

Abstract
We take for granted the ability to fall asleep or to snap out of sleep into wakefulness, but these changes in behavioral state require specific switching mechanisms in the brain that allow well-defined state transitions. In this review, we examine the basic circuitry underlying the regulation of sleep and wakefulness, and discuss a theoretical framework wherein the interactions between reciprocal neuronal circuits enable relatively rapid and complete state transitions. We also review how homeostatic, circadian, and allostatic drives help regulate sleep state switching, and discuss how breakdown of the switching mechanism may contribute to sleep disorders such as narcolepsy.
Introduction
We spend nearly one-third of our lives asleep, and many mammals, including small laboratory rodents, spend half or more of their existence in this state (Savage and West, 2007;Siegel, 2009). Because sleeping animals are inherently more vulnerable, it is necessary for an animal to be able to awaken quickly so it can flee or defend itself. Conversely, it is common experience that one can fall asleep over just a few seconds or minutes. These state transitions involve dramatic alterations in easily observed physiological variables, including eye closure, breathing, arousability, and muscle tone. We measure the changes in cortical activity and muscle tone, respectively, by recording the electroencephalogram (EEG) and electromyogram (EMG), and the actual transitions in electrophysiologically monitored state occur over just a few seconds (Takahashi et al., 2010). Similarly, during the sleep period, animals and people rapidly transition between rapid eye movement (REM) and non-REM (NREM) sleep states. Recent advances in understanding the brain circuitry underlying the waking and sleeping states have given rise to models that may explain these transitions. The principles that govern these models for state transitions may ultimately apply to many other state changes, such as emotional responses, sexual arousal, or cognitive state changes such as reorienting attention. Hence the mechanisms for wake-sleep state transitions potentially have broad implications for a variety of behavioral states.
As an individual falls asleep, the EEG initially transitions from a state of high frequency, low voltage waves in the waking state to higher voltage, slower waves representing NREM sleep. These changes take place over a few seconds or less in rodents but may take 10 sec to a minute in humans (Takahashi et al., 2010;Wright. et al., 1995). The EEG then progressively slows during NREM sleep until it is dominated by high voltage, slow wave (0.5–4 Hz) activity, after which the slow waves progressively diminish, a typical bout lasting from 40 minutes to an hour or more in humans. In rodents, this process is much shorter, with slow waves established within seconds of entering NREM sleep, and the entire NREM bout generally lasting three to five minutes, although occasionally they may extend to 20 minutes or more. Across species, wake bout lengths follow a power law distribution (the log of probability of a bout of a certain length and the log of the bout length forming a linear relationship) whereas the durations of sleep bouts follow an exponential distribution (Lo et al., 2004;Phillips et al., 2010). In each case, though, the transitions between NREM sleep and wakefulness typically take less than 1% of the duration of an average NREM bout. The EEG then makes another abrupt transition over a few seconds from NREM into REM sleep, with lower voltage, higher frequency activity. In rodents, the EEG recorded from the cortical surface during REM sleep is dominated by 5–8 Hz theta activity generated by the underlying hippocampus; in humans, theta activity is present during REM sleep, particularly in the hippocampus, but the dominant cortical frequencies are faster and lower voltage. During REM sleep, there is almost complete loss of tone in skeletal muscles (except those used for breathing and eye movements), accompanied by rapid eye movements that give the state its name. Humans report active dreams during REM sleep but less lively mentation during NREM sleep. Over the sleep period, an individual may switch back and forth from NREM to REM sleep, with occasional transitions to periods of wakefulness. The duration of the NREM, REM, and wake bouts varies with the species, age, and health of the individual, but the electrographic transitions between these states are relatively rapid in comparison to bout duration.
Researchers first began mapping the general circuitry that controls wakefulness and sleep over 50 years ago, and in the last 10–20 years, much has been learned about the specific systems that regulate these states. Progress over the last few years has been especially rapid, leading to an improved understanding of the neurochemicals, pathways, and firing patterns that regulate NREM and REM sleep. Other new work has examined the ways in which behavioral drives, including homeostatic, circadian, and allostatic influences, may affect these switching mechanisms. We will first review these advances, and place them into the context of a model we have proposed for sleep/wake state transitions based upon mutually inhibitory circuits, as are seen in electronic flip-flop switches (Saper et al., 2001, 2005). We will then explore recently proposed mathematical models based on this circuitry that can explain many of the features of natural sleep and state transitions. Finally, we will examine how this circuitry can explain many of the features of the sleep disorder narcolepsy, an example of state instability in which the circuitry that stabilizes switching is damaged.
Networks supporting sleep and wakefulness
Wake-promoting networks
Current models of the ascending arousal system are still generally based on the observations by Moruzzi, Magoun and their coworkers that electrical stimulation of the paramedian reticular formation, particularly within the midbrain produces EEG desynchronization consistent with arousal (Moruzzi and Magoun, 1949). Subsequent studies identified a slab of tissue at the junction of the rostral pons and caudal midbrain as critical for maintaining the waking state (Lindsley et al., 1949). Although the neurons responsible for arousal were initially thought to be part of the undifferentiated reticular formation, subsequent studies showed that the cell groups at the mesopontine junction that project to the forebrain mainly consist of monoaminergic and cholinergic neurons that reside in specific cell groups rather than the “reticular core” (Figure 1) (see (Saper, 1987) for review).
Figure 1. The wake-sleep switch.

Many wake-promoting projections arise from neurons in the upper brainstem (A). Cholinergic neurons (aqua) provide the major input to the thalamus, whereas monoaminergic and (presumably) glutamatergic neurons (dark green) provide direct innervation of the the hypothalamus. basal forebrain, and cerebral cortex. The orexin neurons in the lateral hypothalamus (blue) reinforce activity in these brainstem arousal pathways and also directly excite the cerebral cortex and BF. The main sleep-promoting pathways (magenta in B) from the ventrolateral (VLPO) and median (MnPO) preoptic nuclei inhibit the components of the ascending arousal pathways in both the hypothalamus and the brainstem (pathways that are inhibited are shown as open circles and dashed lines). However, the ascending arousal systems are also capable of inhibiting the VLPO (C). This mutually inhibitory relationship of the arousal- and sleep-promoting pathways produces the conditions for a “flip-flop” switch, which can generate rapid and complete transitions between waking and sleeping states. Abbreviations: DR, dorsal raphe nucleus (serotonin); LC, locus coeruleus (norepinephrine); LDT, laterodorsal tegmental nucleus (acetylcholine); PB, parabrachial nucleus (glutamate); PC, precoeruleus area (glutamate); PPT, pedunculopontine tegmental nucleus (acetylcholine); TMN, tuberomammillary nucleus (histamine); vPAG, ventral periaqueductal gray (dopamine).
Cholinergic neurons that project to the forebrain are found in the pedunculopontine and laterodorsal tegmental nuclei (PPT and LDT). They provide the main innervation from the mesopontine junction to the thalamic relay nuclei, but also innervate the intralaminar and reticular thalamic nuclei, as well as the lateral hypothalamus, basal forebrain, and prefrontal cortex (Hallanger et al., 1987;Satoh and Fibiger, 1986). Many neurons in the PPT and LDT fire most rapidly during wakefulness and rapid eye movement (REM) sleep, and most slowly during non-REM (NREM) sleep, suggesting that they help drive cortical activation (El Mansari et al., 1989;Steriade et al., 1993). These nuclei are heterogeneous, but extracellular recordings combined with juxtacellular labeling confirm that cholinergic neurons in the LDT fire during cortical activation, usually increasing their firing rates just before the transition from cortical slow waves to faster frequencies (Boucetta and Jones, 2009).
The monoaminergic cell groups at the mesopontine level which project to the forebrain include the noradrenergic locus coeruleus (LC) and the serotoninergic dorsal and median raphe nuclei (Aston-Jones and Bloom, 1981;Dahlstrom and Fuxe, 1964;Kocsis et al., 2006), as well as dopaminergic neurons adjacent to the dorsal raphe nucleus (Lu et al., 2006a). Histaminergic neurons in the tuberomammillary nucleus (TMN) have similar projection targets and firing patterns (Panula et al., 1989;Steininger et al., 1999). Axons from these cell groups predominantly target the lateral hypothalamus, basal forebrain and cerebral cortex, where they terminate extensively, particularly in the prefrontal cortex. Each of these monoaminergic systems also sends smaller, but important populations of axons to the thalamus, where they largely target the intralaminar and reticular nuclei. Generally, neurons in these cell groups fire most actively during wakefulness, decrease activity during non-REM sleep, and fall silent during REM sleep (Aston-Jones and Bloom, 1981;Kocsis et al., 2006;Steininger et al., 1999;Takahashi et al., 2006;Takahashi et al., 2010).
Another source of arousal influence from the rostral pons may be glutamatergic neurons in the parabrachial nucleus and the adjacent precoeruleus area (PC, the lateral corner of the rostral pontine periventricular gray matter, just rostral to the main body of the LC), which have been found to send major projections to the lateral hypothalamus, basal forebrain, and cerebral cortex (Hur and Zaborszky, 2005;Lu et al., 2006b;Saper, 1987;Saper and Loewy, 1980). The activity patterns of these glutamatergic neurons have not yet been studied, but recordings in this area in cats and Fos studies in rats have shown predominantly wake- and REM sleep-active neurons (Chu and Bloom, 1973;Lu et al., 2006b;Saito et al., 1977). Tests of the role of these neurons in wakefulness would be of great interest.
Several forebrain neuronal systems also support wakefulness. The importance of these forebrain regions is underscored by the observation that over a period of weeks to months after acute lesions of the brainstem arousal system, animals and humans eventually recover wake-sleep cycles (Adametz, 1959;Posner et al., 2008). Thus, while these forebrain areas appear to depend upon the brainstem arousal influence in the intact individual, they apparently can reorganize to support cortical arousal even without input from the brainstem.
One of these forebrain arousal systems is found in the posterior half of the lateral hypothalamus. Just dorsal and rostral to the histaminergic neurons of the TMN, the lateral hypothalamus contains neurons producing the orexin neuropeptides (orexin-A and –B, also known as hypocretin-1 and 2). Many of the orexin neurons also contain glutamate, and nearly all also contain the neuropeptide dynorphin (Chou et al., 2001;Torrealba et al., 2003). They send axons to the entire cerebral cortex, as well as to the brainstem and basal forebrain, with particularly intense input to the TMN and the LC (Peyron et al., 1998). There is also less intense orexin innervation of the intralaminar nuclei of the thalamus, as well as the anteroventral thalamic nucleus. There are two known orexin receptors, both of which are G-protein coupled receptors with excitatory membrane effects (Sakurai et al., 1998). Orexin neurons receive afferents from many components of the ascending arousal system, including the LC, DR, and parabrachial nucleus, as well as from cortical (medial prefrontal) and amygdaloid (central nucleus) sources associated with arousal and ventral tegmental sites associated with reward (Yoshida et al., 2006). They fire predominantly during wakefulness, and fire particularly briskly during active exploration of the environment or during motivated behaviors (Lee et al., 2005;Mileykovskiy et al., 2005). Orexin neurons are also driven by low glucose (Moriguchi et al., 1999), and may play an important role in motivating foraging behaviors in hungry animals as well as in reward and drug seeking behaviors (Harris et al., 2005;Yamanaka et al., 2003). Selective activation of the orexin neurons with a light-sensitive sodium channel awakens mice from sleep, suggesting that the orexin neurons are capable of driving arousal from sleep (Adamantidis et al., 2007;Carter et al., 2009). Most importantly, selective destruction of the orexin neurons with a genetically targeted toxin results in the symptoms of narcolepsy (Hara et al., 2001), which will be discussed in a separate section below. Overall, the orexin neurons are thought to sustain wakefulness and suppress REM sleep.
On the other hand, large lesions of the posterior lateral hypothalamus (Gerashchenko et al., 2003;Nauta, 1946;Ranson, 1939;Swett and Hobson, 1968) produce much more extensive sleepiness than can be explained by elimination of just orexin and histamine transmission (Gerashchenko, 2003). This suggests the presence of other important arousal-producing neurons in the posterior lateral hypothalamus. There is an additional population of neurons in the supramammillary region and extending laterally to the subthalamic nucleus, which is a known source of projections to the cerebral cortex and basal forebrain (Grove, 1988;Saper, 1985). Many neurons in this region express the vesicular glutamate transporter 2 (Hur and Zaborszky, 2005;Ziegler et al., 2002), but whether these glutamatergic neurons promote arousal remains to be determined.
The most rostral population of arousal-promoting subcortical neurons is located in the basal forebrain. Many of these neurons contain either acetylcholine or GABA, and a small number contain glutamate (Manns et al., 2001; Hur and Zaborszky, 2005). Basal forebrain cholinergic neurons innervate and both directly and indirectly activate cortical pyramidal cells, and likely augment cortical activation and EEG desynchronization (Jones, 2004). GABAergic basal forebrain neurons innervate and presumably inhibit cortical GABAergic interneurons and deep layer pyramidal cells (Freund and Meskenaite, 1992;Henny and Jones, 2008), both of which most likely result in disinhibition of cortical circuits. Many of these basal forebrain neurons are wake-active and fire in bursts correlated with specific EEG rhythms. Small ibotenic acid lesions of the basal forebrain result in modest slowing of the EEG without changing the amount of wake or sleep, while specific lesions of basal forebrain cholinergic neurons reduce wakefulness transiently, without affecting the EEG frequency spectrum (Kaur et al., 2008). On the other hand, acute inactivation of the basal forebrain with the anesthetic procaine produces deep NREM sleep, whereas activation with glutamatergic agonists causes wakefulness (Cape and Jones, 2000). A definitive understanding of the roles of the basal forebrain cell groups in arousal awaits studies that differentially eliminate the GABAergic population.
The thalamic relay nuclei (such as the anterior, ventral and lateral thalamic cell groups; medial and lateral geniculate nuclei; mediodorsal nucleus; and pulvinar), are the most important and abundant sources of subcortical glutamatergic afferents to the cerebral cortex, and the intralaminar and midline nuclei provide a diffuse source of cortical input (Jones and Leavitt, 1974). Surprisingly, there is little evidence that these inputs play a major role in producing wakefulness. Early electrical stimulation studies suggested that the midline and intralaminar thalamic nuclei might constitute a diffuse, “non-specific”, cortical activating system (Morison and Dempsey, 1942;Steriade, 1995), but lesions of the midline and intralaminar nuclei did not prevent cortical activation (Moruzzi and Magoun, 1949;Starzl et al., 1951). Extensive ablation of the thalamus in cats actually caused an increase in wakefulness (Villablanca and Salinas-Zeballos, 1972), but in rats has been reported to have little if any effect on wakefulness or EEG waveforms, other than to eliminate sleep spindles (Buzsaki et al., 1988;Vanderwolf and Stewart, 1988). When overall arousal is impaired with thalamic lesions in humans, close examination of the pathology has shown that there is also damage to the paramedian midbrain or underlying hypothalamus (Posner et al., 2008). Conversely, patients with bilateral thalamic damage are often in a persistent vegetative state, with preserved wake-sleep cycles, but without retained cognitive content (Kinney and Samuels, 1994), and patients with fatal familial insomnia have thalamic degeneration and sleep loss (Montagna et al., 2003). It is difficult to reconcile these observations with the thalamus playing a role in promoting overall cortical arousal.
On the other hand, the thalamus may be important for selecting aspects of the environment for attention, and in this regard may interact with the arousal system. Selective activation of specific cortical areas is thought to be regulated by the reticular nucleus of the thalamus, which covers the rostral and lateral surface of the thalamus and has a major inhibitory influence over the thalamic relay nuclei. The reticular nucleus consists of GABAergic neurons, which sample thalamocortical traffic, and inhibit thalamic relay neurons, resulting in targeted modulation of thalamo-cortical transmission. Thus, selective inhibition of thalamic reticular neurons may be a critical mechanism for selective attention, and a major function of the arousal system. Inputs to the reticular nucleus arise from cholinergic (Levey et al., 1987;Parent and Descarries, 2008), noradrenergic (Asanuma, 1992), serotoninergic, and histaminergic (Manning et al., 1996) arousal systems, along with pyramidal neurons of the frontal cortex (Zikopoulos and Barbas, 2007), and GABAergic neurons of the basal forebrain (Asanuma, 1989;Asanuma and Porter, 1990;Bickford et al., 1994). These likely represent important mechanisms through which the brainstem, basal forebrain and frontal cortex modulate activity within thalamo-cortical circuits.
Finally, the telencephalon is not just a target of the arousal system (as measured by EEG and behavioral activation), but itself contributes to regulation of arousal. All components of the arousal system intensively innervate the prefrontal cortex, in particular the medial prefrontal region, which in turn sends descending projections back to the basal forebrain, hypothalamus, and brainstem components of the arousal system (Aston-Jones and Cohen, 2005;Hurley et al., 1991). Reciprocal excitation might allow the medial prefrontal cortex to rapidly escalate arousal when a behaviorally important stimulus is present.
The presence of such a large number of cell groups that are thought to promote arousal raises the question of how they interact in this process. It is interesting that drugs that block transmission for one or another of these pathways (e.g., muscarinic antagonists, H1-histamine antagonists, or α2-adrenergic agonists) cause acute sleepiness, but chronic ablation of the basal forebrain cholinergic neurons (Kaur et al., 2008), tuberomammillary histaminergic neurons (Gerashchenko et al., 2004), or the LC and pontine cholinergic neurons (Lu et al., 2006a;Shouse and Siegel, 1992;Webster and Jones, 1988), or combinations of these structures (Blanco-Centurion et al., 2007) have minimal effects on the amount of wakefulness. One possible reason for this puzzling result is that the arousal system may contain sufficient redundancy that remaining wake-promoting systems may be able to compensate for the chronic (but perhaps not acute) loss of one or even a few components, e.g., by increasing activity or receptor sensitivity in intact arousal systems.
