

Abstract
The anti-hypertensive drug captopril is used commonly to reduce blood pressure of patients with severe forms of Chagas disease, a cardiomyopathy caused by chronic infection with the intracellular protozoan Trypanosoma cruzi. Captopril acts by inhibiting angiotensin-converting enzyme (ACE), the vasopressor metallopeptidase that generates angiotensin II and promotes the degradation of bradykinin (BK). Recent studies in mice models of Chagas disease indicated that captopril can potentiate the T helper type 1 (Th1)-directing natural adjuvant property of BK. Equipped with kinin-releasing cysteine proteases, T. cruzi trypomastigotes were shown previously to invade non-professional phagocytic cells, such as human endothelial cells and murine cardiomyocytes, through the signalling of G protein-coupled bradykinin receptors (B2KR). Monocytes are also parasitized by T. cruzi and these cells are known to be important for the host immune response during infection. Here we showed that captopril increases the intensity of T. cruzi infection of human monocytes in vitro. The increased parasitism was accompanied by up-regulated expression of ACE in human monocytes. While T. cruzi infection increased the expression of interleukin (IL)-10 by monocytes significantly, compared to uninfected cells, T. cruzi infection in association with captopril down-modulated IL-10 expression by the monocytes. Surprisingly, studies with peripheral blood mononuclear cells revealed that addition of the ACE inhibitor in association with T. cruzi increased expression of IL-17 by CD4+ T cells in a B2KR-dependent manner. Collectively, our results suggest that captopril might interfere with host–parasite equilibrium by enhancing infection of monocytes, decreasing the expression of the modulatory cytokine IL-10, while guiding development of the proinflammatory Th17 subset.
Keywords: captopril, Chagas disease, monocytes, Th17, Trypanosoma cruzi
Introduction
Human infection with Trypanosoma cruzi results in Chagas disease, a parasitic disease that affects approximately 8 million individuals in Latin America [1]. The chronic phase of Chagas disease is either asymptomatic or may lead to cardiac and digestive system pathology. Chagas heart disease is a potentially fatal dilated cardiomyopathy that develops in 30% of T. cruzi-infected individuals [2] and is responsible for the largest number of deaths among chagasic patients. Clinical treatment of chagasic cardiomyopathy-associated hypertension in chagasic patients includes sodium restriction and additional treatment with digitalis, diuretics or angiotensin-converting enzyme (ACE) inhibitors, such as captopril [3,4]. As true for other ACE inhibitors, captopril has also been reported to reduce heart inflammation and fibrosis [5].
ACE has a dual role in vascular homeostasis. Acting primarily in the renin–angiotensin system, ACE processes the inactive intermediate angiotensin I (Ang I), generating the vasopressor octapeptide angiotensin II (Ang II). Although Ang II may bind to different subtypes of G protein coupled receptors, excessive formation of this agonist may increase intracellular volume, peripheral vascular resistance and blood pressure [5]. ACE inhibitors such as captopril exert their anti-hypertensive effects by inhibiting ACE-dependent formation of the vasopressor Ang II and by attenuating ACE (kininase II)-dependent degradation of bradykinin (BK) or lysyl-bradykinin (LBK) [6]. Termed collectively as ‘kinins’, BK/LBK are short-lived peptides liberated from an internal moiety of high or low molecular weight kininogens by the action of specialized proteases of host [7] or microbial origin [8,9]. Once released, BK/LBK exert their vasodilating function by triggering endothelium BK2R, a constitutively expressed G-protein coupled receptor (GPCR) [10]. Alternatively, the released kinins undergo processing by kininase I, generating arginine-truncated metabolites (des-Arg-kinin) that activate BK1R, an inducible subtype of kinin receptor up-regulated in inflamed tissues [11], while losing affinity for BK2R.
Studies on cruzipain, a lysosomal cysteine protease characterized previously as a kinin-releasing enzyme of T. cruzi[12], provided the first evidence that pathogen uptake is driven by the activation of kinin receptors (BK2R and BK1R) [13,14]. Whether involving human endothelial cells or murine cardiomyocytes, these in vitro studies revealed that addition of captopril to the interaction medium potentiated parasite invasion via the kinin signalling pathway [13,14]. More recently, it was reported that BK/LBK induces the maturation of dendritic cells (DCs) through the signalling of BK2R [15,16]. Further underscoring the importance of kinins and ACE to pathogenic outcome, Monteiro and co-workers [17] demonstrated that ACE inhibitors (single-dose administration) potentiated paw oedema evoked by trypomastigotes through mechanisms involving co-operation between Toll-like receptor (TLR)-2 and BK2R. Using the same subcutaneous model of infection, these workers demonstrated that captopril potentiated anti-parasite T helper type 1 (Th1) responses through the up-regulation of the BK2R/interleukin (IL)-12 innate pathway [17]. In another study, involving oral administration of captopril to A/J mice infected acutely with T. cruzi, Leon and co-workers reported that the acute myocarditis was ameliorated by prolonged treatment with this anti-hypertensive drug [3].
Although captopril is administrated routinely to hypertensive patients with chagasic cardiomyopathy, the immunological effects of this ACE inhibitor were not investigated systematically in humans. Our results revealed that ACE inhibitors potentiate T. cruzi infection of human monocytes, decreases the expression of the modulatory cytokine IL-10 while inducing Th17 cells. These studies suggest that anti-hypertensive therapy based on captopril administration potentially alters the host–parasite balance and might influence the outcome of Chagas disease.
Materials and methods
Human samples
The donors included in our studies were non-chagasic individuals (n = 6) from the state of Minas Gerais, Brazil, with average ages ranging between 25 and 32 years. We excluded from our study individuals with any chronic inflammatory disease, diabetes, heart and circulatory illnesses (including hypertension) or bacterial infections. All individuals included in this work were volunteers. This study is part of an extended project evaluating cardiac risk factors in Chagas disease and has the approval of the Ethical Committee of Universidade Federal de Minas Gerais in accordance with the Declaration of Helsinki.
