

Abstract
In traumatic brain injury (TBI), severe disruptions occur in the choroid plexus (CP)–cerebrospinal fluid (CSF) nexus that destabilize the nearby hippocampal and subventricular neurogenic regions. Following invasive and non-invasive injuries to cortex, several adverse sequelae harm the brain interior: (i) structural damage to CP epithelium that opens the blood–CSF barrier (BCSFB) to protein, (ii) altered CSF dynamics and intracranial pressure (ICP), (iii) augmentation of leukocyte traffic across CP into the CSF–brain, (iv) reduction in CSF sink action and clearance of debris from ventricles, and (v) less efficient provision of micronutritional and hormonal support for the CNS. However, gradual post-TBI restitution of the injured CP epithelium and ependyma, and CSF homeostatic mechanisms, help to restore subventricular/subgranular neurogenesis and the cognitive abilities diminished by CNS damage. Recovery from TBI is faciltated by upregulated choroidal/ependymal growth factors and neurotrophins, and their secretion into ventricular CSF. There, by an endocrine-like mechanism, CSF bulk flow convects the neuropeptides to target cells in injured cortex for aiding repair processes; and to neurogenic niches for enhancing conversion of stem cells to new neurons. In the recovery from TBI and associated ischemia, the modulating neuropeptides include FGF2, EGF, VEGF, NGF, IGF, GDNF, BDNF, and PACAP. Homeostatic correction of TBI-induced neuropathology can be accelerated or amplified by exogenously boosting the CSF concentration of these growth factors and neurotrophins. Such intraventricular supplementation via the CSF route promotes neural restoration through enhanced neurogenesis, angiogenesis, and neuroprotective effects. CSF translational research presents opportunities that involve CP and ependymal manipulations to expedite recovery from TBI.
Keywords: CSF homeostasis, Subventricular zone, Blood–CSF barrier permeability, Periventricular lesions, Hydrocephalus, Leukocyte traffic, Neurogenesis, Intracranial pressure, Ischemia, Traumatic brain injury models, Dentate gyrus, Hippocampus, Cerebrospinal formation and drainage, CSF dynamics, Neuropeptides
Introduction
Following physical damage to brain, a number of homeostatic processes initiate recovery. Several regions and cell types in the central nervous system (CNS) aid in the repair of traumatic brain injury (TBI). Control of astrocyte proliferation, microglia activation, and neuroinflammation is key to restoring the integrity of blood–brain barrier (BBB) and cerebral parenchyma (Cernak et al. 2005; Li et al. 2009). TBI models have extensively focused on damage to brain capillaries and vasogenic edema. Systematic analyses of neural injuries have delineated the breakdown and reactivity of the BBB in TBI (Lenzlinger et al. 2001; Morganti-Kossmann et al. 2007; Pun et al. 2009; Unterberg et al. 2004).
In contrast, few investigations have addressed TBI phenomena at the other major transport interface: the choroid plexus (CP) or blood–cerebrospinal fluid barrier (BCSFB) (Davson and Segal 1996). Subtle changes in function of CP, a small but complex organ that dominates CSF composition, can have wide-ranging effects on brain (Emerich et al. 2005). CP is vulnerable to head injury, even to the non-penetrating type of trauma (Kaur et al. 1996). Third ventricle CP is sensitive to frontal blows, whereas the lateral ventricle plexuses suffer from lateral hits to brain (Maxwell et al. 1992). In view of the multitasking performed by CP (Johanson et al. 2008; Redzic et al. 2005), insight is needed on compensatory actions of CSF in reconstituting injured cortex (Johanson et al. 2005a; Walter et al. 1999). By providing trophic proteins to trauma sites and supportive stem cell niches, the CP stabilizes the physiologically perturbed brain.
This review analyzes several aspects of CP–CSF involvement in TBI. First, the CP itself undergoes structural distortion and functional changes even when the primary physical insult has been inflicted on region(s) considerably removed from the ventricular system (Ghabriel et al. 2010). Time course of BCSFB recovery is important in re-establishing brain homeostasis. CP must metabolically stabilize itself post-injury to retain regulatory capacities. Second, CP epithelial cells are bioreactive in a homeostatic sense. That is, in reaction to brain injury and ICP elevation, the epithelium in BCSFB is sensitive to neurohormonal signals that home in to CP epithelial cells (Chodobski and Szmydynger-Chodobska 2001; Johanson et al. 2006). Responding to stressful states, the CP epithelium releases fluid-regulatory neuropeptides into CSF (Johanson et al. 1999a; Shore et al. 2004; Yang et al. 2010). Third, immune and inflammatory phenomena involving the CP are a significant part of post-TBI sequelae. These include increased activity of epiplexus or Kolmer cells at the BCSFB (Maxwell et al. 1992) and increased paracellular movement of leukocytes into CSF (Szmydynger-Chodobska et al. 2009). Such immune phenomena in CP probably impart mixed effects on brain recovery. Fourth, another factor affecting recovery from central trauma and stroke is the state of CSF bulk flow or volume transmission (Johanson et al. 2003). Nascent neurons and migrating neuroblasts, that are important cellular elements in recovery processes for TBI, depend on CSF orderly flow which they follow (Sawamoto et al. 2006). Moreover, continually flowing CSF provides sink action that facilitates removal of catabolites and injury products from brain. When CSF flow is curtailed in CNS injuries or neurodegeneration, there is less efficient clearance of potentially toxic brain waste materials (Silverberg et al. 2001). And finally, the CSF produced by CP contains growth factors (Knuckey et al. 1996) and neurotrophins that are conveyed to the injury locus (Walter et al. 1999), and to neural stem cells in periventricular regions (Schanzer et al. 2004). These peptidergic factors likely promote healing at the injury locus and stimulate neurogenesis for stabilizing cognitive processes.
The above mentioned five factors dealing with TBI in the context of the CP–CSF-brain nexus (Johanson et al. 2010b) constitute the topics of this review: (i) morphological damage to CP, (ii) homeostatic response of CP to brain fluid/pressure imbalance, (iii) immune system interaction with CP–CSF, (iv) sink action and debris clearance by CSF, and (v) provision of growth factors and neurotrophins by CP to brain for revitalization. These thematic points are expanded following presentation below of: the baseline physiology of CP–CSF (Johanson 2008; Spector 2010; Wolburg and Paulus 2010), and the features of brain injury models that reveal CP–CSF mechanisms of damage and recovery (Chodobski et al. 2003; Ghabriel et al. 2010; Kaur et al. 1996; Palm et al. 1995; Sharma and Johanson 2007b; Sharma et al. 2010; Szmydynger-Chodobska et al. 2009).
Physiology of CP, CSF, and periventricular brain
Elevated intracranial pressure (ICP) following TBI can be life threatening. The rising ICP post-TBI is due to fluid retention in brain and CSF cavities. In several neurodegeneration models, including TBI, vasogenic edema at the brain injury site and breakdown of the blood–CSF barrier (BCSFB) in CP both contribute to ventriculomegaly and edematous fluid accumulation in CNS (Johanson et al. 2010a; Sharma et al. 2006, 2010). Fluid and pressure buildup in brain may be exacerbated by debris (e.g. erythrocytes) being transferred away from the injury site via bulk flow, and subsequently interfering with CSF reabsorption at arachnoidal drainage sites. Therefore, the BBB, CP–CSF, and arachnoid are all potentially significant factors affecting ICP and fluid retention after trauma. Of these abovementioned items contributing to CNS destabilization post-TBI, it is the CP of the BCSFB that is emphasized in this review.
Anatomically, the BCSFB resides within the ventricular system in the form of CP tissues within the four ventricles. Compartmentally, the relationship between BCSFB and BBB is schematized in Fig. 1. CSF is the intervening medium between CP and brain. Therefore, CP uses CSF as a distribution medium to transfer anabolites into, and catabolites out of, the cerebral parenchyma. The CP–CSF-mediated supply and removal of substances is vital for fetal brain development, adult maintenance, and the repair of injured brain (including stimulated neurogenesis to sustain cognition). Strategically located, the CP furnishes trophic molecules to nearby neurogenic niches in the subventricular zone (SVZ) and dentate gyrus (DG). CP–CSF thus serves as a source (solute supply) and sink (solute removal) for brain.
Fig. 1.
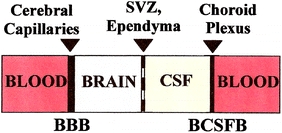
Anatomical relationships among the BCSFB, CSF, and BBB: neurons receive supplies from two sources: the nearby cerebral capillaries or BBB (mainly glucose, amino acids and free fatty acids) and the more distant CP or BCSFB (mainly vitamins, growth factors, neurotrophins, and hormones). Both the BBB and BCSFB reabsorb neural waste products and unneeded proteins into blood. The brain microvessel endothelium and CP epithelium both have tight junctions that restrict diffusion of small water-soluble molecules: therefore ion and organic solutes are actively transported via membrane carriers (structural proteins) across the barriers. Ependyma is a single-cell epithelial layer with highly permeable gap junctions that allows free exchange of solutes paracellularly between brain ISF and ventricular CSF
Physical injury to brain leads to impairment of many tasks performed by the BCSFB. Broadly speaking, three major functions of CP epithelium are altered by TBI sequelae: (a) the barrier role of choroid epithelial tight junctions, (b) the dynamics of CSF production and flow, and (c) homeostatic activities involving molecular transport (into and out of ventricles) to maintain the neurochemical composition of brain ISF and CSF.
Impermeability of BCSFB
Cerebrospinal fluid (CSF), like brain, is protected by a barrier system. Situated in the epithelium of CP, the BCSFB structurally consists of two components: the tight junctions (zonulae occludentes) that weld neighboring epithelial cells at their apical poles, and the basolateral membrane that abuts the choroidal ISF (Smith et al. 2004). Tight junctions in CP are slightly more permeable than counterparts in BBB (Thomas and Segal 1998). Accordingly, there is normally slow penetration of plasma proteins into CSF at a rate inversely proportional to the square root of the molecular weight of diffusing molecules. This choroidal leakage of macromolecules is greater than at the BBB. Still, the CP screens macromolecules to the extent that the steady-state concentration of proteins in CSF is only 0.01–0.001 that in plasma. Molecular sieving (restricted diffusion) at the BCSFB occurs to hydrophilic molecules as small as urea (mw 60). Such sieving of non-electrolytes occurs at the basolateral membrane (Johanson and Woodbury 1978). Together the tight junctions and basolateral membrane impede diffusion of water-soluble agents, small and large, from plasma to CSF.
Epithelial damage in ischemia, arterial hypertension, hyperthermia, and TBI structurally disrupts the BCSFB, allowing increased permeation of solutes across CP into ventricular CSF (Murphy and Johanson 1985; Palm et al. 1995; Sharma and Johanson 2007a; Sharma et al. 2010). These plasma-derived cytokines and peptides, once in CSF, can eventually access the microenvironment of chemically sensitive neurogenic regions under the ependymal wall. Additionally, the CNS-inward movement of protein macromolecules increases CSF osmotic pressure. Elevated osmotic pressure in CSF draws systemic water into the ventricles causing ventriculomegaly (Krishnamurthy et al. 2009).
Modulation of CSF production by CP
Most well known of the CP functions is CSF secretion. Human CSF production is about 500 ml per day. Acetazolamide (a carbonic anhydase inhibitor) and the cardiac glycosides (Na–K-ATPase inhibitors) suppress CSF production (Pollay et al. 1985), thereby reducing ICP. Several neurotransmitters (norepinephrine, acetylcholine, serotonin, and dopamine) and fluid-regulating neuropeptides (arginine vasopressin, angiotensin II, atrial natriuretic peptide, and basic fibroblast growth factor) decrease CSF formation rate (Johanson et al. 2008; Nilsson et al. 1992). Enhanced sympathetic stimulation of CP from the superior cervical ganglia, occurring in certain stresses/injuries to CNS, reduces CSF output by CP (Lindvall et al. 1978).
Neuroendocrine modulation of CSF, particularly the downregulated formation rate in hydrocephalus/↑ICP, involves participation of so-called “dark” epithelial cells in CP (Weaver et al. 2004). Increasing frequency of neuroendocrine-like dark cells is usually associated with states of reduced CSF formation (Johanson et al. 1999a, 1999b). Therefore, CP dark cells become more common when CP is exposed to agents that curtail CSF formation rate. Dark cells also appear in high-pressure hydrocephalus (Shuman and Bryan 1991) when CP epithelial cells shrink (become more electron dense in micrographs) and the width of the intercellular space expands. With TBI-induced decrease of CSF production to protect brain against elevated ICP, one expects to see morphological evidence of altered CP epithelium as described above. Less fluid formation by CP leads to dwindling of CSF bulk flow to the subarachnoid space (SAS), a convective process that normally extrudes injury products and excess proteins out of CNS (Pollay 2010).
Cerebrospinal fluid (CSF) formation is evidently decreased soon after TBI but, in later stages of the injury, may undergo an increase. To prevent ICP from rising to a dangerous level, it is therapeutically important to counter the vasogenic edema buildup in brain by appropriately curtailing CSF production, or by increasing water withdrawal from brain (Harbaugh et al. 1979). Tracking the changes in CSF dynamics and volume during reactive phases of TBI injury is important since these factors critically set the ICP.
