

Abstract
Objective
To report the epidemiology and ocular phenotype of retinal capillary hemangioblastomas associated with von Hippel-Lindau (VHL) disease in a large cohort of patients and to correlate patient and ocular characteristics to visual morbidity in this population.
Design
Prospective consecutive case series.
Participants
In 220 unrelated pedigrees, 335 patients affected with VHL disease and retinal capillary hemangioblastomas (RCHs) in at least 1 eye.
Methods
Demographics of the patient population were recorded and the ocular phenotype of each patient was obtained with a comprehensive ocular examination.
Main Outcome Measures
The patient population was characterized and the ocular phenotype described in relationship to tumor location, number, and extent of retinal involvement. Correlations between patient demographics, ocular phenotype, and visual function were analyzed.
Results
RCHs were detected unilaterally in 42.1% and bilaterally in 57.9% of patients. No correlation was detected between the age, gender, and laterality of involvement. Eighty-seven (86.6%) percent of involved eyes had tumors that could be individually visualized; of these, tumors were commonly found in the peripheral retina (84.7%) only, and less commonly in the juxtapapillary area (15.3%). The tumor count in the periphery averaged 2.5±1.8 per eye, with 25.2% of eyes having greater than 1 quadrant of retinal involvement. Thirteen (13.4%) percent of involved eyes were enucleated or pre-phthsical; approximately 1 in 5 patients had one or more eyes so affected. Severe visual impairment (visual acuity worse or equal to 20/160) in affected eyes were more likely to be associated with increasing age, the presence of juxtapapillary lesions, and an increasing number and extent of peripheral lesions.
Conclusions
This large cohort of VHL patients with RCHs has enabled a systematic and quantitative characterization of the demographics, ocular features, and visual function in VHL disease. Clinical correlations between the visual morbidity and ocular features of the disease were also performed, producing measures that can help clinicians better estimate visual prognoses based on the ocular phenotype of the disease.
Von Hippel-Lindau (VHL) disease (Online Mendelian Inheritance in Man™ (OMIM) Number 193300) is an inherited, autosomal dominant, multi-system cancer syndrome that leads to the development of both benign and malignant tumors 1. Tumor types include hemangioblastomas in the central nervous system (retina, cerebellum, spinal cord, brainstem), renal cysts and clear-cell carcinomas, pheochromocytomas, pancreatic cysts, and cystadenomas in the epididymis and broad ligament. Retinal capillary hemangioblastomas (RCHs) are among the most frequently occurring and earliest presenting tumors in this group with many patients with VHL disease requiring ophthalmic care and continuous follow-up 2.
RCHs in VHL disease are benign tumors that have a characteristic orange-red color, a globular shape, and typically dilated feeding and draining vessels. Histologically, they are comprised of capillary-like vascular channels surrounded by large vacuolated stromal cells, and also smaller “tumorlet” cells that express stem cell markers 3. RCHs can be highly variable in their number, size, and location. Although often slow-growing in nature and benign in their effect, they can sometimes result in vision loss and structural disruption to the integrity of the globe through exudative and tractional effects on the retina 4.
The diagnosis of VHL disease is often performed clinically, using criteria involving a family history of the disease and the detection and characteristic tumors in various organ systems 5. Genetic testing for VHL disease is now available and mutations in the VHL gene can be detected and characterized at a very high rate 6. Owing to the high penetrance of the disease, almost all patients testing positive genetically will develop clinically definite disease with time 7.
The factors that influence the phenotype of RCH in VHL and the visual function in affected eyes are not well understood. The purpose of this study is to report the findings of a large population of patients with clinically confirmed VHL disease with ocular involvement in order to characterize in a quantitative way their demographics, ocular phenotype, and visual function. The population in the present study is one of the largest assembled; enrollment was based not on visual symptoms but to the basis of systemic VHL disease. Each patient had been examined with a multi-system evaluation at a single institution (National Institute of Health) and the diagnosis of VHL confirmed by clinical exam. Almost all (98.1%) of the patients in this population were also found on genotype analysis to have mutations in the VHL gene (unpublished data). The nature of the ascertainment of this population likely provides an accurate profile of ocular involvement in VHL disease. This, together with the large number of patients, enables us to establish statistically reliable clinical correlations between the demographic features, ocular characteristics, and visual function in VHL disease.