Source of parasites
Tissue-culture derived trypomastigotes (TCT) of the Y strain of T. cruzi were isolated from infected monolayers of Vero cells, as described previously [18]. Briefly, Vero cells were infected using five TCT/host cells and kept in RPMI-1640 enriched with 5% fetal calf serum (FCS), supplemented with antibiotics (penicillin at 500 U/ml and streptomycin at 0·5 mg/ml). After approximately 5 days, the TCT were collected from the supernatant, washed once by centrifugation with phosphate-buffered saline (PBS) pH 7·2 at 1000 g for 10 min at 4°C and resuspended in RPMI-1640 to a concentration of 5 × 107 TCT/ml.
Cell preparations
Peripheral blood mononuclear cells (PBMC) were purified as performed previously by us [18]. Briefly, heparinized blood was diluted 1:1 with PBS and applied over a Ficoll gradient. The mixture was centrifuged for 40 min at 600 g and PBMC were collected at the interface between the plasma and the Ficoll. Cells were washed three times by centrifugation with PBS and resuspended in RPMI-1640 supplemented with antibiotic/anti-mycotic (0·25 µg of amphotericin B/ml, 200 U of penicillin/ml, 0·1 mg of streptomycin/ml) and 1 mm l-glutamine at a concentration of 107 cells/ml. To obtain adherent cells, 2 × 106 PBMC/well were plated on 13-mm round coverslips in RPMI-1640 supplemented with 10% FCS and cultured in 24-well plates for 1 h at 37°C, 5% CO2. Non-adherent cells were removed by washing the wells with RPMI-1640 supplemented with 1% antibiotic/anti-mycotic (previously left at 37°C for 30 min), and the adherent cells (monocytes) were used in infection experiments as described below.
Infection of adherent cells and immunofluorescence
Monocyte infection was performed over coverslips, using a total volume of 0·5 ml of RPMI-1640 supplemented with 1% antibiotic/anti-mycotic, 1% 1 mm l-glutamine and 10% FCS. The monocytes were incubated with either medium alone or medium + 50 µm of captopril. Parasites were added immediately at a ratio of 5:1 TCT/monocytes and incubated for 3, 48 or 96 h at 37°C, 5% CO2. The monolayers were washed three times with PBS to remove free parasites. Infection was evaluated by two methods: light and confocal microscopy. For the light microscopy, preparations were incubated with Giemsa dye for 15 min, washed and analysed using a Nikon light microscope (Melville, NY, USA). We analysed 15 field/samples using a power magnification of ×600, and the frequencies of adherent cells infected were expressed as percentage of positive cells in relation to the total cell count. For confocal microscopy analysis, immunofluorescence was carried out by staining with 4′,6′-diamidino-2-phenylindole (DAPI), as follows. Coverslips were incubated DAPI diluted 1:300 in PBS supplemented with 2% bovine serum albumin (BSA) for 10 min and mounting using anti-fade medium. Slides were kept at 4°C and protected from light until acquisition. Confocal analyses were performed using a Meta-510 Zeiss laser scanning confocal system running LSMix software (Oberkochen, Germany) coupled to a Zeiss microscope using an oil immersion Plan-Apochromat objective (63X, 1·2 numerical aperture, Oberkochen, Germany). We performed six independent experiments and analysed 15 fields per sample.
Infection of total PBMC in suspension and flow cytometry
Infection of monocytes in suspension was assessed by flow cytometry using 5 and 6-carboxyfluorescein diacetate succinimidyl ester (CFSE)-labelled TCT, as performed previously by us [18]. Parasites were incubated with 5 µm CFSE for 10 min at 37°C, 5% CO2. Labelled parasites were washed three times with PBS by centrifugation and used for infection of adherent cells treated or not with captopril, as described above. Cells were then stained with anti-CD14-phycoerythrin (PE) monoclonal antibodies by incubation for 15 min at 4°C. Samples were washed and fixed for 20 min with a 4% formaldehyde solution. Stained cells were acquired in a Becton Dickinson fluorescence activated cell sorter (FACScan, Franklin Lakes, NJ, USA). Intensity of infection was evaluated by CFSE fluorescence intensity in gated CD14+CFSE+ cells. Infection with unlabelled parasites and incubation of infected cells with mouse immunoglobulin G1 (IgG1)-PE-labelled isotype control were used as parameters to set markers. A minimum of 30 000 gated events from each sample were collected and analysed using FlowJo software (Ashland, OR, USA). Two independent experiments were performed, with three individuals in each experiment.
Analysis of expression of surface molecules and cytokines by PBMC using flow cytometry
Human PBMC were infected with T. cruzi TCT, as described above. In individual wells, we added captopril (50 µm), captopril + bradykinin (10 nm) or HOE-140 (BK2R antagonist; 200 µm) + bradykinin (10 nm) for a period of 18 h. After incubation, cells were immunostained using fluorochrome-associated antibodies against CD143, CD4, CD8 or CD14. Intracellular cytokine expression was evaluated using PE-labelled antibodies against IL-12, IL-10, tumour necrosis factor (TNF)-α, interferon (IFN)-γ and IL-17. For surface molecule expression analysis, cells were incubated with antibodies for 15 min at 4°C, washed with PBS supplemented with 1% BSA and fixed by 20-min incubation with 4% formaldehyde solution. For intracellular staining, cells were cultured for approximately 18 h. During the last 4 h of culture, brefeldin A (1 µg/ml) was added to each well to prevent cytokine secretion. Cells were then labelled for surface molecules as described above. After removing the fixing solution, cells were permeabilized by incubation for 10 min with a 0·5% saponin solution. Then, cells were incubated with anti-cytokine monoclonal antibodies for 30 min at room temperature, washed twice with 0·5% saponin solution, resuspended in PBS and examined using a FACScan. A total of 30 000 events were acquired and the parameters were analysed in the monocytes or lymphocytes population by gating the region occupied classically by those cells in a size versus granularity plot.
