

Abstract
The precise biological mechanisms that caused the TGN1412 clinical trial tragedy (also known as ‘The Elephant Man Clinical Trial’) in March 2006 remain a mystery to this day. It is assumed widely that the drug used in this trial (TGN1412) bound to CD28 on T lymphocytes and following activation of these cells, a massive ‘cytokine storm’ ensued, leading ultimately to multi-organ failure in all recipients. The rapidity of this in vivo response (within 2 h), however, does not fit well with a classical T lymphocyte response, suggesting that other ‘faster-acting’ cell types may have been involved. In this study we have activated purified human peripheral blood leucocyte populations using various clones of mouse monoclonal anti-CD28 presented to cells in the form of a multimeric array. Cytokines were measured in cell-free supernatants at 2 h, and specific mRNA for tumour necrosis factor (TNF)-α, thought to be the initiator of the cytokine storm, was also measured in cell lysates by reverse transcription–polymerase chain reaction (RT–PCR). Monocytes were the only cell type found to show significant (P < 0·05) up-regulation of TNF-α at 2 h. Eleven other monocyte cytokines were also up-regulated by anti-CD28 within this time-frame. It therefore seems likely that monocytes and not T cells, as widely believed, were probably responsible, at least in part, for initiating the cytokine storm. Furthermore, we propose that a multimeric antibody array may have formed in vivo on the vascular endothelium via an interaction between TGN1412 and CD64 (FcγRI), and we provide some evidence in support of this hypothesis.
Keywords: CD28, clinical trial, cytokine storm, monocyte, TGN1412
Introduction
In March 2006, eight healthy male volunteers took part in a Phase 1 clinical trial at Northwick Park Hospital, London. The drug under study, TGN1412, was a humanized (IgG4κ) monoclonal antibody specific for CD28; an important signalling molecule regarded widely as T cell-specific. Preclinical testing, both in vitro and in vivo, predicted that this antibody would stimulate the release of anti-inflammatory cytokines and expand regulatory T cells (Treg). For this reason TGN1412 was thought to have potential therapeutic value in the treatment of various autoimmune disorders and chronic lymphocytic leukaemia [1].
Unlike conventional anti-CD28 mouse monoclonal antibodies, TGN1412 binds to a particular domain (C′′D loop) close to the T cell membrane and appears to bypass the usual requirement for co-stimulatory signals and results in direct cell activation, i.e. a ‘superagonistic’ effect [2,3].
The clinical trial proved to be a disaster, with all six recipients of TGN1412 developing symptoms rapidly consistent with an acute systemic inflammatory response [4]. Two recipients of placebo were unaffected. It was concluded retrospectively that intravenous infusion of TG1412 had triggered a ‘cytokine storm’ initiated by the rapid release (within 1–2 h) of tumour necrosis factor (TNF)-α[4].
Following an official investigation [5], studies to determine causality reported that addition of TGN1412 in suspension over a wide range of concentrations to whole human blood or to isolated peripheral blood mononuclear cells (PBMCs) in vitro did not result in T cell activation, nor did it stimulate production of proinflammatory cytokines, including TNF-α[6]. Only when peripheral blood mononuclear cells (PBMCs) were exposed to TGN1412 that had been immobilized (‘dry-linked’) onto a plastic surface, or alternatively bound indirectly via an anti-human immunoglobulin G (IgG)-Fc region capture method, was there T cell activation and production of proinflammatory cytokines; but even then, only within 16–24 h [6]. TGN1412 therefore appears to be partly effective only when presented to cells in the form of a multimeric array (MMA). Precisely how such an array could form in vivo is not known.
The cell type(s) involved in triggering the inflammatory response in this trial have not been defined clearly. Because CD28 is found predominantly on the surface of T cells it has been assumed widely that these cells were the main target for TGN1412 and the source of proinflammatory cytokines. However, the swift onset of clinical symptoms and rapid rise in plasma cytokine levels, in particular TNF-α, has cast doubt on the central role of T cells, known to respond relatively slowly to a foreign stimulus [7]. It has been proposed, therefore, that other cell types may have been involved in the initiation of the cytokine storm [7]. Monocytes and granulocytes, for example, are known to synthesize many cytokines actively, including TNF-α, and granulocytes have been shown to have preformed stores of many cytokines, thus allowing for rapid responses [8–11].
The aims of this study, therefore, were to answer the following questions.
Which cell type(s), interacting with anti-CD28 in the form of a multimeric array, was responsible for the rapid release of TNF-α thereby initiating the cytokine storm?
How could TGN1412 possibly form an immunostimulatory multimeric array in vivo?
To answer these questions we developed an in vitro model system using purified human peripheral blood lymphocytes, granulocytes and monocytes that were incubated for 2 h with anti-CD28 antibody linked to plastic. Furthermore, we added anti-CD28 opsonized leucocytes to a substrate of human vascular endothelial cells in an attempt to cross-link CD28 in a more natural system. In order to achieve this, we used one superagonist anti-CD28 (clone ANC28.1) that behaves in an identical manner to TGN1412 in vitro[12], and for comparison we used two non-superagonistic murine anti-CD28 clones (204.12 and L293).
Materials and methods
Isolation of human peripheral blood leucocytes
Mononuclear cells (lymphocytes + monocytes)
Whole heparinized blood was obtained, with signed informed ethical consent (Greater Glasgow and Clyde NHS West Ethics Committee – Ref. no. 09/S0709/16), from 10 healthy subjects: four male and six female (mean age = 40) years. Mononuclear cells were isolated using a standard lymphoprep method and suspended in Iscove's Dulbecco's modified medium (IDMM) +l-glutamine, +25 mm HEPES (Gibco BRL, Invitrogen, Paisley, Scotland, UK) culture medium supplemented with 20% heat-inactivated (56°C for 30 min) fetal calf serum (I + S medium) at a final concentration of 5 × 106 cells/ml. Cells were then added to plastic tissue culture plates (Falcon 24-well plates) using 1 ml/well and incubated at 37°C for 2 h in 5% CO2. Non-adherent cells (lymphocytes) were washed free (observed using an inverted microscope) leaving adherent cells (monocytes) attached to plastic. The purity of the lymphocyte preparations (97–100%) was confirmed by flow cytometry immunophenotyping [Becton-Dickinson fluorescence activated cell sorter (FACScalibur, Becton Dickinson (UK) Ltd, Oxford, England, UK)]. Cell surface and cytoplasmic staining was conducted as described previously [13]. The following antibodies were used for phenotyping: from Dako Ltd (Cambridgeshire, UK), Dual-Tag CD45-fluorescein isothiocyanate (FITC)/CD14-RPE, FITC-conjugated anti-CD66abce (clone Kat4c) and CD70 (clone HNE.51); from Caltag Medsystems (Towcester, UK), FITC-conjugated anti-CD4 (clone S3·5), CD16 (clone 3G8), CD27 (clone CLB27/1), CD28 (clone 15E8), CD64 (clone 10.1), CD80 (clone MEM 233), CD86 (clone BU63) and major histocompatibility complex (MHC) class II(DR) (clone TU36); from BD Pharmingen UK (Oxford, UK), FITC-anti-CD152 (clone BN13); and from Serotec (Oxford, UK), FITC-anti-CD32 (clone AT10).