METHODS
Ascertainment of patients with VHL disease and ocular evaluation
All patients referred to the study were enrolled into a study protocol at the National Cancer Institute (NCI, National Institute of Health, Bethesda, Maryland) from 1988 to 2005. Institutional Review Board (IRB) approval was obtained. A systemic multi-system evaluation was performed at this single center with history and physical examination, laboratory evaluation, and radiographical (CT or MRI) studies of the abdomen, pelvis, brain, and spine, and clinical diagnosis of VHL disease was made on the basis of these findings5.
All patients also underwent an ophthalmologic evaluation with slit lamp examination and indirect ophthalmoscope funduscopy performed at the National Eye Institute (NEI) clinical center. Inclusion into the study was based on 1) a positive clinical diagnosis of VHL disease and 2) an ophthalmoscopic detection of a typical RCH. In the case of juxtapapillary lesions or subtle ophthalmoscopic findings, fluorescein angiography was performed to ascertain the vascular nature of the tumor. Three hundred and thirty-five VHL patients with evidence of RCH in at least one eye were identified and included in the present study.
History of VHL ocular disease including the date of onset, and the various therapies used were obtained. The comprehensive ocular examination included a best-corrected visual acuity using the ETDRS visual acuity chart, slit lamp examination, intraocular pressure measurement, and a fundus evaluation. For eyes in which the posterior pole can be clearly visualized by indirect ophthalmoscopy, the location and number of RCHs were recorded. The term “ocular phenotype” refers to the following characteristics as defined here. The location of tumors was described as either “optic nerve tumors” if they were located on or within 1/2 disk diameter of the optic nerve, or “peripheral” if they were located peripheral to the area around the optic nerve. The number of tumors was recorded only for peripheral RCHs and not for the juxtapapillary RCHs. The extent of peripheral retinal involvement was also scored in terms whether the total involved area spanned greater or less than one quadrant of peripheral retina. For eyes in which the posterior pole examination cannot be perforned (such as in enucleated eyes and phthisical eyes), the ophthalmic features were not recorded, but these eyes were identified as severely affected. Patients were followed longitudinally, and the patient’s most recent visit was designated as the study visit for this cross-sectional evaluation.
Statistical Methods
In our study we conducted analyses at the level of individual patients and at the level of individual eyes to assess the relationships between patient demographics, ocular phenotype and visual function. To evaluate the effect of gender on laterality, a chi-square test was used, and to evaluate the association of gender, laterality and severity of ocular disease with age as outcome, a t-test was used. Age-adjusted logistic regression was used to analyze the effect of laterality on severe ocular involvement. Analysis of variance was used to assess the relationship of visual acuity in the best and worse-seeing eye with age in quartiles, and gender adjusted for age in quartiles. The above were patient-level analyses.
All eye-level analyses were done using generalized estimating equations, a method that takes into account the correlation between both eyes within a person. A binomial link function was specified for the following dichotomous outcomes: number of peripheral RCHs of 3 or more, number of peripheral RCHs of 5 or more, extent of retinal involvement of greater than 1 quadrant, visual impairment of worse than 20/40, visual impairment of worse than 20/160, and the comparison of severe vision (worse than 20/160, legal blindness) versus good vision (better than 20/20). The analyses where visual acuity was the outcome employed a normal link function.
Since visual acuity was found to significantly decrease with increasing age, all visual function analyses were adjusted by age in quartiles.
RESULTS
Patient population characteristics
890 patients with clinically definite VHL disease were screened with a comprehensive ocular exam. 335 patients were found to have ocular involvement with RCHs in one or both eyes. This subgroup of patients originated from 220 unrelated pedigrees (mean number of patients per pedigree = 1.5±1.2; range = 1 - 13). The mean age of patients was 36.9 ±13.7 years (range 8.6-84.3 years) (Fig. 1). 55% of these patients were female. The majority of patients were white; the self-reported racial distribution is shown in Table 1. The ages of patients with RCHs did not vary significantly according to gender (p=0.407). The correlation of age to racial category was not computed as there was only a small fraction of non-white patients in the population.
Figure 1.