A related issue is which of these wake-promoting cell groups participate in the switching between sleep and wakefulness, as opposed to the maintenance of the waking state. This issue will be taken up in the section of this review on switching circuitry.
NREM sleep-promoting networks
During the epidemic of encephalitis lethargica around the time of the First World War, von Economo reported that patients with lesions in the preoptic region around the rostral end of the third ventricle demonstrated profound insomnia (Von Economo, 1930). Experimental lesions of the preoptic-basal forebrain region reduced sleep in rats and cats (McGinty and Sterman, 1968;Nauta, 1946), but the exact population of sleep-promoting neurons was unknown. Sherin and colleagues subsequently identified a population of neurons in the ventrolateral preoptic nucleus (VLPO) that innervate the histaminergic TMN and which express Fos protein selectively during sleep, but not wakefulness (Sherin et al., 1996). VLPO neurons, containing the inhibitory neurotransmitters GABA and galanin, innervate other components of the ascending arousal system as well, including the LC, raphe system, periaqueductal gray matter, parabrachial nucleus, and lateral hypothalamic area (Sherin et al., 1998). The VLPO was found to consist of a dense core of sleep-active, galanin-positive neurons that project heavily to the TMN. However, this is surrounded dorsally and medially by a more diffuse population of sleep-active, galanin-positive neurons, the extended VLPO, which more extensively targets the dorsal raphe and LC (Lu et al., 2000;Sherin et al., 1998). As is true for many cell groups in the hypothalamus that are defined on the basis of common neurotransmitter, connections, and physiology, the VLPO neurons are mixed in among other cell types. In addition, while there are other galaninergic neurons laterally in the basal forebrain and medially in the preoptic area, none of these are sleep-active or project to the TMN, LC, or dorsal raphe (Sherin et al., 1998;Gaus et al., 2002). These complexities make it difficult to interpret single unit recording studies, which necessarily record from only one cell at a time, but rarely identify their chemical phenotype. Nevertheless, such recordingsin rats demonstrated that many neurons in the VLPO region fire at about 1–2 Hz during wakefulness, about 2–4 times faster during NREM sleep, and about twice as fast again during deep NREM sleep after 12 hrs of sleep deprivation (Szymusiak et al., 1998). However, some of the neurons were found to fire fastest during REM sleep. Similar observations have been made in mice (Takahashi et al., 2009). These observations suggest that VLPO neurons constitute a sleep-promoting pathway from the preoptic area that inhibits many arousal systems during sleep. However, there are also some wake-active neurons mixed in with the VLPO cells (Szymusiak et al., 1998; Modirrousta et al., 2004; Takahashi et al., 2009) whose function with respect to wake-sleep regulation is not known. To test the net effect of the neurons in the VLPO region on sleep regulation, sleep recordings were done in animals with cell-specific lesions of the VLPO, and these showed a decrease in NREM, REM, and total sleep by up to 50% (Lu et al., 2000). Cell loss in the VLPO core correlated most closely with loss of NREM sleep, while loss of REM sleep was more closely correlated with loss of neurons in the extended VLPO (Lu et al., 2000).
The preoptic area and basal forebrain near the VLPO also contain other populations of sleep-active neurons (Lee et al., 2004;Szymusiak and McGinty, 1986; Modirrousta et al, 2004; Hassani et al., 2009; Takahashi et al., 2009), however the ability of these cell groups to cause sleep, as opposed to simply firing during sleep, is less clear. The best studied of these is a population of neurons in the median preoptic nucleus (MnPO). Like the VLPO, the MnPO contains many neurons that produce Fos during sleep and contain GABA (although they do not contain galanin) (Gong et al., 2004). About 75% of MnPO neurons fire faster during sleep (Suntsova et al., 2002), although only about 10% are differentially more active in NREM or REM. Unlike VLPO neurons, whose firing increases at just about the same time as sleep onset (Szymusiak et al., 1998;Takahashi et al., 2009), MnPO neurons often fire in advance of sleep, suggesting a role in accumulating sleep pressure. This hypothesis has been strengthened by the observation that MnPO neurons also express Fos during sleep deprivation, while the VLPO neurons only express Fos during sleep (Gvilia et al., 2006). The MnPO provides a major input to the VLPO (Chou et al., 2002;Uschakov et al., 2007), which may allow it to drive VLPO activity. Other projections from the MnPO target the lateral hypothalamic area, the dorsal raphe, the LC, and the midbrain periaqueductal gray matter, but not the cholinergic PPT and LDT nuclei or the TMN (Uschakov et al., 2007). It is not known whether the neurons that contribute to these projections are the same ones that are sleep-active, GABAergic neurons. Studies of the effects of selective MnPO lesions on sleep would be of great value in defining its contribution to sleep regulation.
A key property of VLPO neurons is that they receive reciprocal inputs from many regions implicated in arousal, including the TMN, dorsal raphe nucleus and adjacent ventral periaqueductal gray matter, parabrachial nucleus, and LC (Chou et al., 2002; Lu et al., 2006a). Slice recordings of identified VLPO neurons show that they are inhibited by acetylcholine, norepinephrine, dopamine, and serotonin (Gallopin et al., 2000;Gallopin et al., 2004). While VLPO cells are not inhibited by histamine, the TMN neurons also contain the mu-opioid peptide endomorphin, which inhibits VLPO neurons (Greco et al., 2008). MnPO neurons receive only sparse inputs from the LC and periaqueductal gray matter, and little if any from the dorsal or median raphe nuclei or from the TMN (Saper and Levisohn, 1983). The effects of these inputs on the MnPO sleep-active neurons remain unknown.
Because even rats with very large VLPO lesions still sleep about 50% as much as normal animals, it is likely that the sleep promoting system in the brain is distributed, with other components in addition to the VLPO that may contribute to the inhibition of the arousal systems during sleep. These may include other sleep-active neurons in the MnPO and basal forebrain (Modirrousta et al., 2004;Takahashi et al., 2009), but evidence that these cells promote sleep is lacking. Recent studies on lesions of the striatum and globus pallidus have reported substantial increases in wakefulness and sleep fragmentation (Qiu et al., 2010). The descending projections from both the nucleus accumbens and globus pallidus are largely GABAergic and include the basal forebrain and lateral hypothalamus (Baldo et al., 2004;Kim et al., 1976;Swanson and Cowan, 1975). In addition, a population of cortical neurons has been described that express cFos during sleep and are immunoreactive for both nitric oxide and neuropeptide Y (Gerashchenko et al., 2008). However, their role in producing sleep states, or in state switching, remains to be studied.
Thus, although it is likely that other sleep-promoting neurons participate in the induction and maintenance of sleep, the VLPO neurons appear to play a particularly important role in this process, as VLPO lesions can substantially reduce sleep for months (Lu et al., 2000). Therefore, in our model for behavioral state switching, we will focus on the interactions of the VLPO with wake-promoting systems.
REM sleep-promoting networks
After the discovery of REM sleep in the 1950’s, its regulation became a major focus of research. Much work has indicated that neurons in the pons play an essential role as REM sleep is disrupted by transections of the pons or large excitotoxic lesions of this region (Jouvet, 1962;Webster and Jones, 1988). In addition, just prior to and during REM sleep, high voltage EEG waves occur in the pons, lateral geniculate, and occipital cortex (hence “PGO waves”) in cats. These PGO waves are time locked to bursts of firing by “PGO burst neurons” in the region of the cholinergic PPT and LDT nuclei at the junction of the midbrain and the pons (El Mansari et al., 1989;Sakai and Jouvet, 1980). Conversely, firing of neurons in the LC, dorsal raphe nucleus, and TMN was found to slow almost to a halt during REM sleep (Aston-Jones and Bloom, 1981;Hobson et al., 1975;Steininger et al., 1999;Takahashi et al., 2010;Trulson et al., 1981). This reciprocal relationship suggested that the PPT and LDT might interact with monoaminergic neurons to regulate the alternation of NREM and REM sleep.
McCarley and Hobson produced a predator-prey model of this NREM-REM sleep mechanism using Lottka-Volterra equations (Hobson et al., 1975;McCarley and Hobson, 1975;Pace-Schott and Hobson, 2002). In a predator-prey model, an increase in the prey population allows an increase in the predator population, but this then reduces the prey population, resulting in a subsequent decrease in the predators, thus permitting another cycle in which the prey population expands again. In the Hobson-McCarley model, the cholinergic neurons act like prey, in that an increase in their firing during REM sleep excites monoamine cells. However, the monoaminergic neurons are like predators, in that an increase in their firing inhibits the cholinergic neurons, and terminates the REM period. The oscillations in a predator-prey model require a substantial time delay between the expansion of the prey and predator populations. While this delay is maintained by breeding cycles in the predator-prey model, it is not clear how this model would apply to neurons, which communicate with each other in a matter of milliseconds.
The model of cholinergic-monoaminergic interactions regulating REM sleep was supported by the observations that application of cholinergic drugs to the mesopontine tegmentum can cause REM sleep, whereas drugs that increase monoaminergic signaling inhibit REM sleep (Luppi et al., 2006). On the other hand, the importance of the PPT/LDT and LC in the control of REM sleep has been challenged by studies in which lesions of these nuclei had surprisingly little effect on REM sleep. Electrolytic lesions of the PPT in cats mildly reduced the number of transitions into REM sleep, but lesions of the LDT and LC had no effect (Shouse and Siegel, 1992;Webster and Jones, 1988). More limited lesions of the PPT or LDT in rats have demonstrated minimal effects on REM sleep (Lu et al., 2006b). Even quite complete lesions of the LC using specific toxins showed no lasting changes in REM sleep (Blanco-Centurion et al., 2007;Lu et al., 2006b). Thus, the available evidence suggests that the cholinergic and monoaminergic systems are potent modulators of REM sleep, but are not likely to participate in its switching mechanism.
What then are the cell groups in the pons that regulate REM sleep? To identify populations of REM-promoting neurons, subsequent studies have examined Fos expression during periods of augmented REM sleep (Boissard et al., 2002;Lu et al., 2006b). While these studies have found relatively few Fos immunoreactive neurons in the PPT or LDT, Fos expression was elevated in three slightly more caudal cell groups: the sublaterodorsal nucleus (SLD), which is ventral and caudal to the LDT; the pre-coeruleus region (PC), which lies just dorsal to the SLD and caudal to the LDT; and the medial parabrachial nucleus (MPB) which is just dorsolateral to the SLD (Figure 2).
Figure 2. The REM-NREM sleep switch.
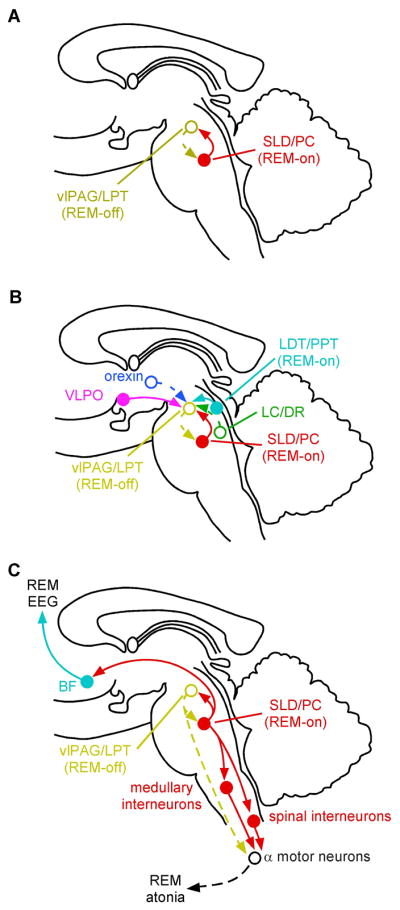
Two populations of mutually inhibitory neurons in the upper pons form a switch for controlling transitions between REM and NREM sleep (A). GABAergic neurons in the ventrolateral periaqueductal gray matter and the adjacent lateral pontine tegmentum (vlPAG/LPT; shown in gold) fire during non-REM states to inhibit entry into REM sleep. During REM sleep, they are inhibited by a population of GABAergic neurons in the sublaterodorsal region (SLD, red) that fire during REM sleep. This mutually inhibitory relationship produces a REM- NREM “flip-flop” switch, promoting rapid and complete transitions between these states. The core REM switch is in turn modulated by other neurotransmitter systems (B). Noradrenergic neurons in the LC and serotoninergic neurons in the DR (green) inhibit REM sleep by actions on both sides of the “flip-flop” switch (exciting REM-off and inhibiting REM-on neurons), and during REM sleep they are silent (dashed lines); whereas cholinergic neurons (aqua) promote REM sleep by having opposite actions on the same two neuronal populations. The orexin neurons inhibit entry into REM sleep by exciting neurons in the REM-off population (and by presynaptic effects that excite monoaminergic terminals), whereas the VLPO neurons promote the entry into REM sleep by inhibiting this same target. During REM sleep (C), a separate population of glutamatergic neurons in the SLD (red) activates a series of inhibitory interneurons in the medulla and spinal cord, which inhibit motor neurons, thus producing the atonia of REM sleep. Withdrawal of tonic excitatory input from the REM-off regions may also contribute to the loss of muscle tone. At the same time, ascending projections from glutamatergic neurons in the PB and PC activate forebrain pathways that drive EEG desynchronization and hippocampal theta rhythms, thus producing the characteristic EEG signs of REM sleep.
The role of the SLD in producing REM sleep has been studied by injecting it with bicuculline, a GABA antagonist, which disinhibits the SLD neurons and elicits REM sleep-like behavior (Boissard et al., 2002). Lesions in the SLD region of cats, also called the subcoeruleus area, have been known since the 1970’s to disrupt atonia during REM sleep such that animals appear to act out their dreams (Hendricks et al., 1982;Sastre and Jouvet, 1979;Shouse and Siegel, 1992). However, lesions of the SLD in rats have more profound effects, fragmenting and reducing the amount of REM sleep (Lu et al., 2006b).
Injections of retrograde tracers into the SLD identified major GABAergic inputs from the ventrolateral periaqueductal gray matter (vlPAG) and adjacent lateral pontine tegmentum (LPT) (Boissard et al., 2003;Lu et al., 2006b). This same region receives convergence of inputs from the extended VLPO and the orexin neurons in the lateral hypothalamus (Lu et al., 2006b). Because the extended VLPO neurons promote REM sleep but are inhibitory, and the orexin neurons prevent REM sleep and are excitatory, the vlPAG-LPT region would be expected to prevent REM sleep. As neurons in the vlPAG-LPT that project to the SLD are GABAergic, they would be expected to fire when REM sleep is inhibited (i.e., to show a REM-off firing pattern). Indeed, inhibition of the vlPAG and LPT with GABA agonists increases REM sleep (Crochet et al., 2006;Sapin et al., 2009;Sastre et al., 1996), and lesions increase REM sleep, particularly during the dark phase (Lu et al., 2006b).
Injections of retrograde tracers into the vlPAG and LPT demonstrate retrogradely labeled GABAergic neurons in the SLD, and anterogradely labeled axons from the SLD are found in close apposition to GABAergic neurons in the vlPAG and LPT (Lu et al., 2006b). These findings suggest that the vlPAG-LPT and the SLD have a mutually inhibitory relationship that may govern switching in and out of REM sleep, much like the relationship between the VLPO and the ascending arousal systems, which we hypothesize is the basis for switching between sleep and wake states.
Glutamatergic neurons that are mixed in with the REM-on GABAergic neurons give rise to long projections that activate the principle components of the REM state (Lu et al., 2006b;Luppi et al., 2004;Luppi et al., 2006;Shouse and Siegel, 1992;Webster and Jones, 1988). Many of these neurons in the SLD contain Fos protein during REM sleep, and they send descending projections to brainstem and spinal inhibitory systems that ultimately hyperpolarize motor neurons and cause atonia (Lu et al., 2006b;Luppi et al., 2004;Vetrivelan et al., 2009). In addition, mixed in with the REM-off GABAergic neurons is a REM-off glutamatergic population with spinal projections that may support motor tone during NREM sleep. Inhibition of these neurons during REM may withdraw motor tone, contributing to atonia in at least some motor neuron pools (Burgess et al., 2008). Other glutamatergic REM-on neurons in the parabrachial nucleus and precoeruleus area project to the forebrain and cause the EEG phenomena that characterize REM sleep (Lu et al., 2006b). Because these REM effector neurons are in isolated pools, they can be regulated independently. In a healthy brain this rarely occurs, but in the absence of sufficient inhibition from the orexin system, the components of the REM switch can become unstable and independent (see section on narcolepsy below).
As with the regulation of wakefulness, the lateral and posterior hypothalamus contains a large number of neurons that influence REM sleep. Neurons producing the peptide melanin-concentrating hormone (MCH) are mixed in with the orexin neurons and innervate many of the same targets. Interestingly, the MCH neurons fire mainly during REM sleep (Hassani et al., 2009;Verret et al., 2003). MCH inhibits target neurons, and many of the MCH neurons contain the inhibitory amino acid transmitter, GABA (Elias et al., 2001). This gives them the exact opposite activity profile and neurotransmitter action as the orexin neurons, inhibiting the same targets during sleep that the orexin neurons activate during wakefulness. Intraventricular injection of MCH increases REM sleep (Verret et al., 2003) and an MCH antagonist decreases REM sleep (Ahnaou, 08). Still, it remains unclear whether the MCH neurons are truly necessary for REM sleep as mice lacking MCH or the MCH1 receptor have no clear decrease in the daily amount of REM sleep (Adamantidis et al., 2008;Willie et al., 2008).