Statistical analysis
We compared our results among different treatments and between infected and not infected cells using Tukey's multiple comparison or paired t-test. All analyses were performed using GraphPad Prism Software (La Jolla, CA, USA). We considered statistically different results with P < 0·05.
Results
Captopril increased the intensity of T. cruzi infection in human monocytes
Previous studies demonstrated that addition of captopril to the interaction medium potentiates BK2R-dependent pathways of T. cruzi (Dm28 strain) invasion of human endothelial cells and murine cardiomyocytes [13,14]. These observations were seen in human primary umbilical vein endothelial cells (HUVECs) and in Chinese hamster ovary (CHO) cells. Here we determined if the addition of captopril could similarly modulate parasite infection of human monocytes. To this end, we incubated TCT with adherent monocytes or with monocytes kept as cell suspensions. Adherent cells were infected with T. cruzi for 3, 48 or 96 h in the presence or absence of captopril. The results depict extent of intracellular infection as measured by confocal microscopy (DAPI+ parasite's nuclei) or light microscopy (Giemsa staining) (Fig. 1a and b, respectively). Incubation of adherent cells with T. cruzi for 3 h in the absence of captopril led to a significantly higher infection rate (54·1% ± 3, P < 0·05) compared to 48 (38·9% ± 6) and 96 (45·2% ± 7) h of incubation (Fig. 1b). After captopril treatment, T. cruzi infection was also more effective when the monocyte cells were incubated with the parasite for 3 h, compared to 48 and 96 h of incubation (53·6% ± 3 versus 38·9% ± 4 and 48·1% ± 9, respectively). Our results showed that the percentage of infected monocytes did not change upon treatment with captopril, as the percentages of infection were similar when comparing captopril-treated with untreated cultures. Moreover, these results allowed for the selection of the 3-h time-point to evaluate the extent of parasite internalization in monocyte suspensions, using CFSE-labelled T. cruzi as the read-out. Our flow cytometry results (Fig. 1c and d) showed that intensity of CFSE fluorescence in infected CD14+ cells increased 27% in monocyte suspensions supplemented with captopril compared to untreated monocytes (1552·3 versus 1128·4; Fig. 1c and d). Collectively, these data indicate that captopril increased markedly the extent of parasite uptake per host cell and, although it did not affect the proportion of infected monocytes, it favoured the penetration of a higher number of parasites per cell.
Fig. 1.
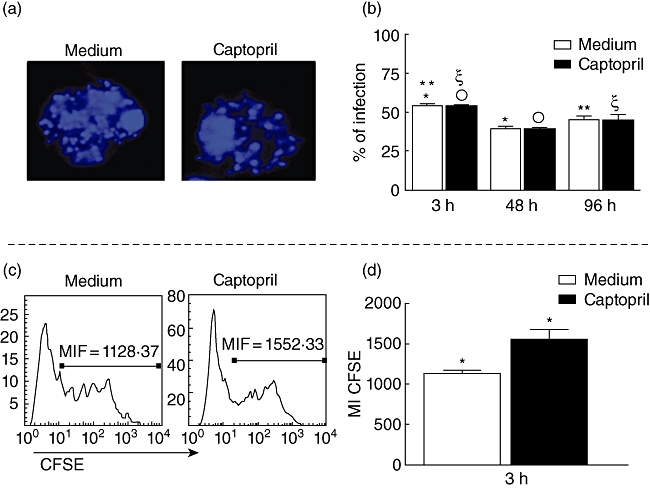
Determination of infectivity in monocytes by Y strain trypomastigotes, treated or not with captopril. (a) Representative confocal microscope analysis image, showing 4′,6′-diamidino-2-phenylindole (DAPI+) parasite's nuclei inside monocytes. Cells were treated or not with captopril, infected with five parasites/cell, preparations were stained with DAPI, and read on a confocal microscope, as described in Material and methods; magnification ×62; (b) determination of the percentage of infected cells after 3, 48 and 96 h of infection in cultures treated or not with captopril, using confocal microscopy analysis; cells were prepared as described above, acquired by confocal microscopy, and the percentage of infected cells was determined by counting the total number of cells and the parasite+ cells; results are expressed as average ± standard deviation. (c) Representative histogram of peripheral blood mononuclear cells exposed to Y strain trypomastigotes labelled with 5- and 6-carboxyfluorescein diacetate succinimidyl ester (CFSE), to determine the intensity of CFSE in cultures treated or not with captopril, as described in Material and methods. Briefly, cells were infected with five CFSE-labelled parasites/cell and incubated for 3 h; after this period, samples were read by fluorescence activated cell sorter and the fluorescence mean intensity was analysed. (d) Determination of the mean intensity of expression of CFSE fluorescence as a measure of intensity of infection in cells from cultures treated or not with CFSE; results are expressed as average ± standard deviation. Identical symbols indicate P < 0·05 between groups marked with the same symbol.