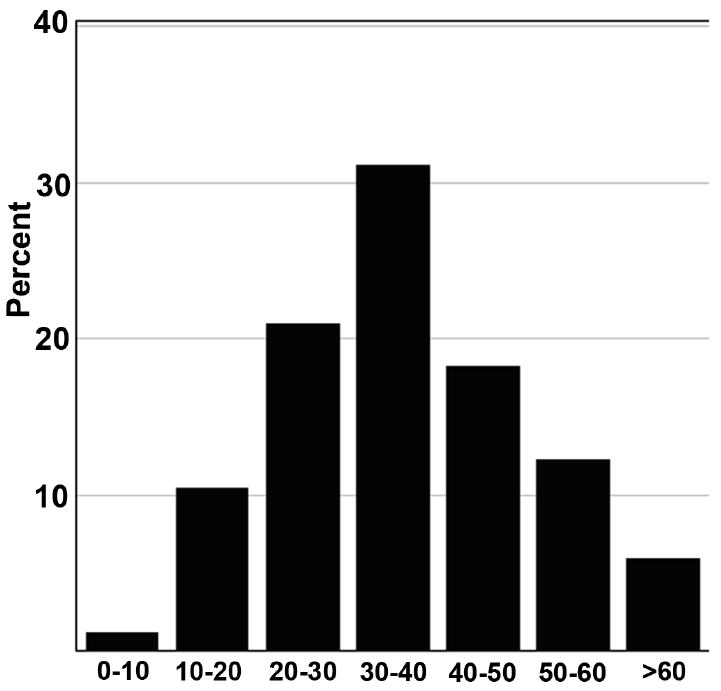
Age distribution of 335 patients with retinal capillary hemangioblastomas (RCHs) related to von Hippel-Lindau disease.
Table 1.
Distribution of Patients with Unilateral and Bilateral Retinal Capillary Hemangioblastomas (RCH) according to Gender and Racial Categories
| Unilateral RCH Number (% of column total) | Bilateral RCH Number (% of column total) | Row Total (% of column total) | |
|---|---|---|---|
| Gender | |||
| Male | 56 (39.7) | 95 (49.0) | 151 (45.1) |
| Female | 85 (60.3) | 99 (51.0) | 184 (54.9) |
| Total | 141 (100) | 194 (100) | 335 (100) |
| Racial category | |||
| Asian | 3 (2.1) | 5 (2.6) | 8 (2.4) |
| Black | 3 (2.1) | 3 (1.5) | 6 (1.8) |
| Hispanic | 9 (6.4) | 7 (3.6) | 16 (4.8) |
| White | 126 (89.4) | 179 (92.3) | 305 (91.0) |
| Total | 141 (100) | 194 (100) | 335 (100) |
Ocular Phenotype
Bilateral RCHs were found in 193 of 335 patients (57.9%). Unilaterality versus bilaterality of RCH occurrence was not found to be statistically associated with gender (p=0.093). The age distributions of patients with unilateral and bilateral RCH involvement were also similar and the mean ages of patients with unilateral (36.2 ±14.3 years) and bilateral involvement (37.3 ±13.3 years) were also not statistically distinct (p=0.476).
Each of the 670 eyes in 335 patients was classified according to its ocular phenotype (Fig 2). No detectable RCHs were found in 141 out of 670 eyes (21.0%) eyes. These comprised of unaffected fellow eyes of the 141 patients with RCHs in only one eye. Of the remaining 529 eyes with ocular involvement, the RCH phenotype of 71 eyes (10.6%) could not be described. In some cases, this was due to prior enucleation resulting from complications of RCH. 42 enucleated eyes (6.3%) were found in 41 patients, with 1 patient having both eyes enucleated. In an additional 29 (4.3%) eyes (of 28 patients), individual RCHs could not be directly visualized by indirect funduscopy due to structural disruption of the eye from phthisis, extensive retinal detachments, or anterior segment opacities secondary to complications from retinal complications.
Figure 2.

Schematic diagram showing the subcategorization of 670 eyes of 335 patients with retinal capillary hemangioblastomas (RCHs) related to von Hippel-Lindau disease according to ocular phenotype.
Eyes that are either enucleated or structurally disrupted due to angiomatous disease are classified as having “severe ocular involvement” (Table 2). Approximately 1 in 5 patients (20.3%) were found to have at least one eye with this degree of severity of disease. The prevalence of having at least one eye with severe ocular involvement was correlated with increasing age of the patient (p=0.001) and with bilateral RCH involvement (p=0.0003, adjusted for age).
Table 2.