A “flip-flop” model of sleep state transitions
The “flip-flop” switch model
As outlined above, one of the most remarkable features of these state control systems is that both the wake- and sleep-promoting neurons, like the REM-on and REM-off neurons in the pons, appear to be mutually inhibitory. We propose that this mutually antagonistic relationship can give rise to behavior similar to that seen with a flip-flop switch (Saper et al., 2001; Mano and Kime, 2004). These types of switches are incorporated into electrical circuits to ensure rapid and complete state transitions. In the brain, because the neurons on each side of the circuit inhibit those on the other side, if either side obtains a small advantage over the other, it turns the neurons off on the other side, thus causing a rapid collapse in activity and a switch in state. Although an electronic flip-flop switch contains a single element on either side and acts almost instantly, in the brain the mutual antagonism is between large populations of neurons, numbering in the thousands on each side, which may also be responding to other inputs. As a result, the transitions occur over seconds to minutes (depending upon the species being studied), but result in clearcut changes in behavioral and EEG states. Recordings in a wide range of species show that the transitions typically take less than 1% of bout length (Takahashi et al., 2010;Wright et al., 1995). Once a state boundary is crossed, the firing of the counterpoised population is suppressed. In practical terms, this should produce stable wake and sleep, preventing an individual from falling asleep during a boring activity, or waking up during the night with every small sound in the house.
Although the concept of mutual inhibition causing relatively rapid and complete state transitions is analogous to an electronic flip-flop switch in some ways, the changes in behavioral state are not instantaneous and generally take place over a few seconds in rodents or a few minutes in humans. Individual neurons in the VLPO, LC, and TMN of rodents change their firing rates over less than a second when transitioning from wake to NREM, or from NREM to wake (Takahashi et al., 2006;Takahashi et al., 2009;Takahashi et al., 2010) (Figure 3), but not all of the neurons in a population will switch at the same instant. Thousands of sleep- and wake-promoting cells must shift their activity, and the emergent behavioral state most likely reflects the summated activity across all these neurons. The time it takes for one population of neurons to overcome the resistance of the other population and the stability of the state once that transition point is crossed may vary with the size and complexity of the brain. This may explain why bout durations and transition state durations vary in a similar proportion across a wide range of mammals (Lo et al., 2004;Phillips et al., 2010) On the other hand, the rate of change in firing of the two populations is maximal near the inflection point (the half-way point in the transition), so that the behavioral state changes often appear to occur rather rapidly.
Figure 3. Reciprocol firing patterns between sleep-promoting neurons in the preoptic area and wake-promoting neurons in the LC, TMN, and basal forebrain.

The top panel shows changes in firing rate during the transition from NREM sleep to wake and the bottom panel shows firing rates during the transition from wake into light NREM sleep. Note that the firing rates of some cell groups, such as the LC, begin to increase or decrease 1–2 sec in advance of awakening or falling asleep, suggesting that they may help drive the transition. In contrast, neurons in the TMN begin to fire only after the transition to wake, suggesting these cells may play more of a role in maintenance of wakefulness. Recordings were made in unanesthetized, head-restrained mice. Adapted with permission from (Takahashi et al., 2010).
The REM-off and REM-on neuronal populations in the mesopontine tegmentum are also configured in a mutually inhibitory circuit (Lu et al., 2006b;Luppi et al., 2004;Luppi et al., 2006;Sapin et al., 2009;Sastre et al., 1996;Verret et al., 2005). Each population is a mixture of both GABAergic neurons and glutamatergic neurons. The GABAergic neurons in each cluster innervate and inhibit both the GABAergic and glutamatergic neurons in the other side of the switch. The result is that transitions into and out of REM sleep are rapid and complete. As would be predicted from this arrangement, lesions of either the REM-on or REM-off population respectively reduce or increase the time spent in REM sleep, but both NREM and REM sleep become fragmented.
Mathematical modeling of the “flip-flop” switch hypothesis
Mathematical modeling of these mutually inhibitory circuits can generate simulated sleep/wake behavior with temporal properties very similar to those seen in natural sleep-wake transitions. Phillips and Robinson (Phillips and Robinson, 2007) used a model based upon the mutual inhibitory interactions of the VLPO and the monoaminergic systems, and included both homeostatic and circadian influences (see next section). By varying the time constant for the buildup of homeostatic sleep drive and the mean drive to the VLPO, they could produce sleep patterns that mimicked those seen in a wide range of mammals, from rodents to humans (Phillips et al., 2010). The same group also modeled the effects of sleep deprivation and produced estimates of sleep debt and recovery in good agreement with experimental data (Phillips and Robinson, 2008). They then added an arousing stimulus to their model, in the form of a simulated auditory tone that provided a sensory input to activate the monoaminergic systems (Fulcher et al., 2008). Their modeling of arousal threshold and its variation across the night closely approximates responses seen in clinical studies.
Rempe and colleagues used coupled oscillator equations to implement a similar model that also incorporated both the wake-sleep and REM-NREM circuitry, and integrated them with models of circadian and homeostatic influences (Rempe et al., 2009). Their model produced simulated behavior that agreed well with experimental data in intact individuals and demonstrated increased sleep and wake fragmentation in individuals with loss of orexin neurons, as is seen in narcolepsy (see final section). Diniz Behn and colleagues also used coupled oscillator equations to incorporate the influence of the orexin neurons into the “flip-flop” switch model, showing how these neurons stabilize behavioral state by prolonging the duration of both waking and sleeping bouts (Diniz Behn et al., 2008). They have also been able to use this model to reproduce accurately the effects of pharmacological agents on sleep and wakefulness (Diniz Behn and Booth, 2010).
Flip-flop models for neuronal circuitry have recently been proposed to explain rapid and complete state transitions in a variety of functions as diverse as alternating zigzag turns in silkworm moths; visual perceptual rivalry in the brains of primates; and Parkinsonian tremor in humans (Burne, 1987;Iwano et al., 2010;Lankheet, 2006). In fact, mutually inhibitory relationships may be a common motif in a wide variety of neural circuits that require rapid and complete state transitions. This property is critical for wake-sleep circuitry because, as we will discuss below, homeostatic and circadian drives for sleep and wake accumulate slowly over many hours. In the absence of a switching mechanism, an individual would drift slowly back and forth between sleep and wakefulness over the course of the day, spending much of the time somewhere in between in a twilight state. Clearly, a half-asleep state would be a liability in finding food or avoiding predation. When conditions of external threat demand sudden state changes (an allostatic input, see next section), the flip-flop mechanism ensures that the transition is accomplished rapidly.
Experimental evidence bearing on the “flip-flop” model derives from three lines of work. First, direct recordings from neurons in the cell groups that constitute the model show that their behavior is very close to what the model would predict. Recordings from VLPO neurons in both rats and mice show a sharp increase in firing just before or at the transition from waking to NREM sleep, and a sharp decrease in firing just before the transition from NREM or REM to waking (Szymusiak et al., 1998; Takahashi et al., 2009). Individual VLPO neurons differ some in their onset of firing relative to the onset of NREM sleep presumably because the individual cells differ slightly in their inputs and responses. A neural network model of these neurons permitted the 2000 neurons on each side of the switch to have independent behavior, and this arrangement demonstrated a similar variability in the onset of firing compared to the actual state transition (Chou, 2003). A key feature in both the modeled neuronal behavior and the actual recordings was the bistable nature of the firing, with abrupt transitions between rapid and slow firing right around the actual state transitions. Another interesting aspect of this system is the time relationship between changes in VLPO neuron firing and cortical activity. The onset of firing began about 200 msec before the EEG synchronization and did not reach a peak until 300 msec after the transition, whereas the fall in firing occurred over about 200 msec beginning just before the loss of EEG synchronization (Takahashi et al., 2009). The neural network model (Chou, 2003) predicts this behavior, and suggests that it underlies the hysteresis in the response of the brain to homeostatic sleep drive, as suggested by Borbely and Achermann (Borbely and Achermann, 1999). Thus the threshold at which homeostatic drive triggers sleep is higher than the threshold at which falling homeostatic sleep drive terminates sleep. This property may arise from a key aspect of the mutually inhibitory sleep-wake circuitry: sleep-promoting VLPO neurons can only be activated during wakefulness by stimuli that overcome their inhibition by wake-promoting neurons, but during sleep, when VLPO neurons are not inhibited by wake-promoting neurons, they can be activated by relatively weak stimuli such as low levels of homeostatic sleep drive.
The activity of LC and TMN neurons also anticipates state transitions. The firing of LC neurons slows many seconds before sleep onset, and then gradually increases 1–2 seconds prior to wake onset (Aston-Jones and Bloom, 1981;Takahashi et al., 2010). The firing of TMN neurons also slows about 1 sec prior to EEG signs of NREM sleep, but unlike the LC, TMN neurons only start to fire about 1 second after wake is established (Takahashi et al., 2006). These observations suggest that the inputs to these different populations of wake- and sleep-promoting neurons are distinct, and that each may contribute differentially at distinct time points during state transitions. Similar recordings from the MnPO sleep-related neurons would be of great interest in this context, as the Fos studies suggest that they might fire with buildup of homeostatic sleep drive, a property that VLPO neurons lack (Gong et al., 2004).
Second, lesions of sleep- and wake-regulating cell groups produce alterations in wake and sleep that are generally consistent with the “flip-flop” model. Lesions of the VLPO not only reduce the amount of time spent asleep, but also reduce the stability in both sleep and wake, resulting in more frequent transitions (Lu et al., 2000). Similarly, lesions of REM-off population in the ventrolateral periaqueductal gray matter also produce not only increased REM sleep, but also fragmentation of sleep (Kaur et al., 2009;Lu et al., 2006b), and lesions of the REM-on neurons in the SLD cause decreased and fragmented REM sleep, as the “flip-flop” model predicts.
Interestingly, lesions of monoaminergic or cholinergic cell groups on the arousal side of the switch, either alone or in combination, have been far less effective, either at causing a change in overall amounts of sleep, or in sleep-wake fragmentation (Blanco-Centurion et al., 2007;Lu et al., 2006b). On the other hand, the effects on wake and sleep were measured after recovery from the lesions, which may have permitted surviving neuronal systems to compensate for the loss of the injured components (e.g., upregulation of receptors for other wake-promoting neurotransmitters). However, the prominent loss of sleep and increase in sleep fragmentation, which lasts for months after VLPO lesions (Lu et al., 2000), suggests that the VLPO neurons represent a central and irreplaceable component of the sleep-promoting system.
Finally, state space analysis of the EEG power spectrum has recently been used to map the dynamic changes in behavioral states over time (Figure 4) (Diniz Behn et al., 2010). This method uses principal components analysis of the EEG to generate a “state space” map (Gervasoni et al., 2004), in which wake, REM, and NREM sleep are reflected as three clusters of points. This analysis allows the examination of second-by-second variations in sleep and wakefulness as shown by a moving point traveling from one state cluster to another as the animal’s EEG characteristics shift. This approach shows that within states such as wake or NREM sleep, the EEG changes fairly slowly over time, but during transitions between states, the EEG rapidly switches into a new pattern. This property underscores the relatively rapid changes in neural activity that occur at the boundaries between states, as predicted by the “flip-flop” model.
Figure 4. Space state analysis of the EEG enables visualization of distinct sleep/wake states and switching between states.

By segmenting the EEG power spectrum into ranges, and comparing ratios of EEG power in different ranges, it is possible to produce graphs that separate the different wake-sleep states spatially. Higher values on the X-axis represent greater amounts of theta activity (6.5 to 9 Hz) which is characteristic of active wake and REM sleep. Higher values on the Y-axis reflect greater amounts of slow EEG activity as is characteristic of NREM sleep. In these graphs, the ratios of power in different EEG bands are computed for each second of EEG activity, which is plotted as a separate point. The EEG and EMG are then scored by experimenters, who designate each second as wake (blue), NREM sleep (red), REM sleep (green), or cataplexy (magenta). In a wild type mouse (A), the clusters of EEG activity associated with each state are distinct, but in a narcoleptic mouse lacking orexin peptides (B), the wake and NREM sleep clusters are closer together with more periods spent in the region between these states. Panel C shows the average densities of EEG activity as a function of low frequency EEG power (a projection of the cluster plots above into the Y axis) in groups of wild type mice (n=6) and orexin knockout mice (n=7). Wild type mice (blue line) spend most of their time (high densities) in well-defined wake and NREM sleep, but orexin knockout mice (green line) spend more time in the transitional region between wake and NREM sleep, in part due to the greater number of state transitions in these mice.
The hands on the switch: Homeostatic, circadian, and allostatic influences on sleep and wakefulness
Changes in internal physiology and the external environment influence transitions from one behavioral state to another. Over time, these forces may change slowly, but as noted previously, the shifts in behavioral state are relatively rapid and complete. It is the job of the switching mechanisms we have described to respond to these slowly accumulating influences, integrate them over time, and convert their influence into sharp transitions in behavioral state. The neurons that regulate switching between behavioral states receive inputs from a wide range of different sources (e.g., see (Chou et al., 2002;Yoshida et al., 2006), and the circuitry that mediates specific types of influences on state transitions will be reviewed briefly.
One of the most widely recognized properties of NREM and REM sleep is that they are homeostatically regulated (Achermann and Borbely, 2003;Borbely and Tobler, 1985). In other words, if an individual is deprived of sleep for some period of time, there will be a subsequent increase in the amount of sleep to compensate. However, the neurochemical factors and neuronal mechanisms that drive these homeostatic responses are the subject of ongoing and intense investigation.
Over one hundred years ago, Pieron and Ishimori independently discovered that the cerebrospinal fluid of sleep-deprived dogs contains a sleep-promoting factor (Ishimori, 1909;Legendre and Pieron, 1913). Much recent work has focused on adenosine, which may accumulate extracellularly as a rundown product of cellular metabolism at least in some parts of the brain (Benington and Heller, 1995;Huang et al., 2005;Porkka-Heiskanen et al., 1997;Radulovacki et al., 1984;Strecker et al., 2000). Astrocytes are the main site of energy storage in the brain, in the form of glycogen granules that are depleted during prolonged waking (Kong et al., 2002). As these energy stores run down, astrocytes may cause an increase in extracellular adenosine that then promotes sleep. This phenomenon was nicely demonstrated in a recent study in which genetic deletion that blocked the rise in adenosine mediated by astrocytes prevented rebound recovery sleep after sleep deprivation (Halassa et al., 2009).
There are two major classes of adenosine receptors in the brain. Adenosine A1 receptors are predominantly inhibitory, while A2a receptors are excitatory. Signaling through A1 receptors, which are diffusely distributed in the brain, may directly inhibit neurons in arousal systems such as the locus coeruleus, tuberomammillary nucleus, and orexin neurons via the A1 receptor (Liu and Gao, 2007;Oishi et al., 2008;Pan et al., 1995; Strecker et al., 2000). On the other hand, A2a receptors are highly enriched in the striatum and in the meningeal cells underlying the VLPO (Svenningsson et al., 1997). We focus here on the A2a receptors near the VLPO, although it is possible that A2a receptors in the striatum, or at other sites not yet known to play a role in sleep state switching, may also be involved (Qiu et al., 2010). Application of an A2a agonist to the subarachnoid space underlying the VLPO causes sleep and induces Fos in the VLPO and the underlying meninges (Scammell et al., 2001a). In addition, the wake-promoting drug caffeine is believed to act through blockade of A2a receptors, as caffeine loses its wake-promoting effect in A2a receptor knockout mice (Huang et al., 2005). Given the absence of A2a mRNA in the VLPO, these findings suggest that adenosine may cause the meningeal cells to produce a second messenger that activates the VLPO. Whole cell patch clamp studies of the effects of adenosine on VLPO neurons in hypothalamic slices have produced conflicting results. Two studies using patch clamp intracellular recordings reported that adenosine disinhibited VLPO neurons by reducing presynaptic inhibitory inputs (Chamberlin et al., 2003;Morairty et al., 2004;Strecker et al., 2000), but another study using extracellular recordings found that adenosine reduced firing of VLPO neurons via a direct A1 effect, but increased it via an A2a effect (Gallopin et al., 2005). It is not known whether these hypothalamic slices may have retained the basal meninges, but future work should probably make note of this. These effects of adenosine on VLPO neurons may bias the switch toward increased activity, and thus increase the likelihood of it flipping into a sleep state. Models of the “flip-flop” switch under conditions of high sleep pressure on the VLPO, indicate that it may become more unstable (Fulcher et al., 2010), thus perhaps accounting for microsleep episodes and lapses in attention seen in human subjects during sleep deprivation (Van Dongen et al., 2003). Still, it is unlikely that adenosine alone can explain the homeostatic drive for sleep, and much ongoing work focuses on additional sleep-promoting factors (Krueger, 2008).
