionnaire. J Subst Abuse Treat. 2003;25:287–292. doi: 10.1016/s0740-5472(03)00118-1. [DOI] [PubMed] [Google Scholar]