

Abstract
In statistical modelling, the effects of single-nucleotide polymorphisms (SNPs) are often regarded as time-independent. However, for traits recorded repeatedly, it is very interesting to investigate the behaviour of gene effects over time. In the analysis, simulated data from the 13th QTL-MAS Workshop (Wageningen, The Netherlands, April 2009) was used and the major goal was the modelling of genetic effects as time-dependent. For this purpose, a mixed model which describes each effect using the third-order Legendre orthogonal polynomials, in order to account for the correlation between consecutive measurements, is fitted. In this model, SNPs are modelled as fixed, while the environment is modelled as random effects. The maximum likelihood estimates of model parameters are obtained by the expectation–maximisation (EM) algorithm and the significance of the additive SNP effects is based on the likelihood ratio test, with p-values corrected for multiple testing. For each significant SNP, the percentage of the total variance contributed by this SNP is calculated. Moreover, by using a model which simultaneously incorporates effects of all of the SNPs, the prediction of future yields is conducted. As a result, 179 from the total of 453 SNPs covering 16 out of 18 true quantitative trait loci (QTL) were selected. The correlation between predicted and true breeding values was 0.73 for the data set with all SNPs and 0.84 for the data set with selected SNPs. In conclusion, we showed that a longitudinal approach allows for estimating changes of the variance contributed by each SNP over time and demonstrated that, for prediction, the pre-selection of SNPs plays an important role.
Keywords: EM algorithm, Legendre polynomials, Longitudinal data, Maximum likelihood, Prediction, Single-nucleotide polymorphism
Introduction
A great majority of phenotypes observed in animals, plants and humans are both quantitative and longitudinal, i.e. they attribute variation on a continuous scale and can be recorded several times during an individual’s lifetime or physiological cycle. A classical example of such a trait is growth. The evidence of variation of genetic effects over time is observed in experimental organisms (Leips et al. 2006), as well as in livestock (e.g. Schaeffer and Dekkers 1994). In the former study, time-dependent quantitative trait loci (QTL) affecting fecundity in Drosophila melanogaster are identified, while the latter study provides evidence that the additive genetic variance underlying milk production traits in dairy cattle is not constant over time.
Most statistical applications aiming to model growth traits either assume that the underlying genetic background is constant in time (e.g. Corva and Medrano 2001), or that the changes of all gene effects are the same throughout the whole growth period (e.g. Jaffrézic et al. 2004). The functional relationship between genetic parameters and time-dependent variables are described by orthogonal polynomials, which were first introduced by Schaeffer and Dekkers (1994) to model a joint additive effect of all genes (a so-called polygenic effect) for milk yield in dairy cattle.
Modelling effects of particular genes as variable over time is known in the literature as functional gene mapping. The changes of a gene effect over time are described by the logistic function (Ma et al. 2002), Legendre orthogonal polynomials (Yang et al. 2006, 2007) or B-splines (Yang et al. 2009). However, none of the applications tackles the problem of the prediction of future trait values.
The major goal of this study is an application of a model in which effects of single-nucleotide polymorphisms (SNPs) representing genetic factors, as well as a permanent environmental effect, are assumed to be variable over time. Such modelling is especially useful for association studies in the situation when genetic effects underlying an SNP affect only some, but not all, time periods of a longitudinal trait. When a model with time-independent effects is applied to such a genetic background, the SNP effect is averaged over the whole time period and may, thereby, be difficult, or even impossible, to be detected. The longitudinal estimators optimally use the information contained in the whole analysed period and are, thus, robust towards the variation of genetic effects over time.
Materials and methods
Statistical model
For the detection of an association between SNPs and a longitudinal trait, a single SNP random regression model proposed by Yang et al. (2006) is used. In this model, all time-dependent parameters (both fixed and random) are described by Legendre polynomials of order 3. This random regression model is an extension of the following single SNP linear mixed model:
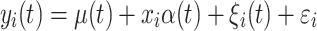 |
1 |
where y
i(t) is a phenotypic trait of individual i at time t, μ(t) is a population mean at time t, x
i ∈ {−1, 0, 1} is an SNP genotype indicator variable for the ith individual and α(t) is a fixed additive SNP effect at time t. It is assumed that ξ
i(t) is a time-dependent random permanent environmental effect with 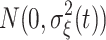 distribution and ε
i is a time-independent residual term with N(0, σ
2) distribution. The genetic variance at time t for model 1 is expressed as:
distribution and ε
i is a time-independent residual term with N(0, σ
2) distribution. The genetic variance at time t for model 1 is expressed as:
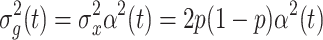 |
2 |
where p is the frequency of allele “1” for a given SNP. The total phenotypic variance at time t has the form:
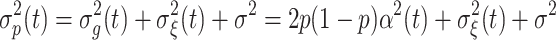 |
3 |
Note that it is a single SNP model, so the genetic and phenotypic variances are calculated separately for each SNP.