Homeostatic transporters in CSF-facing membrane of CP
A little-appreciated function of CP epithelium is the active array of reabsorptive transporters at the apical membrane. These transporters clear away catabolites, injury-breakdown products (from the dismantling of erythrocytes leaking into ventricles), and proteins/cytokines that penetrate CSF in traumatized CNS. By actively removing organic solutes, the transporters normally cleanse CSF of impurities. A key question is whether these reabsorptive transporters, in conjunction with lysosomal disposal of substrates taken up by the epithelium, are overwhelmed in massive injuries. Severe disruption of CP epithelial cells and CSF in CNS disorders greatly taxes the lysosomal/Golgi system (Johanson et al. 2010b). Extensive vacuole generation in CP epithelium following TBI (Kaur et al. 1996; Palm et al. 1995) reflects intense lysosomal activity that digests injury products translocated at the BCSFB. Excessive vacuolation, induced by xenobiotic agents, causes CP swelling. Swollen CP exerts an impeding mass effect within the ventricles leading to interrupted CSF flow. Clearly, the coupled performance of CP clearance transporters and lysosomal digestive systems in diseases (hydrocephalus) and disorders (TBI) requires further examination in terms of CSF pathology and therapeutics.
How CP transport failure can affect the CSF–brain environment is seen in Kearns-Sayre syndrome (Spector and Johanson 2010a), a genetic disorder. A regular supply of ATP from mitochondria is needed to energize the active transport systems. In the Kearns-Sayre disorder, the mitochondria do not provide sufficient ATP to adequately drive the active transporters in CP that remove organic anions from CSF. Consequently the concentration of CSF homovanillic acid, a brain catabolite needing elimination by the Oatp transporter, is augmented due to less efficient removal of anion from the ventricles (Serrano et al. 2010). By functional analogy, the dwindling ATP production in CP at early stages of ischemia or TBI likely results in less effective homeostatic transport at the BCSFB (Palm et al. 1995). This destabilizes CSF composition and delays brain recovery.
When brain is injured, homeostatic transporters in CP apical membrane are set into action in an attempt to restore CSF composition by reabsorbing injury debris from the ventricles. Extravasated proteins and peptide fragments associated with TBI undergo diffusion and convection from the injury site through cortical interstices to CSF for excretion. Two primary CSF excretory mechanisms operate: one is by bulk flow (non-selective) clearance of organic compounds along arachnoidal drainage sites, and the other via active reabsorption by specific apical membrane transporters in CP. The organic anion-transporting polypeptide (Oatp) system plays a major role. CP apical membrane transporters including Oatp, along with the potentially toxic substances they reabsorb from CSF, have been extensively documented (Ghersi-Egea et al. 2009; Spector 2010; Zhang et al. 2010). In regard to reabsorption from CSF of products generated by the breakdown of erythrocytes and hemoglobin, the PEPT-2 transporter clears the potentially toxic heme precursor, 5-aminolevulinic acid, from the ventricles (Hu et al. 2007). Disruption of parenchyma in TBI releases Cl and K into extracellular fluid. Buffering of these ions is accomplished by transport activities of the apically located Na–K–Cl cotransporter and Na pump in CP that homeostatically regulate CSF Cl and K concentrations (Keep et al. 1994; Parmelee et al. 1991). CSF and ISF ion concentrations significantly affect CNS excitability and therefore need to be exquisitely regulated by glia and barrier cells.
Models of TBI for studying CSF components of injury
A variety of techniques have been used to inflict mechanical injury to brain. Several methodological approaches are compiled in Table 1. They involve both invasive TBI (brain stab and weight drop) as well as non-invasive TBI (movement acceleration and blast wave). Analysis of damage was performed on CP tissues and/or CSF at various stages or sequelae of the pathophysiology development post-trauma. Readers are referred to systematic reviews on TBI that cover a wide spectrum of pathophysiology and means for inducing neural trauma (Albert-Weissenberger and Siren 2010; Hicks et al. 2010; Thompson et al. 2005). This review analyzes several experimental approaches revealing how TBI affects the distributional nexus involving several connected compartments (Fig. 2) in the CNS interior: CP, CSF, ependyma, SVZ/neurogenic niches, and periventricular brain tissue bordering CSF (Johanson et al. 2010b). In many studies, the pathophysiological response to TBI was evaluated at several points from hours to weeks. Such time-course analyses can track up- and down-regulation of molecules involved in compensatory or restitution responses; and monitor the reversibility (or not) of structural distortions.
Table 1.
Alterations in lateral ventricle choroid plexus structure and function secondary to traumatic brain injury induction in animals
| Species | TBI model | Injury course | Experimental observations | Interpretation/comments | Investigators |
|---|---|---|---|---|---|
| Baboon | Lateral head movement acceleration (diffuse injury) | 20 min, 1, 3, 4, 6 12 h; 7 and 28 days | Enlarged intercellular spaces between choroidal cells; plasma and erythrocyte passage through tears in vessels and epithelium | Epiplexus cells have major role to clear red cells from CP–CSF; phagocytosis of debris was complete at 1 month | Maxwell et al. (1992) |
| Rat (male Wistar) | Single blast, non-penetrating | 1, 7, 14, 21 and 28 days post-blast | Widened intercellular clefts; increased number/activity of epiplexus cells; paracellular monocytes; apical cytoplas-mic protrusions into CSF; ↑ in OX-42 positive Kolmer cells at 7 & 14 days after blast | Disrupted paracellular pathway allows monocyte permeation; augmented epiplexus (Kolmer) cell activity is likely due to phagocytosis of erythrocytes | Kaur et al. (1996) |
| Rat (female Sprague-Dawley) | Penetrating injury (4-mm deep incision in cerebral cortex) | 0, 2, 5, 7 and 15 days post-lesion | In early recovery (1–7 dpl), IGF-II and IGFBP-2 mRNA ↑ in CP, and their proteins ↑ in CSF | During the acute phase recovery, IGF-II and IGFBP-2 are sent from CP, via CSF bulk flow, to the wound site where they modulate healing of the injury | Walter et al. (1999) |
| Rat (male Sprague-Dawley) | Weight drop (25 g/8 cm ht.) on fronto-parietal cortex | 4, 8 h and 1, 2, 4, 6 and 14 days | At 1 day post-TBI, neutrophils (CD11b) accumulated in CP of injured side, but not intact side (no OX-2 immunostain) | CP on injured side appeared to facilitate migration of neutrophils from blood to ventricular CSF, then to SAS & injury site | Chodobski et al. (2003) |
| Rat (male Wistar) | Stab injury to parietal cortex: 3 × 5 mm long | 5 h | Seven-fold ↑ in uptake by CSF of radioiodine in plasma (10-min volume of distribution) | TBI-induced ↑ permeability of BCSFB was consistent with microscopy-observed damage to the lateral ventricle CP (Fig. 6) | Sharma et al. (2010) |
| Rat (male Sprague-Dawley) | Impact acceleration to cortex (450 g weight, from a height of 2 m) | 5 h and 7, 14, 21 and 28 days | Epithelial swelling/vacuoles; intercellular cleft widening; dark cells in CP (7–14 days); hypertrophic epiplexus cells | Hydropic vacuolation reflects ↑ lysosomal activity; dark cells and wider clefts suggest altered CSF formation; apical macrophages remove debris from ventricular CSF | Ghabriel et al. (2010) |
Fig. 2.
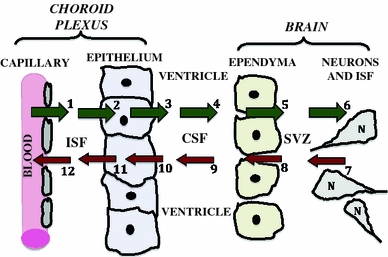
Central role of CP–CSF in exchanging materials with brain: ions, water, and organic molecules filter passively out of choroidal capillaries (arrow #1) into interstitial fluid (ISF). This is the first step in the material flow by distributional nexus to the brain (green arrows). Solutes diffuse through ISF up to basolateral membrane of epithelium. Active mechanisms in membranes transfer solutes sequentially across the basolateral (#2) and apical membranes (#3), into ventricular CSF. As CSF flows (#4) from ventricles to cisterna magna, a small fraction of the CSF-borne substances diffuse across ependyma (#5) into periventricular brain or are taken up by specialized ependymal cells. Ependyma-penetrating substances diffuse through brain ISF (#6) for transport into neurons (N); BBB, not depicted, is interspersed among neurons. Material inflow to the CNS interior thus sequentially involves CP, CSF, ependyma and brain. In the opposite direction, there is a reverse nexus (red arrows) for catabolites or injury products (as in TBI) released by neurons/glia into brain ISF. Accordingly, cerebral catabolites such as homovanillic acid diffuse through ISF (arrow #7), and down a transependymal concentration gradient into CSF (#8). By bulk flow of CSF (#9), catabolites are convected to subarachnoid space (not depicted) or to CP for active removal from the ventricles (#10), and then extrusion transport across basolateral membrane (#11). Therefore, some endogenous solutes or injury products end up being cleared passively into blood of microvessels (#12) and venules draining CP. Overall CSF is simultaneously a source (#1 to #6) and a sink (#7 to #12) for distributing molecules, depending on prevailing concentration gradients between ventricular CSF and brain ISF. As such, the CSF and bordering cells constitute a nexus for mediating trophic (CSF to brain) and excretory (brain to CSF) fluxes
One might think that the CP location, buried deep within the CNS and suspended in shock-absorbing CSF, would confer protection against a mechanical or physical impetus delivered against the brain’s external surface. This is not always the case. CP is sensitive to and injured by physical perturbations created by blast waves or other TBI-induction procedures (Table 1). The diffuse injury model, associated with sudden/intense lateral movement of the head, creates shearing forces that tear choroidal vessels and epithelium; the resulting hemorrhaging into the ventricles provokes inflammation and can increase ICP (Maxwell et al. 1992). Another non-invasive TBI approach exposes experimental animals to a single blast meant to simulate the military situation of soldiers exposed to nearby explosions. Although a non-penetrating model of TBI, the blast condition physically disrupts the epithelium of CP and its intercellular spaces. This markedly affects BCSFB physiology, including activation of phagocytotic systems such as the epiplexus cells or macrophages that physically intertwine with CP apical membrane (Kaur et al. 1996).
Other TBI-induction procedures are partially or fully invasive. Rats have been commonly used in TBI modeling (Table 1). Weight drop is useful for reproducibly applying a certain force to a circumscribed area on the cortical surface. It is usually applied to one hemisphere. Thereby, the contralateral intact hemisphere and lateral ventricle CP can be used to control for the injured ipsilateral side. A craniectomy exposes the dural membrane, upon which a calibrated weight is dropped. Experimental parameters vary considerably. Weights ranging from 25 to 450 g are dropped on the dura from a height of 8 cm–2 m (Chodobski et al. 2003; Ghabriel et al. 2010). Injury to CP is severe enough to provoke an inflammatory response. This involves leukocyte permeation of the inter-epithelial paracellular pathway and recruitment/activation of epiplexus cells that affix to the CSF-facing membrane.
Finally, the fully invasive approach to TBI induction involves an incisive lesion or stab wound inflicted deep in rat cortex (3–4 mm from dural surface). This penetrating lesion does not directly, physically traumatize CP. Because the lesioning cut or stab is made on one side of brain, it is feasible to use the other cerebral hemisphere and lateral CP as non-injured controls. Stab wounding is used to study functional aspects of BCSFB (permeability) and the role of CSF composition/bulk flow in homeostatically mediating recovery at injury loci (Sharma et al. 2010; Walter et al. 1999).
Pathophysiologic disruption of CP–CSF in TBI: effects on CNS
Diverse homeostatic functions of CP normally benefit the brain in many ways. Therefore, it is important that BCSFB damage brought about by TBI be stabilized quickly. Some changes occurring in CP post-TBI are homeostatically reactive, while others are pathologic or physiologically disabling. Even as CP is repairing itself, it is also homeostatically responding to needs of the injured brain. Sorting out these complex actions and reactions is challenging. However, multiple time-course data generated from several TBI models have been used to assess recovery of CP as well as revitalization of brain injury. It is important to consider acute adaptations (over days) versus longer chronic adjustments (over weeks). Five aspects of CP–CSF structure–function in TBI are now treated.
Morphological damage to BCSFB and permeability alteration
Structural alterations often precede functional changes. It is intriguing that external forces of the non-penetrating genre (e.g., blast) physically disrupt CP epithelium situated far from the brain surface receiving the explosion impact. In findings reported below, all rats were active and appeared physically normal after the blast. Following a single, non-penetrative blast there was widening of the intercellular cleft between epithelial cells (Fig. 3) (Kaur et al. 1996). This cleft response occurred as early as 1 day post-blast. A dilated paracellular pathway is not a favorable sign. It is reminiscent of the enlarged intercellular spaces of CP in hydrocephalus, a disease associated with shrunken choroidal epithelium and reduced formation of CSF (Weaver et al. 2004). As the initial step in CSF formation, several ion transporters at the basolateral membrane of CP (located at the edge of the paracellular pathway) receive Na, Cl, and HCO3 from ISF (Fig. 2, arrow #2) (Johanson et al. 2008). Normal CSF formation depends upon the integrity of fluid balance (volume and composition) between the epithelial cytoplasm and ISF in the intercellular cleft. Cleft widening reflects altered ion/water distribution; and it likely compromises CSF formation efficiency.