Regardless of what constitutes the homeostatic sleep factors, there is much evidence that prolonged wakefulness results in more intense slow waves in the EEG during NREM sleep and that these decrease over the sleep period (Achermann and Borbely, 2003). This relationship suggests that the slow wave activity is homeostatically controlled and reflects sleep drive (Vyazovskiy et al., 2009). Slow waves during NREM sleep represent the summation of synaptic potentials onto cortical neurons, which are hyperpolarized and silent (in a down state) during the troughs of the waves and fire bursts of action potentials (in an up state) during the peaks. The duration and frequency of the down periods correlates strongly with the intensity of slow wave activity during spontaneous sleep and recovery sleep. Prolonged wakefulness increases the firing rates of cortical neurons (Vyazovskiy et al., 2009), and cortical areas that recently have been especially active have local increases in slow waves during subsequent NREM sleep, suggesting that the slow wave activity may be homeostatically driven (Huber et al., 2004;Vyazovskiy et al., 2000). It has been proposed that the slow wave activity may reflect synaptic reorganization during sleep in response to recent activity (Vyazovskiy et al., 2009), but it is also possible that the increased metabolic activity may elevate levels of adenosine and other sleep promoting factors that drive slow wave activity (Bjorness et al., 2009;Halassa et al., 2009).
Recordings from VLPO neurons across the wake-sleep cycle show that their firing rates increase with EEG slow waves (Szymusiak et al., 1998). During recovery sleep after twelve hours of sleep deprivation, the amount of slow waves and the firing of VLPO neurons both approximately double. On the other hand, the firing of VLPO neurons does not increase during prolonged wakefulness. Thus as homeostatic sleep drive accumulates, it may influence other neurons in the brain, such as the median preoptic neurons which provide input to the VLPO (Chou et al., 2002;Gvilia et al., 2006), but VLPO neurons do not fire until the state transition itself (Takahashi et al., 2009). This fundamental property of VLPO neurons is consistent with their role in causing rapid and complete state transitions.
A second major influence on sleep state switching is the input from the circadian system (Achermann and Borbely, 2003;Borbely and Tobler, 1985). In mammals, daily rhythms are driven by the suprachiasmatic nucleus (SCN) in the hypothalamus, a key pacemaker that influences the timing of a wide range of behaviors and physiological events. SCN neurons are intrinsically rhythmic and drive behavioral responses with a roughly 24 hour period, even in complete darkness. This rhythmicity is generated by a network of transcriptional/translational/post-translational feedback loops that regulate the expression of clock genes (Jin et al., 1999;Reppert and Weaver, 2002). The clock genes are themselves transcription factors that regulate the expression of hundreds if not thousands of other genes. The activity of the SCN is entrained to the daily light-dark cycle by inputs from intrinsically photosensitive retinal ganglion cells that express the photopigment melanopsin (Gooley et al., 2001;Hattar et al., 2002). Lesions of the SCN, or disruption of expression of key clock genes, results in loss of most circadian rhythms (Bunger et al., 2000;Edgar et al., 1993;Moore and Eichler, 1972).
Surprisingly, the SCN has very little direct output to either the wake or sleep regulatory systems (Watts et al., 1987). Instead, the bulk of its projections run into the subparaventricular zone, a region just dorsal and caudal to the SCN. Cell body-specific lesions of the ventral subparaventricular zone nearly eliminate the circadian rhythms of sleep and wakefulness, suggesting that neurons in this region are necessary for conveying these output signals (Lu et al., 2001). However, the ventral subparaventricular neurons have few direct outputs to either wake or sleep networks. Instead, they send axons to the dorsomedial nucleus of the hypothalamus (Chou et al., 2003;Deurveilher and Semba, 2005). The dorsomedial nucleus contains GABAergic neurons that heavily innervate the VLPO and glutamatergic neurons that innervate the lateral hypothalamic area, including the orexin neurons (Chou et al., 2003;Thompson et al., 1997). Cell body-specific lesions of the dorsomedial nucleus nearly eliminate the circadian rhythms of sleep and wakefulness, as well as feeding, locomotor activity, and corticosteroid secretion (although some rhythms, such as body temperature and melatonin secretion persist because they take a different pathway) (Chou et al., 2003;Saper et al., 2005). This three-stage pathway from the SCN to the subparaventricular zone and then to the dorsomedial nucleus appears necessary for conveying circadian information to the neurons that control wake-sleep state switching, yet it still allows some flexibility for altering the timing of sleep and wakefulness depending upon seasonal changes and the timing of food availability (Fuller et al., 2008;Gooley et al., 2006;Mieda et al., 2006).
In the absence of the dorsomedial nucleus, wake-sleep cycles become ultradian, with 7–8 sleep-wake cycles per day. In mice that are arrhythmic due to clock gene deletions, activity patterns likewise become ultradian (Bunger et al., 2000). However, there is a paucity of information concerning whether the wake-sleep cycles of individual animals become ultradian as well because the few reports on sleep behavior in such mice provide only graphs that summate across groups of animals, which obscures whether ultradian cycles (which are not synchronized across animals) were present (Laposky et al., 2005;Wisor et al., 2002).
Like lesions of the SCN in primates, lesions of the dorsomedial nucleus in rats, or deletions of certain clock genes (such as cryptochromes 1 and 2 or Bmal1) which cause loss of circadian cycling of the SCN in mice reduce the total amount of wakefulness (Chou et al., 2003;Edgar et al., 1993;Laposky et al., 2005;Wisor et al., 2002). These observations suggest that the circadian system mainly promotes wakefulness during the active period, which is consistent with the main outputs of the dorsomedial nucleus being to inhibit the VLPO and excite lateral hypothalamic neurons.
Finally, animals often encounter conditions in their environment that require urgent alterations of specific physiological responses, including wake-sleep states. These would include stressful situations, such as confronting a predator or a hostile conspecific, but also situations such as encountering a potential mate, seasonal changes, or the need for migration that may require an adjustment of wake-sleep behavior (Palchykova et al., 2003;Rattenborg et al., 2004). These situations have been called allostatic loads by McEwen and colleagues (McEwen, 2000), and they require additional circuitry for modifying wake-sleep cycles.
One common stressor in the wild is a lack of food, and in small animals that can carry minimal energy reserves the effects of food deprivation on sleep are dramatic. Food deprived mice have marked increases in wakefulness and locomotor activity, probably reflecting a strong drive to forage for food. However, mice lacking the orexin-producing neurons show very little arousal or increase in locomotion when food deprived, suggesting that these cells are required for the arousing effects of hunger (Yamanaka et al., 2003). In contrast, mice lacking MCH show just the opposite response to food deprivation, with exaggerated increases in locomotion, more wakefulness, and much less REM sleep than normal mice (Willie et al., 2008). Most likely, both the orexin and MCH neurons respond to the stress of insufficient food but with quite opposite effects on sleep/wake pathways.
Another common allostatic load is behavioral stress, which frequently causes insomnia. For example, mice exposed to foot shock or restraint stress have increased levels of CRF that may cause arousal by exciting the orexin neurons through CRF-R1 receptors (Winsky-Sommerer et al., 2005). In another study, Cano and colleagues examined stress-induced insomnia by placing a male rat early in the sleep period into a cage previously occupied by another male rat (Cano et al., 2008). The stressed rat took twice as long to fall asleep as control animals placed into a clean cage, and then had disturbed sleep for the remainder of the next six hours, sleeping only about 50% of the time (instead of the usual 70–80%) of the fifth and sixth hours after cage exchange. At the end of this period, the insomniac animals expressed Fos in a surprising pattern: both the VLPO and some of the arousal systems (LC and TMN) were active. This dual activation of both the wake and sleep circuitry suggests that the VLPO was activated by both homeostatic and circadian sleep drives, while the LC and tuberomammillary nuclei were driven by the allostatic stress. Thus stress-induced insomnia may represent an unusual state in which neither side of the wake- and sleep-regulating circuitry is able to overcome the other, due to both receiving strong excitatory stimuli.
These stressed animals also expressed Fos in the infralimbic cortex, the central nucleus of the amygdala, and the bed nucleus of the stria terminalis (Cano et al., 2008). These corticolimbic sites project to the LC and TMN, as well as the areas in the upper pons that regulate REM sleep switching (Dong et al., 2001;Hurley et al., 1991;Van Bockstaele et al., 1999). The infralimbic cortex also provides a major input to the VLPO (Chou et al., 2002). These inputs may be important in maintaining a waking state during periods of high behavioral arousal, such as an emergency that occurs during the normal sleep period. Their activation by residual stress or anxiety may contribute to inability to sleep in stress-induced insomnia. Lesions of the infralimbic cortex reduce Fos expression in the LC and the TMN, and restore NREM but not REM sleep in animals with experimental stress-induced insomnia (Cano et al., 2008). Lesions of the extended amygdala, including the bed nucleus of the stria terminalis, also quieted both arousal systems, as well as the infralimbic cortex, and restored both REM and NREM sleep. Thus, it appears that the activation of medial prefrontal and amygdaloid circuitry can drive arousal circuitry even in the face of VLPO activity (a state that has been called hyperarousal). These stressed rats also had excessive high frequency EEG activity during NREM sleep, consistent with cortical activation, even during periods of slow wave generation. Increased activation of these corticolimbic sites has also been demonstrated in human subjects with insomnia (Nofzinger et al., 2004). This co-activation may also contribute to the excessive high frequency EEG activity seen during NREM sleep in people with insomnia and the sensation in some insomniacs that they are awake even when the EEG appears to be NREM sleep, a condition known as “sleep state misperception”.
One common feature of stressful situations is the need to maintain vigilance. The medial prefrontal cortex, in addition to providing the major cortical source of inputs to the wake-sleep system, plays a critical role in determining which stimuli in the environment require attention (Aston-Jones and Cohen, 2005). It therefore may provide an important wake-promoting input when attention to the environment is required. In humans who have been sleep deprived, there is decreased activation of the medial prefrontal cortex during tasks that require sustained attention (Chee and Choo, 2004;Chuah et al., 2006). Thus, excessive homeostatic drive for sleep may ultimately degrade the ability of the allostatic system to maintain wakefulness
Switches gone bad: The example of narcolepsy
The common sleep disorder narcolepsy is an excellent clinical example of how sleep switches can become destabilized by loss of a single component of the sleep/wake circuitry. Narcolepsy was first described over 125 years ago (Gelineau, 1880;Westphal, 1877), but only in the last 10 years has the underlying neurobiology become clear. People with narcolepsy often have severe sleepiness that makes it a struggle to stay awake at school or remain alert while driving. In addition, these individuals frequently have what appear to be fragments of REM sleep that intrude into wakefulness: vivid, often frightening dream-like hallucinations as they drift off to sleep; and cataplexy, brief episodes of muscle paralysis triggered by strong emotions. These symptoms typically begin abruptly in the teens or young adulthood and then persist for life. For years, researchers suspected that narcolepsy was caused by some dysfunction of the hypothalamus or REM sleep-regulating pathways, but the fundamental neuropathology had remained a mystery until the last few years.
Substantial research has now established that narcolepsy is caused by a selective loss of orexin signaling in the brain. This connection was first made in 1999, when Lin and colleagues found that dogs with inherited narcolepsy had an exon-skipping mutation in the type 2 orexin receptor gene (Lin et al., 1999). At the same time, Chemelli and colleagues reported that mice with a deletion of the gene coding for the orexin peptides displayed severe sleepiness and cataplexy-like events (Chemelli et al., 1999). The following year, two groups of investigators found that patients who had narcolepsy with cataplexy had a 90% or greater loss of the orexin-producing neurons (Peyron et al., 2000;Thannickal et al., 2000). This finding was quite selective, as the MCH neurons, which are intermingled with the orexin cells, were completely spared, and it probably represented cell loss rather than downregulation of orexin expression, as there was concomitant loss of other markers (dynorphin and neuronal activity-related pentraxin) of the orexin cell population (Crocker et al., 2005). The loss of orexins is not due to a simple genetic abnormality, as orexin deficiency is acquired during young adulthood and the vast majority of people with narcolepsy do not have mutations of the genes encoding the orexin peptides or their OX1 or OX2 receptors (Olafsdottir et al., 2001;Peyron et al., 2000). However, because about 90% of people with narcolepsy have human leukocyte antigen DQB1*0602 (Mignot et al., 2001), researchers have hypothesized that the loss of orexin neurons may be immune-mediated (Lim and Scammell, 2010;Scammell, 2006) It has recently been proposed that at least in some individuals, an autoimmune attack on the orexin neurons may be related to antibodies to Tribbles homolog-2, a protein produced by the orexin neurons and other cells in the brain (Cvetkovic-Lopes et al., 2010;Kawashima et al., 2010).
Several models have been proposed to explain how loss of the orexin neurons results in severe sleepiness. One popular hypothesis is that individuals with narcolepsy may be more sensitive to homeostatic sleep drive as, after a period of sleep deprivation, they fall asleep faster than normal (Tafti et al., 1992a;Tafti et al., 1992b). Mice lacking orexins also tend to fall asleep very quickly after being deprived of sleep, but they recover the lost sleep at a normal rate and to the same extent as wild type mice (Mochizuki et al., 2004). Thus, orexin deficiency hastens the transition to sleep, but the accumulation and expression of homeostatic sleep drive appears normal in mice and people with narcolepsy (Khatami et al., 2008;Mochizuki et al., 2004). Another potential explanation is that circadian waking drive is impaired in narcolepsy. However, this too seems unlikely as mice lacking orexins have normal circadian rhythms of wake and NREM sleep when housed in constant darkness (Kantor et al., 2009;Mochizuki et al., 2004).
A better explanation may be that impaired orexin signaling causes behavioral states to become unstable (Figure 5). In fact, this idea was first raised by Broughton over 20 years ago as narcoleptic people and animals have great difficulty remaining awake, but they also have fragmented sleep and many more transitions between all states (Broughton et al. 1986).
Figure 5. Summary of the cascading wake-sleep and REM-NREM “flip-flop” switches, and how they are both stabilized by orexin neurons.

The populations of wake- and sleep-promoting neurons are shown as components of a counterpoised switch at the upper left, and the REM-on and REM-off populations at the lower right. (Red arrows indicate inhibitory projections, green arrows excitatory ones.) The monoaminergic arousal neurons that inhibit the VLPO during wakefulness also inhibit the REM-on and excite the REM-off neurons in the REM switch, thus making it nearly impossible for normal individuals to transition directly from wakefulness to a REM state. On the other hand, when there is loss of orexin signaling in narcolepsy, both switches become destabilized, and their normal cascading relationship is disrupted, so that it is possible for individuals with narcolepsy to enter fragmentary components of REM sleep (cataplexy, sleep paralysis, hypnagogic hallucinations) directly from the waking state. The clinical phenomena encountered in narcolepsy when each population of wake-, sleep-, or REM-promoting neurons fires at the wrong time is identified in parentheses.
This breakdown in the ability to produce cohesive wake and sleep states is consistent with a destabilized switching mechanism. Across 24 hours, people and animals with narcolepsy have essentially normal amounts of wake and sleep, but they have many more transitions between states. Under normal conditions, the sleep/wake switch resists switching until a sufficiently strong stimulus such as homeostatic sleep drive accumulates to a critical level. In contrast, most individuals with narcolepsy can rapidly doze off at any time of day, especially when they are sedentary. Narcoleptic mice also transition quickly and frequently from well-established wake into NREM sleep (Diniz Behn et al., 2010;Kantor et al., 2009;Mochizuki et al., 2004).
At the same time, because orexins activate REM sleep-suppressing neurons, loss of the orexin neurons permits more frequent transitions into REM sleep. Patients may enter REM sleep after only brief periods of NREM sleep, and REM sleep can occur at any time of day (Dantz et al., 1994;Rechtschaffen et al., 1963). In addition, people and animals with narcolepsy often enter into partial REM sleep-like states, such as cataplexy, in which strong, generally positive emotions activate the REM sleep atonia pathways in the midst of wakefulness. At other times, the atonia of REM sleep can persist for a minute or two upon awakening (sleep paralysis) or vivid, dream-like hypnagogic hallucinations can occur around the onset of sleep. These phenomena rarely occur in healthy, well-rested individuals because the orexin neurons reinforce the activity of the monoaminergic neurons in the LC and dorsal raphe nucleus (Bourgin et al., 2000;Kohlmeier et al., 2008), which in turn activate REM-off neurons and inhibit REM-on neurons, thus locking the individual out of REM sleep and its component behaviors during wakefulness. We propose that these frequent transitions between states, odd mixtures of states, and poor control of REM sleep are consistent with destabilization of the “flip-flop” switches that regulate REM-NREM and wake-sleep transitions due to the loss of orexin signaling.
Several lines of research are now beginning to identify how orexin deficiency destabilizes the wake-sleep switch. In normal animals, the orexin neurons are active during wake, especially during active exploration of the environment (Estabrooke et al., 2001;Lee et al., 2005;Mileykovskiy et al., 2005), and they provide excitatory tone to wake-promoting monoaminergic and cholinergic cell groups. In the absence of this activity, these key arousal systems may have reduced or inconsistent activity which would manifest as sleepiness and frequent transitions into sleep. Orexins have no direct effects on VLPO neurons, but may increase presynaptic inhibition of VLPO neurons (Eggermann et al., 2001;Methippara et al., 2000). Both of these mechanisms are supported by mathematical modeling (Diniz Behn et al., 2008;Rempe et al., 2009). Thus, reduced activity in wake-promoting neurons and less inhibition of the VLPO could destabilize the sleep/wake switch.
Cataplexy is probably due to transient activation of the REM sleep atonia pathways during wakefulness, but how these events are triggered by strong positive emotions, such as laughter and joking, remains a mystery. Feeling weak with laughter is common in many cultures, and even in normal individuals laughter briefly reduces muscle tone (Overeem et al., 1999). However, in people with narcolepsy, positive emotions can trigger partial or generalized atonia. In fact, as cataplexy develops, people often have intermittent lapses in tone that then develop into sustained paralysis lasting a minute or two. This intermittent atonia strongly suggests instability in the brainstem switch controlling atonia. As suggested by Nishino, it seems likely that cataplexy is caused by “increased sensitivity in the pathways that link emotional input and spinal motor inhibition” (Nishino et al., 2000). The “flip-flop” switch model predicts that in normal individuals, even though laughter may inhibit the motor tone-producing system, orexins may prevent transitions into full atonia. In the absence of orexins, these emotionally triggered signals may be unopposed, permitting full activation of the atonia pathways.
