

Abstract
Alzheimer’s disease (AD) is the most common cause of dementia worldwide. AD is a member of a broad range of neurodegenerative diseases characterized pathologically by the conformational change of a normal protein into a pathological conformer with a high β-sheet content that renders it neurotoxic. In the case of AD the normal soluble amyloid β (sAβ) peptide is converted into oligomeric/fibrillar Aβ. The oligomeric forms of Aβ have been hypothesized to be the most toxic, while fibrillar Aβ becomes deposited as amyloid plaques and congophilic angiopathy, which both serve as neuropathological markers of the disease. In addition the accumulation of abnormally phosphorylated tau as soluble toxic oligomers and as neurofibrillary tangles is a critical part of the pathology. Numerous therapeutic interventions are under investigation to prevent and treat AD. Among the most exciting and advanced of these approaches is vaccination. Immunomodulation is being tried for a range of neurodegenerative disorders with great success being reported in most model animal trials; however, the much more limited human data has shown a more modest clinical success so far, with encephalitis occurring in a minority of patients treated with active immunization. The immunomodulatory approaches for neurodegenerative diseases are targeting a self-protein, albeit in an abnormal conformation; hence, effective enhanced clearance of the disease associated conformer has to be balanced with the potential risk to stimulate excessive toxic inflammation within the CNS. The design of future immunomodulatory approaches that are more focused is dependent on addressing a number of questions such as: When is the best time to start immunization? What are the most appropriate targets for vaccination? Is amyloid central to the pathogenesis of AD or is it critical to target tau related pathology also? In this review we discuss the past experience of vaccination for AD and the development of possible future directions that target both amyloid β and tau related pathologies.
Keywords: amyloid β, tau, vaccination, immunomodulation, Alzheimer’s disease, transgenic mice
Alzheimer’s disease is the most common cause of dementia worldwide, affecting approximately 37 million people currently. In the USA, AD is the 6th leading cause of death, with an estimated 5.3 million Americans having AD. By 2050, according to some estimates, 1:85 persons worldwide will be affected by AD (1). Currently available treatments for AD provide largely symptomatic relief with only minor effects on the course of the disease. There is an urgent need for better therapeutic interventions. Besides immunomodulation, numerous other approaches are being studied, which include anti-Aβ aggregation agents, secretase inhibitors/modulators blocking Aβ production, tau aggregation blockers, agents targeting mitochondria, stem cell therapies and various neuroprotective strategies (2). Perhaps the greatest hope for an intervention that shall significantly impact disease progression in the near future comes from the vaccination approaches (3, 4). Certainly in AD Tg mouse models Aβ directed immunization has been spectacularly successful using a wide variety of methods. However significant unanswered questions remain for the current and future human trials as to what is the best design of a vaccine, what is the best target and when should therapy start? A key issue which needs to be addressed is the targeting of both amyloid β (Aβ) and tau related pathology.
PATHOGENESIS OF FAMILIAL AND SPORADIC ALZHEIMER’S DISEASE
The pathological hallmarks of AD are the accumulation of Aβ as neuritic plaques and congophilic angiopathy, as well as accumulation of abnormally phosphorylated tau in the form of neurofibrillary tangles (NFTs). Missense mutations in APP or in the presenilin genes PRES 1 and 2 can cause early onset, familial forms of AD (FAD) affecting <4% of AD patients. The most common form of AD is sporadic and late-onset. The dominant theory for the causation of AD has been the amyloid cascade hypothesis (5, 6). This theory currently suggests that accumulation of Aβ peptides particularly in a highly toxic oligomeric form is the primary pathogenic driver, that downstream leads to tau hyperphosphorylation, NFT formation and ultimately to synaptic and neuronal loss. Extensive evidence supports this hypothesis in FAD patients and in models of FAD: 1) Inherited forms of AD linked with mutations in the APP gene or in the PRES1 or 2 genes are associated with changes in APP processing that favor over production of sAβ or production of more aggregation prone forms of sAβ such as Aβ1-42 (7). 2) Down’s syndrome, where there is an extra copy of the APP gene due to trisomy 21, is associated with AD related pathology at a very early age (8). 3) In transgenic and other models of co-expressed amyloid β and tau, amyloid β oligomer formation precedes and accentuates tau related pathology, consistent with the hypothesis that NFT formation is downstream from Aβ aggregation (9-11). 4) In transgenic mouse models of mutant APP over-expression (where there is no tau pathology) therapeutic prevention and/or removal of Aβ is associated with cognitive benefits in experimental mice (12-15). Importantly, in transgenic mouse models of both mutant APP and tau over-expression (with both amyloid and tau related pathology) prevention of Aβ pathology leads to both amelioration of cognitive deficits and tau related pathology (16-18). However, evidence proving that Aβ is central in the common late-onset sporadic form of AD is more limited: 1) A correlation has been shown between biochemically extracted Aβ peptides species from sporadic AD brains with cognitive decline (19). 2) Isolated Aβ peptide dimers/oligomers from sporadic AD brains have been documented to impair synaptic structure and function (20). 3) Aβ extracted from sporadic AD patients has been shown to induce amyloid deposits when injected into transgenic mice (21). A significant problem for the amyloid cascade hypothesis comes from the autopsy data from the initial human active vaccination trial, which is further discussed below. Post-mortem analysis was available from nine subjects in the active immunization arm (22). All these individuals showed some degree of plaque removal and reduced Aβ load compared to comparable non-immunized controls. Despite this, there were no differences between placebo and active immunization groups in terms of long-term survival outcome, time to severe dementia and in outcome measures such as ADAS-Cog, MMSE or DAD. This may be related to immunization having begun too late in the disease process; alternatively, one can use this data to suggest that the amyloid cascade hypothesis is an oversimplification. A number of investigators have suggested alternative theories, whereby accumulation of Aβ and tau hyperphosphorylation are dual pathways both downstream from a common upstream pathogenic deficit (which remains to be identified) (23-25). In such a scenario it is essential for immunotherapy to address both of these pathologies to be highly effective.