Fig. 3.
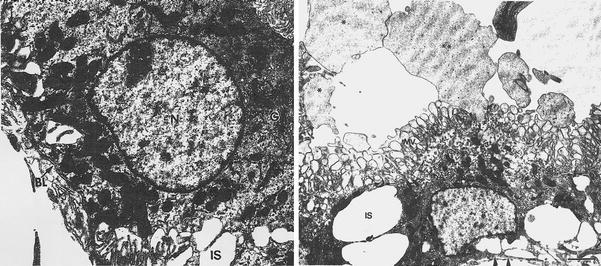
Dilated intercellular spaces in choroid plexus after blast injury: rat choroidal epithelium 7 days post-blast reveals dilated clefts or intercellular spaces (IS). The single blast condition is described in text. Microvilli (MV) are dilated. Ventricular lumen contains pedunculated cytoplasmic protrusions released from epithelium(asterisks). Basal lamina is demarcated by arrowheads. Scale bar is 2 μm. Reprinted from Kaur et al. 1996
Non-penetrative blasting, even a single episode, severely distorts and ruptures the CP apical membrane. One week after blasting, rat microvilli were dilated. And cytoplasmic protusions with their inclusions exuded into the ventricular lumen (Fig. 3). The apical microvillous surface, having a great area for transport, contains numerous ion and solute transporters that effect CSF formation by extruding fluid into the ventricles (Damkier et al. 2010). At the apical membrane are positioned many organic solute transporters, such as LRP-1, PEPT-2, and OATS (Angeletti et al. 1997; Smith et al. 2004; Spector and Johanson 2010b). These apical transporters remove excess anions/acids and peptides/fragments from CSF (Fig. 2, arrow # 10). Reabsorption of potentially toxic substances from CSF is important post-injury to remove material that has leaked into ventricles across a compromised BCSFB.
Choroid plexus (CP) epithelial heterogeneity is manifested by differential response of various cells to the blast. Electron microscopy reveals instances where one epithelial cell is severely damaged but the adjacent neighbor is intact (Fig. 4). This observation implies that certain cells in the epithelial lining of BCSFB are more vulnerable than others to stress. A similar phenomenon was found in a model of acute transient forebrain ischemia (TFI), in which some but not all CP epithelial cells were physically devastated (Johanson et al. 2000). In the experimental TFI, a destroyed epithelial cell was often flanked by morphologically intact neighboring epithelium (Palm et al. 1995). In the blast studies by Kaur et al., there was epithelial restoration by healing or replacement at 2 week. Remarkably in the TFI experiments, the choroid epithelial cells were largely recovered (or replaced?) 24 h after the ischemia insult (10 min of carotid occlusion and hypotension) and reperfusion (Palm et al. 1995). The ability of severely damaged CP epithelium to recover relatively rapidly from both physical and neurochemical disturbances intimates a fundamental role of BCSFB in servicing the brain.
Fig. 4.
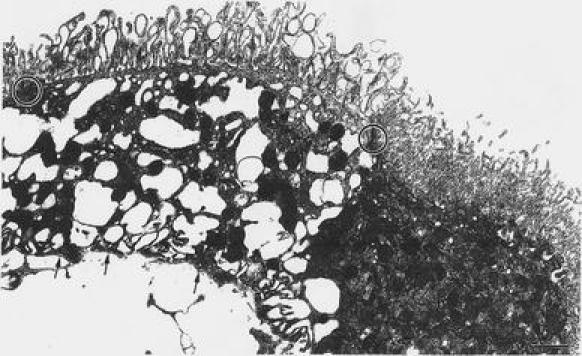
Neighboring epithelial cells in choroid plexus with and without damage: electron micrograph shows adjacent epithelium in rat CP 14 days after a single blast injury. Cell on left has a great number of hydropic vacuoles, likely reflecting enhanced activity of lysosomes (Johanson et al. 2010b) processing/digesting debris generated from injury. Microvilli in injured cell are more dilated than counterparts in the relatively intact epithelial cell on right. Basal lamina is indicated by arrows. Tight junctions are encircled. Scale bar is 1.5 μm. Reprinted from Kaur et al. 1996
For the BCSFB to maintain a barrier function of screening plasma macromolecules from entering CSF, it is essential that tight junctions and external plasma membranes (basolateral and apical) be intact. Breaching of BCSFB is expected when structural components of the epithelium are grossly distorted, as in traumatic injury to brain. Sharma and colleagues have quantified BCSFB permeability in rat TBI caused by stabbing parietal cortex (Fig. 5). Radioactive iodine, administered systemically and allowed to distribute for 5 min, was assayed in CSF at 5 h post-TBI induction. Greater uptake of tracer iodine by CSF of injured rats, versus intact controls, pointed to a more permeable BCSFB. This functional outcome was consistent with morphological damage to CP caused by TBI (Fig. 6). Opening of BCSFB in TBI, with consequent influx of plasma proteins/cytokines into CSF, places the ventricles and periventricular brain regions at risk for edema and other disability (Fig. 7). Ventriculomegaly, observed in TBI and hyperthermia, is adversely caused in part by protein buildup in CSF when CP fails to restrict diffusion of plasma-derived macromolecules (Sharma et al. 2006).
Fig. 5.
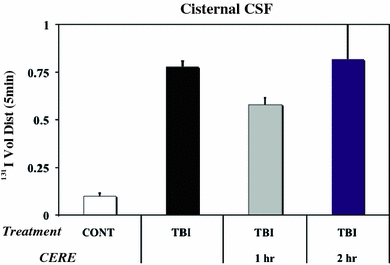
Enhanced permeability of BCSFB after TBI in adult rat: Breakdown of CP epithelial barrier was evaluated by rapid uptake (over 5 min) of radioiodine in plasma into CSF (sampled from cisterna magna). At 5 h of TBI, the permeability to 131I increased significantly by 7-fold, compared to non-lesioned controls (P < 0.01, ANOVA followed by Dunnet’s test). Bars are means + SD for 5 animals. Permeability change was blunted if Cerebrolysin (CERE), a mixture of growth factors and trophic peptides, was administered 1 h (P < 0.05) but not at 2 h post-induction of stab wound to parietal cortex (see Table 1). CONT control (sham surgery, no lesioning)
Fig. 6.
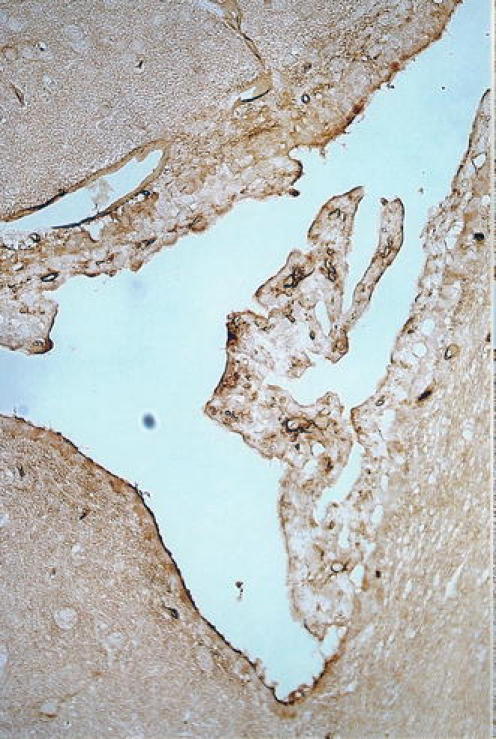
TBI damage to CP: micrograph shows damage to CP in the right lateral ventricle 5 h after stab injury to the ipsilateral cerebral cortex (parietal) in the rat. The choroidal epithelium appears shrunken. Disruption to the epithelial cells manifests as the uptake of serum albumin (brown reaction product). This indicates damage to the normally restrictive BCSFB (Johanson and Sharma, unpublished observation)
Fig. 7.
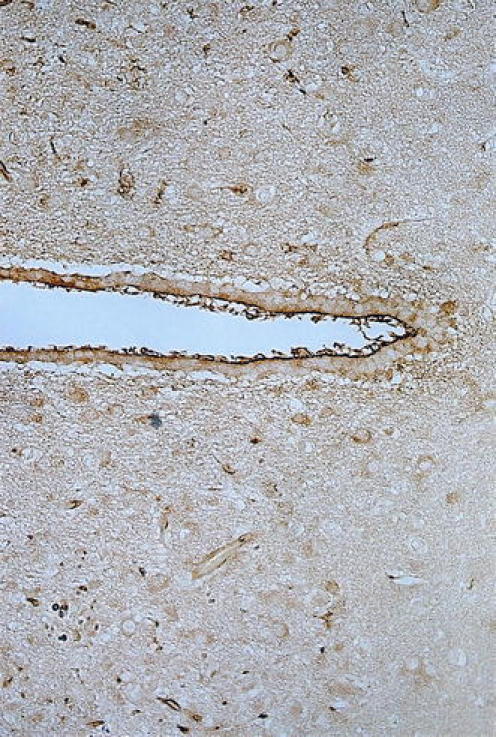
TBI damage to ependyma/periventricular regions: Image shows damage to periventricular ependymal cells in the third ventricle and the surrounding neuropil 5 h after TBI in the right parietal cortex of the rat. Uptake of albumin marker (from plasma) is seen as the brown reaction product visible in the ependymal lining. Also vacuolation, edema, and sponginess around the periventricular neuropil are apparent. In the neuropil, the albumin-labeled neurons reflect widespread disruption of the BBB (and indirectly, the BCSFB) after TBI to cerebral cortex (Johanson and Sharma, unpublished observation)
Homeostatic response of CP to brain fluid/pressure imbalance
A consistent feature of severe TBI is fluid retention by the brain–CSF nexus. Consequently, ICP escalates. Passive and active physiologic mechanisms counter the intracranial hypertension. Passively, the rising pressure in CSF exerts an opposing force on blood flow to CP. Reduced perfusion of the plexus eventually lowers substrate availability to the epithelium for fluid production. On the active side of compensation, fluid-regulating peptides are released into CSF to downregulate fluid turnover into ventricles. Atrial natriuretic peptide (ANP) concentration in human CSF increases in proportion to augmented ICP (Yamasaki et al. 1997). In an animal model, exogenous ANP presented intraventricularly curtails CSF formation (Steardo and Nathanson 1987) and reduces ICP. ANP increases the number of neuroendocrine-like dark epithelial cells in CP (Preston et al. 2003). This is usually a state associated with diminished CSF production (Weaver et al. 2004). Consequently, as an inhibitory modulator of CSF formation, ANP is regarded putatively as a regulator of ICP (Johanson et al. 2006).
Dark epithelial cells appear in CP, in vitro and in vivo, following administration of ANP, arginine vasopressin (AVP), and basic fibroblast growth factor; all of these peptides inhibit CSF formation (Johanson 2008). Dark cells in CP also become more numerous in high-pressure hydrocephalus (Shuman and Bryan 1991), a condition in which epithelial deformation is accompanied by expanded intercellular clefts (Weaver et al. 2004). As noted above, some TBI models also display widening of intercellular clefts. A related question: Is there an increased number of CP dark cells in TBI? Yes, electron dense, dark cytoplasm occurs in many epithelial cells in experimental TBI (Ghabriel et al. 2010); whether these dark cells reflect pathology or a compensatory response awaits resolution. Systematic TBI studies in the future should inform on how ICP changes correlate with CSF titers of ANP and CP epithelial dark cells. Assessing the CP’s role in regulating ICP, during acute versus chronic stages of TBI, would expedite models for brain fluid dyshomeostasis and its correction.
Immune system interaction with CP–CSF and brain
Immune cell access to CP and CSF substantially increases during recovery phases of TBI. Phagocytosis of dismantled structural elements and extracellular debris in injured CP is essential for restoring BCSFB functional integrity. Hemorrhaging associated with brain trauma brings erythrocytes and other blood cells into the CP interstitium, dilated paracellular pathways, and ventricular CSF. Within hours of head acceleration injury in a non-human primate, monocytes are observed in the intercellular clefts of CP (Maxwell et al. 1992). Some peripheral monocytes and other leukocytes that gain access to ISF (Fig. 2, arrow #1) and the impaired epithelium evidently differentiate into macrophages such as epiplexus (Kolmer) cells. Even a week after head acceleration, there was transepithelial migration of monocytes/macrophages into CSF where they evidently differentiated into active Kolmer cells (Maxwell et al. 1992). Similarly, after a cerebral penetrant lesion, the epiplexus cells or intraventricular macrophages became activated by 6 h. Activation was characterized by conversion from a smooth to ruffled appearance, and by loss of filopodia concurrent with a morphing into a spherical body (Maxwell and McGadey 1988).