Captopril in association with T. cruzi infection reduces IL-10 expression by monocytes
Antigen-presenting cells (APC) play a key role in the induction of immune responses, and it is well established that monocytes are able to present major histocompatibility complex (MHC)-restricted epitopes to T cells [19]. ACE was found in tissue macrophages as well as in cultured monocytes [20]. Due to its dipeptidyl carboxydipeptidase activity, ACE enhances the presentation of endogenously synthesized peptides to MHC class I by generation of optimally sized class I-binding peptides from a larger protein fragment [21]. In order to determine if ACE expression is altered by T. cruzi infection and/or captopril treatment, we stained PBMC with anti-CD14 (monocyte marker) and anti-CD143 (ACE) antibodies after 3 h of incubation with T. cruzi in the presence or absence of captopril. We found that T. cruzi infection led to an increase in the frequency of CD14+CD143+ cells in relation to non-infected cells (8·05% ± 2 versus 3% ± 1; Fig. 2a). The same results were observed in infected cells upon treatment with captopril: we observed an increase in CD14+CD143+ cells in cultures treated with the ACE inhibitor compared to captopril-treated, but non-infected cultures (7·7% ± 2 versus 3·3% ± 1; Fig. 2a). Thus, our results indicated that captopril by itself was not able to induce alterations in ACE expression either in infected or non-infected monocytes, as the percentage of expression of CD143 was similar in captopril-treated or untreated cultures (Fig. 2a). T. cruzi induction of CD143 expression by CD14+ cells is consistent with the role of ACE in intracellular antigen presentation [21]. In addition to antigen presentation, monocytes participate in immunoregulatory functions via cytokine production. We then evaluated if the expression of IL-10 and IL-12 by monocytes was altered by T. cruzi infection and/or by captopril treatment (Fig. 2b and c). T. cruzi infection increased significantly the expression of IL-10 by monocytes compared to uninfected cells (9·5% versus 4·5%; Fig. 2b). Interestingly, we found that T. cruzi infection in association with captopril led to a reduction of IL-10 expression by monocytes in comparison to infection in absence of captopril (6·4% versus 9·5%; Fig. 2b). In the absence of T. cruzi, the captopril did not alter the expression of IL-10 by monocytes compared to non-treated cultures (4·5% ± 2 versus 4·6% ± 2 Fig. 2b). Our results showed that IL-12 staining was not modulated by T. cruzi infection or by treatment with captopril (Fig. 2c).
Fig. 2.
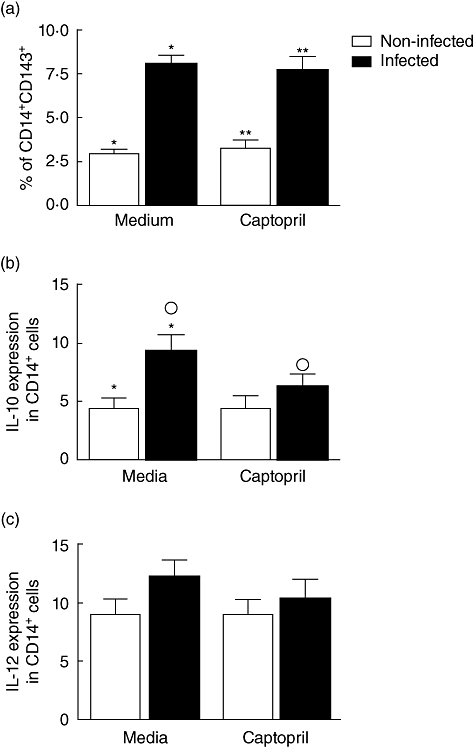
Expression of angiotensin-converting enzyme (ACE) and immunoregulatory cytokines by monocytes exposed to Y strain trypomastigotes and treated or not with captopril. Expression of ACE (a), interleukin (IL)-10 (b) and IL-12 (c) by CD14+ monocytes was determined by fluorescence activated cell sorter (FACS), as described in Material and methods. Briefly, cells were treated or not with captopril and exposed or not to five parasites/cell for 3 h. After treatments and infection, samples were stained with fluorescein isothiocyanate-labelled anti-CD14 monoclonal antibodies, in association with phycoerythrin-labelled anti-cytokine or anti-ACE monoclonal antibodies; stained cells were acquired by FACS and analysed to determine the percentage of expression of each marker in the monocyte population. Results are expressed as mean percentage ± standard deviation of double-positive cells, as indicated. The symbols indicate P < 0·05 between groups marked with the same symbol.
T. cruzi infection in association with captopril leads to an increase in the frequency of CD4+ T cells expressing ACE and enhances frequency of Th17 cells
ACE has been identified as a membrane-bound enzyme in several types of cells, including lymphocytes and macrophages [22]. We sought to evaluate whether T. cruzi infection in the presence or absence of captopril alters ACE expression in T lymphocytes. T. cruzi infection led to an increase in the frequency of CD4+CD143+ cells in non-treated cultures, compared with uninfected non-treated cultured cells (0·87% versus 0·54%; Fig. 3a). The frequency of CD4+CD143+ lymphocytes was increased further when we associated parasites and captopril, compared to uninfected monocytes treated with captopril alone (1·2% versus 0·56%; Fig. 3a). T. cruzi infection associated with captopril led to an elevation of the frequency of CD4+CD143+ cells in comparison with infection alone, in the absence of captopril (1·2 versus 0·87%; Fig. 3a). The percentage of CD8+CD143+ cells was not altered by T. cruzi infection or captopril, neither alone nor in combination (Fig. 3b). Because we observed that T. cruzi infection and captopril selectively modified CD143 expression by CD4+ T lymphocytes, we sought to determine if infection and captopril treatment would have an effect on the cytokine expression by CD4+ T cells or CD8+ T lymphocytes. Our results showed that T. cruzi infection or captopril treatment did not change IL-10 and TNF-α expression by CD4+ T cells (not shown). Notably, T. cruzi infection led to an increase in IFN-γ expression by CD4+ but not CD8+ T cells, compared to non-infected cultures (Fig. 4a and b). In contrast, captopril did not alter IFN-γ expression by CD4+ or CD8+ lymphocytes, whether associated or not with trypomastigote infection (Fig. 4a and b). We then evaluated IL-17 expression by the CD4+ and CD8+ T cell populations (Fig. 4c and d). T. cruzi infection alone did not alter IL-17 expression significantly by CD4+ T cells (Fig. 4c). Surprisingly, however, the association of captopril with TCT led to a 69% increase in the frequency of IL-17+ CD4+ T cells (Fig. 4c). T. cruzi infection alone increased the percentage of IL-17+ CD8+ T cells by 62%, compared to non-infected cultures (Fig. 4d). Conversely, captopril acted over CD8+ T cells infected with T. cruzi, decreasing the frequency of IL-17-expressing cells by 46% in relation to non-infected captopril-treated cultures (Fig. 4d).
Fig. 3.
