

INTRODUCTION
A relationship between sympathetic nerve dysfunction and cardiovascular morbidity and mortality has been recognized for decades yet there has only recently been interest in applying sympathetic nerve imaging to the clinical management of patients with heart disease. With the increasing availability of nuclear tracers of norepinephrine it is likely that in the near future non-invasive imaging of sympathetic function will be translated from the realm of research to the clinical armamentarium. This brief review will focus on sympathetic nerve dysfunction in the setting of coronary artery disease with particular attention to pathophysiology, pre-synaptic molecular imaging, and potential clinical applications. A discussion of technologies for imaging the cardiac sympathetic nervous system can be found in the recent review by Link and Caldwell,1 and a detailed review of PET tracers and receptor ligands has been previously published by Bengel and Schwaiger.2
PATHOPHYSIOLOGY OF DYSINNERVATED BUT VIABLE MYOCARDIUM
There are various pathophysiological explanations for ischemically mediated sympathetic nerve dysfunction in the setting of viable myocardium that are analogous to the continuum of physiologies that occur with ischemia-induced myocardial dysfunction. These can range from complete anatomic denervation following transmural myocardial infarction3 to transient sympathetic nerve dysfunction in association with acute myocardial stunning.4 In this review, we will use “dysinnervation” or sympathetic nerve “dysfunction” as general terms when the relative contribution of reversible neural stunning is unknown, reserving the term “denervation” for situations in which there is an anatomic loss of sympathetic nerves. These distinctions are likely to be clinically important since the relative extent of neural stunning versus denervation may influence the potential for recovery (and time-course of recovery) and may affect any clinical correlates associated with the dysinnervated but viable myocardium (as will be discussed in more detail below).
Myocardial Infarction Causes Sympathetic Denervation
Prolonged ischemia that results in myocardial infarction is invariability associated with irreversible sympathetic nerve dysfunction. Not only does sympathetic denervation affect the area of infarction, but also for decades it has been recognized that the extent of denervation exceeds that of infarction following coronary artery occlusion (Figure 1). This is the result of down stream denervation from irreversible injury of the sympathetic nerves traversing the area of infarction as they course from the base of the heart toward the apex. For example, Zipes and colleagues3 showed that coronary embolization of a diagonal branch in dogs resulted in functional denervation not only in the area of infarction, but also to the area apical to the infarct parallel to the left anterior descending coronary artery (LAD). Quantification of norepinephrine content and norepinephrine fluorescence confirmed that the sympathetic dysfunction was due to anatomic denervation.3 More recent studies have shown that denervation also occurs with nontransmural myocardial infarction but, in contrast to transmural infarction, there is subepicardial viability that may preserve sympathetic nerve fibers, and also limit the extent of apical denervation of viable myocardium.5
Figure 1.
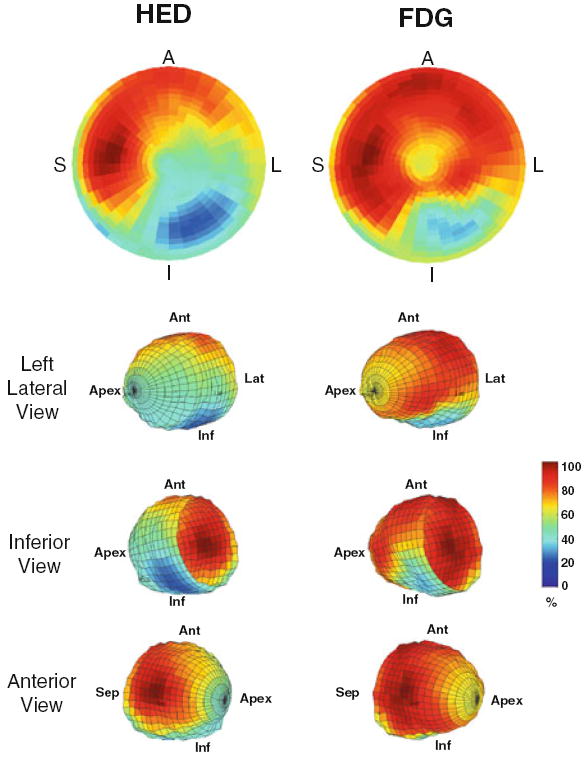
Viable, denervated myocardium after myocardial infarction. These PET polar tomograms and wire diagrams of the left ventricle from a subject in the PAREPET study51 illustrate the relative uptake of the sympathetic nerve tracer 11C-meta-hydroyxephedrine (HED, left images) and the viability tracer 18F-2-deoxyglucose (FDG, right images). Tracer retention is color coded from maximum activity in red to minimum activity in blue. The extent of sympathetic denervation extends beyond that of the inferolateral infarct. In addition to denervation at the periphery of the infarct, there is apical extension due to interruption of the sympathetic nerves that course across the heart from the base to the apex. A and Ant, anterior wall; S and Sep, interventricular septum; L and Lat, lateral wall; I and Inf, inferior wall.
Subsequent studies by Zipes and colleagues have shown that denervated but viable myocardium adjacent to an area of infarction may be particularly arrhythmogenic.6 These regions are hypersensitive to catecholamine infusion (denervation supersensitivity or hypersensitivity), and programmed electrical stimulation in these areas has been shown to induce ventricular fibrillation more frequently than in sham-operated controls.6 This response could be amplified by sympathetic stimulation (norepinephrine infusion or stellate ganglion stimulation), while it was attenuated by infusion of the β-adrenergic receptor antagonist propranolol.6
These and other pivotal studies support the sympathetic model of arrhythmogenesis as championed by Zipes and colleagues.7,8 The principle substrate of this hypothesis is regional heterogeneity in sympathetic nerve function classically the result of infarction, but now also known to occur with reversible myocardial dysfunction (as will be discussed below). Denervated but viable myocardium can develop denervation hypersensitivity with an upregulation of β-adrenergic receptors6 and increased sympathetic nerve density due to nerve sprouting,9 both of which will lead to exaggerated responses during sympathetic stimulation. In addition, abnormalities in neurotransmitter release and re-uptake can cause electrical remodeling of cardiac myocytes. Taken together, these factors culminate in regional heterogeneity in action potential duration which is accentuated during sympathetic activation and facilitates the initiation of ventricular tachyarrhythmias that result in sudden death.