From the CSF side of CP, the Kolmer cells affix their processes and bind intricately to the apical membrane glycocalyx. Well known for phagocytosing ability, the Kolmer cells also have antigen-presenting capabilities. They monitor immune status of CSF and presumably effect adjustments to rid CSF of contaminating plasma proteins and cytokines. Epiplexus cells increase in both number and activation state in response to the broken-down BCSFB in TBI. Kolmer macrophages remain active until CSF is cleansed of contaminating plasma and erythrocyte elements. When residual blood has been cleared from the ventricles (at 1 month post-injury in the head acceleration model, and by 2 week in the cerebral penetrant model), the Kolmer cells become deactivated (Maxwell et al. 1992; Maxwell and McGadey 1988). In the non-penetrative blast model, the epiplexus cells remained activated (enhanced immunostaining with monoclonal antibodies OX-42, OX-18, and OX-6) until 3–4 weeks after injury induction at which time CP epithelium regained normal ultrastructure (Kaur et al. 1996). Collectively, the information indicates that 2–4 weeks are needed by the CP-attached Kolmer cells to completely remove residual blood elements from CSF after TBI induction.
Increased trafficking of leukocytes across CP in TBI also impacts brain wound healing. Neutrophils penetrated choroidal blood vessels (Fig. 2, arrow #1) 4 to 48 h after brain trauma (Carlos et al. 1997). Within 4 h of injury by weight drop to rat cortex there was increased penetration of neutrophils across BCSFB into CSF, apparently facilitated by the ipsilateral CP (Chodobski et al. 2003). Migrating neutrophils were probably carried by CSF bulk flow from the lateral ventricle to SAS overlying the fronto-parietal cortex. There, the neutrophils secreted vascular endothelial growth factor (VEGF) into extracellular matrix at the wound locus for gradual release from heparin sulfate proteoglycan binding sites during healing. Unfortunately, VEGF delivered via CP–CSF to injured cortex exacerbates edema in TBI via enhanced angiogenesis and microvessel permeability. In the relapse phase of another disorder, multiple sclerosis (MS), there is increased migration of leukocytes through CP to CSF. This CSF penetration is followed by convective distribution of the leukocytes to periventricular and perivascular sites of the MS lesion. Therapeutically, there is the need to exert finer control of neutrophil migration across BCSFB in certain diseases as well as trauma.
Sink action and clearance of CSF injury products
Consequent to TBI, the central extracellular fluid becomes contaminated with blood cells, cytokines, and cellular breakdown products. Macrophages, particularly the epiplexus cells that are physically entangled with CP epithelium, primarily digest erythrocytes that have extravasated into CNS. Due to potential toxic effects of organic acids, excess proteins and peptide fragments created by neuronal disintegration, there is urgency for eliminating toxious waste products. Long appreciated for its great ability as a reabsorbing organ, the CP was once regarded as being primarily a tissue for clearing noxious waste substances from CSF (Case 1959). Current thinking recognizes that the plexus performs multiple functions in addition to its extensive reabsorptive capabilities (Johanson et al. 2008).
Following uptake from CSF (Fig. 2, arrow #10), harmful molecules are disposed in part by enzymatic breakdown within CP epithelium. This is manifested by an abundance of vesicles, inclusion bodies, and lysosomes (Johanson et al. 2010b). Another component of disposal involves transporters at the plasma-facing membrane. There, the basolateral exporters Mrp-1, Mrp-4, Oatp2, and Ent1 (Spector and Johanson 2010b) translocate solutes from cytoplasm into choroidal interstitium (Fig. 2, arrow #11). From ISF, substances that are being cleared out of CNS diffuse into venous blood for systemic elimination (Fig. 2, arrow #12).
Before waste solutes are acted upon by CP lysosomal breakdown or basolateral transporter extrusion, they first must be taken up from CSF (Fig. 2, arrow #10). Because CNS lacks true lymphatic vessels, many catabolites and proteinaceous materials are necessarily reabsorbed by active transporters at the BCSFB and BBB. In concert with active reabsorptive transporters at the abluminal (ISF-facing) membrane of cerebral capillaries, the CP apical membrane–CSF interface has a major work role in eliminating toxicants from CSF. An array of solute transporters at the CP apical membrane, including Oatp-D, Oatp3, Pgt, LTC4, and Cnt3 (Spector and Johanson 2010b), actively removes organic anions, peptide fragments, prostaglandins, leukotrienes, and nucleosides that diffuse transependymally into the ventricles from brain. Upon reaching large-cavity CSF, the waste or superfluous products are cleared actively by CP. Additional clearance takes place by CSF bulk flow via arachnoidal drainage into lymph and venous blood (Grzybowski et al. 2006; Johnston et al. 2004; Pollay 2010). The nature, selectivity, affinity, and capacity of efflux/reabsorptive transporters at the BCSFB have been delineated (Spector 2010; Spector and Johanson 2010b). In TBI, quick removal of residual blood in CSF by phagocytosis is essential for restoring efficient reabsorption by organic solute transporters in CP.
By convecting molecules of all sizes (and even residual erythrocytes), the CSF bulk flow through the ventriculo-subarachnoid spaces acts like a quasi-lymphatic excretory system. Indeed, a considerable volume of CSF drains through nasal mucosa into cervical lymphatic glands (Johnston et al. 2004; Szentistvanyi et al. 1984). By exerting sink action on brain extracellular molecules, the flowing CSF in transit to drainage sites gathers excess water, protein debris, and organic catabolites (Hochwald et al. 1976; Parandoosh and Johanson 1982). Effective sink action depends on continual CSF formation and drainage to maintain the appropriate transependymal concentration gradients that promote net diffusion of molecules into the ventricles (Fig. 2, arrow #8). Consequently, if CSF formation by injured CP is curtailed early in the TBI pathophysiologic cascade, then the CSF clearance of waste products would be less efficient.
Optimal CSF secretion depends upon adequate CP blood flow, a clean interstitium free of excessive immune cells and hemorrhage products, a patent paracellular pathway and restrictive tight junctions, and efficient epithelial transport supported by adequate ATP availability (Spector and Johanson 2010a). Therefore, in early stages of TBI when CP structure and function are debilitated, and sympathetic tone to the epithelium may be increased as part of the stress response, one expects less CSF flow into and through the ventricles (Lindvall et al. 1978). Decreased CSF formation in the initial phase of TBI fits the observation of an increased number of dark epithelial cells and widened intercellular clefts (Ghabriel et al. 2010). As recovery from TBI proceeds, however, by 3–4 weeks the BCFSB/epithelial integrity is restored. Re-establishment of CSF formation would restore CSF sink action, assuming that arachnoidal drainage routes are patent. At later stages of TBI, however, the CSF and brain fluids may still be imbalanced as manifested by ICP instability. Additional ICP data for TBI models at various stages will provide a clearer picture of relations between CP and CSF pathology and CSF dynamics throughout recovery.
Provision of growth factors and neurotrophins by CP to brain
As prelude to discussing the role of CSF-delivered peptides in repairing TBI, we now address various levels (or sources) of peptidergic support for homeostatically restoring the injury site. Level one is the primary compensatory (reactive) expression of neurotrophic and regenerative factors by parenchymal/vascular cells in and around the wound. This is exemplified by a 5-fold increase in nerve growth factor (NGF) mRNA in cortex 1 day after TBI in a controlled-contusion model (DeKosky et al. 1994). Also, transcripts for neuropeptides IGF-1 and cholecystokinin are upregulated up to 10-fold in cerebral cortex adjacent to injury 1–3 day post-contusion (Sandberg Nordqvist et al. 1996). The second level is represented by peptides putatively secreted from CP, in homeostatic response to TBI, and distributed via CSF to the traumatized region (Chiaretti et al. 2008; Walter et al. 1999). This is endogenous boosting of brain repair processes by neuroendocrine conveyance of CP-derived peptides over a considerable distance by volume transmission (rather than via local autocrine/paracrine processes). If CP-BCSFB function is severely compromised, however, as in early-stage TBI, additional extrinsic support enhances neuroregenerative capacity of traumatized brain. Accordingly, level three, the one emphasized in this review section, deals with exogenous or pharmacologic augmentation of CSF peptide levels. Although performed in animals by intraventricular delivery of test agent, the experimental information generated for accelerated recovery may be useful for strategizing translational therapy of human TBI. The discussion below and the compilation in Table 2 treat levels 2 and 3, emphasizing the latter.
Table 2.
Enhancement of neurogenesis, cognition or neuroprotection post-TBI by pharmacologic boosting of neuropeptides in CSF
| Species | TBI model | Injury time course and analyses | CSF peptide boosting | Experimental observations | Interpretation and comments | Authors |
|---|---|---|---|---|---|---|
| Rat (adult Sprague–Dawley) | Fluid percussion pulse | 5 days and 5 weeks, followed by Morris water maze | Bovine S100B infused i.c.v. (osmotic pump) for 0, 2, 5 or 7 days post-TBI | Hippocampal progenitor cells (DG) proliferation; Spatial learning memory improved 4–5 weeks post-injury to cortex | Enhanced hippocampal neurogenesis associated with improved cognition after TBI | Kleindienst et al. (2005) |
| Rat (adult Sprague–Dawley) | Fluid percussion pulse | 1 and 4 weeks, followed by Morris water maze | rFGF2 (human) infused i.c.v. (osmotic pump) for 1 week post-TBI | Augmentation of cell proliferation (BrdU) in SVZ and DG at 1 week; bFGF increased the survival of new neurons at 4 weeks | Hippocampal-dependent tasks (water maze) were improved at 2–25 days post-TBI, suggesting a link between cognition recovery and neurogenesis | Sun et al. (2009) |
| Mouse (adult) | Weight drop on fronto-parietal cortex | 1 week | VEGF i.c.v. (osmotic pump) to lateral ventricle | Increased proliferation of cells in SVZ on side of lesion; most new cells were glia, not neurons | Lesion volume decreased. Recovery rates and functional outcomes were improved | Thau-Zuchman et al. (2010) |
| Rat (adult Sprague–Dawley) | Pendulum: lateral fluid percussion | 4 weeks | rVEGF to ipsilateral ventricle for week 0 to 2 | ↑ BrdU +/Prox1 + new neurons; ↓ TUNEL + cells in dentate gyrus | CSF-delivered VEGF activated hippocampal neurogenesis after TBI and ↑ surviving granule neurons | Lee and Agoston (2010) |
| Rat (adult Sprague–Dawley) | Cortical impact, pneumatic cylinder | 1 week, followed by Morris water maze | NGF (2.5S) via osmotic pump to lateral ventricle |
NGF ↓ spatial memory deficits caused by TBI. It reversed post-TBI ↓ in scopolamine-evoked extracellular acetylcholine in hippocampus |
Cognitive deficits after TBI are due to acetylcholine deficiency, and can be countered by exogenous NGF delivered to CSF | Dixon et al. (1997) |
| Rat (adult Sprague–Dawley) | Fluid percussion pulse | 1 and 4 weeks, followed by Morris water maze | EGF infused by osmotic pump for 1 week post-TBI | At 1 week of TBI, infused rats had ↑ BrdU + cells and EGF receptors in SVZ and hippocampus | At 4 week post-injury, EGF did not support long-term survival of new neurons, but cognition was better | Sun et al. (2010) |
| Rat (adult Sprague–Dawley males) | Controlled cortical impact | 1 week | GDNF infused icv by pump for 1 week post-TBI. | GDNF infusion in CSF decreased CA2 and CA3 neuronal loss 1 week after TBI by 58% and 51%, respectively | CSF delivery of GDNF for 1 week led to substantial neuroprotection of hippocampal regions | Kim et al. (2001) |
| Rat (adult Wistar males) | Impact acceleration (axonal injury) | 2 h | PACAP given i.c.v. by pump at 0.5 h or 1 h post-trauma | PACAP treatment did not yield different numbers of RMO-14 + axonal profiles, post-injury versus normals | Authors conclude that PACAP findings justify further experimentation and possible consideration for clinical TBI trials | Tamas et al. (2006) |
Peptide secretions by CP into CSF (Fig. 2, arrow #3) promote recovery from TBI and other neurodegeneration. A plethora of growth factors and neurotrophic proteins synthesized by CP epithelium is released into CSF (Chodobski and Szmydynger-Chodobska 2001; Johanson et al. 2000). These peptidergic agents then flow (Fig. 2, arrow #4) to a myriad of parenchymal targets in brain (arrow #6). A popular pharmacologic approach amplifies the endogenous level of a CSF peptide by injecting/infusing the test agent into a lateral ventricle. This is done to evaluate hypothesized neuroregenerative and neuroprotective actions (Johanson et al. 1999b). CSF bulk flow carries growth factors and neurotrophins from CP to nearby neurogenic niches (SVZ and DG), and to more distant regions such as fronto-parietal cortex injured by trauma. This neuroendocrine function of CSF (Kozlowski 1986; Scott et al. 1977) involves convection-delivered peptides to modulate neurogenesis and facilitate healing. Ischemia has been widely studied in this context (Johanson et al. 2003), but TBI has received less attention for CSF pharmacologic manipulation. TFI and TBI have common pathogenetic mechanisms. Ischemia findings for CSF growth factor- and neurotrophin-promoted neurogenesis enhance this discussion (Kernie and Parent 2010; Matsuoka et al. 2003; Tureyen et al. 2005) because post-traumatic cerebral ischemia is reputedly the first cause of neuronal death after head injury (Chiaretti et al. 2008).