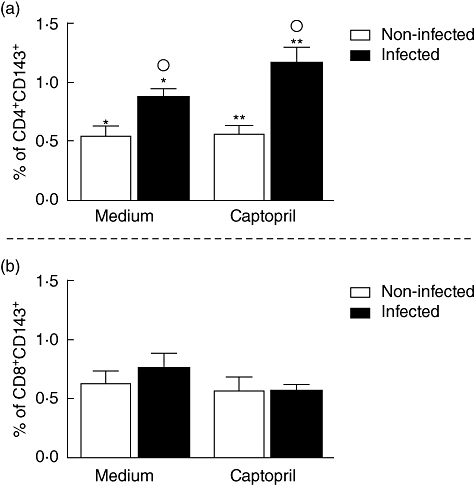
Expression of angiotensin-converting enzyme (ACE) by CD4+ (a) and CD8+ (b) T cells after in vitro exposure of peripheral blood mononuclear cells to Y strain trypomastigotes in the presence or absence of captopril. Expression of ACE was determined by fluorescence activated cell sorter (FACS), as described in Material and methods. Briefly, cells were treated or not with captopril and exposed or not to five parasites/cell for 3 h. After treatments and infection, samples were stained with fluorescein isothiocyanate-lablelled anti-CD4 or CD8 monoclonal antibodies, in association with phycoerythrin-labelled anti-ACE antibodies; stained cells were acquired by FACS and analysed to determine the percentage of expression of ACE by CD4 or CD8 lymphocytes. Results are expressed as mean percentage ± standard deviation of double-positive cells, as indicated. The symbols indicate P < 0·05 between groups marked with the same symbol.
Fig. 4.
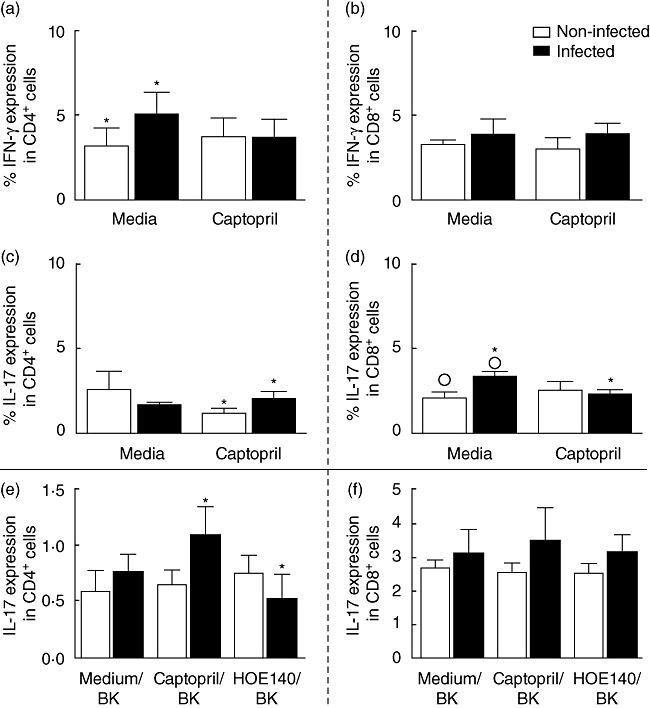
Expression of interferon (IFN)-γ and interleukin (IL)-17 by CD4+ (a,c) and CD8+ (b,d) T cells after in vitro exposure of peripheral blood mononuclear cells (PBMC) with Y strain trypomastigotes and treated or not with captopril; expression of IL-17 by CD4+ (e) and CD8+ (f) T cells after in vitro exposure of PBMC to Y strain trypomastigotes, in the presence or absence of captopril and/or HOE-140. Expression of IFN-γ and IL-17 was determined by fluorescence activated cell sorter (FACS), as described in Material and methods. Briefly, cells were treated or not with captopril and/or HOE-140 and exposed or not to five parasites/cell for 3 h. After treatments and infection, samples were stained with fluorescein isothiocyanate-labelled anti-CD4 or CD8 monoclonal antibodies, in association with phycoerythrin-labelled anti-cytokine antibodies; stained cells were acquired by FACS and analysed to determine the percentage of expression of the cytokines by CD4 or CD8 lymphocytes. Results are expressed as mean percentage ± standard deviation of double-positive cells, as indicated. The symbols indicate P < 0·05 between groups marked with the same symbol.
Captopril induced increase in the frequency of Th17 cells requires B2R kinin receptor
Considering that captopril potentiates the signalling effects of BK/LBK on BK2R, we then checked if HOE 140 (a specific B2R antagonist) could block modulation of cytokine expression. Our results showed that non-infected cells treated with captopril and HOE 140 did not present alterations in IL-17 expression by CD4+ T cells compared to untreated cultures (Fig. 4e). However, upon infection, HOE-140 treatment reduced by twofold the frequency of IL-17+ CD4+ T cells compared to infected untreated cultures (Fig. 4e). In contrast, no differences were seen in IL-17+ CD8+ T cells under the different conditions (Fig. 4f). These data suggest that IL-17 expression by CD4+, but not CD8+ T cells, might be under the influence of kinin pathway.
Discussion
Whether resulting from destruction of parasitized heart cells by cytotoxic lymphocyte (CTL)-mediated attack or other means, the release of intracellular parasites into the interstitial spaces of the myocardium is probably a sporadic event during the chronic phase of Chagas disease, as the presence of pseudocysts are found rarely in myocardial tissues. Thus, we may predict that the extracellular trypomastigotes, once released in interstitial tissues, may either infect neighbouring heart cells or invade blood-borne macrophages as soon as these phagocytes reach the inflammatory foci. Recent studies by our group have underscored the beneficial roles that IL-10-producing macrophages play in the pathogenesis of human Chagas disease [18,23]. In the present study we examined the influence of captopril on macrophage function in the presence/absence of trypomastigotes because this drug is prescribed commonly to patients with Chagas heart disease who suffer from hypertension [24]. At the cellular level, there are at least three reasons to investigate the influence of captopril on the interaction of human monocytes/macrophages with T. cruzi: (i) it is well known that (resting) macrophages express ACE on their surface [16]; (ii) macrophage-like cells of human origin (U-937) were shown recently to assemble a fully active kinin system on their surface [25]; and (iii) studies performed with kinin-releasing strains of T. cruzi revealed that captopril potentiates pathogen-uptake by non-phagocytic cells expressing kinin receptors, such as cardiomyocytes or endothelial cells [13,14].