Granulocyte preparations were contaminated by a small proportion of mononuclear cells consisting of a mixture of monocytes, T cells, B cells and natural killer (NK) cells. Peripheral blood lymphocyte preparations were virtually 100% pure. Monocytes were adherent to plastic and all non-adherent lymphocytes were removed by extensive washings. Purity (> 97%) was confirmed by morphological appearance [haematoxylin and eosin (H&E) stain] and by immunocytochemistry, using anti-CD14 as a marker.
Appropriate mouse IgG isotype controls obtained from the same manufacturers were used to set background thresholds.
Granulocytes
Granulocytes were recovered from the bottom of lymphoprep gradients using a standard dextran sedimentation protocol [13]. Contaminating erythrocytes were removed by hypotonic lysis. Granulocytes were 97% pure, as confirmed by immunophenotyping using a monoclonal antibody specific for CD66abce (Dako Ltd).
Activation of leucocytes by cross-linking (X-L) cell surface CD28
This method was conducted as described by Woerly et al. [14]. Wells of Falcon (24-well) plastic plates were pretreated with 125 µl goat F(ab')2 anti-mouse IgG at 40 µg/ml in phosphate-buffered saline (PBS) (GAM; AbD Serotec, Oxford, UK) for 2 h at 37°C, and following two washes in PBS were treated with mouse monoclonal [anti-CD28 – clone 204.12 from Caltag Medsystems or ANC28.1/5D10 from Axxora (UK) Ltd, Nottingham, UK] at 10 µg/ml PBS for a further 2 h at 37°C and washed twice in PBS prior to addition of cells in I + S culture medium. Granulocytes and lymphocytes were added at a concentration of 2 × 106 cells/well in 1 ml medium. For monocytes, the PBMCs were added at 5 × 106/well and following incubation (37°C/2 h), free cells (lymphocytes) were removed by repeated washings, leaving only adherent monocytes (confirmed by morphological observation using an inverted microscope) attached to the plastic surface.
Flow cytometry dot-plots of forward- versus side-scatter also confirmed that cells recovered from plastic plates were depleted of all monocytes. Cells incubated on plastic alone (presaturated at 37°C/2 h with I + S medium alone) were used to provide a negative control for wells coated with anti-CD28. Furthermore, wells coated with goat anti-mouse IgG (GAM) alone, or GAM + mouse IgG1 or IgG2a (Sigma; mouse myeloma protein at 100 µg/ml) in place of specific anti-CD28 monoclonal antibodies, provided additional negative controls. Following incubation on a plastic surface at 37°C in 5% CO2, cells (granulocytes or lymphocytes) were removed by repeated washings and processed for flow cytometry. Cell surface expression of CD28 and CD66 was measured relative to time zero control. Similarly, mRNA specific for CD28 and TNF-α was quantified by reverse transcription–polymerase chain reaction (RT–PCR). Cytospin preparations of these cells were stained with haematoxylin and eosin (H&E) for morphology studies and for fluorescence microscopy using FITC-anti-CD66 or FITC-anti-CD28. Sections were mounted in Vectashield mounting medium (Vector Laboratories, Peterborough, UK) supplemented with 4′,6-diamidino-2-phenylindole (DAPI; 0·2 mg/ml) and the coverslips sealed with nail varnish. Cells were then inspected using an Olympus IX51 inverted fluorescence microscope at a magnification of ×600.
Adherent monocytes were treated directly with Trizol and RNA extracted as described. Supernatants from these cell cultures were retained and centrifuged at 400 g for 5 min (to remove any cells) and the level of soluble TNF-α measured by enzyme-linked immunosorbent assay (ELISA).
Multiplex cytokine assay
The concentrations of mediators were quantified simultaneously using a sensitive cytokine thirty-plex antibody bead kit (lot no. 497124) from Invitrogen Ltd (Paisley, UK). This is a validated kit with sensitivity levels available online on the kit information sheet (catalogue no LHC6003). The assay was a standard Luminex fluoroimmunoassay, and activity was measured by Bio-Plex System platform (Bio-Rad Laboratories Ltd, Hemel Hempstead, UK). Samples were tested in duplicate and concentrations were calculated by interpolation into a standard curve of known values using National Institute for Biological Standards and Control (NIBSC) calibration standards.
Mediators were subdivided broadly into four groups based on the known functions of these molecules.
Proinflammatory: interleukin (IL)-1β, IL-5, IL-6, IL-12p40/70, IL-13, IL-15, IL-17, TNF-α, interferon (IFN)-γ.
Regulatory: IL-1RA, IL-2R, IL-4, IL-10, IFN-α.
Chemokines: IL-8(CXCL8), macrophage inflammatory protein (MIP)-1α(CCL3), MIP-1β(CXCL4L1), IFN-γ-induced protein (IP)-10(CXCL10), monokine induced by IFN-γ (MIG)(CXCL9), eotaxin(CCL11), regulated upon activation normal T cell expressed and secreted (RANTES)(CCL5), monocyte chemoattractant protein (MCP)-1(CCL2).
Growth factors: IL-2, IL-7, vascular endothelial growth factor (VEGF), granulocyte–macrophage colony-stimulating factor (GM-CSF), G-CSF, EGF, fibroblast growth factor (FGF)-basic and hepatocyte growth factor (HGF).
Standard ELISA
ELISA for TNF-α protein in cell-free supernatants was performed using Biosource human TNF-α CytoSet™ according to the manufacturer's instructions. Samples were added in duplicate and optical densities were compared to a standard curve produced from recombinant human TNF-α to obtain protein concentrations.
Detection of mRNA by quantitative RT–PCR
Design of primers
Primer sequences (sense and anti-sense) were designed using the Universal Probe library assay designer on the Roche Applied Science website.