Sympathetic Nerve Dysfunction Can Be Reversible Following Acute Ischemia
Reversible myocardial ischemia sufficient to result in prolonged contractile dysfunction or “stunning” after reperfusion also results in impaired cardiac sympathetic nerve function.4 Some investigators have suggested that sympathetic nerves are even more sensitive to ischemia than cardiac myocytes, and have concluded that sympathetic dysfunction in the setting of viable myocardium is the result of denervation.2,10,11 However, basic investigations have clearly shown that denervation is not necessarily present. For example, despite the fact that stellate ganglion stimulation failed to augment the function of stunned myocardium in dogs, function could be improved by stimulating the pre-synaptic release of norepinephrine with bretylium.4 Likewise, while vasoconstrictor responses to stellate ganglion stimulation were abolished in stunned myocardium, they could be restored by stimulating pre-synaptic norepinephrine release using bretylium or tyramine.12 Interestingly, neural stunning could be induced independently of ischemia by the administration of adenosine, suggesting that it was the mediator of reversible sympathetic nerve function.13,14
Nuclear imaging data also support the concept of reversible sympathetic nerve dysfunction in the setting of myocardial ischemia. Dae et al15 performed 30-minute intracoronary balloon occlusion in dogs and assessed sympathetic nerve function by the uptake and washout of the norepinephrine analog 123I-meta-iodobenzylguanidine (MIBG). Even though this duration of ischemia produced some degree of subendocardial necrosis, there were variable effects on MIBG imaging 1-2 weeks later. Persistent abnormalities in MIBG uptake were strongly associated with sympathetic denervation and more severe subendocardial necrosis.15 Conversely, animals with normal MIBG imaging had little myocardial necrosis and relatively preserved tissue norepinephrine levels.15 In summary, while these basic studies suggest that sympathetic nerve function may be very sensitive to brief ischemia, sympathetic nerve dysfunction may be transient (neural stunning), and not necessarily indicative of anatomic denervation.
Hibernating Myocardium Results in Partial Sympathetic Denervation
Chronic repetitive ischemia leads to resting contractile dysfunction (chronically stunned and hibernating myocardium) with concomitant regional sympathetic nerve dysfunction. Using a porcine model of chronic hibernating myocardium, we have shown that there is a progression in the degree of sympathetic nerve dysfunction that parallels progression in the physiological significance of the coronary stenosis. Mild dysfunction in the LAD territory at 1-month after instrumentation was associated with a modest reduction in regional coronary flow reserve with normal resting perfusion (stunned myocardium).16 At this time, there were no significant defects in sympathetic nerve function when imaged with the positron emitting norepinephrine analog 11C-meta-hydroxyephedrine (HED, Figure 2).17 As stenosis severity progressed and coronary flow reserve was further reduced (2 months after instrumentation) regional function deteriorated, yet resting perfusion remained normal (chronic stunning).16 At this point, however, the frequency and/or severity of ischemia resulted in regional pre-synaptic sympathetic nerve dysfunction as reflected by regionally reduced HED uptake (Figure 2).17
Figure 2.
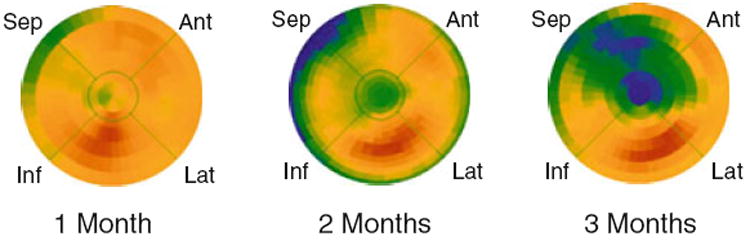
Progression of HED defects during the development of hibernating myocardium in chronically instrumented pigs. Preserved HED uptake at 1 month confirmed that LAD instrumentation did not affect regional sympathetic nerve function.16 The polar maps at 2 and 3 months after instrumentation were serially obtained in the same animal. These studies show a clear progression from a small, localized defect in the apex at 2 months (relative uptake, LAD/normal = .76) to a larger and more severe defect 3 months after instrumentation (relative uptake = .50). The increasing size and severity of the HED defects parallels the changes in stenosis severity as this model progresses from chronically stunned to hibernating myocardium.16 Modified from Luisi et al.17 Reprinted with permission of the Society of Nuclear Medicine, Inc. Ant, anterior; Lat, lateral, Inf, inferior; Sep, septum.
Three months after initial instrumentation, regional flow reserve in the LAD distribution became critically reduced, resulting in a secondary downregulation in resting perfusion but no histological evidence of myocardial necrosis (hibernating myocardium).18,19 Even in the absence of infarction, sympathetic dysinnervation was extensive with large and dense defects in HED uptake (Figures 2 and 3). Physiological evaluation showed moderately impaired pre-synaptic sympathetic nerve function with submaximal increases in regional function in response to stellate ganglion or tyramine stimulation.20 Subsequent molecular analyses have confirmed that the sympathetic dysinnervation in pigs with hibernating myocardium is the result of partial denervation, with moderate (25-50%) reductions in regional tissue norepinephrine levels, norepinephrine uptake-1 transport protein, tyrosine hydroxylase, and sympathetic nerve density (Figure 4).21
Figure 3.
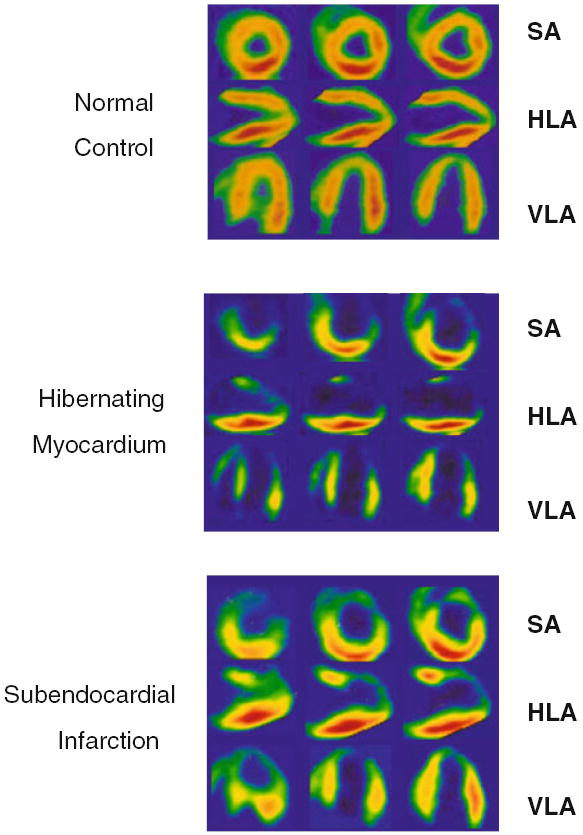
HED images in pigs with normal, hibernating, and subendocardially infarcted myocardium. Standard imaging views demonstrate homogeneous HED uptake in a normal control animal. In contrast, there is a severe and extensive HED defect involving the LAD distribution in a pig with hibernating myocardium, consistent with partial sympathetic denervation. In an animal with subendocardial infarction in the LAD distribution, the defect in HED uptake is actually less severe and extensive than that in hibernating myocardium. Modified from Luisi et al.17 Reprinted with permission of the Society of Nuclear Medicine, Inc. SA, short axis; HLA, horizontal long axis; VLA, vertical long axis).
Figure 4.