Eyes with Retinal Capillary Hemangiomas Resulting in Severe Ocular Involvement
| Structural Disruptions* | Enucleations | Total | |
|---|---|---|---|
| Eyes affected | 29 | 42 | 71 (10.6% of 670 eyes) |
| Patients affected | |||
| In one eye only | 27 | 40 | |
| In both eyes† | 1 | 1 | |
| Total patients with at least one eye severely affected | 28 | 41 | 68 (20.3% of 335 patients) |
| Patients at least one eye with severe ocular involvement | Patients without severe ocular involvement | ||
| Mean age ± s.d.¥ | 41.7 ± 15.0 years | 35.6 ± 13.2 years | p=0.0011 |
| Percentage with bilateral retina hemangiomas | 77.9% | 52.8% | p=0.0002 |
Structural disruptions: eyes in which angiomatous involvement had resulted in an obscured view of the posterior pole in which individual retinal hemangiomas cannot be individually visualized. These include eyes with phthisis, extensive retinal detachments, and anterior segment structural disorganization in end-stage disease.
One patient in the population had one eye that was structurally disrupted and the other eye enucleated.
s.d. = standard deviation
In the remaining 458 (68.4%) affected eyes, the RCHs could be visualized by funduscopy in 2 locations; in the juxtapapillary area, and the peripheral retina (defined as being located beyond the vascular arcades and beyond twice the fovea-disc distance from the optic nerve). No RCHs were detected in the macula. In 37 (8.1%) eyes, RCHs were found only in the juxtapapillary location, compared to 388 (84.7%) eyes that had RCHs only in the peripheral retina. 33 (7.2%) eyes had RCHs in both the juxtapapillary location and the peripheral retina.
In the 421 eyes with RCHs in the periphery, the number of RCHs ranged from 1 to 11 (mean 2.53±1.81, mode = 1) (Fig 3). The extent of peripheral retina occupied by these RCHs was also estimated to affect more than 1 quadrant of the peripheral retina in 105 (25.2%) eyes. Because of the potential to develop new RCH, over time, we examined the relationship between the age and the number of peripheral RCHs found per eye. No statistical significant association was found between age and the number of peripheral RCHs found (p=0.605) or between age and extent of peripheral involvement (scored as greater or less than 1 quadrant of peripheral retina were involved by RCHs) (p=0.695).
Figure 3.
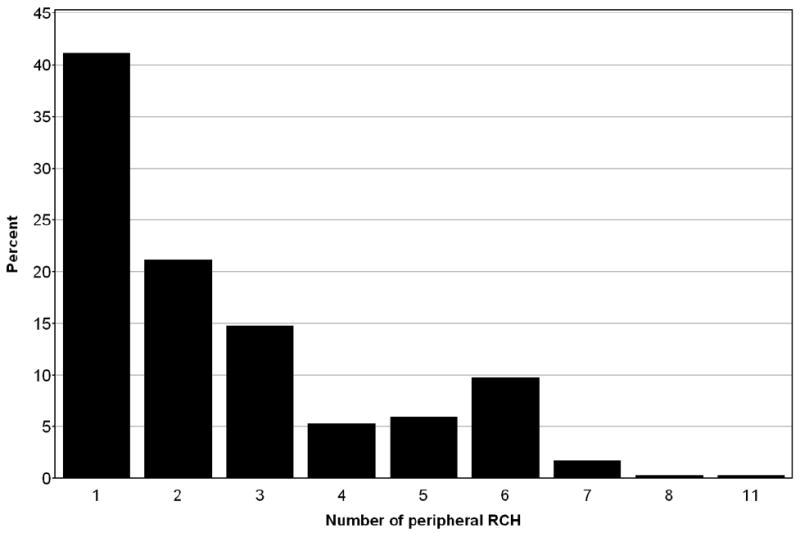
Distribution of peripherally-located retinal capillary hemangioblastomas (RCHs) found in 421 eyes depicted as number of RCHs per eye.
Visual Function
Best-corrected visual acuity, measured bilaterally, was assessed in analyses of “better-seeing eye” or the “worse-seeing eye”. The distributions of visual acuities for the better-seeing eye and in the worse-seeing eye of all 335 patients are shown in Figure 4A, with the distribution was skewed to the left and 77.3% had visual acuities better or equal to 20/20 or better (scoring ≥ 84 letters). However, the visual acuities in the worse-seeing eye had a distinct bimodal distribution, with about one-third of eyes (36.4%) having visual acuities 20/20 or better (≥ 84 letters), and a similar fraction of eyes (31.9%) on the other side of the distribution having with poor visual acuities of worse than 20/160 (0-38 letters). These data show that the visual functional impairment found in a cross-section of patients with ocular VHL disease is usually monocular in nature. Using the cut-off of worse than 20/40 vision as a measure of impairment, only 42.1% of patients have vision in this category in the worse-seeing eye. The prevalence of severe binocular visual impairment is low in this population, with only about 1 in 19 patients (5.7%) having a visual acuity of worse than 20/160 in the better-seeing eye.