Primer sequences used in this study were as follows:
| Specific mRNA | Primer | Sequence | Probe |
|---|---|---|---|
| Housekeeping beta-actin | Sense | ATTGGCAATGAGCGGTTC | 11 |
| Reverse | GGATGCCACAGGACTCCAT | ||
| TNF-α | Sense | CAGCCTCTTCTCCTTCCTGAT | 29 |
| Reverse | GCCAGAGGGCTGATTAGAGA | ||
| CD28 exon 3 | Sense | ATTTCCCGGACCTTCTAAGC | 28 |
| Reverse | CTATAGCAAGCCAGGACTCCA |
Extraction of RNA and production of cDNA
RNA was extracted from purified leucocytes using a standard protocol. Cells were lysed in 1 ml Trizol (containing phenol; Sigma) and stored at −80°C until required. RNA was extracted using chloroform followed by chloroform/phenol (pH6·6) extraction and precipitation with isopropanol at −20°C. The dried nucleic acids were resuspended in molecular grade water. DNA was removed from the samples using DNA-free kit (Ambion, Warrington, England, UK), and the RNA concentration quantified using NanoDrop. cDNA was synthesized in 20 µl reactions from 1 µg RNA using transcriptor reverse transcriptase (Roche Applied Science, West Sussex, England, UK), 1 µg random primers (Invitrogen), 10 mm deoxyribonucleotide triphosphate (dNTPs) (Invitrogen Ltd, Paisley, Scotland, UK), RNase OUT (Invitrogen) and 0·6 µl dimethylsulphoxide (DMSO) (Sigma). cDNA was stored at −20°C until required for RT–PCR assay.
RT–PCR assay
RT–PCR was performed using a standard Roche Universal probe library assay. In each well, 1 µl of cDNA was added to 1 µl of each primer (7·2 µm, sense and reverse), 1 µl of corresponding probe (2 µm), 10 µl of Roche Taq LC480 mastermix and water to 20 µl. Sealed plates were placed in a Roche Light Cycler. Results for the expression of each gene were reported as percentage of beta-actin. Negative controls were included where no enzyme (RT) was added.
Human umbilical vein endothelial cells (HUVECs)
HUVECs were obtained from Lonza Pharmaceuticals (Basel, Switzerland) and cultured according to the manufacturer's instructions. These cells were pooled originally from 10 donors.
Granulocytes and PBMCs were incubated separately with HUVECs adherent to 12-well tissue plastic culture plates (Costar®) at 37°C for 2 h and 18 h. Untreated cells were compared with cells preincubated with mouse monoclonal anti-CD28 (clone 204.12) or with an appropriate isotype control (mouse IgG2a) at the same concentration. Supernatants were collected for cytokine analysis.
We chose clone 204.12 for these experiments because it is an IgG2a class murine antibody, and would bind to human CD64 much more effectively than the mouse IgG1 class ANC28.1.
Human CD64 binds human IgG3>IgG1>IgG4>>>IgG2
Human CD64 binds murine IgG3>IgG2a>>>IgG1, IgG2b http://www.sciencegateway.org/resources/prow/guide/1336793022_g.htm
Statistical analysis
For the purposes of this study, a non-parametric Wilcoxon's two-sample test was used in all comparisons, with a P-value of < 0·05 regarded as significant. In the multiplex assay the level of each cytokine was analysed separately relative to the control values. A cytokine was considered to be up-regulated significantly when the test value was elevated relative to the appropriate isotype control value. The level of significance was set at P < 0·05; no correction was made for multiple testing.
Results
Cross-linking leucocyte CD28 induces cytokine release
Highly purified (> 98%) lymphocytes, granulocytes and monocytes were isolated and incubated separately at 37°C for 2 h with mouse monoclonal anti-CD28 (clones ANC28.1, L293 and 202.12) bound to tissue-culture wells as described in Methods. Thirty mediators were measured in cell-free supernatants using a multiplex assay.
Monocytes showed significantly elevated levels of TNF-α, IL-1β, IL-17, IL-2R and MIP-1α (CCL3) within 2 h relative to background isotype control values with all three clones of anti-CD28 (Table 1). In addition, clones ANC28.1 ‘superagonist’ and L293 both up-regulated monocyte IL-8(CXCL8) and clone ANC28.1 up-regulated monocyte IL-4, IL-6 and MIP-1β, while clone L293 additionally up-regulated IL-12, IL-15 and RANTES(CCL5). Taken together, a total of 12 of 22 proinflammatory or regulatory cytokines or chemokines were up-regulated by monocytes in response to an anti-CD28 stimulus within 2 h. No significant increase in any of the eight growth factors measured was observed – see Supplementary Table S1a.
Table 1.
Cytokines produced by monocytes (from 1 × 106 cells) following cross-linking (X–L) of CD28 at 37°C for 2 h
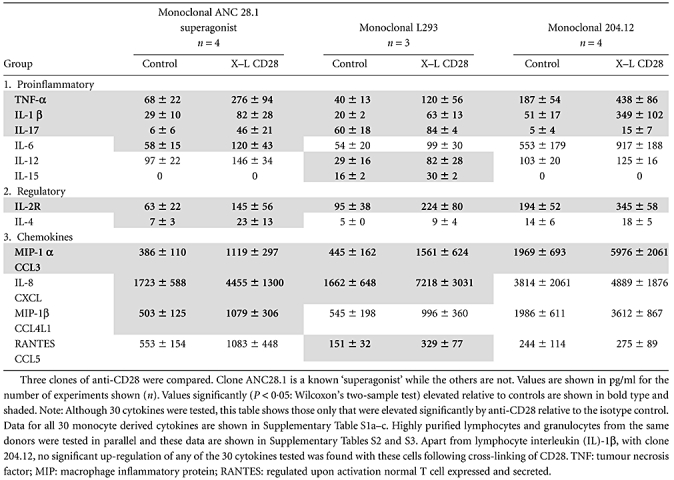 |
In parallel experiments using highly purified lymphocytes or granulocyes from the same donors no significant up-regulation of any of the 30 mediators tested was observed – see Supplementary Table S1b,c. Furthermore, in some experiments we used PBMCs and peripheral blood leucocytes (PBL) in parallel from the same donors, and it was evident that cytokines were produced from PBMCs but not from purified PBL (data not shown). The increase in cytokine levels relative to control antibody treatment and the difference between cytokine levels produced by PBMC and monocyte-depleted PBL make it unlikely that any preactivation during monocyte preparation could explain the differences seen.
To validate the multiplex TNF-α results, this experiment was repeated and the TNF-α protein and mRNA was measured in parallel by ELISA and RT–PCR. As shown in Table 2, significant up-regulation of TNF-α, protein and mRNA was observed only with monocytes.
Table 2.
Up-regulation of tumour necrosis factor (TNF)-α by anti-CD28: comparison of protein and mRNA
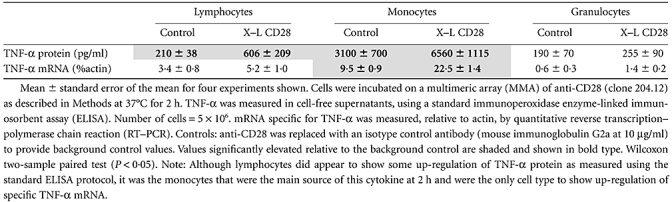 |
Monocytes therefore appear to have the capacity to actively synthesize TNF-αin vitro at equivalent concentrations and within a similar 2-h time-frame of the clinical trial, the only caveat being that anti-CD28 had to be presented to these cells in the form of a multimeric array.