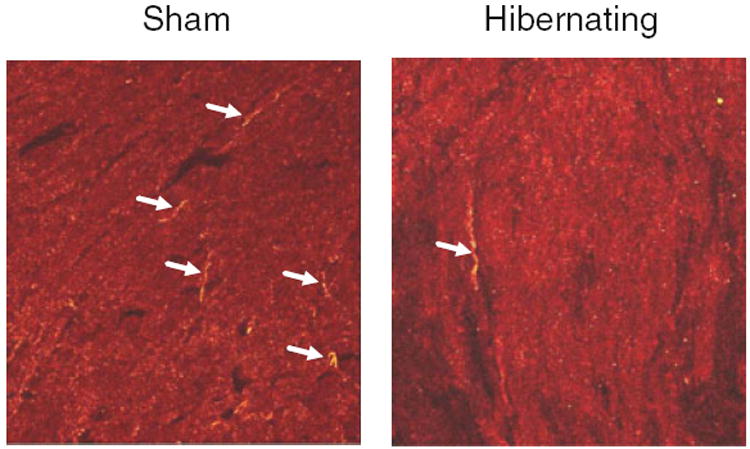
Partial sympathetic denervation in porcine hibernating myocardium. These photomicrographs illustrate the immunohistochemical staining of tyrosine hydroxylase (yellow) in a sham control pig (left) and an animal with hibernating myocardium (right). Sympathetic nerves (arrows) are present in hibernating myocardium, although they are not as abundant as in control myocardium. These images are consistent with the reduced (but not absent) functional responses to sympathetic stimulation20 and candidate sympathetic nerve protein expression,21 confirming partial sympathetic denervation in pigs with hibernating myocardium.
After the development of hibernating myocardium in this chronic porcine model, there is a remarkable stability in physiology with no subsequent changes in regional perfusion, flow reserve, or function.22,23 In spite of this adaptation to ischemia, there is a cumulative 50% risk of spontaneous sudden death due to ventricular tachycardia degenerating into ventricular fibrillation that occurs in the absence of acute infarction.24 While the stability in regional HED retention after the development of hibernating myocardium17 would also suggest a sympathetic nerve adaptation to ischemia, we have recently identified regional upregulation of growth-associated protein-43 which is associated with sympathetic nerve sprouting.21 We have also found upregulation of nerve growth factor in hibernating myocardium, which may serve as the stimulus for new nerve growth.21 As discussed above, nerve sprouting has been hypothesized to play a role in arrhythmic sudden death by exacerbating regional heterogeneity in sympathetic nerve function associated with infarct-related denervation. Our findings suggest that a similar mechanism may contribute to arrhythmogenesis in the setting of viable, partially denervated myocardium without infarction.
We have examined the impact of interventions that improve regional function in hibernating myocardium on sympathetic nerve function. While both percutaneous coronary intervention25,26 and pravastatin27 improve regional and global function in pigs with hibernating myocardium, neither therapy altered HED uptake, retention, defect size, or defect severity after 1 month (Figure 5).28 These results suggest that there is no significant component of reversible neural stunning and further reinforces the notion that sympathetic nerve dysfunction in hibernating myocardium is the result of partial sympathetic denervation.
Figure 5.
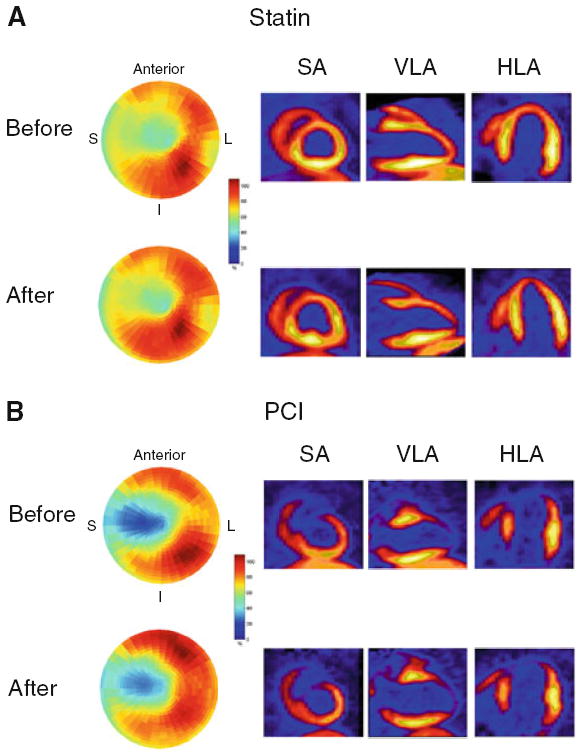
HED imaging before and after therapeutic intervention in pigs with hibernating myocardium. Each pair of images is from a pig with hibernating myocardium serially imaged before and 1 month after therapy with either pravastatin (A) or percutaneous coronary intervention (PCI, B). The polar tomograms illustrate that the size and severity of the HED defects remained remarkably stable between the two studies, despite the fact that each therapy resulted in significant improvement in regional and global function in pigs with hibernating myocardium.25-27 Representative reconstructed short axis (SA), vertical long axis (VLA), and horizontal long axis (HLA) views are also shown. Reprinted from Fallavollita et al.28 S, interventricular septum; L, lateral wall; I, inferior wall.
Clinical Translation of Sympathetic Dysinnervation in Viable Myocardium
This brief review of animal models of myocardial ischemia clearly shows that sympathetic dysinnervation can arise from a number of different mechanisms in reversible and irreversible ischemia, and range from complete anatomic denervation3 to transient reversible sympathetic nerve dysfunction consistent with “neural stunning.”4 Sympathetic denervation in patients with myocardial infarction has been extensively documented,2,29 but the potential role of reversible sympathetic nerve dysfunction is less widely appreciated. For example, in a study of patients with mild coronary artery disease and no exercise-induced perfusion abnormalities, initial MIBG uptake was regionally normal consistent with normal sympathetic nerve density.30 However, the subsequent rate of MIBG washout (after 4-5 hours) was increased, reflecting abnormal sympathetic nerve function. Sympathetic nerve dysfunction immediately following acute ischemia has also been clearly demonstrated in patients with acute coronary syndromes.31 The MIBG defects were nearly identical to the area at risk of ischemia which included both reversibly and irreversibly injured myocardium (Figure 6). Although reversibility of these defects was not documented, another small study showed that five of eight patients without restenosis after percutaneous intervention had qualitative improvement in their MIBG defects, whereas three of four patients with restenosis showed an increase in MIBG defect size.32
Figure 6.
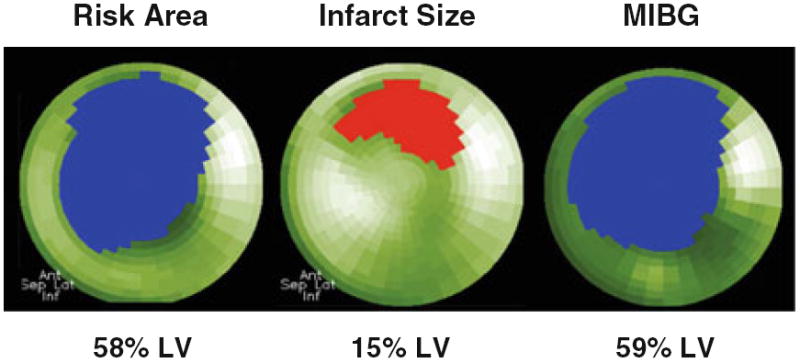
Extent of the MIBG defect correlates with area at risk during acute coronary occlusion. These polar tomograms were obtained from a patient with an acute anterior myocardial infarction. The risk area was quantified with 99mTc-sestamibi prior to reperfusion with percutaneous coronary intervention, and infarct size was documented from repeat imaging 1 week later.31 The defect in sympathetic nerve function assessed with MIBG was significantly larger than the area of infarction and was almost identical to the original extent of myocardial ischemia. Figure source: Dr. Markus Schwaiger. Ant, anterior; Lat, lateral; Inf, inferior; Sep, septum.