Figure 4.
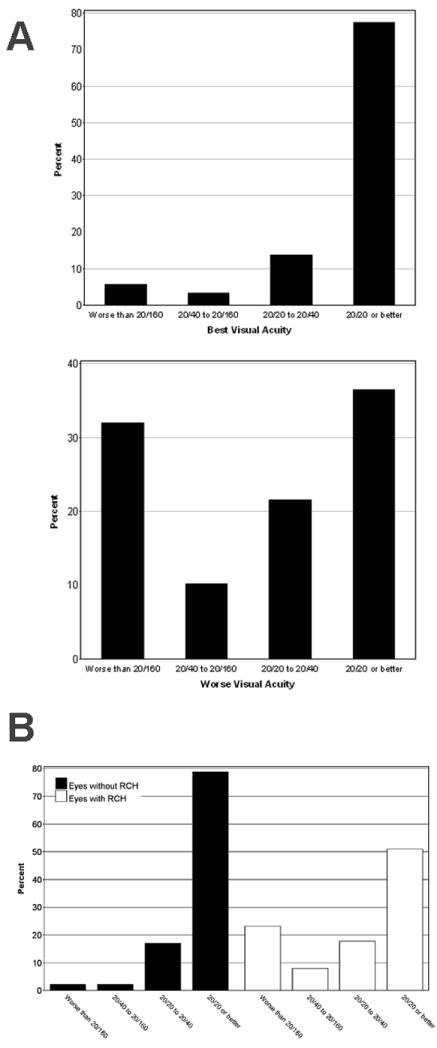
Visual acuities in all 335 patients with retinal capillary hemangioblastomas(RCHs) related to von Hippel-Lindau disease.
A. Distribution of visual acuities of 335 patients in their better-seeing eye (top panel) and worse-seeing eye (bottom panel). B. Distribution of visual acuities of all 670 eyes in 335 patients according to eyes without angiomatous involvement (dark bars, left, n = 141) and eyes with angiomatous involvement (white bars, right, n = 529).
We also examined the contribution of RCH to visual acuity by comparing eyes with at least one RCH (n=529) to unaffected eyes (n = 141) in this population. The distributions of visual acuities for all eyes with and without any RCHs are shown in Figure 4B, illustrating the effect of RCHs in increasing the proportion of eyes with poor vision. The odds ratio for moderate vision loss (worse than 20/40) in eyes with RCHs (versus eyes without RCHs) was 9.1 (4.1–20.2, 95% confidence interval,). The odds ratio for severe vision loss (worse than 20/160) for these 2 groups was 12.8 (95% confidence interval, 4.4–37.7). These data indicate that while the prevalence of severe vision loss in affected VHL patients may be low, on the level of the individual eye, the presence of at least one RCH markedly increases the risk of visual impairment. Only 6 out of 141 eyes without RCHs had visual acuities worse than 20/40 in our population; these decreases were due to intracranial or retrobulbar tumors secondary to VHL disease (n = 2), amblyopia (n =1), epiretinal membrane (n = 1), ocular histoplasmosis (n = 1), and senile cataract (n = 1).
The analysis of relationship between age and visual acuity showed a significant decrease in visual acuity with increasing age (highest vs. lowest age quartile) in both the better-seeing eye (p=0.041) and the worse-seeing eye (p=0.012). When comparing visual acuity between the lowest and highest age quartiles, analysis of variance estimated a difference of 6.1 letters (95% CL=0.3–11.9) in the better-seeing eye and of 14.7 (95% CL=3.3–26.1) in the worse-seeing eye.
The effects of other demographical categories on visual function showed no effect of gender on visual acuity in the worse-seeing eye (p=0.552), while the visual acuity in the better-seeing eye was slightly higher in males than females (84.18 ±15.14 vs. 79.48±21.76 letters, p=0.031). The relationship with race was not calculated owing to the small fraction of non-white patients.