In this work, we investigated the effects of captopril on the extent of monocyte infection with tissue culture-derived trypomastigotes of T. cruzi and evaluated the functional consequences of such in vitro interactions. Our results showed that although captopril did not affect the percentage of monocytes infected by the parasite, assays performed with cell suspensions revealed that the ACE blocker increased significantly the extent of parasite uptake by monocytes. Although our work involved a different T. cruzi strain (Y), the data are in agreement with studies showing that captopril potentiates the infectivity of Dm28 T. cruzi trypomastigotes in assays performed with non-phagocytic cells expressing BK2R (CHO-BK2R or HUVECs) [13].
Intriguingly, we found that addition of captopril to monocyte cultures exposed to Y strain trypomastigotes led to a reduction of IL-10 expression by monocytes. Furthermore, assays performed with PBMC exposed to trypomastigotes revealed that captopril increased frequencies of IL-17 expressing CD4+ T cells. Of further interest, assays performed in cultures supplemented with exogenous BK and/or HOE-140 suggested that the increased frequency of Th17 cells is, at least in part, dependent upon the activation of the B2R kinin receptor. Previous studies in A/J mice infected acutely with the Brazil strain showed that captopril administrated orally improves cardiac function [26]. Although not excluding the beneficial roles that ACE inhibitors bring to cardiac patients, our in vitro findings raise the possibility that, depending upon the T. cruzi strain and genetic make-up of the host, the administration of captopril may induce immunological changes that could aggravate chagasic myocardiopathy. Although our in vitro findings cannot be extrapolated readily to the in vivo settings, the finding that captopril reduced the frequency of IL-10-producing macrophages and increased IL-17-producing cells might aggravate T cell-dependent immunopathology.
Among PBMC, monocytes are the host cells invaded preferentially by Y strain T. cruzi trypomastigotes [18]. It is well established that these APCs are able to process and present peptide antigens in a MHC-restricted manner, and along with DCs contribute to the initiation of adaptive immunity through the up-regulation of co-stimulatory molecules and enhanced cytokine production [18]. Highly expressed in the endothelium lining, ACE plays an important role in blood pressure regulation [27]. APCs express ACE (CD143), and its expression is induced during the differentiation of human monocytes [28,29]. Evidence exists that ACE may play an immunomodulatory role by generating Ang II and/or by swiftly degrading BK agonists generated by kallikrein or microbial protease [30]. ACE 10/10 mice present macrophages overexpressing ACE and display exuberant immune responses, which has been associated with the enhanced presentation of MHC class I-peptides to CD8+ T cells observed in these mice [21]. It was proposed that these effects were due, at least in part, to ACE's ability to modify the C termini of peptides for presentation by MHC class I molecules [21,31]. In another interesting finding, we observed that the addition of captopril to monocyte suspensions translated into increased expression of ACE (CD143), whereas IL-10 expression is decreased reciprocally. Previous studies by our group and by other investigations have linked IL-10 expression to protection of Chagas heart disease [18,23]. Thus, it is conceivable that chagasic patients treated with captopril could present enhanced CD8+ T cell response in an environment lacking immunomodulatory mechanisms, given the decrease in IL-10 expression, which could lead to an aggravation of cardiac disease.
The anti-inflammatory property of captopril has been associated with suppression of the synthesis of proinflammatory cytokines [30,31]. Other studies have shown that captopril inhibition of activation-induced apoptosis in T cell hybridomas and Fas-induced apoptosis in human peripheral T cells [32,33] supports the idea that captopril may hamper clonal deletion and interfere with maintenance of self-tolerance, thereby causing autoimmunity. Recently, a subset of IL-17-producing T cells (Th17) distinct from Th1 or Th2 cells has been described and shown to be crucial in induction of autoimmune tissue injury [34]. Th17 response has been linked to the pathogenesis of diseases such as multiple sclerosis, psoriasis, rheumatoid arthritis, colitis, autoimmune encephalitis [35] and leishmaniasis [36]. Although a recent study has suggested a protective role for IL-17 in experimental T. cruzi infection [37], considering the pathogenic nature of this cytokine in human diseases, it is possible that it plays a role in Chagas disease-associated pathology. In our study we observed that captopril, in the presence of T. cruzi, increased the frequency of CD4+IL-17+ T cells and that this effect was impaired when cells were treated with HOE-140, a B2R antagonist. Interestingly, infection in association with captopril led to a decrease of IL-17 expression by CD8+ T cells, which was not affected by treatment with HOE-140. Considering that IL-17 expression by CD4+, but not CD8+ T cells, is impaired by HOE-140 in our model, we may surmise that BK2R is probably involved in IL-17 induction by captopril. Of interest in this context, studies in BALB/c mice infected by the periodontal pathogen Porphyromonas gingivalis linked Th17 and Th1 responses to pathogen-induced activation of the BK2R pathway [38]. In a myosin-induced experimental autoimmune myocarditis, A/J mice were immunized and treated orally with captopril, which ameliorated autoimmune myocarditis as measured by the reduction in cardiac hypertrophy and the incidence and severity of inflammation, necrosis and fibrosis [26]. Captopril also reduced in vivo cell-mediated inflammatory responses based upon the observed reduction of myosin-specific delayed-type hypersensitivity in antigen-immunized mice. However, these effects were not due to a direct effect on T cells as these cells proliferated normally and the level of secreted cytokines was unaltered [26]. Of note, however, IL-17 levels were not evaluated in that study.