Since phenotypic values for each individual are expressed as a longitudinal trait, measured at n + 1 time points, t = [t 0, t 1,..., t n], Legendre orthogonal polynomials can be used for describing time-dependent effects. These polynomials are defined on an interval [−1, 1], therefore, the original time points t are recoded into τ using the following formula:
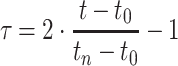 |
4 |
where t
0 and t
n are the extreme time values. For modelling three time-dependent effects (μ(τ), α(τ), ξ
i(τ)) in model 1, Legendre polynomials of order 3 are used. Namely, a time-dependent parameter μ(τ) can be described as a linear combination of Ψ(τ)μ, where  are coefficients of the polynomial and
are coefficients of the polynomial and 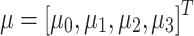 is a vector of the time-independent population means. The polynomial coefficients have the following form:
is a vector of the time-independent population means. The polynomial coefficients have the following form:
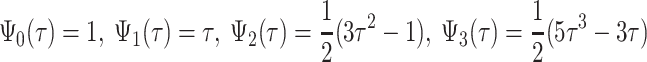 |
5 |
The remaining time-dependent fixed and random terms (α(τ) and ξ
i(τ)) are described in the same way, using linear combinations of Ψ(τ)α and Ψ(τ)ξ
i. The third order of polynomials is chosen because linear or quadratic curves (polynomial order ≤2) usually exhibit very poor fit to growth curves. On the other hand, since, for each individual, several observations are available, it would be impossible to fit a higher order of polynomials. It is assumed that random regression coefficients for a permanent environmental effect 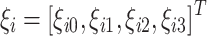 are normally distributed with mean 0 and covariance matrix Σ. Under the above parameterisation, the model 1 can be transformed into a random regression model, which has the following form:
are normally distributed with mean 0 and covariance matrix Σ. Under the above parameterisation, the model 1 can be transformed into a random regression model, which has the following form:
 |
6 |
The genetic variance for model 6 is given by:
 |
7 |
and the total phenotypic variance is expressed as:
 |
8 |
The estimation of all unknown parameters (μ, α, Σ, σ 2) in model 6 is based on the expectation–maximisation (EM) algorithm as described by Yang et al. (2006).
Hypothesis testing
For testing the association between an SNP and a phenotype, the likelihood ratio test can be used. The corresponding hypotheses are: H
0: α = 0 vs. H
1: ∃ i ∈ {0, 1, 2, 3} (α
i ≠ 0). Under H
0, the test statistic asymptotically follows the  distribution. Since a single SNP is tested at a time, a multiple testing problem arises. Our SNP selection approach involves two criteria. First, we applied the Bonferroni correction of the nominal p-values. Second, we used a pairwise linkage disequilibrium measure between all SNP pairs (r
2) for dropping one of the two SNPs, which remained in strong linkage disequilibrium.
distribution. Since a single SNP is tested at a time, a multiple testing problem arises. Our SNP selection approach involves two criteria. First, we applied the Bonferroni correction of the nominal p-values. Second, we used a pairwise linkage disequilibrium measure between all SNP pairs (r
2) for dropping one of the two SNPs, which remained in strong linkage disequilibrium.
Prediction of future yields
The model for the prediction of future yields at time point t* > t n is based on the linear regression, where the population mean (μ) and the fixed additive effects of SNPs (α) are described using Legendre polynomials of order 3. First, estimators of (μ, α) based on the data set consisting of phenotyped animals are obtained using the following model:
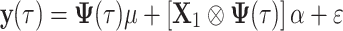 |
9 |
where y(τ) is a vector of trait values for all phenotyped individuals at time point τ, X
1 is an incidence matrix of SNP genotypes for phenotyped animals and 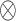 is the Kronecker product. Then, using
is the Kronecker product. Then, using 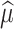 and
and 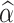 , future yields at time point t* for unphenotyped animals are calculated using:
, future yields at time point t* for unphenotyped animals are calculated using:
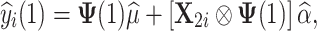 |
10 |
where 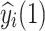 is the predicted future phenotypic value for the ith unphenotyped animal at time point t* and X
2i is an incidence matrix of SNP genotypes for the ith unphenotyped animal. Note, now, that n + 2 (not n + 1) time points are available and the corresponding formula for the transformation of time points t into τ has the following form:
is the predicted future phenotypic value for the ith unphenotyped animal at time point t* and X
2i is an incidence matrix of SNP genotypes for the ith unphenotyped animal. Note, now, that n + 2 (not n + 1) time points are available and the corresponding formula for the transformation of time points t into τ has the following form:
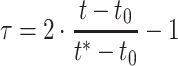 |
11 |
Note that prediction is performed for two data sets: the nominal data set with all available markers and the selected data set with only SNPs significant for model 6.