If monocytes express very low levels of cell surface CD28 this may be sufficient to achieve cross-linking when presented to anti-CD28 in the form of a multimeric array and may also explain why anti-CD28, when added to cells in suspension, fails to elicit a cytokine response in vitro, i.e. insufficient densities for cross-linking to occur [6].
For this reason we therefore conducted flow cytometric analysis to determine whether or not monocytes have CD28 on the cell surface. Furthermore, we also looked for evidence for intracellular expression of CD28, the rationale being that rapid translocation from cryptic ‘intracellular stores’ onto the cell surface, following cell activation, could amplify the opportunity for binding of anti-CD28 and consequently boost cytokine synthesis.
Detection of leucocyte cell surface and cytoplasmic CD28 by flow cytometry and by fluorescence microscopy
CD28 was detected by flow cytometry before and after fixation and permeabilization of human peripheral blood leucocytes, obtained from 10 healthy donors, to demonstrate surface and cytoplasmic expression, respectively. The presence of cytoplasmic CD28 was demonstrated by a significant increase in net mean fluorescence intensity (MFI) following permeabilization relative to the net surface MFI.
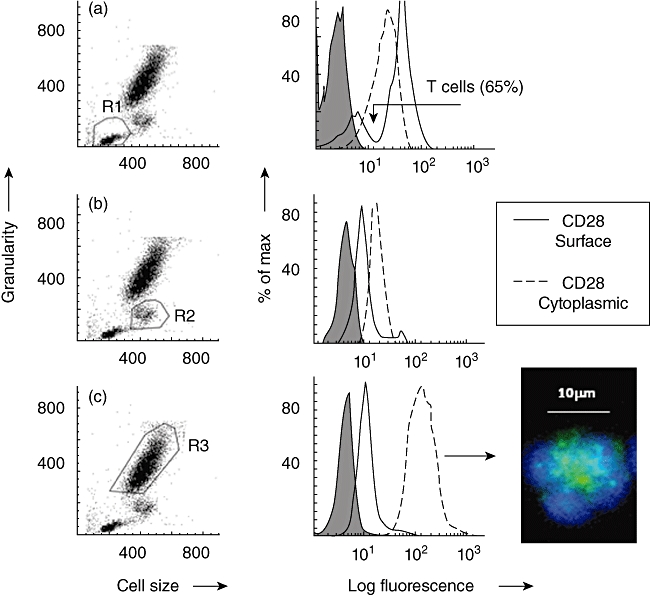
From flow cytometry dot-plots of cell size versus granularity, lymphocytes, monocytes and granulocytes were gated as regions 1, 2 and 3, respectively (R1–R3) (Fig. 1).
Fig. 1.
Demonstration of surface and cytoplasmic staining for CD28 by flow cytometry. Typical examples are shown as histogram overlays with an appropriate isotype control (shown as a grey infill) used to provide background values. (a) Dot-plot of forward-scatter (cell size) versus side-scatter (granularity) showing lymphocytes gated as region 1 (R1). The histogram overlay shows that CD28 was expressed on a distinct subpopulation of lymphocytes as indicated by the arrow i.e. T cells. (b) Dot-plot of forward- versus side-scatter showing monocytes gated as region 2 (R2). (c) Dot-plot of forward- versus side-scatter showing neutrophils gated as region 3 (R3). Cytoplasmic staining: cell surface staining (solid line) of viable cells was compared to staining following fixation and permeabilization of cells (dotted line). These experiments were conducted on leucocytes obtained from 10 healthy donors. Net mean fluorescence intensity (MFI) are shown below. A significant (Wilcoxon two-sample test) increase in net MFI following fixation and permeabilization, relative to surface values, indicates the presence of cytoplasmic ‘stores’ of CD28. Inspection of permeabilized granulocytes, showing distinctive polymorphonuclear morphology, by fluorescence microscopy confirmed that cytoplasmic CD28 (green) was confined to granules. Nuclear staining (blue) was demonstrated using 4′,6-diamidino-2-phenylindole (DAPI). No significant ‘stores’ were detected within T cells or monocytes. Data obtained from 10 healthy donors are shown below. Average MFI ± standard error of the mean shown
| CD28 net MFI for 10 donors | Lymphocytes (T cells) | Monocytes | Granulocytes |
|---|---|---|---|
| Control (surface) | 29·2 ± 5·2 | 6 ± 1·2 | 7·3 ± 1·2 |
| Fixed & Permeabilized (cytoplasmic) | 14·1 ± 3·4 | 9 ± 1·9 | 153 ± 37 P < 0·05 |
Mononuclear cells
CD28 was detected by flow cytometry using FITC-conjugated antibody (clone 15E8), as illustrated in the parallel histograms shown in Fig. 1. In agreement with the published literature, a distinct lymphocyte subpopulation expressing CD28 was identified in all 10 healthy donors with an average net MFI value [± standard error of the mean (s.e.m.)] of 29·2 ± 5·2, range 9–57. In contrast, monocytes (R2) showed very weak surface expression of CD28 (net MFI = 6 ± 1·2, range 2–13). Following cell permeabilization, no significant increase in CD28 staining was observed, either by flow cytometry or by fluorescence microscopy, suggesting that mononuclear cells do not appear to have cytoplasmic stores of CD28 (see Fig. 1a and b).
Granulocytes
Granulocytes were gated as region 3 (R3), as shown in Fig. 1c. These cells also showed very weak surface staining for CD28, with an average net MFI of 7·3 ± 1·2, range 5–31. Following fixation and permeabilization of granulocytes, a significant increase in CD28 detection was observed (net MFI = 153 ± 37, range 35–425, P < 0·05) (see Fig. 1c). Visualization by fluorescence microscopy confirmed that CD28 was located within neutrophil cytoplasmic granules (Fig. 1, panel).
This observation of cytoplasmic CD28 within granulocytes was unexpected, as CD28 is regarded as T cell-specific, and for this reason was investigated further.
Following cross-linking of CD28, granulocytes showed a rapid translocation of cytoplasmic CD28 onto the cell surface in the form of large heterogeneous clusters. This was not observed with lymphocytes from the same donors (Fig. 2). Kinetic studies showed that increased surface expression of CD28 occurred rapidly and was accompanied by increased surface expression of the granulocyte activation marker CD66 (Fig. 3). Granulocyte activation, induced by CD28 cross-linking, was also noted to produce a striking change in neutrophil nuclear morphology, known as the pseudo-Pelger–Huët anomaly, which manifests as a reduction in the number of nuclear lobes (Fig. 4). These in vitro observations are important, because this nuclear anomaly was observed in all recipients of TGN1412 [4].