Hence in this review we will summarize the preclinical and clinical data for both Aβ and phosphorylated tau reduction immunotherapeutic approaches.
PAST ACTIVE IMMUNIZATION HUMAN EXPERIENCE TARGETING AB
Initial data supporting immunotherapy for AD showed that anti-Aβ antibodies could inhibit Aβ peptide fibrillization/oligomerizaton and prevent cell culture based neurotoxicity (26, 27). This lead to vaccination of AD Tg mice with Aβ1-42 or Aβ homologous peptides co-injected with Freund’s adjuvant which demonstrated striking reductions in Aβ deposition and as a consequence elimination of behavioral impairments (12-15, 28, 29). Similar effects on Aβ load and behavior have been demonstrated in AD Tg mice by peripheral injections of anti-Aβ monoclonal antibodies indicating that the therapeutic effect of the vaccine is based primarily on eliciting a humoral response (30, 31). In the initial preclinical studies no toxicity was evident in the treated mice; however, some investigators suggested that use of non-fibrillogenic, non-toxic Aβ homologous peptides along with approaches that stimulate primarily humoral, Th-2 immunity, in contrast to a primary Th-1 cell mediated response might reduce potential toxicity (32-34). The dramatic biological effect of vaccination in preclinical testing encouraged Elan/Wyeth in April 2000 to launch a randomized, multiple-dose, dose-escalation, double-blind Phase I clinical trial with a vaccine designated as AN1792, which contained pre-aggregated Aβ1-42 and QS21 as an adjuvant. This type of vaccine design was aimed to induce a strong cell mediated immune response, since QS21 is known to be a strong inducer of Th-1 lymphocytes (35). The initial trial was conducted in the UK and involved 80 patients with mild to moderate AD (36). This trial was designed to assess the antigenicity and the toxicity of multiple dose immunization with the full length Aβ1-42 peptide with the QS21. 53% of patients developed an anti-Aβ humoral response. During the later stages of the phase I trial, the emulsifier polysorbate 80 was added causing a greater shift from a Th2 biased response to a proinflammatory Th1 response (37). In the subsequent phase IIa trial, begun in October 2001, 372 patients were enrolled with 300 receiving the aggregated Aβ1-42 (AN1792) with QS21 in the polysorbate 80 formulation. This trial was prematurely terminated in January 2002 when 6% of vaccinated patients manifested symptoms of acute meningoencephalitis (18 out of 298 subjects) (35, 38, 39). Autopsies performed on a limited number of trial patients suggested that striking Aβ clearance of parenchymal plaques had occurred, similar to what had been reported in the animal studies, confirming the validity of this approach for amyloid clearance in humans(39-44). In these cases extensive areas of cerebral cortex were devoid of plaques, with residual plaques having a “moth-eaten” appearance or persisting as “naked” dense cores. This amyloid clearance in most cases was in association with microglia that showed Aβ immunoreactivity, suggesting phagocytosis. Additional striking features were the persistence of amyloid in cerebral vessels, as well as unaltered tau immunoreactive NFTs and neuropil threads in regions of cerebral cortex where plaque clearing had apparently occurred, compared to regions without clearing (42-44). Hence, this initial vaccination approach did not address vascular amyloid or NFT related pathology. Some cases also showed a deleterious T-cell reaction surrounding some cerebral vessels, suggestive of an excessive Th-1 immune response. It appeared that the immune reaction triggered by AN1792 was a double-edge sword, where the benefits of a humoral response against Aβ were overshadowed in some individuals by a detrimental T cell mediated inflammatory response (39, 45). The likely involvement of an excess cell mediated response in mediating toxicity was supported by analysis of peripheral blood mononuclear cells from trial patients, which were stimulated in vitro with the Aβ peptide, followed by quantification of cytokine secretion by enzyme-linked immunosorbent spot assay (37). The cells of most responder trial patients mounted IL-2 and IFN-γ positive responses indicative of a Class II (CD4+) Th-1 type response (37). Not all patients who received AN1792 responded with antibody production. The majority mounted a humoral response and showed a modest but statistically significant cognitive benefit demonstrated as an improvement on some cognitive testing scales comparing to baseline and a slowed rate of disease progression comparing to the patients who did not form antibodies (36, 46). The follow-up data from the Zurich cohort, who are a subset of the Elan/Wyeth trial (46, 47), indicated that the vaccination approach may be beneficial for