Simulation study
The analysed data set was generated using Monte Carlo simulations for the 13th QTL-MAS Workshop (Wageningen, The Netherlands, April 2009). It consists of 2,025 genetically related individuals from two generations: 25 individuals represent the parental generation (20 females and 5 males) and the remaining 2,000 individuals are offspring (100 full sib families, one from each combination of a male and female parent). All individuals have complete marker information consisting of 453 biallelic markers represented by SNPs, which are randomly distributed over five chromosomes, each of approximately 1 Morgan in length. Chromosome two is the most densely covered by SNPs (99 SNPs), while chromosome five is the most sparsely covered (86 SNPs).
Quantitative phenotypes are available for 50 full sib families, while the other 50 full sib families have unknown phenotypes. Phenotypes were generated using a logistic growth curve and recorded at five different time points, denoted by 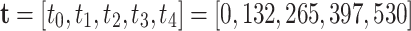 . The asymptotic values of individuals’ yield range from 14 to 66. The phenotyped full sib families are selected such that each female parent has at least 40 phenotyped offspring, while each male parent has 100 phenotyped offspring. There are 18 QTL simulated—three on the first and fifth chromosomes and four on the second, third and fourth chromosomes. The QTL are also generated using a logistic growth curve.
. The asymptotic values of individuals’ yield range from 14 to 66. The phenotyped full sib families are selected such that each female parent has at least 40 phenotyped offspring, while each male parent has 100 phenotyped offspring. There are 18 QTL simulated—three on the first and fifth chromosomes and four on the second, third and fourth chromosomes. The QTL are also generated using a logistic growth curve.
Results
The summary of the effect of the particular SNP is presented in Fig. 1. It was evident that, even after Bonferroni correction was applied, many SNPs significantly affected the trait. In total, 207 SNPs were significant at the 0.05 level (all SNPs above the black line in Fig. 1). Additionally, one of the two SNPs which remain in strong pairwise linkage disequilibrium (r 2 > 0.8) were excluded from the data set. After applying both selection criteria, the final number of selected SNPs was 179. In order to check how well the set of significant SNPs covered the positions of simulated QTL, 95% confidence intervals were constructed for the distances between two neighbouring SNPs. An average distance between two SNPs ranged between 0.999 and 1.206 cM. As a result, 16 out of 18 QTL (88.89%) were located within the confidence intervals.
Fig. 1.

p-values after Bonferroni correction for all 453 single-nucleotide polymorphisms (SNPs). p-values are after logarithm transformation
Figure 2 illustrated an advantage of a longitudinal model, which was capable of describing the genetic background of a longitudinal trait in a more flexible way than models assuming a genetic effect constant over time. Effects of the selected SNPs during the whole growth period changed between the early and the late growth stage. For example, the effect of SNP 48 was low until approximately the 120th day, and then it increased towards the terminal day of the growth period, with an especially rapid change between days 120 and 300. On the other hand, SNP 198 had an opposite effect. Its influence on the genetic variance was observed mainly at the beginning of the growth phase, between days 1 and 200. An example of a locus with a relatively constant effect was SNP 89 (Fig. 3).
Fig. 2.

a Percentage of contributed variance of each significant SNP on chromosome one in five different time points. b Percentage of variance contributed by SN P37
Fig. 3.

Percentage of contributed variance of three different SNPs
The results for the prediction of future yields at time 600 are shown in Fig. 4a for the data set with all SNPs and in Fig. 4b for the data set with the selected 179 SNPs. Note that, when all SNPs were used, some of the individuals had predicted phenotypic values which exceed the trait limit predefined in the simulation. The correlation between predicted and true trait values was 0.73. When only the selected SNPs were used, no individual exceeded the trait limit and the correlation of 0.84 was reached. Phenotypes predicted based on the selected SNP data set were less variable (standard deviation 6.20) than the ones predicted using all SNPs (8.10), while the standard deviation of the true values of trait at time point 600 was 5.03.
Fig. 4a, b.