In summary, our results suggest that captopril might interfere with host–parasite equilibrium by enhancing infection of monocytes, decreasing the expression of the modulatory cytokine IL-10, while guiding development of the proinflammatory Th17 subset. Further studies are under way to investigate the effects of captopril in the immune response of chronic chagasic patients and whether this would influence pathology development.
Acknowledgments
This work was supported by CNPq, INCT-DT and FAPEMIG. C. A. S. M., L. M. D. M., J. S., K. J. G. and W. O. D. are CNPq fellows; J. S. C. S. and F. A. V. are CAPES fellows.
Disclosure
The authors do not have any conflict of interest with the material presented in the paper.
References
- 1.Rassi A, Jr, Rassi A, Marin-Neto JA. Chagas disease. Lancet. 2010;375:1388–402. doi: 10.1016/S0140-6736(10)60061-X. [DOI] [PubMed] [Google Scholar]
- 2.Marin-Neto JA, Cunha-Neto E, Maciel BC, Simoes MV. Pathogenesis of chronic Chagas heart disease. Circulation. 2007;115:1109–23. doi: 10.1161/CIRCULATIONAHA.106.624296. [DOI] [PubMed] [Google Scholar]
- 3.Batlouni M, Barretto AC, Armaganijan D, et al. [Treatment of mild and moderate cardiac failure with captopril. A multicenter study] Arq Bras Cardiol. 1992;58:417–21. [PubMed] [Google Scholar]
- 4.Leon JS, Wang K, Engman DM. Captopril ameliorates myocarditis in acute experimental Chagas disease. Circulation. 2003;107:2264–9. doi: 10.1161/01.CIR.0000062690.79456.D0. [DOI] [PubMed] [Google Scholar]
- 5.Bahk TJ, Daniels MD, Leon JS, Wang K, Engman DM. Comparison of angiotensin converting enzyme inhibition and angiotensin II receptor blockade for the prevention of experimental autoimmune myocarditis. Int J Cardiol. 2008;125:85–93. doi: 10.1016/j.ijcard.2007.04.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Godsel LM, Leon JS, Engman DM. Angiotensin converting enzyme inhibitors and angiotensin II receptor antagonists in experimental myocarditis. Curr Pharm Des. 2003;9:723–35. doi: 10.2174/1381612033455440. [DOI] [PubMed] [Google Scholar]
- 7.Skidgel RA, Erdos EG. Angiotensin converting enzyme (ACE) and neprilysin hydrolyze neuropeptides: a brief history, the beginning and follow-ups to early studies. Peptides. 2004;25:521–5. doi: 10.1016/j.peptides.2003.12.010. [DOI] [PubMed] [Google Scholar]
- 8.Regoli D, Barabe J. Pharmacology of bradykinin and related kinins. Pharmacol Rev. 1980;32:1–46. [PubMed] [Google Scholar]
- 9.Scharfstein J, Schmitz V, Svensjo E, Granato A, Monteiro AC. Kininogens coordinate adaptive immunity through the proteolytic release of bradykinin, an endogenous danger signal driving dendritic cell maturation. Scand J Immunol. 2007;66:128–36. doi: 10.1111/j.1365-3083.2007.01983.x. [DOI] [PubMed] [Google Scholar]
- 10.Chen BC, Yu CC, Lei HC, et al. Bradykinin B2 receptor mediates NF-kappaB activation and cyclooxygenase-2 expression via the Ras/Raf-1/ERK pathway in human airway epithelial cells. J Immunol. 2004;173:5219–28. doi: 10.4049/jimmunol.173.8.5219. [DOI] [PubMed] [Google Scholar]
- 11.Maruo K, Akaike T, Ono T, Maeda H. Involvement of bradykinin generation in intravascular dissemination of Vibrio vulnificus and prevention of invasion by a bradykinin antagonist. Infect Immun. 1998;66:866–9. doi: 10.1128/iai.66.2.866-869.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Murta AC, Persechini PM, Padron Tde S, de Souza W, Guimaraes JA, Scharfstein J. Structural and functional identification of GP57/51 antigen of Trypanosoma cruzi as a cysteine proteinase. Mol Biochem Parasitol. 1990;43:27–38. doi: 10.1016/0166-6851(90)90127-8. [DOI] [PubMed] [Google Scholar]
- 13.Scharfstein J, Schmitz V, Morandi V, et al. Host cell invasion by Trypanosoma cruzi is potentiated by activation of bradykinin B(2) receptors. J Exp Med. 2000;192:1289–300. doi: 10.1084/jem.192.9.1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Todorov AG, Andrade D, Pesquero JB, et al. Trypanosoma cruzi induces edematogenic responses in mice and invades cardiomyocytes and endothelial cells in vitro by activating distinct kinin receptor (B1/B2) subtypes. FASEB J. 2003;17:73–5. doi: 10.1096/fj.02-0477fje. [DOI] [PubMed] [Google Scholar]
- 15.Monteiro AC, Schmitz V, Morrot A, et al. Bradykinin B2 receptors of dendritic cells, acting as sensors of kinins proteolytically released by Trypanosoma cruzi, are critical for the development of protective type-1 responses. PLoS Pathog. 2007;3:e185. doi: 10.1371/journal.ppat.0030185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Monteiro AC, Schmitz V, Svensjo E, et al. Cooperative activation of TLR2 and bradykinin B2 receptor is required for induction of type 1 immunity in a mouse model of subcutaneous infection by Trypanosoma cruzi. J Immunol. 2006;177:6325–35. doi: 10.4049/jimmunol.177.9.6325. [DOI] [PubMed] [Google Scholar]
- 17.Souza PE, Rocha MO, Rocha-Vieira E, et al. Monocytes from patients with indeterminate and cardiac forms of Chagas' disease display distinct phenotypic and functional characteristics associated with morbidity. Infect Immun. 2004;72:5283–91. doi: 10.1128/IAI.72.9.5283-5291.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jensen PE. Recent advances in antigen processing and presentation. Nat Immunol. 2007;8:1041–8. doi: 10.1038/ni1516. [DOI] [PubMed] [Google Scholar]