Prior to pursuing therapies for disease/injury modification, it helps to gain insight on natural homeostatic mechanisms that aid injured brain. Expression of trophic factor transcripts in lateral ventricle CP is sensitive to CNS injuries. Responding to forebrain ischemia, the CP epithelium upregulates TGFβ isoforms (Knuckey et al. 1996); and in reaction to cortical trauma, the plexus increases secretion of IGF-II and IGFBP-2 into CSF (Walter et al. 1999). In rat TBI, IGF-II is conveyed via CSF from CP to cortical injury sites (Fig. 2, arrows #3-6) where the hormone presumably stabilizes parenchyma and interstitium. Such endogenous IGF-II boosting happens relatively early after injury and thus appears to be a rapid response to neural trauma. A key question is how CP, not directly traumatized, receives information from a remote brain injury site to appropriately adjust its expression/secretion of certain peptides to mediate homeostasis. Deserving elucidation is whether injury signals (cytokines?) travel from injury site to CP via ISF–CSF routes (Fig. 2, arrows 7–9), neurogenically, or by a vascular pathway (arrow 1). Such information would advance modeling for homeostatic peptide feedback loops: from injured brain to CP–CSF (afferent limb) and then back to the injury site (efferent).
In addition to analyzing exogenous or pharmacologic peptide modulation of injury loci, it is relevant to determine effects of the same test agent on neurogenic niches (Fig. 2, arrow #5). This is salient because stem cell regions stimulated by peptides spawn newly birthed neurons for migration to injury sites. There, remodeling supposedly replaces dying or dead cells with new neurons for incorporation into circuits. Neurogenesis targets for endogenous choroidal secretions [or for exogenous peptide administered intraventricularly (arrow #4)] are the SVZ (arrow #5) near the ependymal wall in the lateral ventricle and the subgranular region (SGR) of the dentate gyrus–hippocampus. In embryonic-fetal development and for adult neurogenic maintenance, the niche stem cells require extrinsic modulation from factors distributed by CP–CSF (Miyan et al. 2003; Owen-Lynch et al. 2003). Studies of neurodegeneration, including TBI and stroke, provide substantial evidence that growth factors and neurotrophins given exogenously to CSF (i.c.v. by osmotic pump) strengthen the niches and stimulate neurogenesis. Enhanced generation of new neurons secondary to CSF peptide supplementation, under many conditions, reduces cognitive deficits created by TBI. Pharmacologically exploiting the CP–CSF–ependyma/SVZ distributional nexus (Fig. 2, arrows #1-5) may offer therapeutic opportunities to remediate cognition losses sustained in brain injury (Johanson et al. 2005b).
Level 3 studies addressing TBI, in the context of neurogenic peptides delivered from the CSF side of the neurogenic niches, are overviewed in Table 2. Several peptides, when exogenously boosted in CSF, are neuroregenerative and help to restore cognition post-TBI. CSF-administered S100B, FGF2, and NGF all raise or restore neurogenic capacity diminished in TBI (Table 2). Typical experimentation has involved osmotic pump infusion of the test peptide for several days. BrdU was injected at varying time points prior to sacrifice to monitor proliferation (dividing cells) and nascent neuron survival. Intraventricular infusion of peptide was followed by cognition testing, usually by behavior assessment in the Morris water maze. Post-mortem analyses of hippocampus included immunostaining with markers for stem cells, glia, and neurons. Neurogenic and neuroprotective effects of several peptides in CSF are now discussed below.
S100B is a peptide marker that increases in CSF after TBI. As a peptide released from injured astrocytes, S100B in CSF reflects trauma sequelae in brain. S100B has also been tested for neurogenic ability. Intraventricularly infused S100B stimulates neurogenesis and improves spatial memory in rats traumatized by fluid percussion (Table 2). Simultaneous occurrence of increased neurogenesis and improved cognition led to the hypothesis that the two phenomena are linked (Kleindienst et al. 2005). Additional support for this working model, i.e., CSF drug-induced neuroregeneration, has been provided in comparable studies testing other neuropeptides (Table 2).
Basic fibroblast growth factor (FGF2) has mitogenic and neurogenic properties (Kernie and Parent 2010; Mudo et al. 2009; Tureyen et al. 2005). When instilled i.c.v into rats impaired by fluid percussion injury, the exogenous FGF2 in CSF increased cell proliferation in the SVZ and DG (Sun et al. 2009). Cognition reduced by TBI was also improved by FGF2 infusion into a lateral ventricle (Table 2). FGF2 levels in periventricular regions are also elevated by intraventricular infusion of adenovirus that mediates gene transfer of FGF2. This transgenetic manipulation efficiently elevated the FGF2 in CSF and amplified the number of BrdU-positive cells in gerbil SVZ (Matsuoka et al. 2003). That CSF delivery of peptide can provide effective TBI therapy is encouraged by promising findings on intranasally delivered FGF2. Thus, following ischemia induction in rats, the nasal delivery of FGF2 augmented progenitor cell number in SVZ and DG; and improved behavioral performance as judged by modified Neurological Severity Scores (Wang et al. 2008). Intranasal administration of peptides circumvents the BBB, allowing efficient access to CSF and thus neurogenic regions bordering the ventricles (Reger et al. 2008).
Vascular endothelial growth factor (VEGF) instilled intraventricularly holds promise in promoting neurogenesis/angiogenesis and reversing untoward effects of TBI. In both mice and rats, the osmotic pump infusion of VEGF into a lateral ventricle promoted hippocampal and SVZ neurogenesis beneficial for recovery from TBI. In vitro experimentation demonstrated that VEGF directly modulates clonally derived adult rat neural stem cells, which themselves express VEGF and VEGFR-2/Flk-1 kinase receptor (Schanzer et al. 2004). Thus, both intrinsic and extrinsic modulation mechanisms are likely in play in neurogenic niches. Drugs such as simvastatin upregulate VEGF in DG, stimulate neurogenesis, and enhance recovery of spatial learning (Morris water maze) in rats (Wu et al. 2008). A relatively low-dose infusion of VEGF (2.4 ng/day) into a lateral ventricle (to minimize angiogenesis and enhanced vascular permeability) resulted in unchanged proliferation and reduced apoptosis of neurons. This intimates a survival-promoting effect by VEGF on neural progenitors (Schanzer et al. 2004). Strong expression of VEGFR-2 was observed in the ventricular wall near CP, a site of significant VEGF production. As discussed above for modulatory neuropeptide sources, for VEGF, levels 1 (stem cell intrinsic) and 2 (endogenous CSF extrinsic) are evidently operative in the niche. This gives rise to possible level 3 (pharmacologic extrinsic) boosting of CSF VEGF to enhance neurogenesis in treating TBI. However, a potential therapeutic caveat is offered. One must consider possible effects of VEGF/nitric oxide in augmenting CP permeability, already high early in experimental TBI (Sharma et al. 2010) and ischemia (Palm et al. 1995; Sivakumar et al. 2008). Excessive VEGF could place CSF at risk for destabilization by causing hemorrhaging through BCSFB.
Nerve growth factor (NGF) compensatory alteration in brain and CSF, following TBI, has received intense experimental scrutiny. In reaction to CNS trauma, hippocampal NGF upregulates rapidly and displays a biphasic response (DeKosky et al. 2004). Thus, NGF protein was increased at 6 h, then declined by 7 days, but then underwent a second rise at 14 days in rats injured by cortical impact. Changes in brain NGF upregulation after TBI in children are reflected in CSF levels, which also become elevated (Chiaretti et al. 2008). NGF upregulation in hippocampus and NGF delivery to CSF (Table 2), after TBI, helps to reverse cholinergic deficits (acetycholine and choline acetyl transferase) and restore spatial memory (Dixon et al. 1997). Evidence that immune system activity is linked to brain NGF regulation comes from observing that intraventricularly infused interleukin-1 beta, which is elevated after trauma, increases NGF in rat cortex and hippocampus by 2- to 4-fold (DeKosky et al. 1994). Upregulated NGF has a primary role in restoring cholinergic neurotransmission and related cognition in subjects compromised by neural trauma.
Epidermal growth factor (EGF) is a commonly used mitogen for inducing neurogenesis in animal models. EGF, like FGF2, is involved with cholinergic lineage. EGF is sometimes combined with other factors (e.g. NGF) in CSF infusion models to accelerate proliferation of dividing cells in SVZ of both lesioned and non-lesioned animals (Calza et al. 2003). EGF and FGF2, combined in intraventricular infusion, increase post-ischemic neural progenitor cells in adult rat SVZ and hippocampal DG (Tureyen et al. 2005). In TBI modeling (Table 2), the infusion of EGF by osmotic pump into a lateral ventricle for 1 week increased the number of dividing cells (BrdU+) in hippocampus (Sun et al. 2010). However, the EGF-induced neurogenesis was not productive, as manifested by lack of a sustained increase of BrdU+ cells at 4 week after trauma. Still, the improved cognition in rats at 1 month post-TBI, in spite of gliosis, was attributed to a neuroprotective rather than neurogenic effect of EGF.
Glial derived neurotrophic factor (GDNF) exerts neuroprotective effects against cerebral ischemia (Lee et al. 2010). GDNF, administered by injection into lateral ventricle CSF, also confers protection in a 4-h whole body hyperthermia model (Sharma and Johanson 2007b). Improvement by GDNF treatment in the face of acute hyperthermia stress is attributable in part to stabilization (restoration of restricted permeability) of the BBB and BCSFB; consequently, the CSF and brain extracellular milieus are less disrupted. This improved neural microenvironment, due to tighter barriers, improves cognition and enables better motor performance on a battery of behavioral tests (Sharma and Johanson 2007b). In a cortical impact study in rats, GDNF also provided neuroprotection when administered by osmotic pump into CSF; the hippocampal CA2 and CA3 regions were spared neuronal loss (Table 2). Another neurotrophin is brain derived neurotrophic factor (BDNF). Although not neurogenic (Galvao et al. 2008), BDNF is neuroprotective in ischemia and hyperthermia challenges to brain when delivered to CNS via CSF (Sharma and Johanson 2007b).
Pituitary adenylate cylcase activating polypeptide (PACAP) effectively protects the brain against edema (Nakamachi et al. 2010), neuronal apoptosis, and the oxidative stress of hypoxia/ischemia (Stumm et al. 2007). PACAP has also been tested in TBI modeling (Table 2). Post-injury administration of PACAP into CSF reduces traumatically induced axonal injury in rats (Tamas et al. 2006). Immunostaining analyses of damaged axons revealed that PACAP given i.c.v. significantly protected the corticospinal tract in regard to axoplasmic transport and cytoskeletal integrity (Kovesdi et al. 2008). PACAP may be neuroprotective in TFI/TBI through its stimulation of IL-6 secretion into CSF (Shioda et al. 2004). Il-6 has diverse roles in minimizing the adverse effects of ischemia and trauma, and in stimulating neuronal growth. Clearly, yet to be explored and characterized are many CSF neuroendocrine–immune system interactions that promote recovery from TBI.
Vistas for CSF translational research in TBI
Exciting prospects are on the horizon, in regard to how CSF manipulations post-trauma can expedite recovery from the pathologic sequelae of TBI. More information is needed, obtained within the same experiment, on how both the BCSFB and BBB breakdowns in TBI affect the extracellular (interstitial) fluid microenvironment of neuronal networks. During the course of TBI and its pathophysiologic sequelae, there are marked changes in the fluid dynamics (including the distorting effects of cerebral edema) of both the brain ISF and ventricular CSF with which it functionally interacts. Because the BCSFB interface controls CSF dynamics, and the BBB capillaries regulate ISF turnover, it is of great importance to assess in a given animal the effects of simultaneous breakdown of both major barrier systems and the respective fluids regulated by them (Sharma and Johanson 2007a). Neurogenic regions surrounding the ventricles are particularly vulnerable. They are modulated and doubly injured in TBI as the result of the malfunctioning CP (indirectly) and periventricular capillaries (directly) (Sharma et al. 2006). On the other hand, there is opportunity for concurrent pharmacologic resolution of damaged neurogenic niches by delivering therapeutic agents from both the CSF (CP) and plasma (brain capillaries) sides of the SVZ as well as DG. Overall, TBI is a cogent research/therapeutic challenge (physiologically, pathologically and pharmacologically) for application to the complex systems biology approach.
Another investigational requirement is to characterize more thoroughly the CSF composition of neuropeptides, proteins, and hormones as a function of TBI stage and severity; in animals as well as humans. Changes in CSF peptide levels after TBI induction, for example IGF-1 (Walter et al. 1999) and NGF (Chiaretti et al. 2008), provide insight on natural homeostatic peptide systems (perhaps servomechanistic) that can guide the development of therapeutic strategies and clinical trials. Alterations in CSF titers of peptides reflect cellular actions/events occurring in both CP and astrocytes. It is important to distinguish between these two (and other) cellular compartments so that pharmacological targets can be pinpointed.