In this model, orexins may act through several pathways to inhibit cataplexy. First, during cataplexy, LC and dorsal raphe neurons are essentially silent, just as in REM sleep (Wu et al., 1999;Wu et al., 2004). However, the histaminergic neurons of the TMN remain active, possibly accounting for the preservation of consciousness during this state (John et al., 2004). Orexins excite neurons of the LC and dorsal raphe nucleus(Brown et al., 2001;Hagan et al., 1999), and drugs that increase noradrenergic or serotoninergic tone suppress cataplexy (Nishino and Mignot, 1997). Thus, enhancement of monoaminergic tone by orexins may directly increase the activity of motor neurons and inhibit brainstem atonia mechanisms. In addition, orexins may directly and indirectly excite bulbar and spinal motor neurons probably via OX2 receptors (Fung et al., 2001;Greco and Shiromani, 2001;Peever et al., 2003;Volgin et al., 2002;Yamuy et al., 2004).
Looking forward to sleep
We have reviewed some of the current thinking on the regulation of sleep and wakefulness, and how this might be influenced by mutually inhibitory circuitry functioning analogous to electronic flip-flop switches. We recognize that this is a working model that has stimulated active debate and that there are alternative models of sleep state switching (e.g., the Hobson-McCarley model of REM state switching, as discussed above). However, we expect that ongoing and future experimental tests of the model will help resolve the many important questions remain to be addressed. For example, both wake and sleep may be governed by additional brain regions not yet identified, as lesions of the cholinergic or monoaminergic neurons in the brainstem, hypothalamus, or basal forebrain have only minimal effects on the total amounts of sleep or wakefulness. Even lesions of several of these cell groups in combination have only slightly greater effects (Blanco-Centurion et al., 2007). Similarly, even very large and complete lesions of the VLPO at most reduce sleep by about 50%, suggesting that there are other pathways capable of inhibiting the ascending arousal system in the absence of VLPO neurons. Thus, there are likely to be components of our proposed wake-sleep “flip-flop” switch that are as yet unknown, and evidence in support of this switch and its full understanding will require a more complete knowledge of its components. In addition, it will be important to gain a more complete understanding of the interactions of these components of both the proposed wake-sleep and NREM-REM sleep switches, and to test these circuit models rigorously. One challenge in dissecting these circuits and testing their functions is that wake-promoting and sleep-promoting cell populations may overlap spatially in some locations. Fortunately, these neurons have different neurotransmitters and connections, which allow us to introduce novel genetic and transneuronal methods for manipulating them independently. Approaches might include testing the functional roles of these components using optogenetic or perhaps genetically driven, ligand-gated channels such as ivermectin-activated chloride channels (Adamantidis et al., 2007;Lynagh and Lynch, 2010). It would be particularly valuable to record simultaneously from neurons in multiple components of a putative switch, while driving one component, and to examine their firing patterns across wake-sleep or NREM-REM sleep transitions.
We also know little about the mechanisms by which homeostatic sleep drive is either accumulated or discharged. Current evidence points to a role of adenosine, but it is unlikely that this explains the entire process, as adenosine levels in many parts of the brain do not increase with prolonged wakefulness (Strecker et al., 2000) and homeostatic sleep drive persists even in the absence of adenosine receptors. It remains possible that slower, progressive changes as animals rest before sleep may contribute to modulating the “flip-flop” switch, and are not necessarily in conflict with this model. Multiple neuronal dynamics with different time scales are likely to occur within the wake-sleep system. Such slower modulation may contribute to reducing noise and stabilizing the switch, and could help explain how putative flip-flop switches could accommodate noisy, unstable neuronal circuits, and integrate multiple influences into sharp transitions. It will also be important to understand better how motivation and emotions influence the wake-sleep system. As these terms imply, these drivers for animal behavior involve movement, which is the antithesis of quiet sleep. Unraveling how the orexin neurons and other systems impel motivated behaviors, addictions, and reward will be important new vistas for understanding their overall role in the functions of the brain. At the same time, we need to understand how stress, anxiety, and depression can drive insomnia. Finally, the extent to which the wake-sleep circuitry is so deeply embedded within the brain, and intricately related to circuitry controlling movement, motivation, and emotion, suggests that sleep is fundamentally important for normal brain function. Yet the way in which sleep is restorative, and why brain function is impaired in its absence, remain among the most enduring mysteries of neuroscience.
Acknowledgments
The authors thank Dr. K. Sakai for permission to use Fig. 3, and Dr. C. Diniz Behn for the data analysis used to construct Figure 4. This work was supported by USPHS grants NS055367, AG09975, and HL60292.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- Achermann P, Borbely AA. Mathematical models of sleep regulation. Front Biosci. 2003;8:s683–s693. doi: 10.2741/1064. [DOI] [PubMed] [Google Scholar]
- Adamantidis A, Salvert D, Goutagny R, Lakaye B, Gervasoni D, Grisar T, Luppi PH, Fort P. Sleep architecture of the melanin-concentrating hormone receptor 1-knockout mice. Eur J Neurosci. 2008;27:1793–1800. doi: 10.1111/j.1460-9568.2008.06129.x. [DOI] [PubMed] [Google Scholar]
- Adamantidis AR, Zhang F, Aravanis AM, Deisseroth K, de LL. Neural substrates of awakening probed with optogenetic control of hypocretin neurons. Nature. 2007;450:420–424. doi: 10.1038/nature06310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adametz JH. Rate of recovery of functioning in cats with rostral reticular lesions. J Neurosurg. 1959;16:85–98. doi: 10.3171/jns.1959.16.1.0085. [DOI] [PubMed] [Google Scholar]
- Asanuma C. Axonal arborizations of a magnocellular basal nucleus input and their relation to the neurons in the thalamic reticular nucleus of rats. Proc Natl Acad Sci U S A. 1989;86:4746–4750. doi: 10.1073/pnas.86.12.4746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asanuma C. Noradrenergic Innervation of the Thalamic Reticular Nucleus - A Light and Electron Microscopic Immunohistochemical Study in Rats. J Comp Neurol. 1992;319:299–311. doi: 10.1002/cne.903190209. [DOI] [PubMed] [Google Scholar]
- Asanuma C, Porter LL. Light and electron microscopic evidence for a GABAergic projection from the caudal basal forebrain to the thalamic reticular nucleus in rats. J Comp Neurol. 1990;302:159–172. doi: 10.1002/cne.903020112. [DOI] [PubMed] [Google Scholar]
- Aston-Jones G, Bloom FE. Activity of norepinephrine-containing locus coeruleus neurons in behaving rats anticipates fluctuations in the sleep-waking cycle. J Neurosci. 1981;1:876–886. doi: 10.1523/JNEUROSCI.01-08-00876.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aston-Jones G, Cohen JD. Adaptive gain and the role of the locus coeruleus-norepinephrine system in optimal performance. J Comp Neurol. 2005;493:99–110. doi: 10.1002/cne.20723. [DOI] [PubMed] [Google Scholar]
- Baldo BA, Gual-Bonilla L, Sijapati K, Daniel RA, Landry CF, Kelley AE. Activation of a subpopulation of orexin/hypocretin-containing hypothalamic neurons by GABAA receptor-mediated inhibition of the nucleus accumbens shell, but not by exposure to a novel environment. Eur J Neurosci. 2004;19:376–386. doi: 10.1111/j.1460-9568.2004.03093.x. [DOI] [PubMed] [Google Scholar]
- Benington JH, Heller HC. Restoration of brain energy metabolism as the function of sleep. Prog Neurobiol. 1995;45:347–360. doi: 10.1016/0301-0082(94)00057-o. [DOI] [PubMed] [Google Scholar]
- Bickford ME, Gunluk AE, Van Horn SC, Sherman SM. GABAergic projection from the basal forebrain to the visual sector of the thalamic reticular nucleus in the cat. J Comp Neurol. 1994;348:481–510. doi: 10.1002/cne.903480402. [DOI] [PubMed] [Google Scholar]
- Bjorness TE, Kelly CL, Gao T, Poffenberger V, Greene RW. Control and function of the homeostatic sleep response by adenosine A1 receptors. J Neurosci. 2009;29:1267–1276. doi: 10.1523/JNEUROSCI.2942-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco-Centurion C, Gerashchenko D, Shiromani PJ. Effects of saporin-induced lesions of three arousal populations on daily levels of sleep and wake. J Neurosci. 2007;27:14041–14048. doi: 10.1523/JNEUROSCI.3217-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boissard R, Fort P, Gervasoni D, Barbagli B, Luppi PH. Localization of the GABAergic and non-GABAergic neurons projecting to the sublaterodorsal nucleus and potentially gating paradoxical sleep onset. Eur J Neurosci. 2003;18:1627–1639. doi: 10.1046/j.1460-9568.2003.02861.x. [DOI] [PubMed] [Google Scholar]
- Boissard R, Gervasoni D, Schmidt MH, Barbagli B, Fort P, Luppi PH. The rat ponto-medullary network responsible for paradoxical sleep onset and maintenance: a combined microinjection and functional neuroanatomical study. Eur J Neurosci. 2002;16:1959–1973. doi: 10.1046/j.1460-9568.2002.02257.x. [DOI] [PubMed] [Google Scholar]
- Borbely AA, Achermann P. Sleep homeostasis and models of sleep regulation. J Biol Rhythms. 1999;14:557–568. doi: 10.1177/074873099129000894. [DOI] [PubMed] [Google Scholar]
- Borbely AA, Tobler I. Homeostatic and circadian principles in sleep regulation in the rat. In: McGinty DJ, et al., editors. Brain Mechanisms of Sleep. New York: Raven Press; 1985. pp. 35–44. [Google Scholar]
- Boucetta S, Jones BE. Activity profiles of cholinergic and intermingled GABAergic and putative glutamatergic neurons in the pontomesencephalic tegmentum of urethane-anesthetized rats. J Neurosci. 2009;29:4664–4674. doi: 10.1523/JNEUROSCI.5502-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourgin P, Huitron-Resendiz S, Spier AD, Fabre V, Morte B, Criado JR, Sutcliffe JG, Henriksen SJ, de Lecea L. Hypocretin-1 modulates rapid eye movement sleep through activation of locus coeruleus neurons. J Neurosci. 2000;20:7760–7765. doi: 10.1523/JNEUROSCI.20-20-07760.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broughton R, Valley V, Aguirre M, Roberts J, Suwalski W, Dunham W. Excessive daytime sleepiness and the pathophysiology of narcolepsy-cataplexy: a laboratory perspective. Sleep. 1986;9:205–215. doi: 10.1093/sleep/9.1.205. [DOI] [PubMed] [Google Scholar]
- Brown RE, Sergeeva O, Eriksson KS, Haas HL. Orexin A excites serotonergic neurons in the dorsal raphe nucleus of the rat. Neuropharmacology. 2001;40:457–459. doi: 10.1016/s0028-3908(00)00178-7. [DOI] [PubMed] [Google Scholar]
- Bunger MK, Wilsbacher LD, Moran SM, Clendenin C, Radcliffe LA, Hogenesch JB, Simon MC, Takahashi JS, Bradfield CA. Mop3 is an essential component of the master circadian pacemaker in mammals. Cell. 2000;103:1009–1017. doi: 10.1016/s0092-8674(00)00205-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess C, Lai D, Siegel J, Peever J. An endogenous glutamatergic drive onto somatic motoneurons contributes to the stereotypical pattern of muscle tone across the sleep-wake cycle. J Neurosci. 2008;28:4649–4660. doi: 10.1523/JNEUROSCI.0334-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burne JA. Reflex origin of parkinsonian tremor. Exp Neurol. 1987;97:327–339. doi: 10.1016/0014-4886(87)90093-8. [DOI] [PubMed] [Google Scholar]
- Buzsaki G, Bickford RG, Ponomareff G, Thal LJ, Mandel R, Gage FH. Nucleus basalis and thalamic control of neocortical activity in the freely moving rat. J Neurosci. 1988;8:4007–4026. doi: 10.1523/JNEUROSCI.08-11-04007.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cano G, Mochizuki T, Saper CB. Neural circuitry of stress-induced insomnia in rats. J Neurosci. 2008;28:10167–10184. doi: 10.1523/JNEUROSCI.1809-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cape EG, Jones BE. Effects of glutamate agonist versus procaine microinjections into the basal forebrain cholinergic cell area upon gamma and theta EEG activity and sleep-wake state. Eur J Neurosci. 2000;12:2166–2184. doi: 10.1046/j.1460-9568.2000.00099.x. [DOI] [PubMed] [Google Scholar]
- Carter ME, Adamantidis A, Ohtsu H, Deisseroth K, deLecea L. Sleep homeostasis modulates hypocretin-mediated sleep-to-wake transitions. J Neurosci. 2009;29:10939–10949. doi: 10.1523/JNEUROSCI.1205-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlin NL, Arrigoni E, Chou TC, Scammell TE, Greene RW, Saper CB. Effects of adenosine on gabaergic synaptic inputs to identified ventrolateral preoptic neurons. Neuroscience. 2003;119:913–918. doi: 10.1016/s0306-4522(03)00246-x. [DOI] [PubMed] [Google Scholar]
- Chee MW, Choo WC. Functional imaging of working memory after 24 hr of total sleep deprivation. J Neurosci. 2004;24:4560–4567. doi: 10.1523/JNEUROSCI.0007-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chemelli RM, Willie JT, Sinton CM, Elmquist JK, Scammell T, Lee C, Richardson JA, Williams SC, Xiong Y, Kisanuki Y, Fitch TE, Nakazato M, Hammer RE, Saper CB, Yanagisawa M. Narcolepsy in orexin knockout mice: molecular genetics of sleep regulation. Cell. 1999;98:437–451. doi: 10.1016/s0092-8674(00)81973-x. [DOI] [PubMed] [Google Scholar]
- Chou TC. PhD dissertation. MA: Harvard University; 2003. A bistable model of sleep-wake regulation. Regulation of wake-sleep timing: circadian rhythms and bistability of sleep-wake states; pp. 82–99. [Google Scholar]
- Chou TC, Bjorkum AA, Gaus SE, Lu J, Scammell TE, Saper CB. Afferents to the ventrolateral preoptic nucleus. J Neurosci. 2002;22:977–990. doi: 10.1523/JNEUROSCI.22-03-00977.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou TC, Lee CE, Lu J, Elmquist JK, Hara J, Willie JT, Beuckmann CT, Chemelli RM, Sakurai T, Yanagisawa M, Saper CB, Scammell TE. Orexin (hypocretin) neurons contain dynorphin. J Neurosci. 2001;21:RC168. doi: 10.1523/JNEUROSCI.21-19-j0003.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou TC, Scammell TE, Gooley JJ, Gaus SE, Saper CB, Lu J. Critical role of dorsomedial hypothalamic nucleus in a wide range of behavioral circadian rhythms. J Neurosci. 2003;19;23:10691–10702. doi: 10.1523/JNEUROSCI.23-33-10691.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu N, Bloom FE. Norepinephrine-containing neurons: changes in spontaneous discharge patterns during sleeping and waking. Science. 1973;179:908–910. doi: 10.1126/science.179.4076.908. [DOI] [PubMed] [Google Scholar]
- Chuah YM, Venkatraman V, Dinges DF, Chee MW. The neural basis of interindividual variability in inhibitory efficiency after sleep deprivation. J Neurosci. 2006;26:7156–7162. doi: 10.1523/JNEUROSCI.0906-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crochet S, Onoe H, Sakai K. A potent non-monoaminergic paradoxical sleep inhibitory system: a reverse microdialysis and single-unit recording study. Eur J Neurosci. 2006;24:1404–1412. doi: 10.1111/j.1460-9568.2006.04995.x. [DOI] [PubMed] [Google Scholar]