There is also abundant clinical data that sympathetic nerve dysfunction can develop in humans as a result of chronic ischemic heart disease without infarction. This was initially described by Hartikainen et al33 in a series of patients with stable ischemic symptoms and no history of prior infarction. Regional defects in MIBG were found in almost all patients with significant stenoses (>50% diameter), with defect size increasing as a function of stenosis severity. Among those with a severe stenosis (>90% diameter) MIBG defect size was indistinguishable from patients with previous infarction.33 Similar results were subsequently published by Bulow et al34 using HED and PET. Although the underlying pathology is unknown, our data in chronically instrumented pigs would suggest that there is at least some residual sympathetic innervation.
CLINICAL APPLICATION OF SYMPATHETIC NERVE IMAGING
Prognostic Potential of Cardiac Sympathetic Imaging—Global or Regional?
There is now ample evidence to support the contention that the imaging of sympathetic nerve function (primarily with MIBG) provides independent prognostic information in patients with heart disease.35 For example, the recently completed ADMIRE-HF trial enrolled 961 subjects with NYHA Class II or III heart failure and left ventricular ejection fraction ≤35%.36 Their primary analysis confirmed that a low MIBG heart-to-mediastinum ratio predicted the time to first cardiac event. This was largely driven by heart failure progression (increase in one heart failure class in 69% of events), although “potentially life-threatening arrhythmias” and cardiac deaths were also predicted. In another recently published study of 116 subjects undergoing MIBG imaging prior to ICD implantation, a summed, semi-quantitative MIBG defect score obtained 3-4 hours after tracer administration (but not heart-to-mediastinum ratio) was an independent predictor of appropriate ICD therapy.37 Thus, while the global assessment of myocardial sympathetic nerve function predicts subsequent cardiovascular events, there continues to be a paucity of data to address the critical question regarding how cardiac sympathetic imaging should impact patient management.
Multiple lines of evidence suggest that the prognostic potential of dysinnervated but viable myocardium may be even more important than global alterations in cardiac sympathetic function. First, the volume of dysinnervated but viable myocardium provides an index for the extent of myocardium at risk for ischemia,31 which independently predicts risk of subsequent cardiac events. Second, following myocardial infarction in pigs, animals with inducible monomorphic ventricular tachycardia had larger volumes of denervated but viable myocardium than those without arrhythmias.38 Third, using an ovine model, Kramer et al39 showed that the dysfunctional and denervated but viable border region adjacent to an infarct contributes to post-infarct, pathological remodeling. Finally, Chen and colleagues have documented increased density of sympathetic nerves due to nerve sprouting adjacent to areas of infarction. This was correlated with ventricular arrhythmias in dog40 and rabbit41 chronic infarct models as well as in patients with coronary artery disease.42 Therefore, regional quantification of cardiac sympathetic nerve function, especially in conjunction with an assessment of regional viability, may be particularly important for clinical applications.
Interestingly, a semi-quantitative assessment of regional MIBG retention was included in the ADMIRE-HF trial in addition to the global parameters of cardiac sympathetic nerve function.36 However, the regional analysis showed “no consistent contribution to event prediction” even in conjunction with perfusion images (99mTc-tetrofosmin).36 Another recent study demonstrated that the MIBG/99mTc-tetrofosmin mismatch score was a univariate predictor of appropriate ICD therapy, but only ICD indication (secondary vs primary prevention) and late MIBG defect score were independent predictors by multivariate analysis.37 Although these data suggest that the specific substrate of viable dysinnervated myocardium may be less important than global myocardial norepinephrine uptake, assessment of regional MIBG uptake is challenging and has important methodological limitations. For example, in pigs with hibernating myocardium we found a 48 ± 3% regional reduction in HED retention by PET imaging.17 In contrast, the relative difference in MIBG retention quantified with the more sensitive technique of ex vivo tissue counting was only 25 ± 3% (Figure 7).43 Thus, PET imaging of HED appears to provide an improved signal-to-noise ratio over MIBG, with an almost twofold improvement in defect severity that would likely facilitate quantitative regional analysis. In addition, 99mTc-tetrofosmin is an imperfect assessment of myocardial viability as compared to gadolinium late-enhancement MR imaging or even PET imaging with 18F-2-deoxy-glucose (FDG). This would tend to underestimate a mismatch between dysinnervation and viability, especially in hibernating myocardium with reduced resting perfusion. Finally, attenuation correction is clearly superior with PET as compared to SPECT imaging.
Figure 7.
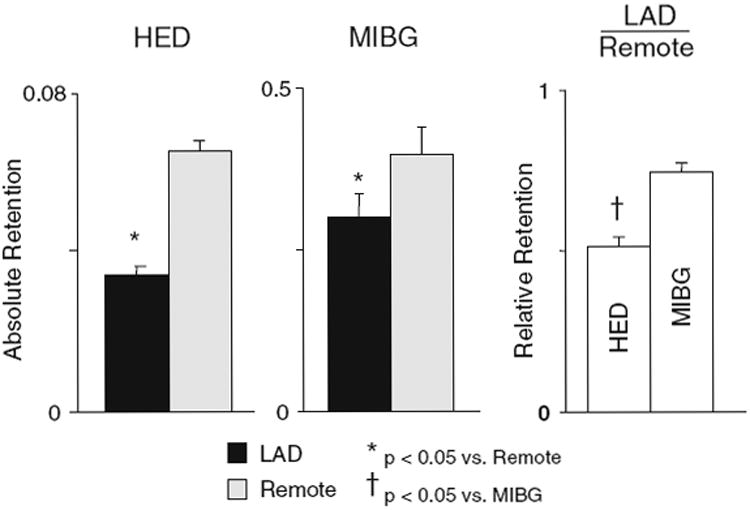
Regional differences in HED are greater than MIBG in hibernating myocardium. PET imaging demonstrated a 48 ± 3% reduction in HED retention in hibernating myocardium (black bars) as compared to the remote, normally perfused region (gray bars) of chronically instrumented pigs.17 In contrast, the regional difference in MIBG retention was only 25 ± 3% despite the fact that activity was quantified by ex vivo counting. This significant difference in relative retention (LAD/remote) was the result of improved specificity of HED due to less non-specific uptake than MIBG. Modified from Luisi et al.17 *P < 0.05 vs remote. †P < 0.05 vs MIBG. Reprinted with permission of the Society of Nuclear Medicine, Inc.