Fig. 2.
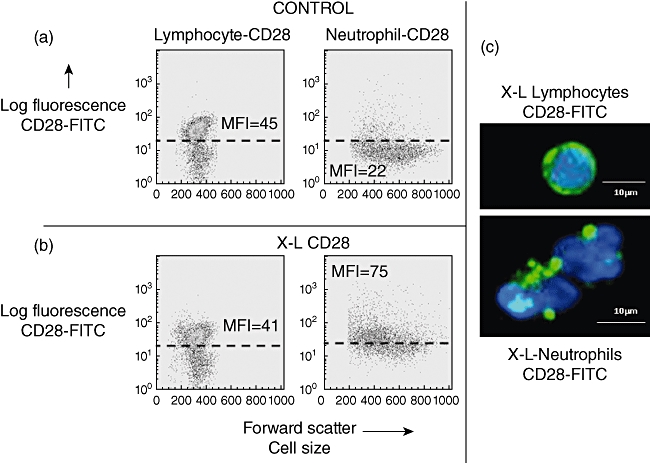
Effect of cross-linking (X–L) CD28 on cell surface expression of CD28. (a) Flow cytometry dot-plots of forward-scatter (cell size) versus log fluorescence [fluorescein isothiocyanate (FITC)-anti CD28, clone15E8] shows baseline values for lymphocytes and neutrophils. CD28 was detected on a distinct subpopulation of cells (T cells) as indicated by the population appearing above the dotted line with a mean fluorescence intensity (MFI) of 45. In contrast, all neutrophils in this example showed very weak surface staining for CD28 (MFI = 22). (b) Flow cytometry dot-plots of forward-scatter (cell size) versus log fluorescence (FITC-anti CD28, clone 15E8) shows the effect of cross-linking CD28 at 37°C for 1 h. No change was observed in the MFI for CD28 on the surface of T cells. There was a striking increase, however, in surface staining for CD28 on all neutrophils (MFI = 75). (c) Cells visualized by fluorescence microscopy using FITC-anti-CD28 (green) and counterstained with 4′,6-diamidino-2-phenylindole (DAPI) (blue). Lymphocytes show surface staining for CD28 in a continuous ring pattern. This was not altered by cross-linking CD28. In contrast, CD28 was not visible by fluorescence microscopy on the surface of control neutrophils but following cross-linking of CD28 at 37°C for 1 h, CD28 was clearly visible in the form of large heterogeneous clusters on the cell surface.
Fig. 3.
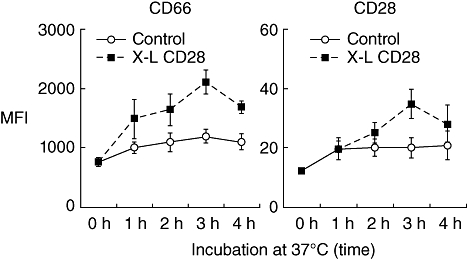
The effect of cross-linking (X–L) granulocyte CD28 on surface expression of CD66 and CD28: kinetic studies. Purified granulocytes were incubated on a multimeric array of anti-CD28 (clone 204.12) or mouse IgG1 antibody as a control, at 37°C for various time-intervals prior to measurement of cell surface CD66 (clone Kat4c) and CD28 (clone 15E8) by flow cytometry using fluorescein isothiocyanate (FITC)-conjugated antibodies. Results are shown as the mean fluorescence intensity (MFI). Mean ± standard error of the mean of three experiments.
Fig. 4.
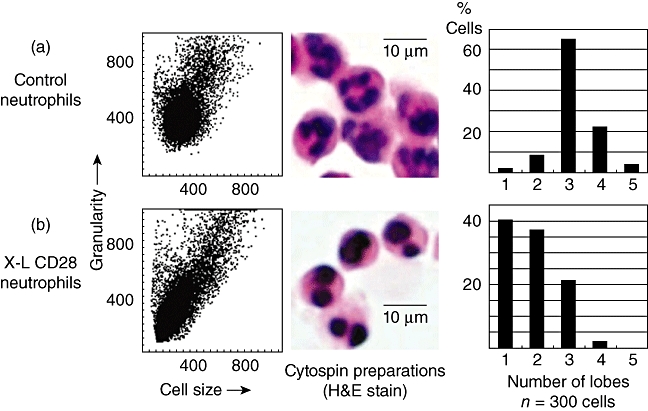
The effect of cross-linking (X–L) CD28 using a multimeric array (clone 204.12) on neutrophil granulocyte nuclear morphology. Cells were incubated at 37°C for 1 h on a multimeric array of anti-CD28 (or isotype control immunoglobulin G) as described in Methods prior to flow cytometry or morphological inspection using cytospin preparations of cells. (a) Dot-plot of forward- versus side-scatter (cell size versus granularity) showing control neutrophils and a haematoxylin and eosin (H&E)-stained cytospin preparation of neutrophils showing the characteristic multi-lobed structure of the nucleus. The bar chart shows counts performed on 300 cells showing that the vast majority of control neutrophils have three or four lobes. (b) Cross-linking of neutrophil CD28 produces a reduction in side-scatter (granularity) and a reduction in nuclear lobes with the vast majority of cells (76%) now showing one or two lobes. This is known as the pseudo-Pelger–Huët anomaly that was observed in all six recipients of TGN1412 [4].
These anti-CD28-activated granulocytes were also shown to significantly up-regulate mRNA specific for CD28 by 18 h, thus providing further evidence that these cells express constitutively and have the capacity to synthesize CD28 (Table S1).
Although these observations are of interest with regard to the complex role of CD28 within the immune system, it is clear that neutrophils do not either release or synthesize TNF-α, or any other of the 30 cytokines tested, within 2 h (see Table 1c). For this reason it seems highly unlikely that activated neutrophils play a direct role in the TGN1412 response, but it reinforces the view that a cell expressing very low levels of surface CD28 can indeed be activated by anti-CD28 as long as it is in the form of a multimeric array.
This raises the crucial question: how could such a multimeric array form in vivo? It has been implied that TGN1412 may have interacted in some way with vascular endothelial cells to form a multimeric array in vivo but no mechanism was proposed for this interaction [6]. We therefore considered the possibility that TGN1412 (a humanized IgG4 class antibody) may bind to CD64 (high-affinity FcγRI) expressed on vascular endothelial cells.
Demonstration of CD64 (FcγRI) on HUVECs
CD64 was demonstrated on the surface of HUVECs by immunofluorescence being distributed on the cell surface in the form of small homogeneous clusters (Fig. 5b). This experiment was repeated on three separate occasions. Similar results were obtained.
Fig. 5.