- Crocker A, Espana RA, Papadopoulou M, Saper CB, Faraco J, Sakurai T, Honda M, Mignot E, Scammell TE. Concomitant loss of dynorphin, NARP, and orexin in narcolepsy. Neurology. 2005;65:1184–1188. doi: 10.1212/01.wnl.0000168173.71940.ab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cvetkovic-Lopes V, Bayer L, Dorsaz S, Maret S, Pradervand S, Dauvilliers Y, Lecendreux M, Lammers GJ, Donjacour CE, Du Pasquier RA, Pfister C, Petit B, Hor H, Muhlethaler M, Tafti M. Elevated Tribbles homolog 2-specific antibody levels in narcolepsy patients. J Clin Invest. 2010;120:713–719. doi: 10.1172/JCI41366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlstrom A, Fuxe K. Evidence for the existence of monoamine-containing neurons in the central nervous system. I. Demonstration of monoamines in the cell bodies of brain stem neurons. Acta Physiol Scand. 1964;62(suppl 232):1–55. [PubMed] [Google Scholar]
- Dantz B, Edgar DM, Dement WC. Circadian rhythms in narcolepsy: studies on a 90 minute day. Electroencephalogr Clin Neurophysiol. 1994;90:24–35. doi: 10.1016/0013-4694(94)90110-4. [DOI] [PubMed] [Google Scholar]
- Deurveilher S, Semba K. Indirect projections from the suprachiasmatic nucleus to major arousal-promoting cell groups in rat: implications for the circadian control of behavioural state. Neuroscience. 2005;130:165–183. doi: 10.1016/j.neuroscience.2004.08.030. [DOI] [PubMed] [Google Scholar]
- Diniz Behn CG, Booth V. Simulating microinjection experiments in a novel model of the rat sleep-wake regulatory network. J Neurophysiol. 2010 doi: 10.1152/jn.00795.2009. epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Diniz Behn CG, Klerman EB, Mochizuki T, Lin SC, Scammell TE. Abnormal sleep/wake dynamics in orexin knockout mice. Sleep. 2010;33:29–306. doi: 10.1093/sleep/33.3.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diniz Behn CG, Kopell N, Brown EN, Mochizuki T, Scammell TE. Delayed orexin signaling consolidates wakefulness and sleep: physiology and modeling. J Neurophysiol. 2008;99:3090–3103. doi: 10.1152/jn.01243.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong HW, Petrovich GD, Watts AG, Swanson LW. Basic organization of projections from the oval and fusiform nuclei of the bed nuclei of the stria terminalis in adult rat brain. J Comp Neurol. 2001;436:430–455. doi: 10.1002/cne.1079. [DOI] [PubMed] [Google Scholar]
- Edgar DM, Dement WC, Fuller CA. Effect of SCN lesions on sleep in squirrel monkeys: evidence for opponent processes in sleep-wake regulation. J Neurosci. 1993;13:1065–1079. doi: 10.1523/JNEUROSCI.13-03-01065.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggermann E, Serafin M, Bayer L, Machard D, Saint-Mleux B, Jones BE, Muhlethaler M. Orexins/hypocretins excite basal forebrain cholinergic neurones. Neuroscience. 2001;108:177–181. doi: 10.1016/s0306-4522(01)00512-7. [DOI] [PubMed] [Google Scholar]
- El Mansari M, Sakai K, Jouvet M. Unitary characteristics of presumptive cholinergic tegmental neurons during the sleep-waking cycle in freely moving cats. Exp Brain Res. 1989;76:519–529. doi: 10.1007/BF00248908. [DOI] [PubMed] [Google Scholar]
- Elias CF, Lee CE, Kelly JF, Ahima RS, Kuhar M, Saper CB, Elmquist JK. Characterization of CART neurons in the rat and human hypothalamus. J Comp Neurol. 2001;432:1–19. doi: 10.1002/cne.1085. [DOI] [PubMed] [Google Scholar]
- Estabrooke IV, McCarthy MT, Ko E, Chou TC, Chemelli RM, Yanagisawa M, Saper CB, Scammell TE. Fos expression in orexin neurons varies with behavioral state. J Neurosci. 2001;21:1656–1662. doi: 10.1523/JNEUROSCI.21-05-01656.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freund TF, Meskenaite V. gamma-Aminobutyric acid-containing basal forebrain neurons innervate inhibitory interneurons in the neocortex. Proc Natl Acad Sci U S A. 1992;89:738–742. doi: 10.1073/pnas.89.2.738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulcher BD, Phillips AJ, Robinson PA. Modeling the impact of impulsive stimuli on sleep-wake dynamics. Phys Rev E Stat Nonlin Soft Matter Phys. 2008;78:051920. doi: 10.1103/PhysRevE.78.051920. [DOI] [PubMed] [Google Scholar]
- Fulcher BD, Phillips AJ, Robinson PA. Quantitative physiologically based modeling of subjective fatigue during sleep deprivation. J Theor Biol. 2010;264:407–419. doi: 10.1016/j.jtbi.2010.02.028. [DOI] [PubMed] [Google Scholar]
- Fuller PM, Lu J, Saper CB. Differential rescue of light- and food-entrainable circadian rhythms. Science. 2008;320:1074–1077. doi: 10.1126/science.1153277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung SJ, Yamuy J, Sampogna S, Morales FR, Chase MH. Hypocretin (orexin) input to trigeminal and hypoglossal motoneurons in the cat: a double-labeling immunohistochemical study. Brain Res. 2001;903:257–262. doi: 10.1016/s0006-8993(01)02318-6. [DOI] [PubMed] [Google Scholar]
- Gallopin T, Fort P, Eggermann E, Cauli B, Luppi PH, Rossier J, Audinat E, Muhlethaler M, Serafin M. Identification of sleep-promoting neurons in vitro. Nature. 2000;404:992–995. doi: 10.1038/35010109. [DOI] [PubMed] [Google Scholar]
- Gallopin T, Luppi PH, Cauli B, Urade Y, Rossier J, Hayaishi O, Lambolez B, Fort P. The endogenous somnogen adenosine excites a subset of sleep-promoting neurons via A2A receptors in the ventrolateral preoptic nucleus. Neuroscience. 2005;134:1377–1390. doi: 10.1016/j.neuroscience.2005.05.045. [DOI] [PubMed] [Google Scholar]
- Gallopin T, Luppi PH, Rambert FA, Frydman A, Fort P. Effect of the wake-promoting agent modafinil on sleep-promoting neurons from the ventrolateral preoptic nucleus: an in vitro pharmacologic study. Sleep. 2004;27:19–25. [PubMed] [Google Scholar]
- Gelineau JB. De la narcolepsie. Gaz Hop (Paris) 1880;53:626f. [Google Scholar]
- Gerashchenko D, Blanco-Centurion C, Greco MA, Shiromani PJ. Effects of lateral hypothalamic lesion with the neurotoxin hypocretin-2-saporin on sleep in Long-Evans rats. Neuroscience. 2003;116:223–235. doi: 10.1016/s0306-4522(02)00575-4. [DOI] [PubMed] [Google Scholar]
- Gerashchenko D, Chou TC, Blanco-Centurion CA, Saper CB, Shiromani PJ. Effects of lesions of the histaminergic tuberomammillary nucleus on spontaneous sleep in rats. Sleep. 2004;27:1275–1281. doi: 10.1093/sleep/27.7.1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerashchenko D, Wisor JP, Burns D, Reh RK, Shiromani PJ, Sakurai T, del I, Kilduff TS. Identification of a population of sleep-active cerebral cortex neurons. Proc Natl Acad Sci U S A. 2008;105:10227–10232. doi: 10.1073/pnas.0803125105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gervasoni D, Lin SC, Ribeiro S, Soares ES, Pantoja J, Nicolelis MA. Global forebrain dynamics predict rat behavioral states and their transitions. J Neurosci. 2004;24:11137–11147. doi: 10.1523/JNEUROSCI.3524-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong H, McGinty D, Guzman-Marin R, Chew KT, Stewart D, Szymusiak R. Activation of c-fos in GABAergic neurones in the preoptic area during sleep and in response to sleep deprivation. J Physiol. 2004;556:935–946. doi: 10.1113/jphysiol.2003.056622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gooley JJ, Lu J, Chou TC, Scammell TE, Saper CB. Melanopsin in cells of origin of the retinohypothalamic tract. Nat Neurosci. 2001;4:1165. doi: 10.1038/nn768. [DOI] [PubMed] [Google Scholar]
- Gooley JJ, Schomer A, Saper CB. The dorsomedial hypothalamic nucleus is critical for the expression of food-entrainable circadian rhythms. Nat Neurosci. 2006;9:398–407. doi: 10.1038/nn1651. [DOI] [PubMed] [Google Scholar]
- Greco MA, Fuller PM, Jhou TC, Martin-Schild S, Zadina JE, Hu Z, Shiromani P, Lu J. Opioidergic projections to sleep-active neurons in the ventrolateral preoptic nucleus. Brain Res. 2008;1245:96–107. doi: 10.1016/j.brainres.2008.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greco MA, Shiromani PJ. Hypocretin receptor protein and mRNA expression in the dorsolateral pons of rats. Brain Res Mol Brain Res. 2001;88:176–182. doi: 10.1016/s0169-328x(01)00039-0. [DOI] [PubMed] [Google Scholar]
- Grove EA. Neural associations of the substantia innominata in the rat: afferent connections. J Comp Neurol. 1988;277:315–346. doi: 10.1002/cne.902770302. [DOI] [PubMed] [Google Scholar]
- Gvilia I, Xu F, McGinty D, Szymusiak R. Homeostatic regulation of sleep: a role for preoptic area neurons. J Neurosci. 2006;26:9426–9433. doi: 10.1523/JNEUROSCI.2012-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagan JJ, Leslie RA, Patel S, Evans ML, Wattam TA, Holmes S, Benham CD, Taylor SG, Routledge C, Hemmati P, Munton RP, Ashmeade TE, Shah AS, Hatcher JP, Hatcher PD, Jones DN, Smith MI, Piper DC, Hunter AJ, Porter RA, Upton N. Orexin A activates locus coeruleus cell firing and increases arousal in the rat. Proc Natl Acad Sci U S A. 1999;96:10911–10916. doi: 10.1073/pnas.96.19.10911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halassa MM, Florian C, Fellin T, Munoz JR, Lee SY, Abel T, Haydon PG, Frank MG. Astrocytic modulation of sleep homeostasis and cognitive consequences of sleep loss. Neuron. 2009;61:213–219. doi: 10.1016/j.neuron.2008.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallanger AE, Levey AI, Lee HJ, Rye DB, Wainer BH. The origins of cholinergic and other subcortical afferents to the thalamus in the rat. J Comp Neurol. 1987;262:105–124. doi: 10.1002/cne.902620109. [DOI] [PubMed] [Google Scholar]
- Hara J, Beuckmann CT, Nambu T, Willie JT, Chemelli RM, Sinton CM, Sugiyama F, Yagami K, Goto K, Yanagisawa M, Sakurai T. Genetic ablation of orexin neurons in mice results in narcolepsy, hypophagia, and obesity. Neuron. 2001;30:345–354. doi: 10.1016/s0896-6273(01)00293-8. [DOI] [PubMed] [Google Scholar]
- Harris GC, Wimmer M, Aston-Jones G. A role for lateral hypothalamic orexin neurons in reward seeking. Nature. 2005;437:556–559. doi: 10.1038/nature04071. [DOI] [PubMed] [Google Scholar]
- Hassani OK, Lee MG, Jones BE. Melanin-concentrating hormone neurons discharge in a reciprocal manner to orexin neurons across the sleep-wake cycle. Proc Natl Acad Sci U S A. 2009;106:2418–2422. doi: 10.1073/pnas.0811400106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattar S, Liao HW, Takao M, Berson DM, Yau KW. Melanopsin-containing retinal ganglion cells: architecture, projections, and intrinsic photosensitivity. Science. 2002;295:1065–1070. doi: 10.1126/science.1069609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendricks JC, Morrison AR, Mann GK. Different behaviors during paradoxical sleep without atonia depend on pontine lesion site. Brain Res. 1982;239:81–105. doi: 10.1016/0006-8993(82)90835-6. [DOI] [PubMed] [Google Scholar]
- Henny P, Jones BE. Projections from basal forebrain to prefrontal cortex comprise cholinergic, GABAergic and glutamatergic inputs to pyramidal cells or interneurons. Eur J Neurosci. 2008;27:654–670. doi: 10.1111/j.1460-9568.2008.06029.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobson JA, McCarley RW, Wyzinski PW. Sleep cycle oscillation: reciprocal discharge by two brainstem neuronal groups. Science. 1975;189:55–58. doi: 10.1126/science.1094539. [DOI] [PubMed] [Google Scholar]
- Huang ZL, Qu WM, Eguchi N, Chen JF, Schwarzschild MA, Fredholm BB, Urade Y, Hayaishi O. Adenosine A2A, but not A1, receptors mediate the arousal effect of caffeine. Nat Neurosci. 2005;8:858–859. doi: 10.1038/nn1491. [DOI] [PubMed] [Google Scholar]
- Huber R, Ghilardi MF, Massimini M, Tononi G. Local sleep and learning. Nature. 2004;430:78–81. doi: 10.1038/nature02663. [DOI] [PubMed] [Google Scholar]
- Hur EE, Zaborszky L. Vglut2 afferents to the medial prefrontal and primary somatosensory cortices: a combined retrograde tracing in situ hybridization study. J Comp Neurol. 2005;483:351–373. doi: 10.1002/cne.20444. [DOI] [PubMed] [Google Scholar]
- Hurley KM, Herbert H, Moga MM, Saper CB. Efferent Projections of the Infralimbic Cortex of the Rat. J Comp Neurol. 1991;308:249–276. doi: 10.1002/cne.903080210. [DOI] [PubMed] [Google Scholar]
- Ishimori K. True cause of sleep: A hypnogenic substance as evidenced in the brain of sleep-deprived animals. Tokyo Igakkai Zasshi. 1909;23:429–457. [Google Scholar]
- Iwano M, Hill ES, Mori A, Mishima T, Mishima T, Ito K, Kanzaki R. Neurons associated with the flip-flop activity in the lateral accessory lobe and ventral protocerebrum of the silkworm moth brain. J Comp Neurol. 2010;518:366–388. doi: 10.1002/cne.22224. [DOI] [PubMed] [Google Scholar]
- Jin X, Shearman LP, Weaver DR, Zylka MJ, De Vries GJ, Reppert SM. A molecular mechanism regulating rhythmic output from the suprachiasmatic circadian clock. Cell. 1999;96:1–20. doi: 10.1016/s0092-8674(00)80959-9. [DOI] [PubMed] [Google Scholar]
- John J, Wu MF, Boehmer LN, Siegel JM. Cataplexy-active neurons in the hypothalamus: implications for the role of histamine in sleep and waking behavior. Neuron. 2004;42:619–634. doi: 10.1016/s0896-6273(04)00247-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones BE. Activity, modulation and role of basal forebrain cholinergic neurons innervating the cerebral cortex. Prog Brain Res. 2004;145:157–69. doi: 10.1016/S0079-6123(03)45011-5. [DOI] [PubMed] [Google Scholar]
- Jones EG, Leavitt RY. Retrograde axonal transport and the demonstration of non-specific projections to the cerebral cortex and striatum from thalamic intralaminar nuclei in the rat, cat and monkey. J Comp Neurol. 1974;154:349–378. doi: 10.1002/cne.901540402. [DOI] [PubMed] [Google Scholar]
- Jouvet M. Research on the neural structures and responsible mechanisms in different phases of physiological sleep. Arch Ital Biol. 1962;100:125–206. [PubMed] [Google Scholar]
- Kantor S, Mochizuki T, Janisiewicz AM, Clark E, Clain E, Nishino S, Scammell TE. Orexin Neurons Are Necessary for the Circadian Control of REM Sleep. Sleep. 2009;32:1127–1134. doi: 10.1093/sleep/32.9.1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur S, Junek A, Black MA, Semba K. Effects of ibotenate and 192IgG-saporin lesions of the nucleus basalis magnocellularis/substantia innominata on spontaneous sleep and wake states and on recovery sleep after sleep deprivation in rats. J Neurosci. 2008;28:491–504. doi: 10.1523/JNEUROSCI.1585-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur S, Thankachan S, Begum S, Liu M, Blanco-Centurion C, Shiromani PJ. Hypocretin-2 saporin lesions of the ventrolateral periaquaductal gray (vlPAG) increase REM sleep in hypocretin knockout mice. PLoS ONE. 2009;4:e6346. doi: 10.1371/journal.pone.0006346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawashima M, Lin L, Tanaka S, Jennum P, Knudsen S, Nevsimalova S, Plazzi G, Mignot E. Anti-Tribbles homolog 2 (TRIB2) autoantibodies in narcolepsy are associated with recent onset of cataplexy. Sleep. 2010;33:869–874. doi: 10.1093/sleep/33.7.869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khatami R, Landolt HP, Achermann P, Adam M, Retey JV, Werth E, Schmid D, Bassetti CL. Challenging sleep homeostasis in narcolepsy-cataplexy: implications for non-REM and REM sleep regulation. Sleep. 2008;31:859–867. doi: 10.1093/sleep/31.6.859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim R, Nakano K, Jayaraman A, Carpenter MB. Projections of the globus pallidus and adjacent structures: an autoradiographic study in the monkey. J Comp Neurol. 1976;169:263–290. doi: 10.1002/cne.901690302. [DOI] [PubMed] [Google Scholar]
- Kinney HC, Samuels MA. Neuropathology of the persistent vegetative state. A review. J Neuropathol Exp Neurol. 1994;53:548–558. doi: 10.1097/00005072-199411000-00002. [DOI] [PubMed] [Google Scholar]