The analyses of the relationship between visual acuity and location of RCH (n=458) are shown in Figure 5A. In general, the visual acuity of an affected eye varied with the location of the RCHs and the mean visual acuities for eyes with RCHs at the (1) periphery only, (2) juxtapapillary only, and (3) in both peripheral and juxtapapillary locations decreased in that order and were respectively 75.93±24.06 letters, 67.11±30.15 letters, and 58.82±34.56 letters. The difference between (1) and (3) was statistically significant at p=0.004 but the difference between (1) and (2) was not. The presence of juxtapapillary RCHs in addition to peripheral RCHs also significantly increased the risk of severe vision loss; the odds ratio of having poor (worse than 20/160) versus good (better than 20/20) vision is significantly greater in the presence of juxtapapillary lesions (Table 3).
Figure 5.

Visual acuities of eyes with retinal capillary hemangioblastomas (RCHs) related to von Hippel-Lindau disease. according to phenotype subcategories. A, Distribution of visual acuities of affected eyes according to the location of documented RCHs. Location categories were: RCH in the peripheral location only (n = 388 eyes), in the juxtapapillary location only (n = 37 eyes), and in both juxtapapillary and peripheral locations (n = 33). B, Distribution of visual acuities according to number of peripheral RCHs in eyes with documented RCHs (n = 421 eyes). Categories were: 1-2 RCHs/per eye (n = 262 eyes), 3-4 RCHs/per eye (n = 84 eyes), >5 RCHs/per eye (n = 75). C, Distribution of visual acuities according to whether RCHs extended to less (n = 316) or greater (n = 105) than one quadrant of peripheral retinal involvement.
Table 3. Impact of Features of Ocular Angiomatosis on Visual Acuity in Eyes with Retinal Capillary Hemangiomas that can be Individually Characterized.
Odds Ratio of Eyes having a visual acuity < 20/160 (letter score 0 – 38) versus a visual acuity of >20/20 (letter score ≥84)
| Odds ratio (95% Wald Confidence Limits) | |
|---|---|
| Fundus location of retinal capillary hemangiomas | |
| Juxtapapillary tumor only versus peripheral tumor/s only | 2.88 (1.12 – 7.41) |
| Juxtapapillary + peripheral tumors versus peripheral tumor/s only | 6.38 (2.12 – 19.18) |
| Number of retinal capillary hemangiomas | |
| ≥ 3 peripheral tumors versus < 3 peripheral tumors | 4.18 (2.15 – 8.12) |
| ≥ 5 peripheral tumors versus < 5 peripheral tumors | 8.27 (3.78 – 18.10) |
| Extent of peripheral retinal involvement | |
| ≥ 1 quadrant of involvement versus < 1 quadrant of involvement | 26.58 (11.93 – 59.25) |
In affected eyes with peripheral RCHs only (n=421), the visual acuity decreased with increasing number of peripheral RCHs (p<0.0001) (Figure 5B) and with an increasing extent of peripheral involvement (p<0.0001). (Figure 5C). The odds ratio of having poor (worse than 20/160) versus good (better than 20/20) vision is also significantly associated with peripheral RCH number and peripheral involvement (p<0.001 in all comparisons made) (Table 3).
DISCUSSION
The present study provides a comprehensive characterization of the demographics, ocular phenotype, and visual function of ocular VHL disease in one of the the largest groups of patients in the literature. The patients referred had been evaluated systemically in a single center and almost all patients (98.1%) clinically diagnosed with VHL disease could also be assigned a germline mutation responsible for VHL disease 7. The comprehensive clinical diagnosis of VHL disease, supported by genotype analysis excludes with certainty patients with sporadic RCHs unrelated to VHL. This is significant as non-VHL-associated RCHs may have a distinct etiology and a separate demographic, phenotype, and visual prognosis profile 9. The population of VHL patients in the present study has been referred as a result of systemic VHL disease, rather than presenting because of ocular symptoms, and is thus more likely to provide an general profile of ocular involvement across all patients with VHL disease.
In our population, RCHs of VHL were found in all racial groups, both genders, and all age groups (1st through 9th decade of life). There was a wide range in the phenotypes exhibited by affected patients. On the whole, RCH occurred bilaterally in about 6 out of 10 patients, about 1 in 5 patients had at least one eye with severe complications resulting in either enucleation or structural disruption of the globe, and only about 1 in 18 patients had vision approximating legal blindness (worse than 20/160 in the better-seeing eye). We did not find any effect of gender on either the laterality or severity of the ocular phenotype, similar to the findings of a previous study 10.