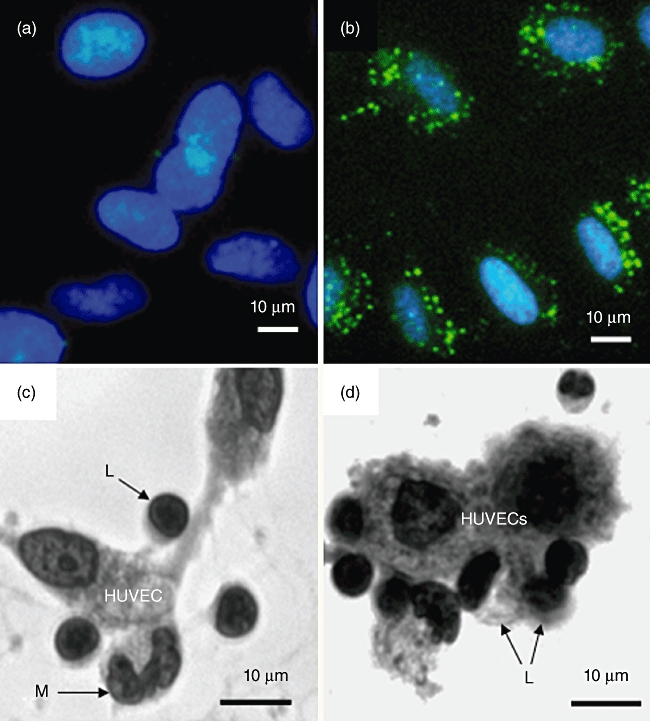
Human umbilical vein endothelial cells (HUVECs) express FcγRI (CD64). (a,b) HUVECs before and after incubation with fluorescein isothiocyanate (FITC)-conjugated mouse anti-human CD64 (FcγRI), clone 10.1 [immunoglobulin (Ig) G1]. Cells were counterstained with 4′,6-diamidino-2-phenylindole (DAPI). Cell surface CD64 appears as punctate staining. (c) Human peripheral blood mononuclear cells (PBMCs), i.e. lymphocytes (L) plus monocytes (M) adhering spontaneously to HUVECs following incubation for 18 h at 37°C. Cells were visualized using an inverted microscope at a magnification of ×400. Examples of adherent cells are indicated by arrows. (d) Increased binding of PBMCs to HUVECS (18 h at 37°C) as a result of preincubating the same number of cells (opsonization) with mouse anti-CD28 (clone 204.12). No increase in binding was observed when cells were preincubated at 4°C for 1 h with mouse IgG2a at the same concentration (4 µg per 106 cells). Based on microscopic inspection of 100 HUVECs the ratio of PBMC : HUVEC was 1:1 for non-opsonized cells and 2:1 for anti-CD28 opsonized cells.
Enhanced binding of anti-CD28 opsonized PBMCs to HUVECs
PBMC that had been pre-opsonized at 4°C for 1 h with anti-CD28 or with an appropriate isotype control antibody at the same concentration were incubated with HUVECs adherent to plastic tissue culture wells. Binding of control PBMCs to HUVECs was observed by 18 h (Fig. 5c), and the number of adherent cells increased considerably when anti-CD28-pre-opsonized PBMCs were used (Fig. 5d).
Binding of anti-CD28 opsonized PBMCs to HUVECs causes cytokine release
We tested for a functional consequence of binding of anti-CD28-opsonized PBMCs to HUVECs resulting in cytokine production. A significant increase in TNF-α, IL-6, MIP-1α and possibly IP-10 (CXCL10) was observed in cell-free supernatants obtained from anti-CD28-opsonized PBMCs + HUVECs. Addition of opsonized granulocytes to HUVECs did not result in up-regulation of any of the 30 mediators tested (Table 3 and Table S3).
Table 3.
Up-regulation of cytokines following an interaction between anti-CD28-opsonized leucocytes and human umbilical vein endothelial cells (HUVECS)
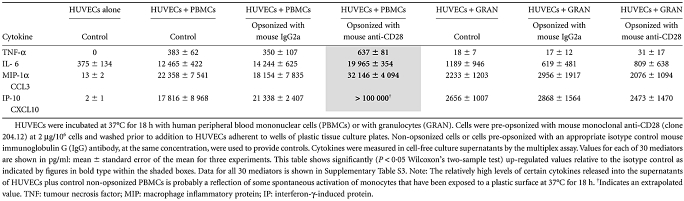 |
As further controls for these experiments, the addition of prefixed PBMC to HUVECs did not result in up-regulation of cytokines, and cross-linking of HUVEC CD64 using unconjugated mouse monoclonal anti-CD64 (clone 10.1 at 10 µg/ml) + GAM (40 µg/ml) did not stimulate cytokine release from these cells (data not shown).
Discussion
This study was designed to address the following two questions.
Which cell type was responsible for the rapid rise in plasma cytokine levels reported in all recipients of TGN1412?
How could an antibody that was innocuous in preclinical trials trigger a cytokine storm initiated by TNF-α?
Studies conducted by the UK National Institute for Biological Standards and Controls showed that TGN1412 could activate human T cells and stimulate cytokine synthesis release in vitro by 18 h, but only when presented to cells in the form of a multimeric array (MMA) [6]. Exactly how such an array may have formed in vivo is not clear.
The assumption that CD28+ T cells were the primary target for TGN1412 has been questioned [7]. The rapid onset of clinical symptoms suggests that other ‘faster-acting’ cell types may have been involved [7]. Furthermore, although T cells can produce TNF-α, this cytokine is associated more commonly with cells of the monocyte/macrophage lineage.
Granulocytes were considered as possible candidates for initiation of the cytokine storm. These cells are known to respond rapidly to activation signals, and low levels of CD28 have been detected on the surface of these cells [14–17]. In addition, cross-linking of granulocyte CD28, by murine monoclonal antibodies in the form of a multimeric array, was shown to result in up-regulation of certain cytokines by 24 h [14–17].
In this study we have confirmed that human granulocytes do indeed express low levels of cell surface CD28 and have shown, for the first time, that they contain significant cytoplasmic ‘stores’ of CD28. Granulocytes appeared to be activated by the anti-CD28-MMA as evidenced by the following:
A rapid increase in surface expression of the activation marker CD66 [18] and CD28 (in clusters).
A distinct change in nuclear morphology (the pseudo-Pelger–Huët anomaly) consistent with the in vivo effect observed in all recipients of TGN1412.
Up-regulation of specific CD28 mRNA.
These findings are of general interest and raise many questions with regard to the function of CD28 within the immune system, but cytokine up-regulation data (shown in Supplementary Table S1c) suggest that activated granulocytes were not involved, at least directly, in the initiation of the TGN1412-induced cytokine storm.