- Kocsis B, Varga V, Dahan L, Sik A. Serotonergic neuron diversity: identification of raphe neurons with discharges time-locked to the hippocampal theta rhythm. Proc Natl Acad Sci U S A. 2006;103:1059–1064. doi: 10.1073/pnas.0508360103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohlmeier KA, Watanabe S, Tyler CJ, Burlet S, Leonard CS. Dual orexin actions on dorsal raphe and laterodorsal tegmentum neurons. J Neurophysiol. 2008;100:2265–2281. doi: 10.1152/jn.01388.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong J, Shepel PN, Holden CP, Mackiewicz M, Pack AI, Geiger JD. Brain glycogen decreases with increased periods of wakefulness: implications for homeostatic drive to sleep. J Neurosci. 2002;22:5581–5587. doi: 10.1523/JNEUROSCI.22-13-05581.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger JM. The role of cytokines in sleep regulation. Curr Pharm Des. 2008;14:3408–3416. doi: 10.2174/138161208786549281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lankheet MJ. Unraveling adaptation and mutual inhibition in perceptual rivalry. J Vis. 2006;6:304–310. doi: 10.1167/6.4.1. [DOI] [PubMed] [Google Scholar]
- Laposky A, Easton A, Dugovic C, Walisser J, Bradfield C, Turek F. Deletion of the mammalian circadian clock gene BMAL1/Mop3 alters baseline sleep architecture and the response to sleep deprivation. Sleep. 2005;28:395–409. doi: 10.1093/sleep/28.4.395. [DOI] [PubMed] [Google Scholar]
- Lee MG, Hassani OK, Jones BE. Discharge of identified orexin/hypocretin neurons across the sleep-waking cycle. J Neurosci. 2005;25:6716–6720. doi: 10.1523/JNEUROSCI.1887-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MG, Manns ID, Alonso A, Jones BE. Sleep-wake related discharge properties of basal forebrain neurons recorded with micropipettes in head-fixed rats. J Neurophysiol. 2004;92:1182–1198. doi: 10.1152/jn.01003.2003. [DOI] [PubMed] [Google Scholar]
- Legendre R, Pieron H. Recherches sur le besoin de commeil consecutif a une veille prolongee. Z Allgem Physiol. 1913;14:235–262. [Google Scholar]
- Levey AI, Hallanger AE, Wainer BH. Cholinergic nucleus basalis neurons may influence the cortex via the thalamus. Neurosci Lett. 1987;74:7–13. doi: 10.1016/0304-3940(87)90042-5. [DOI] [PubMed] [Google Scholar]
- Lim AS, Scammell TE. The trouble with Tribbles: do antibodies against TRIB2 cause narcolepsy? Sleep. 2010;33:857–858. doi: 10.1093/sleep/33.7.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin L, Faraco J, Li R, Kadotani H, Rogers W, Lin X, Qiu X, de Jong PJ, Nishino S, Mignot E. The sleep disorder canine narcolepsy is caused by a mutation in the hypocretin (orexin) receptor 2 gene. Cell. 1999;98:365–376. doi: 10.1016/s0092-8674(00)81965-0. [DOI] [PubMed] [Google Scholar]
- Lindsley DB, Bowden JW, Magoun HW. Effect upon the EEG of acute injury to the brain stem activating system. Electroencephalogr Clin Neurophysiol. 1949;1:475–486. [PubMed] [Google Scholar]
- Liu ZW, Gao XB. Adenosine inhibits activity of hypocretin/orexin neurons by the A1 receptor in the lateral hypothalamus: a possible sleep-promoting effect. J Neurophysiol. 2007;97:837–848. doi: 10.1152/jn.00873.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo CC, Chou T, Penzel T, Scammell TE, Strecker RE, Stanley HE, Ivanov PC. Common scale-invariant patterns of sleep-wake transitions across mammalian species. Proc Natl Acad Sci U S A. 2004;101:17545–17548. doi: 10.1073/pnas.0408242101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Greco MA, Shiromani P, Saper CB. Effect of lesions of the ventrolateral preoptic nucleus on NREM and REM sleep. J Neurosci. 2000;20:3830–3842. doi: 10.1523/JNEUROSCI.20-10-03830.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Jhou TC, Saper CB. Identification of wake-active dopaminergic neurons in the ventral periaqueductal gray matter. J Neurosci. 2006a;26:193–202. doi: 10.1523/JNEUROSCI.2244-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Sherman D, Devor M, Saper CB. A putative flip-flop switch for control of REM sleep. Nature. 2006b;441:589–594. doi: 10.1038/nature04767. [DOI] [PubMed] [Google Scholar]
- Lu J, Zhang YH, Chou TC, Gaus SE, Elmquist JK, Shiromani P, Saper CB. Contrasting effects of ibotenate lesions of the paraventricular nucleus and subparaventricular zone on sleep-wake cycle and temperature regulation. J Neurosci. 2001;21:4864–4874. doi: 10.1523/JNEUROSCI.21-13-04864.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luppi PH, Gervasoni D, Boissard R, Verret L, Goutagny R, Peyron C, Salvert D, Leger L, Barbagli B, Fort P. Brainstem structures responsible for paradoxical sleep onset and maintenance. Arch Ital Biol. 2004;142:397–411. [PubMed] [Google Scholar]
- Luppi PH, Gervasoni D, Verret L, Goutagny R, Peyron C, Salvert D, Leger L, Fort P. Paradoxical (REM) sleep genesis: the switch from an aminergic-cholinergic to a GABAergic-glutamatergic hypothesis. J Physiol Paris. 2006;100:271–283. doi: 10.1016/j.jphysparis.2007.05.006. [DOI] [PubMed] [Google Scholar]
- Lynagh T, Lynch JW. An improved ivermectin-activated chloride channel receptor for inhibiting electrical activity in defined neuronal populations. J Biol Chem. 2010;285:14890–14897. doi: 10.1074/jbc.M110.107789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning KA, Wilson JR, Uhlrich DJ. Histamine-immunoreactive neurons and their innervation of vX isual regions in the cortex, tectum, and thalamus in the primate X Macaca mulatta.X. J Comp Neurol. 1996;373:271–282. doi: 10.1002/(SICI)1096-9861(19960916)373:2<271::AID-CNE9>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Manns ID, Mainville L, Jones BE. Evidence for glutamate, in addition to acetylcholine and GABA, neurotransmitter synthesis in basal forebrain neurons projecting to the entorhinal cortex. Neuroscience. 2001;107:249–263. doi: 10.1016/s0306-4522(01)00302-5. [DOI] [PubMed] [Google Scholar]
- Mano MM, Kime CR. Logic and Computer Design Fundamentals. 3. Pearson Education International; Upper Saddle River, NJ: 2004. p. 283. [Google Scholar]
- McCarley RW, Hobson JA. Neuronal excitability modulation over the sleep cycle: a structural and mathematical model. Science. 1975;189:58–60. doi: 10.1126/science.1135627. [DOI] [PubMed] [Google Scholar]
- McEwen BS. Allostasis and allostatic load: Implications for neuropsychopharmacology. Neuropsychopharmacology. 2000;22:108–124. doi: 10.1016/S0893-133X(99)00129-3. [DOI] [PubMed] [Google Scholar]
- McGinty DJ, Sterman MB. Sleep suppression after basal forebrain lesions in the cat. Science. 1968;160:1253–1255. doi: 10.1126/science.160.3833.1253. [DOI] [PubMed] [Google Scholar]
- Methippara MM, Alam MN, Szymusiak R, McGinty D. Effects of lateral preoptic area application of orexin-A on sleep-wakefulness. Neuroreport. 2000;11:3423–3426. doi: 10.1097/00001756-200011090-00004. [DOI] [PubMed] [Google Scholar]
- Mieda M, Williams SC, Richardson JA, Tanaka K, Yanagisawa M. The dorsomedial hypothalamic nucleus as a putative food-entrainable circadian pacemaker. Proc Natl Acad Sci U S A. 2006;103:12150–12155. doi: 10.1073/pnas.0604189103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mignot E, Lin L, Rogers W, Honda Y, Qiu X, Lin X, Okun M, Hohjoh H, Miki T, Hsu S, Leffell M, Grumet F, Fernandez-Vina M, Honda M, Risch N. Complex HLA-DR and -DQ interactions confer risk of narcolepsy-cataplexy in three ethnic groups. Am J Hum Genet. 2001;68:686–699. doi: 10.1086/318799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mileykovskiy BY, Kiyashchenko LI, Siegel JM. Behavioral correlates of activity in identified hypocretin/orexin neurons. Neuron. 2005;46:787–798. doi: 10.1016/j.neuron.2005.04.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochizuki T, Crocker A, McCormack S, Yanagisawa M, Sakurai T, Scammell TE. Behavioral state instability in orexin knock-out mice. J Neurosci. 2004;24:6291–6300. doi: 10.1523/JNEUROSCI.0586-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modirrousta M, Mainville L, Jones BE. Gabaergic neurons with alpha2-adrenergic receptors in basal forebrain and preoptic area express c-Fos during sleep. Neuroscience. 2004;129:803–810. doi: 10.1016/j.neuroscience.2004.07.028. [DOI] [PubMed] [Google Scholar]
- Montagna P, Gambetti P, Cortelli P, Lugaresi E. Familial and sporadic fatal insomnia. Lancet Neurol. 2003;2:167–176. doi: 10.1016/s1474-4422(03)00323-5. [DOI] [PubMed] [Google Scholar]
- Moore RY, Eichler VB. Loss of a circadian adrenal corticosterone rhythm following suprachiasmatic lesions in the rat. Brain Res. 1972;42:201–206. doi: 10.1016/0006-8993(72)90054-6. [DOI] [PubMed] [Google Scholar]
- Morairty S, Rainnie D, McCarley R, Greene R. Disinhibition of ventrolateral preoptic area sleep-active neurons by adenosine: a new mechanism for sleep promotion. Neuroscience. 2004;123:451–457. doi: 10.1016/j.neuroscience.2003.08.066. [DOI] [PubMed] [Google Scholar]
- Moriguchi T, Sakurai T, Nambu T, Yanagisawa M, Goto K. Neurons containing orexin in the lateral hypothalamic area of the adult rat brain are activated by insulin-induced acute hypoglycemia. Neurosci Lett. 1999;264:101–104. doi: 10.1016/s0304-3940(99)00177-9. [DOI] [PubMed] [Google Scholar]
- Morison RS, Dempsey EW. A study of thalamo-cortical relations. Am J Physiol. 1942;135:281–292. [Google Scholar]
- Moruzzi G, Magoun HW. Brain stem reticular formation and activation of the EEG. Electroencephalogr Clin Neuro. 1949;1:455–473. [PubMed] [Google Scholar]
- Nauta WJH. Hypothalamic regulation of sleep in rats. An experimental study. J Neurophysiol. 1946;9:285–314. doi: 10.1152/jn.1946.9.4.285. [DOI] [PubMed] [Google Scholar]
- Nishino S, Mignot E. Pharmacological aspects of human and canine narcolepsy. Prog Neurobiol. 1997;52:27–78. doi: 10.1016/s0301-0082(96)00070-6. [DOI] [PubMed] [Google Scholar]
- Nishino S, Riehl J, Hong J, Kwan M, Reid M, Mignot E. Is narcolepsy a REM sleep disorder? Analysis of sleep abnormalities in narcoleptic Dobermans. Neurosci Res. 2000;38:437–446. doi: 10.1016/s0168-0102(00)00195-4. [DOI] [PubMed] [Google Scholar]
- Nofzinger EA, Buysse DJ, Germain A, Price JC, Miewald JM, Kupfer DJ. Functional neuroimaging evidence for hyperarousal in insomnia. Am J Psychiatry. 2004;161:2126–2128. doi: 10.1176/appi.ajp.161.11.2126. [DOI] [PubMed] [Google Scholar]
- Oishi Y, Huang ZL, Fredholm BB, Urade Y, Hayaishi O. Adenosine in the tuberomammillary nucleus inhibits the histaminergic system via A1 receptors and promotes non-rapid eye movement sleep. Proc Natl Acad Sci U S A. 2008;105:19992–19997. doi: 10.1073/pnas.0810926105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olafsdottir BR, Rye DB, Scammell TE, Matheson JK, Stefansson K, Gulcher JR. Polymorphisms in hypocretin/orexin pathway genes and narcolepsy. Neurology. 2001;57:1896–1899. doi: 10.1212/wnl.57.10.1896. [DOI] [PubMed] [Google Scholar]
- Overeem S, Lammers GJ, van Dijk JG. Weak with laughter. Lancet. 1999;354:838. doi: 10.1016/s0140-6736(99)80023-3. [DOI] [PubMed] [Google Scholar]
- Pace-Schott EF, Hobson JA. The neurobiology of sleep: genetics, cellular physiology and subcortical networks. Nat Rev Neurosci. 2002;3:591–605. doi: 10.1038/nrn895. [DOI] [PubMed] [Google Scholar]
- Palchykova S, Deboer T, Tobler I. Seasonal aspects of sleep in the Djungarian hamster. BMC Neurosci. 2003;4:9. doi: 10.1186/1471-2202-4-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan WJ, Osmanovic SS, Shefner SA. Characterization of the adenosine A1 receptor-activated potassium current in rat locus ceruleus neurons. J Pharmacol Exp Ther. 1995;273:537–544. [PubMed] [Google Scholar]
- Panula P, Pirvola U, Auvinen S, Airaksinen MS. Histamine-immunoreactive nerve fibers in the rat brain. Neuroscience. 1989;28:585–610. doi: 10.1016/0306-4522(89)90007-9. [DOI] [PubMed] [Google Scholar]
- Parent M, Descarries L. Acetylcholine innervation of the adult rat thalamus: distribution and ultrastructural features in dorsolateral geniculate, parafascicular, and reticular thalamic nuclei. J Comp Neurol. 2008;511:678–691. doi: 10.1002/cne.21868. [DOI] [PubMed] [Google Scholar]
- Peever JH, Lai YY, Siegel JM. Excitatory effects of hypocretin-1 (orexin-A) in the trigeminal motor nucleus are reversed by NMDA antagonism. J Neurophysiol. 2003;89:2591–2600. doi: 10.1152/jn.00968.2002. [DOI] [PubMed] [Google Scholar]
- Peyron C, Faraco J, Rogers W, Ripley B, Overeem S, Charnay Y, Nevsimalova S, Aldrich M, Reynolds D, Albin R, Li R, Hungs M, Pedrazzoli M, Padigaru M, Kucherlapati M, Fan J, Maki R, Lammers GJ, Bouras C, Kucherlapati R, Nishino S, Mignot E. A mutation in a case of early onset narcolepsy and a generalized absence of hypocretin peptides in human narcoleptic brains. Nat Med. 2000;6:991–997. doi: 10.1038/79690. [DOI] [PubMed] [Google Scholar]
- Peyron C, Tighe DK, van den Pol AN, de Lecea L, Heller HC, Sutcliffe JG, Kilduff TS. Neurons containing hypocretin (orexin) project to multiple neuronal systems. J Neurosci. 1998;18:9996–10015. doi: 10.1523/JNEUROSCI.18-23-09996.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips AJ, Robinson PA. A quantitative model of sleep-wake dynamics based on the physiology of the brainstem ascending arousal system. J Biol Rhythms. 2007;22:167–179. doi: 10.1177/0748730406297512. [DOI] [PubMed] [Google Scholar]
- Phillips AJ, Robinson PA. Sleep deprivation in a quantitative physiologically based model of the ascending arousal system. J Theor Biol. 2008;255:413–423. doi: 10.1016/j.jtbi.2008.08.022. [DOI] [PubMed] [Google Scholar]
- Phillips AJ, Robinson PA, Kedziora DJ, Abeysuriya RG. Mammalian sleep dynamics: how diverse features arise from a common physiological framework. PLoS Comput Biol. 2010;6:e1000826. doi: 10.1371/journal.pcbi.1000826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porkka-Heiskanen T, Strecker RE, Thakkar M, Bjorkum AA, Greene RW, McCarley RW. Adenosine: a mediator of the sleep-inducing effects of prolonged wakefulness. Science. 1997;276:1265–1268. doi: 10.1126/science.276.5316.1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posner JB, Saper CB, Schiff ND, Plum F. Plum and Posner’s Diagnosis of Stupor and Coma. New York: Oxford University Press; 2008. pp. 1–37. [Google Scholar]
- Qiu MH, Vetrivelan R, Fuller PM, Lu J. Basal ganglia control of sleep-wake behavior and cortical activation. Eur J Neurosci. 2010;31:499–507. doi: 10.1111/j.1460-9568.2009.07062.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radulovacki M, Virus RM, Djuricic-Nedelson M, Green RD. Adenosine analogs and sleep in rats. J Pharmacol Exp Ther. 1984;228:268–274. [PubMed] [Google Scholar]
- Ranson SW. Somnolence caused by hypothalamic lesions in monkeys. Arch Neurol Psychiatr. 1939;41:1–23. [Google Scholar]
- Rattenborg NC, Mandt BH, Obermeyer WH, Winsauer PJ, Huber R, Wikelski M, Benca RM. Migratory sleeplessness in the white-crowned sparrow (Zonotrichia leucophrys gambelii) PLoS Biol. 2004;2:E212. doi: 10.1371/journal.pbio.0020212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rechtschaffen A, Wolpert TEA, Dement WC, Mitchell SA, Fisher C. Nocturnal sleep of narcoleptics. Electroencephalogr Clin Neurophysiol. 1963;15:599–609. doi: 10.1016/0013-4694(63)90032-4. [DOI] [PubMed] [Google Scholar]
- Rempe MJ, Best J, Terman D. A mathematical model of the sleep/wake cycle. Journal of mathematical biology. 2009;60:616–644. doi: 10.1007/s00285-009-0276-5. [DOI] [PubMed] [Google Scholar]
- Reppert SM, Weaver DR. Coordination of circadian timing in mammals. Nature. 2002;418:935–941. doi: 10.1038/nature00965. [DOI] [PubMed] [Google Scholar]
- Saito H, Sakai K, Jouvet M. Discharge patterns of the nucleus parabrachialis lateralis neurons of the cat during sleep and waking. Brain Res. 1977;134:59–72. doi: 10.1016/0006-8993(77)90925-8. [DOI] [PubMed] [Google Scholar]
- Sakai K, Jouvet M. Brain stem PGO-on cells projecting directly to the cat dorsal lateral geniculate nucleus. Brain Res. 1980;194:500–505. doi: 10.1016/0006-8993(80)91231-7. [DOI] [PubMed] [Google Scholar]