Therapeutic targets in the traumatized CNS are diverse. They include glia, neurons, and endothelium. Why not also include CP epithelium as a potential target for facilitating the recovery from TBI-generated damage, or at least preventing a greater degree of injury? As the master homeostatic organ in CNS, the CP needs protection from oxidative damage and the noxious substrates that it clears from CSF. Neurotrophic factors secreted by CP modulate distant targets via neuroendocrine CSF bulk flow, and likely also stimulate peptide receptors at the BCSFB by autocrine/paracrine mechanisms. Because CP capillaries are not diffusion-restricting (Johanson et al. 2011), like those at the BBB, a given potentially therapeutic water-soluble agent is readily accessible to epithelial parenchyma of CP from the blood side. This favorable accessibility factor, pharmacokinetically, is expected to increase the number of drug candidates for protecting the BCSFB.
It is now evident that many different peptides in CSF bestow neuroprotective and neuroregenerative effects on brain parenchyma and the cells bordering the ventricles. In the natural setting, this endogenous battery of CSF-borne peptides confers a wide spectrum of benefits to the CNS. The basis for this phenomenon is largely the normal CP secretion of a plethora of growth factors and neurotrophins into CSF that are increased in response to ischemia, TBI, and other neurodegeneration. This physiologic reality prompts the consideration that a pharmacologic mixture, or cocktail, of growth factors and neurotrophins (Sharma et al. 2010) would exogenously provide additive or synergistic effects on neurogenesis, angiogenesis, and neuroprotection. Some animal model approaches have already used this strategy (Tureyen et al. 2005). More experimental information is desirable to evaluate the effects of neuropeptide cocktails, in the context of exogenous CSF supplementation, in neurodegeneration models such as ischemia and TBI.
Promising data have been obtained from animal research, in regard to favorable effects of a wide variety of CSF-delivered neuropeptides to boost CNS function in repair states. Caveats are necessary, however, for the possibility that too much of a dose, or the inappropriate combination, of growth factors could predispose toward oncogenicity or aberrant incorporation of new neurons into circuitry (leading to seizures). Future research can sort out and eliminate unfavorable side effects by selecting the best protocols for CSF neuropeptide supplementation in TBI treatment. Upon procuring TBI recovery data for animals that consistently point to safe/effective regimens for CSF peptide manipulations, there will be numerous venues for expanding translational research: CP transplants (Borlongan et al. 2004; Emerich et al. 2005), the use of capsules or beads (intraventricular or intraparenchymal instillation) to encase cells bioengineered to secrete certain neuropeptides (Heile et al. 2009), cisternal injection of marrow stromal cells (Hu et al. 2005), and perhaps eventually the transgenetic/vector manipulation of CP/BCSFB (Gonzalez et al. 2010; Regev et al. 2010) to control more finely neuropeptide secretion into CSF for the brain’s benefit.
Conclusion
Functional destabilization of the choroid plexuses and ependymal wall in the brain interior following TBI has marked effects on CSF homeostasis and periventricular neurogenic niche viability. Pharmacologic boosting of the impaired ventricular CSF system with appropriate growth factors/neurotrophins may improve neurogenesis and help restore cognitive abilities diminished by neural trauma.
Acknowledgments
NIH support to CEJ and EGS (NIA R01-027910) enabled the development of some of the concepts and modeling presented in this review. The Laerdal Foundation for Acute Medicine, Stavanger, Norway provided support to HSS. We thank N. Johanson, A. Sharma and J. Johanson for support in the construction and processing of this manuscript.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Abbreviations
- ANP
Atrial natriuretic peptide
- BBB
Blood–brain barrier
- BCSFB
Blood–CSF barrier
- BDNF
Brain-derived neurotrophic factor
- CNS
Central nervous system
- CP
Choroid plexus
- CSF
Cerebrospinal fluid
- DG
Dentate gyrus
- FGF
Fibroblast growth factor
- GDNF
Glial cell line-derived neurotrophic factor
- ICP
Intracranial pressure
- IGF
Insulin-like growth factor
- ISF
Interstitial fluid
- NGF
Nerve growth factor
- OATS
Organic anion-transporting polypeptides
- PACAP
Pituitary adenylate cyclase activating polypeptide
- SAS
Subarachnoid space
- SVZ
Subventricular zone
- TBI
Traumatic brain injury
- TFI
Transient forebrain ischemia
- VEGF
Vascular endothelial growth factor
References
- Albert-Weissenberger C, Siren AL. Experimental traumatic brain injury. Exp Transl Stroke Med. 2010;2:16. doi: 10.1186/2040-7378-2-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angeletti RH, Novikoff PM, Juvvadi SR, Fritschy JM, Meier PJ, Wolkoff AW. The choroid plexus epithelium is the site of the organic anion transport protein in the brain. Proc Natl Acad Sci USA. 1997;94:283–286. doi: 10.1073/pnas.94.1.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borlongan CV, Skinner SJ, Geaney M, Vasconcellos AV, Elliott RB, Emerich DF. Intracerebral transplantation of porcine choroid plexus provides structural and functional neuroprotection in a rodent model of stroke. Stroke. 2004;35:2206–2210. doi: 10.1161/01.STR.0000138954.25825.0b. [DOI] [PubMed] [Google Scholar]
- Calza L, Giuliani A, Fernandez M, Pirondi S, D’Intino G, Aloe L, Giardino L. Neural stem cells and cholinergic neurons: regulation by immunolesion and treatment with mitogens, retinoic acid, and nerve growth factor. Proc Natl Acad Sci USA. 2003;100:7325–7330. doi: 10.1073/pnas.1132092100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlos TM, Clark RS, Franicola-Higgins D, Schiding JK, Kochanek PM. Expression of endothelial adhesion molecules and recruitment of neutrophils after traumatic brain injury in rats. J Leukoc Biol. 1997;61:279–285. doi: 10.1002/jlb.61.3.279. [DOI] [PubMed] [Google Scholar]
- Case NM. Hemosiderin granules in the choroid plexus. J Biophys Biochem Cytol. 1959;6:527–530. doi: 10.1083/jcb.6.3.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cernak I, Stoica B, Byrnes KR, Di Giovanni S, Faden AI. Role of the cell cycle in the pathobiology of central nervous system trauma. Cell Cycle. 2005;4:1286–1293. doi: 10.4161/cc.4.9.1996. [DOI] [PubMed] [Google Scholar]
- Chiaretti A, Antonelli A, Genovese O, Pezzotti P, Rocco CD, Viola L, Riccardi R. Nerve growth factor and doublecortin expression correlates with improved outcome in children with severe traumatic brain injury. J Trauma. 2008;65:80–85. doi: 10.1097/TA.0b013e31805f7036. [DOI] [PubMed] [Google Scholar]
- Chodobski A, Szmydynger-Chodobska J. Choroid plexus: target for polypeptides and site of their synthesis. Microsc Res Tech. 2001;52:65–82. doi: 10.1002/1097-0029(20010101)52:1<65::AID-JEMT9>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Chodobski A, Chung I, Kozniewska E, Ivanenko T, Chang W, Harrington JF, Duncan JA, Szmydynger-Chodobska J. Early neutrophilic expression of vascular endothelial growth factor after traumatic brain injury. Neuroscience. 2003;122:853–867. doi: 10.1016/j.neuroscience.2003.08.055. [DOI] [PubMed] [Google Scholar]
- Damkier HH, Brown PD, Praetorius J. Epithelial pathways in choroid plexus electrolyte transport. Physiology (Bethesda) 2010;25:239–249. doi: 10.1152/physiol.00011.2010. [DOI] [PubMed] [Google Scholar]
- Davson H, Segal M. Physiology of the CSF and blood–brain barriers. Boca Raton, FL: CRC; 1996. p. 822. [Google Scholar]
- DeKosky ST, Goss JR, Miller PD, Styren SD, Kochanek PM, Marion D. Upregulation of nerve growth factor following cortical trauma. Exp Neurol. 1994;130:173–177. doi: 10.1006/exnr.1994.1196. [DOI] [PubMed] [Google Scholar]
- DeKosky ST, Taffe KM, Abrahamson EE, Dixon CE, Kochanek PM, Ikonomovic MD. Time course analysis of hippocampal nerve growth factor and antioxidant enzyme activity following lateral controlled cortical impact brain injury in the rat. J Neurotrauma. 2004;21:491–500. doi: 10.1089/089771504774129838. [DOI] [PubMed] [Google Scholar]
- Dixon CE, Flinn P, Bao J, Venya R, Hayes RL. Nerve growth factor attenuates cholinergic deficits following traumatic brain injury in rats. Exp Neurol. 1997;146:479–490. doi: 10.1006/exnr.1997.6557. [DOI] [PubMed] [Google Scholar]
- Emerich DF, Skinner SJ, Borlongan CV, Vasconcellos AV, Thanos CG. The choroid plexus in the rise, fall and repair of the brain. Bioessays. 2005;27:262–274. doi: 10.1002/bies.20193. [DOI] [PubMed] [Google Scholar]
- Galvao RP, Garcia-Verdugo JM, Alvarez-Buylla A. Brain-derived neurotrophic factor signaling does not stimulate subventricular zone neurogenesis in adult mice and rats. J Neurosci. 2008;28:13368–13383. doi: 10.1523/JNEUROSCI.2918-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghabriel MN, Zdziarski IM, Leigh C, Vink R. Changes in the blood-CSF barrier in experimental traumatic brain injury. Acta Neurochir Suppl. 2010;106:239–245. doi: 10.1007/978-3-211-98811-4_45. [DOI] [PubMed] [Google Scholar]
- Ghersi-Egea JF, Gazzin S, Strazielle N. Blood-brain interfaces and bilirubin-induced neurological diseases. Curr Pharm Des. 2009;15:2893–2907. doi: 10.2174/138161209789058147. [DOI] [PubMed] [Google Scholar]
- Gonzalez AM, Podvin S, Leadbeater W, Borboa A, Burg M, Sawada R, Rayner J, Sims K, Terasaki T, Johanson C, Stopa E, Eliceiri B, Baird A (2010) Epidermal growth factor targeting of bacteriophage to the choroid plexus for gene delivery to the central nervous system via cerebrospinal fluid. Brain Res Aug 21 [Epub ahead of print] [DOI] [PMC free article] [PubMed]
- Grzybowski DM, Holman DW, Katz SE, Lubow M. In vitro model of cerebrospinal fluid outflow through human arachnoid granulations. Invest Ophthalmol Vis Sci. 2006;47:3664–3672. doi: 10.1167/iovs.05-0929. [DOI] [PubMed] [Google Scholar]
- Harbaugh RD, James HE, Marshall LF, Shapiro HM, Laurin R. Acute therapeutic modalities for experimental vasogenic edema. Neurosurgery. 1979;5:656–665. doi: 10.1227/00006123-197912000-00002. [DOI] [PubMed] [Google Scholar]
- Heile AM, Wallrapp C, Klinge PM, Samii A, Kassem M, Silverberg G, Brinker T. Cerebral transplantation of encapsulated mesenchymal stem cells improves cellular pathology after experimental traumatic brain injury. Neurosci Lett. 2009;463:176–181. doi: 10.1016/j.neulet.2009.07.071. [DOI] [PubMed] [Google Scholar]
- Hicks RR, Fertig SJ, Desrocher RE, Koroshetz WJ, Pancrazio JJ. Neurological effects of blast injury. J Trauma. 2010;68:1257–1263. doi: 10.1097/TA.0b013e3181d8956d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochwald GM, Wald A, Malhan C. The sink action of cerebrospinal fluid volume flow. Effect on brain water content. Arch Neurol. 1976;33:339–344. doi: 10.1001/archneur.1976.00500050025005. [DOI] [PubMed] [Google Scholar]
- Hu DZ, Zhou LF, Zhu J, Mao Y, Wu XH. Upregulated gene expression of local brain-derived neurotrophic factor and nerve growth factor after intracisternal administration of marrow stromal cells in rats with traumatic brain injury. Chin J Traumatol. 2005;8:23–26. [PubMed] [Google Scholar]