To our surprise, it was monocytes that appeared to be the main cell type responsible for anti-CD28-induced TNF-α synthesis at 2 h. Furthermore, up-regulation of TNF-α was demonstrated using anti-CD28 clone ANC28.1, shown previously to behave in vitro in a similar manner to TGN1412, i.e. a superagonist [12], as well as clones L293 and 204.12: non-superagonists. Triggering of monocyte TNF-α synthesis does not therefore seem to require any particular ‘superagonist’ properties of the antibody. Eleven other monocyte cytokines were also up-regulated significantly by anti-CD28 within 2 h (see Table 1). These findings are surprising, as monocytes are not thought to express any cell surface CD28. Here we have shown, for the first time, that these cells do in fact express very low levels of cell surface CD28. This level is insufficient for direct cross-linking by anti-CD28 when presented to cells in suspension, but sufficient when antibody is presented to cells in the form of a multimeric array.
In the second part of this study we have attempted to address the following question: how could such a multimeric array of anti-CD28 form in vivo?
We speculated that TGN1412 (an IgG4-class antibody) binds to CD64 (FcγRI) [19] on vascular endothelial cells, thereby forming a multimeric array in vivo. However, the evidence that human vascular endothelial cells express FcγRI is controversial [20–23]. In addition, high-affinity FcγRI may be saturated in vivo by plasma IgG [19]. If, however, the antibody bound first to cells expressing CD28 this would effectively create a complex that could competitively displace any bound IgG monomer from CD64 [24], thus allowing cells to bind to the vascular endothelium and permit cross-linking of CD28. Opsonized lymphocytes may also bind to adjacent HUVEC-adherent monocytes via CD64, thereby boosting cytokine production.
Here we have shown that HUVECs do indeed express CD64 and that PBMCs can bind to HUVECs. Addition of pre-opsonized cells to HUVECs up-regulated TNF-α and MIP-1α (CCL3) significantly by 18 h, but no significant increase was observed at 2 h. This failure to detect rapid cytokine release via HUVECs may be a technical problem requiring further optimization of experimental conditions, but may simply reflect the fact that we have used a murine monoclonal anti-CD28 instead of TGN1412 in this model system.
The main aim of the present study was to identify the cell type(s) responsible for producing TNF-α and initiating the cytokine storm, but it has not escaped our notice that our model system does not explain satisfactorily the extremely high levels of IFN-γ(up to 5000 pg/ml) that appeared rapidly following administration of TGN1412 in vivo[4]. It therefore seems likely that another cell type may have been involved in the production of this cytokine (see hypothesis: Fig. 6).
Fig. 6.
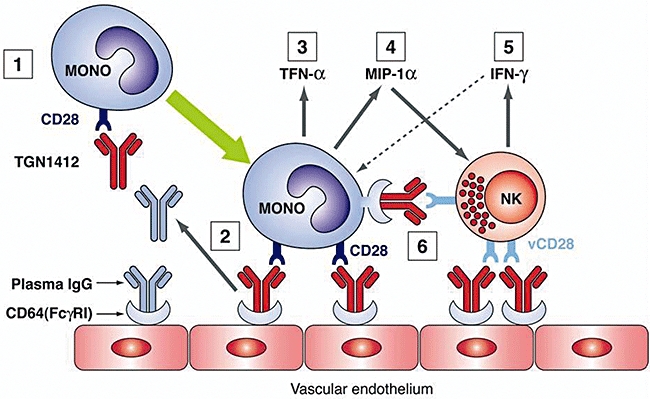
Hypothesis: (1) TGN1412 binds to CD28+ cells, including monocytes, to form a complex. (2) Complex binds to CD64 (FcγRI) on vascular endothelial cells displacing any passively bound plasma immunoglobulin (Ig)G. (3) Cross-linking of monocyte CD28 occurs leading to cell activation and rapid up-regulation of multiple cytokines, including tumour necrosis factor (TNF)-α and macrophage inflammatory protein (MIP)-1α. (4) MIP-1α recruits natural killer (NK) cells. (5) Cross-linking of CD28 on NK cells (known to express a variant form of CD28: vCD28) causes release of other ‘storm’ cytokines including interferon (IFN)-γ, that in turn activate monocytes. (6) Opsonized NK cells could also interact with CD64 on adjacent monocytes to provide a further amplification signal.
The primary source of rapidly produced IFN-γ in large quantities is thought to be the NK cell [25,26]. Some NK cells have been shown to express a variant of CD28 that differs from the molecule expressed on T cells [27]. It is possible that this CD28 variant (vCD28) may have been recognized by TGN1412, thus accounting for the rapid, prolonged elevation of IFN-γ observed in vivo. [4] It may also be relevant that MIP-1α, found to be up-regulated by all three clones of anti-CD28 in the multimeric array and by the interaction between opsonized PBMCs and HUVECs, has been shown to recruit NK cells and to promote IFN-γ delivery at sites of inflammation [28].
In conclusion, in this study we have developed an in vitro model system that suggests strongly that monocytes were the probable source of TNF-α, responsible for initiating the cytokine storm in the TGN1412 clinical trial. However, the in vivo situation is inevitably more complex, and an explanation for the high plasma levels of IFN-γ reported in all recipients requires elucidation by further studies, preferably using TGN1412 itself.
Disclosure
None declared.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Table S1. Cytokines produced by human peripheral blood leucocytes following cross-linking of CD28.
Table S2. Effect of cross-linking (X–L) leucocyte CD28 on mRNA specific for CD28.
Table S3. Up-regulation of cytokines following an interaction between anti-CD28-opsonized leucocytes and human umbilical vein endothelial cells (HUVECS).