- Sakurai T, Amemiya A, Ishii M, Matsuzaki I, Chernelli RM, Tanaka H, Williams SC, Richardson JA, Kozlowski GP, Wilson S, Arch JRS, Buckingham RE, Haynes AC, Carr SA, Annan RS, McNulty DE, Liu WS, Terrett JA, Eishourbahy NA, Bergsma DJ, Yangisawa M. Orexins and Orexin Receptors: A family of Hypothalamic Neuropeptides and G Protein-Coupled Receptors that Regulate Feeding Behavior. Cell. 1998;92:573–585. doi: 10.1016/s0092-8674(00)80949-6. [DOI] [PubMed] [Google Scholar]
- Saper CB. Organization of cerebral cortical afferent systems in the rat. II. Hypothalamocortical projections. J Comp Neurol. 1985;237:21–46. doi: 10.1002/cne.902370103. [DOI] [PubMed] [Google Scholar]
- Saper CB. Diffuse cortical projection systems: Anatomical organization and role in cortical function. In: Plum F, editor. Handbook of Physiology The Nervous System V. American Physiological Society; Bethesda: 1987. pp. 169–210. [Google Scholar]
- Saper CB, Chou TC, Scammell TE. The sleep switch: hypothalamic control of sleep and wakefulness. Trends Neurosci. 2001;24:726–731. doi: 10.1016/s0166-2236(00)02002-6. [DOI] [PubMed] [Google Scholar]
- Saper CB, Levisohn D. Afferent connections of the median preoptic nucleus in the rat: Anatomical evidence for a cardiovascular integrative mechanism in the anteroventral third ventricular (AV3V) region. Brain Res. 1983;288:21–31. doi: 10.1016/0006-8993(83)90078-1. [DOI] [PubMed] [Google Scholar]
- Saper CB, Loewy AD. Efferent connections of the parabrachial nucleus in the rat. Brain Res. 1980;197:291–317. doi: 10.1016/0006-8993(80)91117-8. [DOI] [PubMed] [Google Scholar]
- Saper CB, Lu J, Chou TC, Gooley J. The hypothalamic integrator for circadian rhythms. Trends Neurosci. 2005;28:152–157. doi: 10.1016/j.tins.2004.12.009. [DOI] [PubMed] [Google Scholar]
- Sapin E, Lapray D, Berod A, Goutagny R, Leger L, Ravassard P, Clement O, Hanriot L, Fort P, Luppi PH. Localization of the brainstem GABAergic neurons controlling paradoxical (REM) sleep. PLoS ONE. 2009;4:e4272. doi: 10.1371/journal.pone.0004272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sastre JP, Buda C, Kitahama K, Jouvet M. Importance of the ventrolateral region of the periaqueductal gray and adjacent tegmentum in the control of paradoxical sleepX as studied by muscimol microinjections in the cat. Neuroscience. 1996;74:415–426. doi: 10.1016/0306-4522(96)00190-x. [DOI] [PubMed] [Google Scholar]
- Sastre JP, Jouvet M. [Oneiric behavior in cats] Physiol Behav. 1979;22:979–989. doi: 10.1016/0031-9384(79)90344-5. [DOI] [PubMed] [Google Scholar]
- Satoh K, Fibiger HC. Cholinergic neurons of the laterodorsal tegmental nucleus: efferent and afferent connections. J Comp Neurol. 1986;253:277–302. doi: 10.1002/cne.902530302. [DOI] [PubMed] [Google Scholar]
- Savage VM, West GB. A quantitative, theoretical framework for understanding mammalian sleep. Proc Natl Acad Sci U S A. 2007;104:1051–1056. doi: 10.1073/pnas.0610080104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scammell TE. The frustrating and mostly fruitless search for an autoimmune cause of narcolepsy. Sleep. 2006;29:601–602. doi: 10.1093/sleep/29.5.601. [DOI] [PubMed] [Google Scholar]
- Scammell TE, Gerashchenko DY, Mochizuki T, McCarthy MT, Estabrooke IV, Sears CA, Saper CB, Urade Y, Hayaishi O. An adenosine A2a agonist increases sleep and induces Fos in ventrolateral preoptic neurons. Neuroscience. 2001;107:653–663. doi: 10.1016/s0306-4522(01)00383-9. [DOI] [PubMed] [Google Scholar]
- Sherin JE, Elmquist JK, Torrealba F, Saper CB. Innervation of histaminergic tuberomammillary neurons by GABAergic and galaninergic neurons in the ventrolateral preoptic nucleus of the rat. J Neurosci. 1998;18:4705–4721. doi: 10.1523/JNEUROSCI.18-12-04705.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherin JE, Shiromani PJ, McCarley RW, Saper CB. Activation of ventrolateral preoptic neurons during sleep. Science. 1996;271:216–219. doi: 10.1126/science.271.5246.216. [DOI] [PubMed] [Google Scholar]
- Shouse MN, Siegel JM. Pontine regultaion of REM sllep components in cats--integrity of the penduculopontine tegmentum (PPT) is important for phasic events but unnecessary for atonia during REM sleep. Brain Res. 1992;571:50–63. doi: 10.1016/0006-8993(92)90508-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel JM. Sleep viewed as a state of adaptive inactivity. Nat Rev Neurosci. 2009;10:747–753. doi: 10.1038/nrn2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starzl TE, Taylor CW, Magoun HW. Ascending conduction in reticular activating system, with special reference to the diencephalon. J Neurophysiol. 1951;14:461–477. doi: 10.1152/jn.1951.14.6.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steininger TL, Alam MN, Gong H, Szymusiak R, McGinty D. Sleep-waking discharge of neurons in the posterior lateral hypothalamus of the albino rat. Brain Res. 1999;840:138–147. doi: 10.1016/s0006-8993(99)01648-0. [DOI] [PubMed] [Google Scholar]
- Steriade M. Thalamic origin of sleep spindles: Morison and Bassett (1945) J Neurophysiol. 1995;73:921–922. doi: 10.1152/jn.1995.73.3.921. [DOI] [PubMed] [Google Scholar]
- Steriade M, McCormick DA, Sejnowski TJ. Thalamocortical oscillations in the sleeping and aroused brain. Science. 1993;262:679–685. doi: 10.1126/science.8235588. [DOI] [PubMed] [Google Scholar]
- Strecker RE, Morairty S, Thakkar MM, Porkka-Heiskanen T, Basheer R, Dauphin LJ, Rainnie DG, Portas CM, Greene RW, McCarley RW. Adenosinergic modulation of basal forebrain and preoptic/anterior hypothalamic neuronal activity in the control of behavioral state. Behav Brain Res. 2000;115:183–204. doi: 10.1016/s0166-4328(00)00258-8. [DOI] [PubMed] [Google Scholar]
- Suntsova N, Szymusiak R, Alam MN, Guzman-Marin R, McGinty D. Sleep-waking discharge patterns of median preoptic nucleus neurons in rats. J Physiol. 2002;543:665–677. doi: 10.1113/jphysiol.2002.023085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svenningsson P, Le MC, Kull B, Sunahara R, Bloch B, Fredholm BB. Cellular expression of adenosine A2A receptor messenger RNA in the rat central nervous system with special reference to dopamine innervated areas. Neuroscience. 1997;80:1171–1185. doi: 10.1016/s0306-4522(97)00180-2. [DOI] [PubMed] [Google Scholar]
- Swanson LW, Cwan WM. A note on the connections and development of the nucleus accumbens. Brain Res. 1975;92:324–330. doi: 10.1016/0006-8993(75)90278-4. [DOI] [PubMed] [Google Scholar]
- Swett CP, Hobson JA. The effects of posterior hypothalamic lesions on behavioral and electrographic manifestations of sleep and waking in cat. Arch Ital Biol. 1968;106:270–282. [PubMed] [Google Scholar]
- Szymusiak R, Alam N, Steininger TL, McGinty D. Sleep-waking discharge patterns of ventrolateral preoptic/anterior hypothalamic neurons in rats. Brain Res. 1998;803:178–188. doi: 10.1016/s0006-8993(98)00631-3. [DOI] [PubMed] [Google Scholar]
- Szymusiak R, McGinty D. Sleep-related neuronal discharges in the basal forebrain of cats. Brain Res. 1986;370:82–92. doi: 10.1016/0006-8993(86)91107-8. [DOI] [PubMed] [Google Scholar]
- Tafti M, Rondouin G, Besset A, Billiard M. Sleep deprivation in narcoleptic subjects: effect on sleep stages and EEG power density. Electroencephalogr Clin Neurophysiol. 1992a;83:339–349. doi: 10.1016/0013-4694(92)90069-t. [DOI] [PubMed] [Google Scholar]
- Tafti M, Villemin E, Carlander B, Besset A, Billiard M. Sleep in human narcolepsy revisited with special reference to prior wakefulness duration. Sleep. 1992b;15:344–351. doi: 10.1093/sleep/15.4.344. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Kayama Y, Lin JS, Sakai K. Locus Coeruleus Neuronal Activity During the Sleep-Waking Cycle in Mice. Neuroscience. 2010 doi: 10.1016/j.neuroscience.2010.06.009. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Lin JS, Sakai K. Neuronal activity of histaminergic tuberomammillary neurons during wake-sleep states in the mouse. J Neurosci. 2006;26:10292–10298. doi: 10.1523/JNEUROSCI.2341-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Lin JS, Sakai K. Characterization and mapping of sleep-waking specific neurons in the basal forebrain and preoptic hypothalamus in mice. Neuroscience. 2009;161:269–292. doi: 10.1016/j.neuroscience.2009.02.075. [DOI] [PubMed] [Google Scholar]
- Thannickal TC, Moore RY, Nienhuis R, Ramanathan L, Gulyani S, Aldrich M, Cornford M, Siegel JM. Reduced number of hypocretin neurons in human narcolepsy. Neuron. 2000;27:469–474. doi: 10.1016/s0896-6273(00)00058-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson R, Swanson LW, Canteras N. Organization of Projections From the Dorsomedial Nucleus of the Hypothalamus: A PHA-L Study in the Rat. The Journal of Comparative Neurology. 1997;376:143–173. doi: 10.1002/(SICI)1096-9861(19961202)376:1<143::AID-CNE9>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Torrealba F, Yanagisawa M, Saper CB. Colocalization of orexin a and glutamate immunoreactivity in axon terminals in the tuberomammillary nucleus in rats. Neuroscience. 2003;119:1033–1044. doi: 10.1016/s0306-4522(03)00238-0. [DOI] [PubMed] [Google Scholar]
- Trulson ME, Jacobs BL, Morrison AR. Raphe unit acitivity during REM sleep in normal cats and in pontine lesioned cats displaying REM sleep without atonia. Brain Res. 1981;226:75–91. doi: 10.1016/0006-8993(81)91084-2. [DOI] [PubMed] [Google Scholar]
- Uschakov A, Gong H, McGinty D, Szymusiak R. Efferent projections from the median preoptic nucleus to sleep- and arousal-regulatory nuclei in the rat brain. Neuroscience. 2007;150:104–120. doi: 10.1016/j.neuroscience.2007.05.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Bockstaele EJ, Peoples J, Valentino RJ. A.E. Bennett Research Award. Anatomic basis for differential regulation of the rostrolateral peri-locus coeruleus region by limbic afferents. Biol Psychiatry. 1999;46:1352–1363. doi: 10.1016/s0006-3223(99)00213-9. [DOI] [PubMed] [Google Scholar]
- Van Dongen HP, Maislin G, Mullington JM, Dinges DF. The cumulative cost of additional wakefulness: dose-response effects on neurobehavioral functions and sleep physiology from chronic sleep restriction and total sleep deprivation. Sleep. 2003;26:117–126. doi: 10.1093/sleep/26.2.117. [DOI] [PubMed] [Google Scholar]
- Vanderwolf CH, Stewart DJ. Thalamic control of neocortical activation: A critical re-evaluation. Brain Res Bull. 1988;20:529–538. doi: 10.1016/0361-9230(88)90143-8. [DOI] [PubMed] [Google Scholar]
- Verret L, Fort P, Gervasoni D, Leger L, Luppi P-H. Localization of the neurones potentially responsible for the inhibition of Locus Coeruleus noradrenergic neurones during paradoxical sleep in the rat. J Comp Neurol. 2005;495:573–586. doi: 10.1002/cne.20891. [DOI] [PubMed] [Google Scholar]
- Verret L, Goutagny R, Fort P, Cagnon L, Salvert D, Leger L, Boissard R, Salin P, Peyron C, Luppi PH. A role of melanin-concentrating hormone producing neurons in the central regulation of paradoxical sleep. BMC Neurosci. 2003;4:19. doi: 10.1186/1471-2202-4-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetrivelan R, Fuller PM, Tong Q, Lu J. Medullary circuitry regulating rapid eye movement sleep and motor atonia. J Neurosci. 2009;29:9361–9369. doi: 10.1523/JNEUROSCI.0737-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villablanca J, Salinas-Zeballos ME. Sleep-wakefulness, EEG and behavioral studies of chronic cats without the thalamus: the ‘athalamic’ cat. Arch Ital Biol. 1972;110:383–411. [PubMed] [Google Scholar]
- Volgin DV, Saghir M, Kubin L. Developmental changes in the orexin 2 receptor mRNA in hypoglossal motoneurons. Neuroreport. 2002;13:433–436. doi: 10.1097/00001756-200203250-00014. [DOI] [PubMed] [Google Scholar]
- Von Economo C. Sleep as a problem of localization. J Nerv Ment Dis. 1930;71:249–259. [Google Scholar]
- Vyazovskiy V, Borbely AA, Tobler I. Unilateral vibrissae stimulation during waking induces interhemispheric EEG asymmetry during subsequent sleep in the rat. J Sleep Res. 2000;9:367–371. doi: 10.1046/j.1365-2869.2000.00230.x. [DOI] [PubMed] [Google Scholar]
- Vyazovskiy VV, Olcese U, Lazimy YM, Faraguna U, Esser SK, Williams JC, Cirelli C, Tononi G. Cortical firing and sleep homeostasis. Neuron. 2009;63:865–878. doi: 10.1016/j.neuron.2009.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts AG, Swanson LW, Sanchez-Watts G. Efferent projections of the suprachiasmatic nucleus: I. Studies using anterograde transport of Phaseolus vulgaris leucoagglutinin in the rat. J Comp Neurol. 1987;258:204–229. doi: 10.1002/cne.902580204. [DOI] [PubMed] [Google Scholar]
- Webster HH, Jones BE. Neurotoxic lesions of the dorsolateral pontomesencephalic tegmentum-cholinergic cell area in the cat. II. Effects upon sleep-waking states. Brain Res. 1988;458:285–302. doi: 10.1016/0006-8993(88)90471-4. [DOI] [PubMed] [Google Scholar]
- Westphal C. Eigenthümlichemit einschläfen verbundene anfälle. Arch Psychiat. 1877;7:631–635. [Google Scholar]
- Willie JT, Sinton CM, Maratos-Flier E, Yanagisawa M. Abnormal response of melanin-concentrating hormone deficient mice to fasting: hyperactivity and rapid eye movement sleep suppression. Neuroscience. 2008;156:819–829. doi: 10.1016/j.neuroscience.2008.08.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winsky-Sommerer R, Boutrel B, de LL. Stress and arousal: the corticotrophin-releasing factor/hypocretin circuitry. Mol Neurobiol. 2005;32:285–294. doi: 10.1385/MN:32:3:285. [DOI] [PubMed] [Google Scholar]
- Wisor JP, O’Hara BF, Terao A, Selby CP, Kilduff TS, Sancar A, Edgar DM, Franken P. A role for cryptochromes in sleep regulation. BMC Neurosci. 2002;3:20. doi: 10.1186/1471-2202-3-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright KP, Jr, Badia P, Wauquier A. Topographical and temporal patterns of brain activity during the transition from wakefulness to sleep. Sleep. 1995;18:880–889. doi: 10.1093/sleep/18.10.880. [DOI] [PubMed] [Google Scholar]
- Wu MF, Gulyani SA, Yau E, Mignot E, Phan B, Siegel JM. Locus coeruleus neurons: cessation of activity during cataplexy. Neuroscience. 1999;91:1389–1399. doi: 10.1016/s0306-4522(98)00600-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu MF, John J, Boehmer LN, Yau D, Nguyen GB, Siegel JM. Activity of dorsal raphe cells across the sleep-waking cycle and during cataplexy in narcoleptic dogs. J Physiol. 2004;554:202–215. doi: 10.1113/jphysiol.2003.052134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka A, Beuckmann CT, Willie JT, Hara J, Tsujino N, Mieda M, Tominaga M, Yagami K, Sugiyama F, Goto K, Yanagisawa M, Sakurai T. Hypothalamic orexin neurons regulate arousal according to energy balance in mice. Neuron. 2003;38:701–713. doi: 10.1016/s0896-6273(03)00331-3. [DOI] [PubMed] [Google Scholar]
- Yamuy J, Fung SJ, Xi M, Chase MH. Hypocretinergic control of spinal cord motoneurons. J Neurosci. 2004;24:5336–5345. doi: 10.1523/JNEUROSCI.4812-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida K, McCormack S, Espana RA, Crocker A, Scammell TE. Afferents to the orexin neurons of the rat brain. J Comp Neurol. 2006;494:845–861. doi: 10.1002/cne.20859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler DR, Cullinan WE, Herman JP. Distribution of vesicular glutamate transporter mRNA in rat hypothalamus. J Comp Neurol. 2002;448:217–229. doi: 10.1002/cne.10257. [DOI] [PubMed] [Google Scholar]
- Zikopoulos B, Barbas H. Circuits formultisensory integration and attentional modulation through the prefrontal cortex and the thalamic reticular nucleus in primates. Rev Neurosci. 2007;18:417–438. doi: 10.1515/revneuro.2007.18.6.417. [DOI] [PMC free article] [PubMed] [Google Scholar]