- 19.Friedland J, Setton C, Silverstein E. Induction of angiotensin converting enzyme in human monocytes in culture. Biochem Biophys Res Commun. 1978;83:843–9. doi: 10.1016/0006-291x(78)91471-7. [DOI] [PubMed] [Google Scholar]
- 20.Shen XZ, Lukacher AE, Billet S, Williams IR, Bernstein KE. Expression of angiotensin-converting enzyme changes major histocompatibility complex class I peptide presentation by modifying C termini of peptide precursors. J Biol Chem. 2008;283:9957–65. doi: 10.1074/jbc.M709574200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Costerousse O, Allegrini J, Lopez M, Alhenc-Gelas F. Angiotensin I-converting enzyme in human circulating mononuclear cells: genetic polymorphism of expression in T-lymphocytes. Biochem J. 1993;290:33–40. doi: 10.1042/bj2900033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Costa GC, da Costa Rocha MO, Moreira PR, et al. Functional IL-10 gene polymorphism is associated with Chagas disease cardiomyopathy. J Infect Dis. 2009;199:451–4. doi: 10.1086/596061. [DOI] [PubMed] [Google Scholar]
- 23.Roberti RR, Martinez EE, Andrade JL, et al. Chagas cardiomyopathy and captopril. Eur Heart J. 1992;13:966–70. doi: 10.1093/oxfordjournals.eurheartj.a060301. [DOI] [PubMed] [Google Scholar]
- 24.Barbasz A, Kozik A. The assembly and activation of kinin-forming systems on the surface of human U-937 macrophage-like cells. Biol Chem. 2009;390:269–75. doi: 10.1515/BC.2009.032. [DOI] [PubMed] [Google Scholar]
- 25.Godsel LM, Leon JS, Wang K, Fornek JL, Molteni A, Engman DM. Captopril prevents experimental autoimmune myocarditis. J Immunol. 2003;171:346–52. doi: 10.4049/jimmunol.171.1.346. [DOI] [PubMed] [Google Scholar]
- 26.Hooper NM. Angiotensin converting enzyme: implications from molecular biology for its physiological functions. Int J Biochem. 1991;23:641–7. doi: 10.1016/0020-711x(91)90032-i. [DOI] [PubMed] [Google Scholar]
- 27.Saijonmaa O, Nyman T, Fyhrquist F. Atorvastatin inhibits angiotensin-converting enzyme induction in differentiating human macrophages. Am J Physiol Heart Circ Physiol. 2007;292:H1917–21. doi: 10.1152/ajpheart.00920.2006. [DOI] [PubMed] [Google Scholar]
- 28.Danilov SM, Sadovnikova E, Scharenborg N, et al. Angiotensin-converting enzyme (CD143) is abundantly expressed by dendritic cells and discriminates human monocyte-derived dendritic cells from acute myeloid leukemia-derived dendritic cells. Exp Hematol. 2003;31:1301–9. doi: 10.1016/j.exphem.2003.08.018. [DOI] [PubMed] [Google Scholar]
- 29.Schindler R, Dinarello CA, Koch KM. Angiotensin-converting-enzyme inhibitors suppress synthesis of tumour necrosis factor and interleukin 1 by human peripheral blood mononuclear cells. Cytokine. 1995;7:526–33. doi: 10.1006/cyto.1995.0071. [DOI] [PubMed] [Google Scholar]
- 30.Shen XZ, Li P, Weiss D, et al. Mice with enhanced macrophage angiotensin-converting enzyme are resistant to melanoma. Am J Pathol. 2007;170:2122–34. doi: 10.2353/ajpath.2007.061205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Constantinescu CS, Goodman DB, Ventura ES. Captopril and lisinopril suppress production of interleukin-12 by human peripheral blood mononuclear cells. Immunol Lett. 1998;62:25–31. doi: 10.1016/s0165-2478(98)00025-x. [DOI] [PubMed] [Google Scholar]
- 32.Odaka C, Mizuochi T. Angiotensin-converting enzyme inhibitor captopril prevents activation-induced apoptosis by interfering with T cell activation signals. Clin Exp Immunol. 2000;121:515–22. doi: 10.1046/j.1365-2249.2000.01323.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Deas O, Dumont C, Mollereau B, et al. Thiol-mediated inhibition of FAS and CD2 apoptotic signaling in activated human peripheral T cells. Int Immunol. 1997;9:117–25. doi: 10.1093/intimm/9.1.117. [DOI] [PubMed] [Google Scholar]
- 34.Harrington LE, Hatton RD, Mangan PR, et al. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6:1123–32. doi: 10.1038/ni1254. [DOI] [PubMed] [Google Scholar]
- 35.Tesmer LA, Lundy SK, Sarkar S, Fox DA. Th17 cells in human disease. Immunol Rev. 2008;223:87–113. doi: 10.1111/j.1600-065X.2008.00628.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bacellar O, Faria D, Nascimento M, et al. Interleukin 17 production among patients with American cutaneous leishmaniasis. J Infect Dis. 2009;200:75–8. doi: 10.1086/599380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.da Matta Guedes PM, Gutierrez FR, Maia FL, et al. IL-17 produced during Trypanosoma cruzi infection plays a central role in regulating parasite-induced myocarditis. PLoS Negl Trop Dis. 2010;4:e604. doi: 10.1371/journal.pntd.0000604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Monteiro AC, Scovino A, Raposo S, et al. Kinin danger signals proteolytically released by gingipain induce Fimbriae-specific IFN-gamma- and IL-17-producing T cells in mice infected intramucosally with Porphyromonas gingivalis. J Immunol. 2009;183:3700–11. doi: 10.4049/jimmunol.0900895. [DOI] [PMC free article] [PubMed] [Google Scholar]