Sympathetic Nerve Dysfunction Is Dynamic in Patients with Coronary Artery Disease
Although there is emerging data to support the potential role of sympathetic imaging in ischemic heart disease, it is imperative to remain cognizant of the multiple factors that can affect a single, snapshot view of cardiac sympathetic nerve function. First, the commonly used tracers MIBG and HED are incapable of determining the relative roles of neural stunning versus anatomic denervation in image defects. Although reversible sympathetic nerve dysfunction may also have significant prognostic implications, there will likely be differences in temporal significance and response to therapy (i.e., revascularization) as compared to denervation. Second, although global MIBG uptake in heart failure patients has been shown to improve in response to therapy,44 it is unclear whether this reflects an improvement in cardiac sympathetic nerve function or is secondary to the well-described inverse relationship between MIBG heart-to-mediastinum ratio and plasma norepinephrine levels, which decrease with clinical improvement.45,46 Third, any progression of underlying coronary artery disease will tend to produce new areas of sympathetic dysinnervation. Finally, even anatomic denervation is not permanent as variable degrees of sympathetic re-innervation have been documented months to years after cardiac transplantation,47,48 and in some49,50 but not all studies10 after myocardial infarction.
Future Applications of Sympathetic Nerve Imaging
The ongoing PAREPET trial (Prediction of ARrhythmic Events with Positron Emission Tomography)51 may help to determine the clinical utility of assessing regional sympathetic nerve function and its role as a risk factor for sudden cardiac death. This NIH-sponsored observational cohort study has enrolled over 200 patients with ischemic cardiomyopathy who underwent PET imaging to quantify resting perfusion (13N-ammonia), myocardial viability (insulin-stimulated FDG uptake52), and sympathetic nerve function (HED). Figure 8 shows representative subjects that illustrate the range in myocardial viability and sympathetic nerve function in these patients. There is significant variability in the extent of viable, dysinnervated myocardium, from small borders around areas of infarction to large confluent regions encompassing several myocardial segments. PAREPET will determine the clinical importance of this variability by testing the hypothesis that the presence (≥ 1 segment) or the volume (as a % of the LV) of dysinnervated but viable myocardium will predict sudden death (primary outcome) or cardiac mortality (secondary outcome).51 This study continues in the follow-up phase with results of the primary end-point anticipated within the next year.
Figure 8.
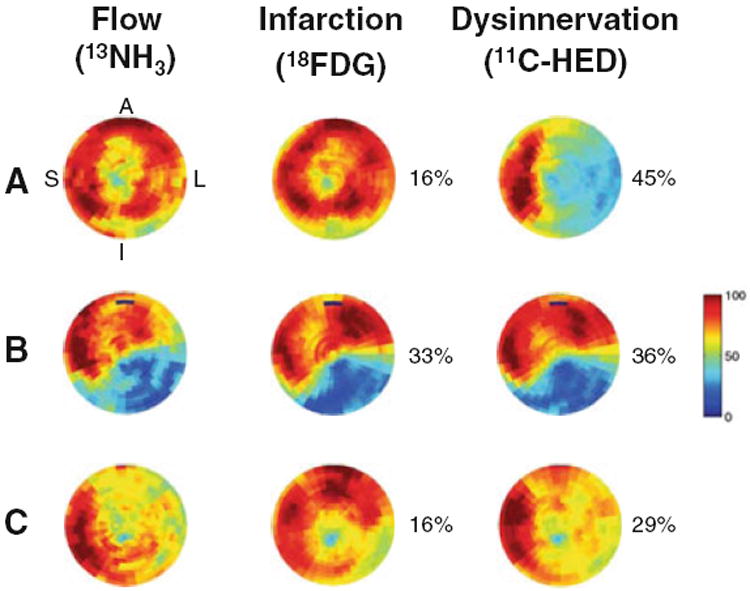
Representative patterns of resting flow, viability, and sympathetic innervation in subjects with ischemic cardio-myopathy. The tomograms within each row are from the same subject. The integrated volumes of infarction and dysinnervation as a percentage of the left ventricle are to the right of each image. The upper row (A) shows a subject with an extensive area of viable dysinnervated myocardium involving the anterior, lateral, and inferior myocardium. In (B), the inferolateral defect in sympathetic innervation is primarily the result of a large infarct with a concomitant reduction in resting flow and metabolic viability. The lower row (C) shows a mixed pattern of abnormal sympathetic innervation involving both viable and nonviable myocardium. Reprinted from Fallavollita et al.51 With permission of Elsevier, copyright 2006. A, anterior; L, lateral; I, inferior, S, septal.
Another very promising approach to identify a substrate associated with accentuated heterogeneity in cardiac sympathetic nerve function that may be particularly arrhythmogenic8 is the detection of a mismatch between pre-synaptic and post-synaptic sympathetic functions. This has been non-invasively imaged in patients with ischemic heart disease using the mismatch between HED and the β-adrenergic receptor tracer 11C-CGP 12177.53,54 Although no definitive prognostic data are available, Caldwell et al1,54 have suggested that an increase in the β-receptor density to norepinephrine transport ratio (i.e., a CGP–HED mismatch) portends a worse prognosis than a matched reduction in patients with ischemic cardiomyopathy (Figure 9). Additional studies will be necessary to determine the clinical application of this approach.
Figure 9.
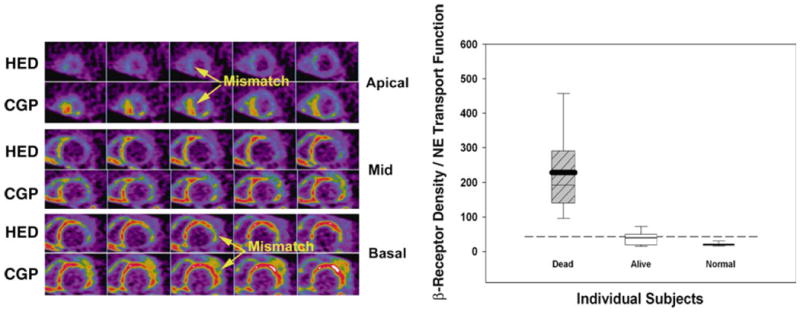
Mismatch between pre-synaptic and post-synaptic sympathetic functions may predict mortality. These short-axis PET images of pre-synaptic norepinephrine transport function (HED) and post-synaptic β-adrenergic receptor density (11C-CGP 12177) were obtained in a patient with NYHA Class III ischemic heart failure. Extensive mismatch between the two tracers is most evident in the apical and basal segments. The box and whisker plot (right graph) shows the ratio between β-receptor density and norepinephrine (NE) transport function for patients with ischemic heart failure (N = 13) as compared to normal controls (N = 19). The four patients who died within 15 months of imaging had significantly greater mismatch ratios than the nine survivors. The boxes span the 25th to 75th percentiles with the 95% confidence intervals delineated by the error bars. Modified from Link and Caldwell1 and reprinted with permission of MacMillan Publishers Ltd. Nature Clinical Practice Cardiovascular Medicine, copyright 2008.