Among the main demographic variables, only age was found to be associated with ocular phenotype but only in some respects. The biology of VHL disease has suggested that the accumulation of the tumor phenotype may be age-dependent. All somatic cells in patients with inherited VHL disease bear a single mutated copy of the VHL gene. Based on the two-hit model for tumorigenesis proposed by Knudson 11, it is postulated that a RCH arises when a susceptible cell in the retina acquires a second mutation in the remaining VHL gene, resulting in a loss of VHL tumor suppression function and subsequently the transformation of the cell into the tumor phenotype. This potential for cells to acquire a second “hit” and become transformed is often thought to be life-long in nature; as a result, new ocular tumors arise separately in susceptible tissues over time and increase in number with age. However, in our cross-sectional study, we did not find a significant association between increasing age and either the number of peripheral RCHs per eye, the extent of peripheral retina involved, or the laterality of ocular angiomatosis. This suggests that the susceptibility for the formation of new RCHs in the retina may not constant over time and may be determined early in life. Webster and colleagues 10 also failed to find an age-dependence to tumor number. Dollfus and colleagues 12 documented increases in RCH number and the rate of bilateral involvement from the start to the end of their study, but a relationship with age per se was not statistically measured.
Visual acuity of an individual eye, however, was found to generally decrease with increasing age in our study. Likewise, the prevalence of severe ocular involvement that results in enucleation or structural disruption also significantly increased with age. These findings suggest that visual morbidity and structural disruptions due to ocular angiomatosis may be secondary to the effect of the development of secondary complications as part of the natural history of RCHs 2 (combined with the additional aging effects that occur independently of VHL disease, eg. cataracts, epiretinal membrane) rather than a direct result of increasing RCH number.
We found in our population that ocular VHL is relatively uncommon as a cause of poor visual acuity. This can be attributed to the finding that more than 42% of patients have only one eye affected, and the severity of involvement is highly variable. Nevertheless, the impact of RCH on the visual function of the individual eye is real and significant. Our statistical analyses comparing eyes with RCHs with those lacking RCHs in the same population show a markedly increased risk of severe vision loss (vision <20/160) and structural disruption for eyes containing RCHs. Although vision loss may result from manifestations of VHL disease that are independent of the effects of RCHs (such as retinal neovascularization and resulting epiretinal membrane formation or intracranial and retrobulbar hemangioblastomas), these are relatively infrequent, causing poor vision (<20/160) in only 3 out of 141 eyes that lack RCHs in our population.
Among these eyes with RCHs, the nature of angiomatosis in the form of RCH distribution, number of peripheral tumors, and extent of peripheral retinal involvement were all found to affect visual function as measured by visual acuity. We found that the presence of juxtapapillary RCHs had a statistically significant negative impact on visual function than eyes with only peripheral RCHs. This may be attributed to the closer proximity of juxtapapillary tumors to the macula, increasing the likelihood that exudation from this source impacts central vision 13. Additionally, the lack of efficacious therapy for juxtapapillary tumors may be the key reason for this resulting decrease in vision. We also found a significant relationship between decreasing visual acuity and increasing peripheral RCHs number and extent; this is likely to originate from larger exudative effects, a poorer response to treatment, and an increased rate of tractional retinal detachments from fibrovascular proliferation that are seen with increasing peripheral involvement 2,14.
Our study did not include measurements of visual field function. As some eyes with RCHs have extensive peripheral involvement, visual field compromise in the affected areas is likely to be present, either as a result of the tumor or the treatment directed to it. In addition, structurally disrupted eyes will be expected to have both poor visual acuity and compromised visual fields. An analysis of the impact of ocular VHL disease on visual field function specifically was outside the scope of this study but will be an interesting and relevant area of future study.
In summary, our findings provide a comprehensive, quantitative, and likely “population-based” profile of ocular angiomatosis in all-comers with VHL disease, regardless of visual symptoms. Ophthalmologists evaluating patients with a clinical diagnosis of VHL disease may use the results in the following ways: VHL patients with RCHs may be counseled that while their overall risk of bilateral poor visual acuity is low (1 out of 18), on the level of an affected eye, the presence of RCHs does statistically increase the risks of vision loss, structural disruption, and enucleation in affected eyes (overall, 1 in about 4 affected eyes have visual acuities < 20/160). The risk of visual loss in an eye with RCHs increases with the presence of an RCH in a juxtapapillary location, and with the number of peripheral lesions and the extent of peripheral involvement. The total number of RCHs per se does not necessarily increase with age as may be imagined but the risk of vision loss tends to increase statistically as patients get older.