- Hu Y, Shen H, Keep RF, Smith DE. Peptide transporter 2 (PEPT2) expression in brain protects against 5-aminolevulinic acid neurotoxicity. J Neurochem. 2007;103:2058–2065. doi: 10.1111/j.1471-4159.2007.04905.x. [DOI] [PubMed] [Google Scholar]
- Johanson C. Choroid plexus-CSF circulatory dynamics: impact on brain growth, metabolism and repair. In: Conn P, editor. Neuroscience in medicine. Totowa, NJ: The Humana Press; 2008. pp. 173–200. [Google Scholar]
- Johanson CE, Woodbury DM. Uptake of [14C]urea by the in vivo choroid plexus–cerebrospinal fluid–brain system: identification of sites of molecular sieving. J Physiol. 1978;275:167–176. doi: 10.1113/jphysiol.1978.sp012183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johanson CE, Preston JE, Chodobski A, Stopa EG, Szmydynger-Chodobska J, McMillan PN. AVP V1 receptor-mediated decrease in Cl-efflux and increase in dark cell number in choroid plexus epithelium. Am J Physiol. 1999;276:C82–C90. doi: 10.1152/ajpcell.1999.276.1.C82. [DOI] [PubMed] [Google Scholar]
- Johanson CE, Szmydynger-Chodobska J, Chodobski A, Baird A, McMillan P, Stopa EG. Altered formation and bulk absorption of cerebrospinal fluid in FGF-2-induced hydrocephalus. Am J Physiol. 1999;277:R263–R271. doi: 10.1152/ajpregu.1999.277.1.R263. [DOI] [PubMed] [Google Scholar]
- Johanson CE, Palm DE, Primiano MJ, McMillan PN, Chan P, Knuckey NW, Stopa EG. Choroid plexus recovery after transient forebrain ischemia: role of growth factors and other repair mechanisms. Cell Mol Neurobiol. 2000;20:197–216. doi: 10.1023/A:1007097622590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johanson C, McMillan P, Palm D, Stopa E, Doberstein C, Duncan JA. Volume transmission-mediated protective impact of choroid plexus-CSF growth factors on forebrain ischemic injury. In: Sharma H, Westman J, editors. Blood-spinal cord and brain barriers in health and disease. San Diego: Academic Press; 2003. pp. 361–384. [Google Scholar]
- Johanson C, Duncan J, Baird A, Stopa E, McMillan P. Choroid plexus: a key player in neuroprotection and neuroregeneration. Int J Neuroprot Neuroregener. 2005;1:77–85. [Google Scholar]
- Johanson CE, Duncan JA, Stopa EG, Baird A. Enhanced prospects for drug delivery and brain targeting by the choroid plexus-CSF route. Pharm Res. 2005;22:1011–1037. doi: 10.1007/s11095-005-6039-0. [DOI] [PubMed] [Google Scholar]
- Johanson CE, Donahue JE, Spangenberger A, Stopa EG, Duncan JA, Sharma HS. Atrial natriuretic peptide: its putative role in modulating the choroid plexus-CSF system for intracranial pressure regulation. Acta Neurochir Suppl. 2006;96:451–456. doi: 10.1007/3-211-30714-1_92. [DOI] [PubMed] [Google Scholar]
- Johanson CE, Duncan JA, 3rd, Klinge PM, Brinker T, Stopa EG, Silverberg GD. Multiplicity of cerebrospinal fluid functions: new challenges in health and disease. Cerebrospinal Fluid Res. 2008;5:10. doi: 10.1186/1743-8454-5-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johanson CE, Miller M, Stopa E, Sharma HS (2010a) Disruption of the choroid plexus-CSF-ependymal wall nexus in CNS injury and aging models: rescue by i.c.v. peptides. In: 7th congress of the global college of neuroprotection and neuroregeneration, Stockholm, Sweden
- Johanson CE, Stopa E, McMillan PN, Roth DR, Funk J, Krinke G (2010b) The distributional nexus of choroid plexus to CSF, ependyma and brain: toxicologic/pathologic phenomena, periventricular destabilization and lesion spread. Toxicol Pathol (in press) [DOI] [PubMed]
- Johanson CE, Stopa E, McMillan PN. The blood-cerebrospinal fluid barrier: structure and functional significance. In: Nag S, editor. The blood-brain and other neural barriers. New York: Springer; 2011. p. 475. [DOI] [PubMed] [Google Scholar]
- Johnston M, Zakharov A, Papaiconomou C, Salmasi G, Armstrong D. Evidence of connections between cerebrospinal fluid and nasal lymphatic vessels in humans, non-human primates and other mammalian species. Cerebrospinal Fluid Res. 2004;1:2. doi: 10.1186/1743-8454-1-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur C, Singh J, Lim MK, Ng BL, Yap EP, Ling EA. Studies of the choroid plexus and its associated epiplexus cells in the lateral ventricles of rats following an exposure to a single non-penetrative blast. Arch Histol Cytol. 1996;59:239–248. doi: 10.1679/aohc.59.239. [DOI] [PubMed] [Google Scholar]
- Keep RF, Xiang J, Betz AL. Potassium cotransport at the rat choroid plexus. Am J Physiol. 1994;267:C1616–C1622. doi: 10.1152/ajpcell.1994.267.6.C1616. [DOI] [PubMed] [Google Scholar]
- Kernie SG, Parent JM. Forebrain neurogenesis after focal ischemic and traumatic brain injury. Neurobiol Dis. 2010;37:267–274. doi: 10.1016/j.nbd.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleindienst A, McGinn MJ, Harvey HB, Colello RJ, Hamm RJ, Bullock MR. Enhanced hippocampal neurogenesis by intraventricular S100B infusion is associated with improved cognitive recovery after traumatic brain injury. J Neurotrauma. 2005;22:645–655. doi: 10.1089/neu.2005.22.645. [DOI] [PubMed] [Google Scholar]
- Knuckey NW, Finch P, Palm DE, Primiano MJ, Johanson CE, Flanders KC, Thompson NL. Differential neuronal and astrocytic expression of transforming growth factor beta isoforms in rat hippocampus following transient forebrain ischemia. Brain Res Mol Brain Res. 1996;40:1–14. doi: 10.1016/0169-328x(96)00016-2. [DOI] [PubMed] [Google Scholar]
- Kovesdi E, Tamas A, Reglodi D, Farkas O, Pal J, Toth G, Bukovics P, Doczi T, Buki A. Posttraumatic administration of pituitary adenylate cyclase activating polypeptide in central fluid percussion injury in rats. Neurotox Res. 2008;13:71–78. doi: 10.1007/BF03033558. [DOI] [PubMed] [Google Scholar]
- Kozlowski GP. Hormone pathways in cerebrospinal fluid. Neurol Clin. 1986;4:907–917. [PubMed] [Google Scholar]
- Krishnamurthy S, Li J, Schultz L, McAllister JP., 2nd Intraventricular infusion of hyperosmolar dextran induces hydrocephalus: a novel animal model of hydrocephalus. Cerebrospinal Fluid Res. 2009;6:16. doi: 10.1186/1743-8454-6-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C, Agoston DV (2010) Vascular endothelial growth factor is involved in mediating increased de novo hippocampal neurogenesis in response to traumatic brain injury. J Neurotrauma 27:541–553 [DOI] [PubMed]
- Lee HJ, Lim IJ, Lee MC, Kim SU (2010) Human neural stem cells genetically modified to overexpress brain-derived neurotrophic factor promote functional recovery and neuroprotection in a mouse stroke model. J Neurosci Res Sept 3 [Epub ahead of print] [DOI] [PubMed]
- Lenzlinger PM, Morganti-Kossmann MC, Laurer HL, McIntosh TK. The duality of the inflammatory response to traumatic brain injury. Mol Neurobiol. 2001;24:169–181. doi: 10.1385/MN:24:1-3:169. [DOI] [PubMed] [Google Scholar]
- Li B, Mahmood A, Lu D, Wu H, Xiong Y, Qu C, Chopp M. Simvastatin attenuates microglial cells and astrocyte activation and decreases interleukin-1beta level after traumatic brain injury. Neurosurgery. 2009;65:179–185. doi: 10.1227/01.NEU.0000346272.76537.DC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindvall M, Edvinsson L, Owman C. Sympathetic nervous control of cerebrospinal fluid production from the choroid plexus. Science. 1978;201:176–178. doi: 10.1126/science.663649. [DOI] [PubMed] [Google Scholar]
- Matsuoka N, Nozaki K, Takagi Y, Nishimura M, Hayashi J, Miyatake S, Hashimoto N. Adenovirus-mediated gene transfer of fibroblast growth factor-2 increases BrdU-positive cells after forebrain ischemia in gerbils. Stroke. 2003;34:1519–1525. doi: 10.1161/01.STR.0000070840.56414.3B. [DOI] [PubMed] [Google Scholar]
- Maxwell WL, McGadey J. Response of intraventricular macrophages after a penetrant cerebral lesion. J Anat. 1988;160:145–155. [PMC free article] [PubMed] [Google Scholar]
- Maxwell WL, Hardy IG, Watt C, McGadey J, Graham DI, Adams JH, Gennarelli TA. Changes in the choroid plexus, responses by intrinsic epiplexus cells and recruitment from monocytes after experimental head acceleration injury in the non-human primate. Acta Neuropathol. 1992;84:78–84. doi: 10.1007/BF00427218. [DOI] [PubMed] [Google Scholar]
- Miyan JA, Nabiyouni M, Zendah M. Development of the brain: a vital role for cerebrospinal fluid. Can J Physiol Pharmacol. 2003;81:317–328. doi: 10.1139/y03-027. [DOI] [PubMed] [Google Scholar]
- Morganti-Kossmann MC, Satgunaseelan L, Bye N, Kossmann T. Modulation of immune response by head injury. Injury. 2007;38:1392–1400. doi: 10.1016/j.injury.2007.10.005. [DOI] [PubMed] [Google Scholar]
- Mudo G, Bonomo A, Di Liberto V, Frinchi M, Fuxe K, Belluardo N. The FGF-2/FGFRs neurotrophic system promotes neurogenesis in the adult brain. J Neural Transm. 2009;116:995–1005. doi: 10.1007/s00702-009-0207-z. [DOI] [PubMed] [Google Scholar]
- Murphy VA, Johanson CE. Adrenergic-induced enhancement of brain barrier system permeability to small nonelectrolytes: choroid plexus versus cerebral capillaries. J Cereb Blood Flow Metab. 1985;5:401–412. doi: 10.1038/jcbfm.1985.55. [DOI] [PubMed] [Google Scholar]
- Nakamachi T, Ohtaki H, Yofu S, Dohi K, Watanabe J, Mori H, Sato A, Hashimoto H, Shintani N, Baba A, Shioda S. Endogenous pituitary adenylate cyclase activating polypeptide is involved in suppression of edema in the ischemic brain. Acta Neurochir Suppl. 2010;106:43–46. doi: 10.1007/978-3-211-98811-4_6. [DOI] [PubMed] [Google Scholar]
- Nilsson C, Lindvall-Axelsson M, Owman C. Neuroendocrine regulatory mechanisms in the choroid plexus-cerebrospinal fluid system. Brain Res Brain Res Rev. 1992;17:109–138. doi: 10.1016/0165-0173(92)90011-a. [DOI] [PubMed] [Google Scholar]
- Owen-Lynch PJ, Draper CE, Mashayekhi F, Bannister CM, Miyan JA. Defective cell cycle control underlies abnormal cortical development in the hydrocephalic Texas rat. Brain. 2003;126:623–631. doi: 10.1093/brain/awg058. [DOI] [PubMed] [Google Scholar]
- Palm D, Knuckey N, Guglielmo M, Watson P, Primiano M, Johanson C. Choroid plexus electrolytes and ultrastructure following transient forebrain ischemia. Am J Physiol. 1995;269:R73–R79. doi: 10.1152/ajpregu.1995.269.1.R73. [DOI] [PubMed] [Google Scholar]
- Parandoosh Z, Johanson CE. Ontogeny of blood-brain barrier permeability to, and cerebrospinal fluid sink action on, [14C]urea. Am J Physiol. 1982;243:R400–R407. doi: 10.1152/ajpregu.1982.243.3.R400. [DOI] [PubMed] [Google Scholar]
- Parmelee JT, Bairamian D, Johanson CE. Response of infant and adult rat choroid plexus potassium transporters to increased extracellular potassium. Brain Res Dev Brain Res. 1991;60:229–233. doi: 10.1016/0165-3806(91)90051-j. [DOI] [PubMed] [Google Scholar]
- Pollay M. The function and structure of the cerebrospinal fluid outflow system. Cerebrospinal Fluid Res. 2010;7:9. doi: 10.1186/1743-8454-7-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollay M, Hisey B, Reynolds E, Tomkins P, Stevens FA, Smith R. Choroid plexus Na+/K+-activated adenosine triphosphatase and cerebrospinal fluid formation. Neurosurgery. 1985;17:768–772. doi: 10.1227/00006123-198511000-00007. [DOI] [PubMed] [Google Scholar]
- Preston JE, McMillan PN, Stopa EG, Nashold JR, Duncan JA, Johanson CE. Atrial natriuretic peptide induction of dark epithelial cells in choroid plexus: consistency with the model of CSF downregulation in hydrocephalus. Eur J Pediatr Surg. 2003;13(Suppl 1):S40-2. [PubMed] [Google Scholar]