Finally, it is important to recognize that other advances are likely to have significant implications for the clinical application of cardiac autonomic function. These include tracer developments to increase the specificity for imaging sympathetic nerve function, to assess more than simply norepinephrine uptake,2,55 and possibly to image cardiac parasympathetic nerve function56,57 or nerve spouting. In addition, advanced modeling of dynamic image sets may be able to improve the physiologic relevance of tracer parameters.58-60
Acknowledgments
We would like to thank Dr. Markus Schwaiger and Dr. James Caldwell for their contribution of figures, and Anne Coe for her help with the preparation of this manuscript. This study was supported by the National Heart, Lung and Blood Institute (HL-76252 and HL-81722); the Department of Veterans Affairs; and the Albert and Elizabeth Rekate Fund.
References
- 1.Link JM, Caldwell JH. Diagnostic and prognostic imaging of the cardiac sympathetic nervous system. Nat Clin Pract Cardiovasc Med. 2008;5:S79–86. doi: 10.1038/ncpcardio1150. [DOI] [PubMed] [Google Scholar]
- 2.Bengel FM, Schwaiger M. Assessment of cardiac sympathetic neuronal function using PET imaging. J Nucl Cardiol. 2004;11:603–16. doi: 10.1016/j.nuclcard.2004.06.133. [DOI] [PubMed] [Google Scholar]
- 3.Barber MJ, Mueller TM, Henry DP, Felten SY, Zipes DP. Transmural myocardial infarction in the dog produces sympathectomy in noninfarcted myocardium. Circulation. 1983;67:787–96. doi: 10.1161/01.cir.67.4.787. [DOI] [PubMed] [Google Scholar]
- 4.Ciuffo AA, Ouyang P, Becker LC, Levin L, Weisfeldt ML. Reduction of sympathetic inotropic response after ischemia in dogs. Contributor to stunned myocardium. J Clin Invest. 1985;75:1504–9. doi: 10.1172/JCI111854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dae MW, Herre JM, O’Connell JW, Botvinick EH, Newman D, Munoz L. Scintigraphic assessment of sympathetic innervation after transmural versus nontransmural myocardial infarction. J Am Coll Cardiol. 1991;17:1416–23. doi: 10.1016/s0735-1097(10)80156-1. [DOI] [PubMed] [Google Scholar]
- 6.Inoue H, Zipes DP. Results of sympathetic denervation in the canine heart: Supersensitivity that may be arrhythmogenic. Circulation. 1987;75:877–87. doi: 10.1161/01.cir.75.4.877. [DOI] [PubMed] [Google Scholar]
- 7.Tomaselli GF, Zipes DP. What causes sudden death in heart failure? Circ Res. 2004;95:754–63. doi: 10.1161/01.RES.0000145047.14691.db. [DOI] [PubMed] [Google Scholar]
- 8.Rubart M, Zipes DP. Mechanisms of sudden cardiac death. J Clin Invest. 2005;115:2305–15. doi: 10.1172/JCI26381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cao JM, Chen LS, KenKnight BH, Ohara T, Lee MH, Tsai J, et al. Nerve sprouting and sudden cardiac death. Circ Res. 2000;86:816–21. doi: 10.1161/01.res.86.7.816. [DOI] [PubMed] [Google Scholar]
- 10.Allman KC, Wieland DM, Muzik O, DeGrado TR, Wolfe ER, Jr, Schwaiger M. Carbon-11 hydroxyephedrine with positron emission tomography for serial assessment of cardiac adrenergic neuronal function after acute myocardial infarction in humans. J Am Coll Cardiol. 1993;22:368–75. doi: 10.1016/0735-1097(93)90039-4. [DOI] [PubMed] [Google Scholar]
- 11.Joho S, Asanoi H, Takagawa J, Kameyama T, Hirai T, Nozawa T, et al. Cardiac sympathetic denervation modulates the sympat-hoexcitatory response to acute myocardial ischemia. J Am Coll Cardiol. 2002;39:436–42. doi: 10.1016/s0735-1097(01)01770-3. [DOI] [PubMed] [Google Scholar]
- 12.Gutterman DD, Morgan DA, Miller FJ. Effect of brief myocardial ischemia on sympathetic coronary vasoconstriction. Circ Res. 1992;71:960–9. doi: 10.1161/01.res.71.4.960. [DOI] [PubMed] [Google Scholar]
- 13.Pettersen MD, Abe T, Morgan DA, Gutterman DD. Role of adenosine in postischemic dysfunction of coronary innervation. Circ Res. 1995;76:95–101. doi: 10.1161/01.res.76.1.95. [DOI] [PubMed] [Google Scholar]
- 14.Abe H, Morgan DA, Gutterman DD. Role of adenosine receptor subtypes in neural stunning of sympathetic coronary innervation. Am J Physiol Heart Circ Physiol. 1997;272:H25–34. doi: 10.1152/ajpheart.1997.272.1.H25. [DOI] [PubMed] [Google Scholar]
- 15.Dae MW, O’Connell JW, Botvinick EH, Chin MC. Acute and chronic effects of transient myocardial ischemia on sympathetic nerve activity, density, and norepinephrine content. Cardiovasc Res. 1995;30:270–80. [PubMed] [Google Scholar]
- 16.Fallavollita JA, Canty JM., Jr Differential 18F-2-deoxyglucose uptake in viable dysfunctional myocardium with normal resting perfusion: Evidence for chronic stunning in pigs. Circulation. 1999;99:2798–805. doi: 10.1161/01.cir.99.21.2798. [DOI] [PubMed] [Google Scholar]
- 17.Luisi AJ, Jr, Suzuki G, de Kemp R, Haka MS, Toorongian SA, Canty JM, Jr, et al. Regional 11C-hydroxyephedrine retention in hibernating myocardium: Chronic inhomogeneity of sympathetic innervation in the absence of infarction. J Nucl Med. 2005;46:1368–74. [PubMed] [Google Scholar]
- 18.Canty JM, Fallavollita JA. Hibernating myocardium. J Nucl Cardiol. 2005;12:104–19. doi: 10.1016/j.nuclcard.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 19.Fallavollita JA, Perry BJ, Canty JM., Jr 18F-2-deoxyglucose deposition and regional flow in pigs with chronically dysfunctional myocardium: Evidence for transmural variations in chronic hibernating myocardium. Circulation. 1997;95:1900–9. doi: 10.1161/01.cir.95.7.1900. [DOI] [PubMed] [Google Scholar]
- 20.Ovchinnikov V, Suzuki G, Canty JM, Jr, Fallavollita JA. Blunted functional responses to pre- and postjunctional sympathetic stimulation in hibernating myocardium. Am J Physiol Heart Circ Physiol. 2005;289:H1719–28. doi: 10.1152/ajpheart.00273.2005. [DOI] [PubMed] [Google Scholar]