Most of the patients in this study have received the necessary long-term follow-up ophthalmic evaluation and timely standard treatment with laser photocoagulation, cryotherapy, and vitrectomy, and the results in this study should be interpreted in this context. Preliminary studies with anti-angiogenic agents such as pegaptanib sodium and ranibizumab (unpublished data) have not significantly improved visual function in cases not amenable to standard therapy. While future therapies such as anti-angiogenic agents delivered systemically or in combination may have the potential to improve visual outcomes, at the present time, the results of this study are likely to accurately reflect in a quantitative way the visual prognosis and function of treated ocular VHL disease.
Acknowledgments
This research was supported in part by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research, Division of Epidemiology and Clinical Research, National Eye Institute.
References
- 1.Lonser RR, Glenn GM, Walther M, et al. von Hippel-Lindau disease. Lancet. 2003;361:2059–67. doi: 10.1016/S0140-6736(03)13643-4. [DOI] [PubMed] [Google Scholar]
- 2.Singh AD, Shields CL, Shields JA. von Hippel-Lindau disease. Surv Ophthalmol. 2001;46:117–42. doi: 10.1016/s0039-6257(01)00245-4. [DOI] [PubMed] [Google Scholar]
- 3.Chan CC, Chew EY, Shen D, et al. Expression of stem cells markers in ocular hemangioblastoma associated with von Hippel-Lindau (VHL) disease. Mol Vis. 2005;11:697–704. [PMC free article] [PubMed] [Google Scholar]
- 4.Webster AR, Maher ER, Moore AT. Clinical characteristics of ocular angiomatosis in von Hippel-Lindau disease and correlation with germline mutation. Arch Ophthalmol. 1999;117:371–8. doi: 10.1001/archopht.117.3.371. [DOI] [PubMed] [Google Scholar]
- 5.Melmon KL, Rosen SW. Lindau’s Disease. Review of the literature and study of a large kindred. Am J Med. 1964;36:595–617. doi: 10.1016/0002-9343(64)90107-x. [DOI] [PubMed] [Google Scholar]
- 6.Stolle C, Glenn G, Zbar B, et al. Improved detection of germline mutations in the von Hippel-Lindau disease tumor suppressor gene. Hum Mutat. 1998;12:417–23. doi: 10.1002/(SICI)1098-1004(1998)12:6<417::AID-HUMU8>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 7.Maher ER, Iselius L, Yates JR, et al. Von Hippel-Lindau disease: a genetic study. J Med Genet. 1991;28:443–7. doi: 10.1136/jmg.28.7.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Latif F, Tory K, Gnarra J, et al. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science. 1993;260:1317–20. doi: 10.1126/science.8493574. [DOI] [PubMed] [Google Scholar]
- 9.Singh AD, Nouri M, Shields CL, Shields JA, Smith AF. Retinal capillary hemangioma: a comparison of sporadic cases and cases associated with von Hippel-Lindau disease. Ophthalmology. 2001;108:1907–11. doi: 10.1016/s0161-6420(01)00758-8. [DOI] [PubMed] [Google Scholar]
- 10.Webster AR, Richards FM, MacRonald FE, et al. An analysis of phenotypic variation in the familial cancer syndrome von Hippel-Lindau disease: evidence for modifier effects. Am J Hum Genet. 1998;63:1025–35. doi: 10.1086/302037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Knudson AG., Jr Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci U S A. 1971;68:820–3. doi: 10.1073/pnas.68.4.820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dollfus H, Massin P, Taupin P, et al. Retinal hemangioblastoma in von Hippel-Lindau disease: a clinical and molecular study. Invest Ophthalmol Vis Sci. 2002;43:3067–74. [PubMed] [Google Scholar]
- 13.McCabe CM, Flynn HW, Jr, Shields C, et al. Juxtapapillary capillary hemangiomas. Clinical features and visual acuity outcomes. Ophthalmology. 2000;107:2240–8. doi: 10.1016/s0161-6420(00)00422-x. [DOI] [PubMed] [Google Scholar]
- 14.Annesley WH, Jr, Leonard BC, Shields J. Fifteen year review of treated cases of retinal angiomatosis. Trans Sect Ophthalmol Am Acad Ophthalmol Otolaryngol. 1977;83:446–53. [PubMed] [Google Scholar]