- Pun PB, Lu J, Moochhala S. Involvement of ROS in BBB dysfunction. Free Radic Res. 2009;43:348–364. doi: 10.1080/10715760902751902. [DOI] [PubMed] [Google Scholar]
- Redzic ZB, Preston JE, Duncan JA, Chodobski A, Szmydynger-Chodobska J. The choroid plexus-cerebrospinal fluid system: from development to aging. Curr Top Dev Biol. 2005;71:1–52. doi: 10.1016/S0070-2153(05)71001-2. [DOI] [PubMed] [Google Scholar]
- Reger MA, Watson GS, Green PS, Wilkinson CW, Baker LD, Cholerton B, Fishel MA, Plymate SR, Breitner JC, DeGroodt W, Mehta P, Craft S. Intranasal insulin improves cognition and modulates beta-amyloid in early AD. Neurology. 2008;70:440–448. doi: 10.1212/01.WNL.0000265401.62434.36. [DOI] [PubMed] [Google Scholar]
- Regev L, Ezrielev E, Gershon E, Gil S, Chen A. Genetic approach for intracerebroventricular delivery. Proc Natl Acad Sci USA. 2010;107:4424–4429. doi: 10.1073/pnas.0907059107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandberg Nordqvist AC, von Holst H, Holmin S, Sara VR, Bellander BM, Schalling M. Increase of insulin-like growth factor (IGF)-1, IGF binding protein-2 and -4 mRNAs following cerebral contusion. Brain Res Mol Brain Res. 1996;38:285–293. doi: 10.1016/0169-328x(95)00346-t. [DOI] [PubMed] [Google Scholar]
- Sawamoto K, Wichterle H, Gonzalez-Perez O, Cholfin JA, Yamada M, Spassky N, Murcia NS, Garcia-Verdugo JM, Marin O, Rubenstein JL, Tessier-Lavigne M, Okano H, Alvarez-Buylla A. New neurons follow the flow of cerebrospinal fluid in the adult brain. Science. 2006;311:629–632. doi: 10.1126/science.1119133. [DOI] [PubMed] [Google Scholar]
- Schanzer A, Wachs FP, Wilhelm D, Acker T, Cooper-Kuhn C, Beck H, Winkler J, Aigner L, Plate KH, Kuhn HG. Direct stimulation of adult neural stem cells in vitro and neurogenesis in vivo by vascular endothelial growth factor. Brain Pathol. 2004;14:237–248. doi: 10.1111/j.1750-3639.2004.tb00060.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott DE, Krobisch-Dudley G, Paull WK, Kozlowski GP. The ventricular system in neuroendocrine mechanisms III. Supraependymal neuronal networks in the primate brain. Cell Tissue Res. 1977;179:235–254. doi: 10.1007/BF00219799. [DOI] [PubMed] [Google Scholar]
- Serrano M, Garcia-Silva MT, Martin-Hernandez E, O’Callaghan Mdel M, Quijada P, Martinez-Aragon A, Ormazabal A, Blazquez A, Martin MA, Briones P, Lopez-Gallardo E, Ruiz-Pesini E, Montoya J, Artuch R, Pineda M. Kearns-Sayre syndrome: cerebral folate deficiency, MRI findings and new cerebrospinal fluid biochemical features. Mitochondrion. 2010;10:429–432. doi: 10.1016/j.mito.2010.04.001. [DOI] [PubMed] [Google Scholar]
- Sharma HS, Johanson CE. Blood-cerebrospinal fluid barrier in hyperthermia. Prog Brain Res. 2007;162:459–478. doi: 10.1016/S0079-6123(06)62023-2. [DOI] [PubMed] [Google Scholar]
- Sharma HS, Johanson CE. Intracerebroventricularly administered neurotrophins attenuate blood cerebrospinal fluid barrier breakdown and brain pathology following whole-body hyperthermia: an experimental study in the rat using biochemical and morphological approaches. Ann N Y Acad Sci. 2007;1122:112–129. doi: 10.1196/annals.1403.008. [DOI] [PubMed] [Google Scholar]
- Sharma HS, Duncan JA, Johanson CE. Whole-body hyperthermia in the rat disrupts the blood-cerebrospinal fluid barrier and induces brain edema. Acta Neurochir Suppl. 2006;96:426–431. doi: 10.1007/3-211-30714-1_88. [DOI] [PubMed] [Google Scholar]
- Sharma HS, Zimmermann-Meinzingen S, Johanson CE. Cerebrolysin reduces blood-cerebrospinal fluid barrier permeability change, brain pathology, and functional deficits following traumatic brain injury in the rat. Ann N Y Acad Sci. 2010;1199:125–137. doi: 10.1111/j.1749-6632.2009.05329.x. [DOI] [PubMed] [Google Scholar]
- Shioda S, Ohtaki H, Suzuki R, Nakamachi T, Takenoya F, Dohi K, Nakajo S. Prevention of delayed neuronal cell death by PACAP and its molecular mechanism. Nippon Yakurigaku Zasshi. 2004;123:243–252. doi: 10.1254/fpj.123.243. [DOI] [PubMed] [Google Scholar]
- Shore PM, Jackson EK, Wisniewski SR, Clark RS, Adelson PD, Kochanek PM. Vascular endothelial growth factor is increased in cerebrospinal fluid after traumatic brain injury in infants and children. Neurosurgery. 2004;54:605–611. doi: 10.1227/01.neu.0000108642.88724.db. [DOI] [PubMed] [Google Scholar]
- Shuman CS, 3rd, Bryan JH. Comparative quantitative ultrastructural studies of the choroidal epithelium of hydrocephalic (hpy/hpy) and normal mice, and the effect of stress induced by water deprivation. Anat Anz. 1991;173:33–44. [PubMed] [Google Scholar]
- Silverberg GD, Heit G, Huhn S, Jaffe RA, Chang SD, Bronte-Stewart H, Rubenstein E, Possin K, Saul TA. The cerebrospinal fluid production rate is reduced in dementia of the Alzheimer’s type. Neurology. 2001;57:1763–1766. doi: 10.1212/wnl.57.10.1763. [DOI] [PubMed] [Google Scholar]
- Sivakumar V, Lu J, Ling EA, Kaur C. Vascular endothelial growth factor and nitric oxide production in response to hypoxia in the choroid plexus in neonatal brain. Brain Pathol. 2008;18:71–85. doi: 10.1111/j.1750-3639.2007.00104.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DE, Johanson CE, Keep RF. Peptide and peptide analog transport systems at the blood-CSF barrier. Adv Drug Deliv Rev. 2004;56:1765–1791. doi: 10.1016/j.addr.2004.07.008. [DOI] [PubMed] [Google Scholar]
- Spector R. Nature and consequences of mammalian brain and CSF efflux transporters: four decades of progress. J Neurochem. 2010;112:13–23. doi: 10.1111/j.1471-4159.2009.06451.x. [DOI] [PubMed] [Google Scholar]
- Spector R, Johanson CE. Choroid plexus failure in the Kearns-Sayre syndrome. Cerebrospinal Fluid Res. 2010;7:14. doi: 10.1186/1743-8454-7-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spector R, Johanson CE. Vectorial ligand transport through mammalian choroid plexus. Pharm Res. 2010;27(10):2054–2062. doi: 10.1007/s11095-010-0162-2. [DOI] [PubMed] [Google Scholar]
- Steardo L, Nathanson JA. Brain barrier tissues: end organs for atriopeptins. Science. 1987;235:470–473. doi: 10.1126/science.2879355. [DOI] [PubMed] [Google Scholar]
- Stumm R, Kolodziej A, Prinz V, Endres M, Wu DF, Hollt V. Pituitary adenylate cyclase-activating polypeptide is up-regulated in cortical pyramidal cells after focal ischemia and protects neurons from mild hypoxic/ischemic damage. J Neurochem. 2007;103:1666–1681. doi: 10.1111/j.1471-4159.2007.04895.x. [DOI] [PubMed] [Google Scholar]
- Sun D, Bullock MR, McGinn MJ, Zhou Z, Altememi N, Hagood S, Hamm R, Colello RJ. Basic fibroblast growth factor-enhanced neurogenesis contributes to cognitive recovery in rats following traumatic brain injury. Exp Neurol. 2009;216:56–65. doi: 10.1016/j.expneurol.2008.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun D, Bullock MR, Altememi N, Zhou Z, Hagood S, Rolfe A, McGinn MJ, Hamm R, Colello RJ. The effect of epidermal growth factor in the injured brain after trauma in rats. J Neurotrauma. 2010;27:923–938. doi: 10.1089/neu.2009.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szentistvanyi I, Patlak CS, Ellis RA, Cserr HF. Drainage of interstitial fluid from different regions of rat brain. Am J Physiol. 1984;246:F835–F844. doi: 10.1152/ajprenal.1984.246.6.F835. [DOI] [PubMed] [Google Scholar]
- Szmydynger-Chodobska J, Strazielle N, Zink BJ, Ghersi-Egea JF, Chodobski A. The role of the choroid plexus in neutrophil invasion after traumatic brain injury. J Cereb Blood Flow Metab. 2009;29:1503–1516. doi: 10.1038/jcbfm.2009.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamas A, Zsombok A, Farkas O, Reglodi D, Pal J, Buki A, Lengvari I, Povlishock JT, Doczi T. Postinjury administration of pituitary adenylate cyclase activating polypeptide (PACAP) attenuates traumatically induced axonal injury in rats. J Neurotrauma. 2006;23:686–695. doi: 10.1089/neu.2006.23.686. [DOI] [PubMed] [Google Scholar]
- Thau-Zuchman O, Shohami E, Alexandrovich AG, Leker RR (2010) Vascular endothelial growth factor increases neurogenesis after traumatic brain injury. J Cereb Blood Flow Metab 30:1008–1016 [DOI] [PMC free article] [PubMed]
- Thomas SA, Segal MB. The transport of the anti-HIV drug, 2′,3′-didehydro-3′-deoxythymidine (D4T), across the blood-brain and blood-cerebrospinal fluid barriers. Br J Pharmacol. 1998;125:49–54. doi: 10.1038/sj.bjp.0702044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson HJ, Lifshitz J, Marklund N, Grady MS, Graham DI, Hovda DA, McIntosh TK. Lateral fluid percussion brain injury: a 15-year review and evaluation. J Neurotrauma. 2005;22:42–75. doi: 10.1089/neu.2005.22.42. [DOI] [PubMed] [Google Scholar]
- Tureyen K, Vemuganti R, Bowen KK, Sailor KA, Dempsey RJ. EGF and FGF-2 infusion increases post-ischemic neural progenitor cell proliferation in the adult rat brain. Neurosurgery. 2005;57:1254–1263. doi: 10.1227/01.neu.0000186040.96929.8a. [DOI] [PubMed] [Google Scholar]
- Unterberg AW, Stover J, Kress B, Kiening KL. Edema and brain trauma. Neuroscience. 2004;129:1021–1029. doi: 10.1016/j.neuroscience.2004.06.046. [DOI] [PubMed] [Google Scholar]
- Walter HJ, Berry M, Hill DJ, Cwyfan-Hughes S, Holly JM, Logan A. Distinct sites of insulin-like growth factor (IGF)-II expression and localization in lesioned rat brain: possible roles of IGF binding proteins (IGFBPs) in the mediation of IGF-II activity. Endocrinology. 1999;140:520–532. doi: 10.1210/endo.140.1.6463. [DOI] [PubMed] [Google Scholar]
- Wang ZL, Cheng SM, Ma MM, Ma YP, Yang JP, Xu GL, Liu XF. Intranasally delivered bFGF enhances neurogenesis in adult rats following cerebral ischemia. Neurosci Lett. 2008;446:30–35. doi: 10.1016/j.neulet.2008.09.030. [DOI] [PubMed] [Google Scholar]
- Weaver C, McMillan P, Duncan JA, Stopa E, Johanson C. Hydrocephalus disorders: their biophysical and neuroendocrine impact on the choroid plexus epithelium. In: Hertz L, editor. Non-neuronal cells of the nervous system: function and dysfunction. Amsterdam: Elsevier Press; 2004. pp. 269–293. [Google Scholar]
- Wolburg H, Paulus W. Choroid plexus: biology and pathology. Acta Neuropathol. 2010;119:75–88. doi: 10.1007/s00401-009-0627-8. [DOI] [PubMed] [Google Scholar]
- Wu H, Lu D, Jiang H, Xiong Y, Qu C, Li B, Mahmood A, Zhou D, Chopp M. Simvastatin-mediated upregulation of VEGF and BDNF, activation of the PI3K/Akt pathway, and increase of neurogenesis are associated with therapeutic improvement after traumatic brain injury. J Neurotrauma. 2008;25:130–139. doi: 10.1089/neu.2007.0369. [DOI] [PubMed] [Google Scholar]
- Yamasaki H, Sugino M, Ohsawa N. Possible regulation of intracranial pressure by human atrial natriuretic peptide in cerebrospinal fluid. Eur Neurol. 1997;38:88–93. doi: 10.1159/000113166. [DOI] [PubMed] [Google Scholar]
- Yang J, Dombrowski SM, Deshpande A, Krajcir N, Luciano MG. VEGF/VEGFR-2 changes in frontal cortex, choroid plexus, and CSF after chronic obstructive hydrocephalus. J Neurol Sci. 2010;296:39–46. doi: 10.1016/j.jns.2010.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Song YN, Liu WG, Guo XL, Yu LG. Regulation and role of organic anion-transporting polypeptides (OATPs) in drug delivery at the choroid plexus. J Clin Neurosci. 2010;17:679–684. doi: 10.1016/j.jocn.2009.11.001. [DOI] [PubMed] [Google Scholar]