- 21.Ovchinnikov V, Canty JM, Jr, Fallavollita JA. Hibernating myocardium leads to an upregulation in nerve growth factor and partial subendocardial sympathetic denervation. J Am Coll Cardiol. 2007;49:34A. [Google Scholar]
- 22.Fallavollita JA, Logue M, Canty JM., Jr Stability of hibernating myocardium in pigs with a chronic left anterior descending coronary artery stenosis: Absence of progressive fibrosis in the setting of stable reductions in flow, function and coronary flow reserve. J Am Coll Cardiol. 2001;37:1989–95. doi: 10.1016/s0735-1097(01)01250-5. [DOI] [PubMed] [Google Scholar]
- 23.Lynch P, Lee T-C, Fallavollita J, Canty JJM, Suzuki G. Intracoronary administration of AdvFGF-5 (Fibroblast Growth Factor-5) ameliorates left ventricular dysfunction and prevents myocyte loss in swine with developing collaterals and ischemic cardiomyopathy. Circulation. 2007;116:I-71–6. doi: 10.1161/CIRCULATIONAHA.106.681866. [DOI] [PubMed] [Google Scholar]
- 24.Canty JM, Jr, Suzuki G, Banas MD, Verheyen F, Borgers M, Fallavollita JA. Hibernating myocardium: Chronically adapted to ischemia but vulnerable to sudden death. Circ Res. 2004;94:1142–9. doi: 10.1161/01.RES.0000125628.57672.CF. [DOI] [PubMed] [Google Scholar]
- 25.Banas MD, Page B, Young RF, Fallavollita JA, Canty JM., Jr Residual dysfunction after revascularization of hibernating myocardium is independent of fibrosis and secondary to myocyte loss and persistent regional reduction in mitochondrial oxidative enzymes. Circulation. 2006;114:II–66. [Google Scholar]
- 26.Banas MD, Young H, Fallavollita JA, Canty JM., Jr Persistent reductions in flow and function after revascularization of swine with hibernating myocardium. J Am Coll Cardiol. 2006;47:179A. [Google Scholar]
- 27.Suzuki G, Iyer V, Cimato T, Canty JM., Jr Pravastatin improves function in hibernating myocardium by mobilizing CD133+ and cKit+ hematopoietic progenitor cells and promoting myocytes to reenter the growth phase of the cardiac cell cycle. Circ Res. 2009;104:255–64. doi: 10.1161/CIRCRESAHA.108.188730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fallavollita JA, Banas MD, Suzuki G, de Kemp RA, Sajjad M, Canty JM., Jr 11C-meta-hydroxyephedrine defects persist despite functional improvement in hibernating myocardium. J Nucl Cardiol. 2010;17:85–96. doi: 10.1007/s12350-009-9164-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Patel AD, Iskandrian AE. MIBG imaging. J Nucl Cardiol. 2002;9:75–94. doi: 10.1067/mnc.2002.121471. [DOI] [PubMed] [Google Scholar]
- 30.Simula S, Lakka T, Kuikka J, Laitinen T, Remes J, Kettunen R, et al. Cardiac adrenergic innervation within the first 3 months after acute myocardial infarction. Clin Physiol. 2000;20:366–73. doi: 10.1046/j.1365-2281.2000.00278.x. [DOI] [PubMed] [Google Scholar]
- 31.Matsunari I, Schricke U, Bengel FM, Haase HU, Barthel P, Schmidt G, et al. Extent of cardiac sympathetic neuronal damage is determined by the area of ischemia in patients with acute coronary syndromes. Circulation. 2000;101:2579–85. doi: 10.1161/01.cir.101.22.2579. [DOI] [PubMed] [Google Scholar]
- 32.Guertner C, Klepzig H, Jr, Maul FD, Hartmann A, Lelbach S, Hellmann A, et al. Noradrenaline depletion in patients with coronary artery disease before and after percutaneous transluminal coronary angioplasty with iodine-123 metaiodobenzylguanidine and single-photon emission tomography. Eur J Nucl Med. 1993;20:776–82. doi: 10.1007/BF00180908. [DOI] [PubMed] [Google Scholar]
- 33.Hartikainen J, Mustonen J, Kuikka J, Vanninen E, Kettunen R. Cardiac sympathetic denervation in patients with coronary artery disease without previous myocardial infarction. Am J Cardiol. 1997;80:273–7. doi: 10.1016/s0002-9149(97)00345-7. [DOI] [PubMed] [Google Scholar]
- 34.Bulow HP, Stahl F, Lauer B, Nekolla SG, Schuler G, Schwaiger M, et al. Alterations of myocardial presynaptic sympathetic innervation in patients with multi-vessel coronary artery disease but without history of myocardial infarction. Nucl Med Commun. 2003;24:233–9. doi: 10.1097/00006231-200303000-00002. [DOI] [PubMed] [Google Scholar]
- 35.Verberne HJ, Brewster LM, Somsen GA, van Eck-Smit BL. Prognostic value of myocardial 123I-metaiodobenzylguanidine (MIBG) parameters in patients with heart failure: A systematic review. Eur Heart J. 2008;29:1147–59. doi: 10.1093/eurheartj/ehn113. [DOI] [PubMed] [Google Scholar]
- 36.Jacobson AF, Senior R, Cerqueira MD, Wong ND, Thomas GS, Lobez VA, et al. Myocardial iodine-123 Meta-Iodobenzylguanidine imaging and cardiac events in heart failure. J Am Coll Cardiol. 2010;55:2212–21. doi: 10.1016/j.jacc.2010.01.014. [DOI] [PubMed] [Google Scholar]
- 37.Boogers MJ, Borleffs CJ, Henneman MM, van Bommel RJ, van Ramshorst J, Boersma E, et al. Cardiac sympathetic denervation assessed with 123-iodine metaiodobenzylguanidine imaging predicts ventricular arrhythmias in implantable cardioverter-defibrillator patients. J Am Coll Cardiol. 2010;55:2769–77. doi: 10.1016/j.jacc.2009.12.066. [DOI] [PubMed] [Google Scholar]
- 38.Sasano T, Abraham MR, Chang KC, Ashikaga H, Mills KJ, Holt DP, et al. Abnormal sympathetic innervation of viable myocardium and the substrate of ventricular tachycardia after myocardial infarction. J Am Coll Cardiol. 2008;51:2266–75. doi: 10.1016/j.jacc.2008.02.062. [DOI] [PubMed] [Google Scholar]
- 39.Kramer CM, Nicol PD, Rogers WJ, Suzuki MM, Shaffer A, Theobald TM, et al. Reduced sympathetic innervation underlies adjacent noninfarcted region dysfunction during left ventricular remodeling. J Am Coll Cardiol. 1997;30:1079–85. doi: 10.1016/s0735-1097(97)00244-1. [DOI] [PubMed] [Google Scholar]