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- 1.Beyersdorf N, Hanke T, Kerkau T, Hunig T. Superagonistic anti-CD28 antibodies: potential activators of regulatory T cells for therapy of autoimmune diseases. Ann Rheum Dis. 2005;64:91–5. doi: 10.1136/ard.2005.042564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hünig T, Dennehy K. CD28 superagonists: mode of action and therapeutic potential. Immunol Lett. 2005;100:21–8. doi: 10.1016/j.imlet.2005.06.012. [DOI] [PubMed] [Google Scholar]
- 3.Beyersdorf N, Hanke T, Kerkau T, Hunig T. CD28 superagonists put a break on autoimmunity by preferentially activating CD4+CD25+ regulatory T cells. Autoimmun Rev. 2006;5:40–5. doi: 10.1016/j.autrev.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 4.Suntharalingam G, Perry MR, Ward S, et al. Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N Engl J Med. 2006;355:1018–28. doi: 10.1056/NEJMoa063842. [DOI] [PubMed] [Google Scholar]
- 5.Expert Group on Phase One Clinical Trials (Chairman Professor Gordon W. Duff) Expert group on Phase One clinical trials; final report. 2006. The Stationary Office (TSO). Available at: http://www.dh.gov.uk/en/Publicationsandstatistics/Publications/PublicationsPolicyAndGuidance/DH_063117 (accessed 29 September 2010)
- 6.Stebbings R, Findlay L, Edwards C, et al. ‘Cytokine storm’ in the phase I trial of monoclonal antibody TGN1412: better understanding the causes to improve preclinical testing of immunotherapeutics. J Immunol. 2007;179:3325–31. doi: 10.4049/jimmunol.179.5.3325. [DOI] [PubMed] [Google Scholar]
- 7.Puellmann K, Beham AW, Kaminski WE. Cytokine storm and an anti-CD28 monoclonal antibody. N Engl J Med. 2006;355:1018–28. doi: 10.1056/NEJMc062750. [DOI] [PubMed] [Google Scholar]
- 8.Dubravec DB, Spriggs DR, Mannick JA, Rodrick ML. Circulating peripheral blood granulocytes synthesise and secrete tumour necrosis factor-α. Proc Natl Acad Sci USA. 1990;87:6758–61. doi: 10.1073/pnas.87.17.6758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Costa JJ, Matossian K, Resnick MB, et al. Human eosinophils can express the cytokines tumour necrosis factor-α and macrophage inflammatory protein-la. J Clin Invest. 1993;91:2673–84. doi: 10.1172/JCI116506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lacy P, Moqbel R. Eokines: synthesis, storage and release from human eosinophils. Mem Inst Oswaldo Cruz. 1993;92(Suppl. II):125–33. doi: 10.1590/s0074-02761997000800017. [DOI] [PubMed] [Google Scholar]
- 11.Bandeira-Melo C, Weller PF. Mechanisms of eosinophil cytokine release. Mem Inst Oswaldo Cruz. 2005;100:73–81. doi: 10.1590/s0074-02762005000900013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Waibler Z, Sender LY, Merten C, et al. Signaling signatures and functional properties of anti-human CD28 superagonistic antibodies. PLoS ONE. 2008;3:e1708. doi: 10.1371/journal.pone.0001708. doi: 10.1371/journal.pone.0001708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sandilands GP, McCrae J, Hill K, Perry M, Baxter D. Major histocompatibility complex class II (DR) antigen and costimulatory molecules on in vitro and in vivo activated human polymorphonuclear neutrophils. Immunology. 2006;119:562–71. doi: 10.1111/j.1365-2567.2006.02471.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Woerly G, Roger N, Loiseau S, Dombrowicz D, Capron A, Capron M. Expression of CD28 and CD86 by human eosinophils and role in the secretion of type 1 cytokines (interleukin 2 and interferon γ) J Exp Med. 1999;90:487–95. doi: 10.1084/jem.190.4.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Venuprasad K, Banerjee PP, Chattopadhyay S, et al. Human neutrophil-expressed CD28 interacts with macrophage B7 to induce phosphatidylinositol 3-kinase-dependent IFN-γ secretion and restriction of Leishmania growth. J Immunol. 2002;169:920–28. doi: 10.4049/jimmunol.169.2.920. [DOI] [PubMed] [Google Scholar]
- 16.Venuprasad K, Parab P, Prasad DVR, et al. Immunobiology of CD28 expression on human neutrophils. CD28 regulates neutrophil migration by modulating CXCR-1 expression. Eur J Immunol. 2001;31:1536–43. doi: 10.1002/1521-4141(200105)31:5<1536::AID-IMMU1536>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 17.Venuprasad K, Chattopadhyay S, Saha B. CD28 signalling in neutrophil induces T-cell chemotactic factor(s) modulating T-cell response. Hum Immunol. 2003;64:38–43. doi: 10.1016/s0198-8859(02)00689-4. [DOI] [PubMed] [Google Scholar]
- 18.Honig M, Peter HH, Jantscheff P, Grunert F. Synovial PMN show a coordinated up-regulation of CD66 molecules. J Leukoc Biol. 1999;66:429–36. doi: 10.1002/jlb.66.3.429. [DOI] [PubMed] [Google Scholar]
- 19.Van de Winkel JGJ, Anderson CL. Biology of human immunoglobulin G Fc receptors. J Leukoc Biol. 1991;49:511–24. doi: 10.1002/jlb.49.5.511. [DOI] [PubMed] [Google Scholar]
- 20.Devaraj S, Du Clos TW, Jialal I. Binding and internalization of C-reactive protein by Fc gamma receptors on human aortic endothelial cells mediates biological effects. Arterioscler Thromb Vasc Biol. 2005;25:1359–63. doi: 10.1161/01.ATV.0000168573.10844.ae. [DOI] [PubMed] [Google Scholar]
- 21.GrÖger M, Sarmay G, Fiebiger E, Wolff K, Petzelbauer P. Dermal microvascular endothelial cells express CD32 receptors in vivo and in vitro. J Immunol. 1996;156:1549–56. [PubMed] [Google Scholar]
- 22.Silvana V, Virella G, Gorod AJ, Lopes-Virella MF. Chlamydophila pneumoniae infection of human aortic endothelial cells induces the expression of Fc receptor II (FcRII) Clin Immunol. 2002;104:265–73. doi: 10.1006/clim.2002.5237. [DOI] [PubMed] [Google Scholar]
- 23.Alberto MF, Bermejo EI, Lazzari MA. Receptor expression for IgG constant fraction in human umbilical vein endothelial cells. Thromb Res. 2000;97:505–11. doi: 10.1016/s0049-3848(99)00203-0. [DOI] [PubMed] [Google Scholar]
- 24.Guyre CA, Keler K, Swink SL, Vitale LA, Graziano RF, Fanger MW. Receptor modulation by FcγRI-specific fusion proteins is dependent on receptor number and modified by IgG. J Immunol. 2001;167:6303–11. doi: 10.4049/jimmunol.167.11.6303. [DOI] [PubMed] [Google Scholar]
- 25.Schoenborn JR, Wilson CB. Regulation of interferon-gamma during innate and adaptive immune responses. Adv Immunol. 2007;96:41–101. doi: 10.1016/S0065-2776(07)96002-2. [DOI] [PubMed] [Google Scholar]
- 26.Godfrey DI, Hammond KJL, Poulton LD, Smyth MJ, Baxter AG. NKT cells; facts, functions and fallacies. Immunol Today. 2000;21:573–83. doi: 10.1016/s0167-5699(00)01735-7. [DOI] [PubMed] [Google Scholar]
- 27.Galea-Lauri J, Darling D, Gan S-U, et al. Expression of a variant of CD28 on a subpopulation of human NK cells: implications for B-7 mediated stimulation of NK cells. J Immunol. 1999;163:62–70. [PubMed] [Google Scholar]
- 28.Salazar-Mather TP, Hamilton TA, Biron CA. A chemokine-to-cytokine-to chemokine cascade critical in anti-viral defense. J Clin Invest. 2000;105:985–93. doi: 10.1172/JCI9232. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.