Predicted trait value at time point 600 for non-phenotyped animals. The two horizontal lines are asymptotic values of individuals’ yield. a For the data set with all available 453 SNPs. b For the data set with 179 selected significant SNPs
Discussion and conclusions
In the context of interval QTL mapping, using simulation, Yang et al. (2007) observed that models with time-dependent gene effects provide accurate estimates of QTL positions and effects. They allow for a more precise description of the variability of the dependent variable and require only a few parameters for this purpose. The present analysis demonstrates that the parameterisation of each SNP in a longitudinal data context gives a model with a very high flexibility. In contrast to the original model proposed by Yang et al. (2006), in model 6, only additive effects of SNPs were taken into account. Such parsimonious parameterisation allowed us to fit a multiple SNPs model (model 9). The model of Yang et al. (2006) is extended so that it was not only used for QTL detection, but also for the prediction of future yields. This may play a key role in the pre-selection of animals for breeding. The method proposed for the prediction of future yields is based on the linear regression with SNP effects modelled as time-dependent variables. A disadvantage of the proposed prediction approach is that it takes no account of the relationship between individuals. Its advantage lies in its computational speed, which might be a critical issue for practical breeding with large data sets and demand for frequent evaluations (e.g. daily, as it is the case in poultry or pig breeding).
The same simulated data set was also analysed with other approaches. Bayesian methods (Bayes A and Bayes B) provided the best accuracy of prediction at time 600, ranging between 0.86 and 0.95 (Bastiaansen et al. 2010), whereas the lowest accuracy of 0.65 was obtained using a BLUP model. A comparison of these results indicates that the SNP selection approach plays an important role for prediction purposes, even for a quantitative trait determined by only several (18) QTL.
Rapid advances in high-throughput genotyping technologies in livestock make large amounts of genotypic data (thousands of SNPs) available widely with reasonable costs. On the other hand, phenotypic observations are routinely recorded through individuals’ production lifespan. Such rich sources of information can be utilised not only for the prediction of future yields, but also for predicting the genetic value of young selection candidates (Schaeffer 2006). That is why models capable of incorporating time-dependent effects are going to gain importance in practical applications in the near future.
Acknowledgments
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
References
- Bastiaansen JWM, Bink MCAM, Coster A, Maliepaard C, Calus MPL. Comparison of analyses of the QTLMAS XIII common dataset. I: genomic selection. BMC Proc. 2010;4(Suppl 1):S1. doi: 10.1186/1753-6561-4-s1-s1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corva PM, Medrano JF. Quantitative Trait Loci (QTLs) mapping for growth traits in the mouse: a review. Genet Sel Evol. 2001;33(2):105–132. doi: 10.1186/1297-9686-33-2-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffrézic F, Venot E, Laloë D, Vinet A, Renand G. Use of structured antedependence models for the genetic analysis of growth curves. J Anim Sci. 2004;82:3465–3473. doi: 10.2527/2004.82123465x. [DOI] [PubMed] [Google Scholar]
- Leips J, Gilligan P, Mackay TFC. Quantitative Trait Loci with age-specific effects on fecundity in Drosophila melanogaster. Genetics. 2006;172:1595–1605. doi: 10.1534/genetics.105.048520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma CX, Casella G, Wu R. Functional mapping of Quantitative Trait Loci underlying the character process: a theoretical framework. Genetics. 2002;161:1751–1762. doi: 10.1093/genetics/161.4.1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaeffer LR. Strategy for applying genome-wide selection in dairy cattle. J Anim Breed Genet. 2006;123:218–223. doi: 10.1111/j.1439-0388.2006.00595.x. [DOI] [PubMed] [Google Scholar]
- Schaeffer LR, Dekkers JCM (1994) Random regressions in animal models for test-day production in dairy cattle. Proceedings of the 5th World Congress of Genetics Applied to Livestock Production (WCGALP), Guelph, Ontario, Canada, August 1994, vol 18, pp 443–446
- Yang R, Tian Q, Xu S. Mapping Quantitative Trait Loci for longitudinal traits in line crosses. Genetics. 2006;173:2339–2356. doi: 10.1534/genetics.105.054775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang R, Gao H, Wang X, Zhang J, Zeng ZB, Wu R. A semiparametric approach for composite functional mapping of dynamic quantitative traits. Genetics. 2007;177:1859–1870. doi: 10.1534/genetics.107.077321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Wu R, Casella G. Nonparametric functional mapping of quantitative trait loci. Biometrics. 2009;65:30–39. doi: 10.1111/j.1541-0420.2008.01063.x. [DOI] [PubMed] [Google Scholar]