- 40.Zhou S, Chen LS, Miyauchi Y, Miyauchi M, Kar S, Kangavari S, et al. Mechanisms of cardiac nerve sprouting after myocardial infarction in dogs. Circ Res. 2004;95:76–83. doi: 10.1161/01.RES.0000133678.22968.e3. [DOI] [PubMed] [Google Scholar]
- 41.Liu YB, Wu CC, Lu LS, Su MJ, Lin CW, Lin SF, et al. Sympathetic nerve sprouting, electrical remodeling, and increased vulnerability to ventricular fibrillation in hypercholesterolemic rabbits. Circ Res. 2003;92:1145–52. doi: 10.1161/01.RES.0000072999.51484.92. [DOI] [PubMed] [Google Scholar]
- 42.Cao JM, Fishbein MC, Han JB, Lai WW, Lai AC, Wu TJ, et al. Relationship between regional cardiac hyperinnervation and ventricular arrhythmia. Circulation. 2000;101:1960–9. doi: 10.1161/01.cir.101.16.1960. [DOI] [PubMed] [Google Scholar]
- 43.Luisi AJ, Jr, Fallavollita JA, Suzuki G, Canty JM., Jr Spatial inhomogeneity of sympathetic nerve function in hibernating myocardium. Circulation. 2002;106:779–81. doi: 10.1161/01.cir.0000028604.23202.ac. [DOI] [PubMed] [Google Scholar]
- 44.Merlet P, Pouillart F, Dubois-Rande JL, Delahaye N, Fumey R, Castaigne A, et al. Sympathetic nerve alterations assessed with 123I-MIBG in the failing human heart. J Nucl Med. 1999;40:224–31. [PubMed] [Google Scholar]
- 45.de Milliano PA, van Eck-Smit BL, van Zwieten PA, de Groot AC, Tijssen JG, Lie KI. Relationship between cardiac metaiodobenzylguanidine uptake and hemodynamic, functional and neurohormonal parameters in patients with heart failure. Eur J Heart Fail. 2001;3:693–7. doi: 10.1016/s1388-9842(01)00184-2. [DOI] [PubMed] [Google Scholar]
- 46.de Milliano PA, Tijssen JG, van Eck-Smit BL, Lie KI. Cardiac 123 I-MIBG imaging and clinical variables in risk stratification in patients with heart failure treated with beta blockers. Nucl Med Commun. 2002;23:513–9. doi: 10.1097/00006231-200206000-00002. [DOI] [PubMed] [Google Scholar]
- 47.Bengel FM, Ueberfuhr P, Ziegler SI, Nekolla S, Reichart B, Schwaiger M. Serial assessment of sympathetic reinnervation after orthotopic heart transplantation. A longitudinal study using PET and C-11 hydroxyephedrine. Circulation. 1999;99:1866–71. doi: 10.1161/01.cir.99.14.1866. [DOI] [PubMed] [Google Scholar]
- 48.Odaka K, von Scheidt W, Ziegler SI, Ueberfuhr P, Nekolla SG, Reichart B, et al. Reappearance of cardiac presynaptic sympathetic nerve terminals in the transplanted heart: Correlation between PET using 11C-hydroxyephedrine and invasively measured norepinephrine release. J Nucl Med. 2001;42:1011–6. [PubMed] [Google Scholar]
- 49.Hartikainen J, Kuikka J, Mantysaari M, Lansimies E, Pyorala K. Sympathetic reinnervation after acute myocardial infarction. Am J Cardiol. 1996;77:5–9. doi: 10.1016/s0002-9149(97)89125-4. [DOI] [PubMed] [Google Scholar]
- 50.Fallen EL, Coates G, Nahmias C, Chirakal R, Beanlands R, Wahl L, et al. Recovery rates of regional sympathetic reinnervation and myocardial blood flow after acute myocardial infarction. Am Heart J. 1999;137:863–9. doi: 10.1016/s0002-8703(99)70410-2. [DOI] [PubMed] [Google Scholar]
- 51.Fallavollita JA, Luisi JAJ, Michalek SM, Valverde AM, de Kemp RA, Haka MS, et al. Prediction of ARrhythmic events with positron emission tomography: PAREPET study design and methods. Contemp Clin Trials. 2006;27:374–88. doi: 10.1016/j.cct.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 52.Fallavollita JA, Luisi AJ, Jr, Yun E, Dekemp RA, Canty JM., Jr An abbreviated hyperinsulinemic-euglycemic clamp results in similar myocardial glucose utilization in both diabetic and non-diabetic patients with ischemic cardiomyopathy. J Nucl Cardiol. 2010;17:637–45. doi: 10.1007/s12350-010-9228-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.John AS, Mongillo M, Depre C, Khan MT, Rimoldi OE, Pepper JR, et al. Pre- and post-synaptic sympathetic function in human hibernating myocardium. Eur J Nucl Med Mol Imaging. 2007;34:1973–80. doi: 10.1007/s00259-007-0507-1. [DOI] [PubMed] [Google Scholar]
- 54.Caldwell JH, Link JM, Levy WC, Poole JE, Stratton JR. Evidence for pre- to postsynaptic mismatch of the cardiac sympathetic nervous system in ischemic congestive heart failure. J Nucl Med. 2008;49:234–41. doi: 10.2967/jnumed.107.044339. [DOI] [PubMed] [Google Scholar]
- 55.Munch G, Nguyen NT, Nekolla S, Ziegler S, Muzik O, Chakraborty P, et al. Evaluation of sympathetic nerve terminals with [11C]epinephrine and [11C]hydroxyephedrine and positron emission tomography. Circulation. 2000;101:516–23. doi: 10.1161/01.cir.101.5.516. [DOI] [PubMed] [Google Scholar]
- 56.Bucerius J, Joe AY, Schmaljohann J, Gundisch D, Minnerop M, Biersack HJ, et al. Feasibility of 2-deoxy-2-[18F]fluoro-D-glucose- A85380-PET for imaging of human cardiac nicotinic acetylcholine receptors in vivo. Clin Res Cardiol. 2006;95:105–9. doi: 10.1007/s00392-006-0342-6. [DOI] [PubMed] [Google Scholar]
- 57.Mazzadi AN, Pineau J, Costes N, Le Bars D, Bonnefoi F, Croisille P, et al. Muscarinic receptor upregulation in patients with myocardial infarction: a new paradigm. Circ Cardiovasc Imaging. 2009;2:365–72. doi: 10.1161/CIRCIMAGING.108.822106. [DOI] [PubMed] [Google Scholar]
- 58.Caldwell JH, Kroll K, Li Z, Seymour K, Link JM, Krohn KA. Quantitation of presynaptic cardiac sympathetic function with carbon-11-meta-hydroxyephedrine. J Nucl Med. 1998;39:1327–34. [PubMed] [Google Scholar]
- 59.Mongillo M, John AS, Leccisotti L, Pennell DJ, Camici PG. Myocardial pre-synaptic sympathetic function correlates with glucose uptake in the failing human heart. Eur J Nucl Med Mol Imaging. 2007;34:1172–7. doi: 10.1007/s00259-007-0371-z. [DOI] [PubMed] [Google Scholar]
- 60.Link JM, Stratton JR, Levy W, Poole JE, Shoner SC, Stuetzle W, et al. PET measures of pre- and post-synaptic cardiac beta adrenergic function. Nucl Med Biol. 2003;30:795–803. doi: 10.1016/j.nucmedbio.2003.09.002. [DOI] [PubMed] [Google Scholar]