human AD patients. In agreement with the findings in the Zurich cohort, immune responders with high antibody titers in the multi-center cohort scored significantly better in composite scores of memory functions as compared to low- and non-responders or to the placebo group of patients (37). However, it is striking that despite the apparent success in amyloid clearance indicated by the autopsy data, the clinical cognitive benefits were very modest when the active vaccination group was compared to the placebo group (48). No difference between the antibody responders and the placebo group was found on the ADAS-Cog, Disability Assessment for Dementia, Clinical Dementia Rating scale, MMSE or on the Clinical Global Impression of Change. It was only on a nine-item composite NTB that antibody responders had a slight benefit compared to the placebo group. This data can be used to suggest that vaccination in this cohort was started too late; hence, tau related pathology was unaffected by vaccination and thus the cognitive benefits were small. Alternatively it can be suggested that the amyloid cascade hypothesis must be an oversimplification of the pathogenesis of sporadic AD. The latter view is supported by the follow up study of the 80 patients in the initial phase I AN1782 trial, of whom 8 came to autopsy (22). This study showed that despite evidence of very significant amyloid plaque removal in 6 out of the 8 autopsy subjects, which correlated with the anti-Aβ titer, in the overall group there was no evidence of improved survival or an improvement in the time to severe dementia (22).
PAST PASSIVE IMMUNIZATION EXPERIENCE FOR AD
Passive immunization consists of an injection of pre-prepared antibodies to patients, as opposed to active immunization where the immune system is stimulated to produce its own antibodies. Passive transfer of exogenous monoclonal anti-Aβ antibodies appears to be the easiest way to fulfill the goal of providing anti-Aβ antibodies without risk of uncontrolled Th-1 mediated autoimmunity. AD Tg model mice treated this way had a significantly reduced Aβ level and demonstrated cognitive benefit (30, 31). Potential problems with passive immunization include the need for repeated injections in a chronic disease, high cost, proper selection of antigen targets, blood-brain barrier penetration, the risk of hemorrhages and the development of an immune response to the injected antibodies. Several passive immunization trials are underway with the most advanced being the Phase III Bapineuzumab trial begun in Dec 2007 (4). The Phase II trial using this anti-Aβ monoclonal antibody was a randomized, double-blind, placebo controlled trial testing 3 doses in 240 participants. In each of the escalating doses of the antibody, approximately 32 subjects received active agent and 28 placebos. Although the study did not attain statistical significance on the primary efficacy endpoint in the whole study population, in the sub-group of non-apoE4 carriers clinically significant benefits were documented using a number of scales including the Mini Mental State Examination (MMSE) and the Alzheimer’s Disease Assessment Scale Battery, over the 18 month trial period. In addition among non-apoE4 carriers, evaluation of the MRI results showed less loss of brain volume in treated versus control patients. However, it was reported that some patients in the treatment group developed vasogenic edema, a significant adverse reaction. The Phase III trial is targeting to recruit 800 patients and run until December 2010.
Using a somewhat similar approach, IVIg is currently in clinical trial for AD, with the rationale being that IVIg contains some anti-Aβ antibodies. In a pilot, open label study in 8 mild AD patients IVIg was infused over 6 months, discontinued and resumed for another 9 months (49). Following each infusion the plasma Aβ levels increased transiently with CSF Aβ being decreased after 6 months. The MMSE increased an average of 2.5 after 6 months and returned to baseline after washout and remained stable with the subsequent IVIg infusions. These promising initial findings clearly need to be repeated in a larger cohort. The attraction of IVIg use is that there is extensive experience using IVIg safely for multiple neurological disorders; however, it is a very expensive treatment and the percentage of anti-Aβ antibodies in IVIg is extremely low so this is not likely to be neither very specific nor highly effective form of treatment.
A particular concern in association with passive immunization is cerebral microhemorrhage. The mechanism of this hemorrhage is thought to be related to Aβ deposition in the form of congophilic amyloid angiopathy (CAA) that causes degeneration of smooth muscle cells and weakening of the blood vessel wall. A number of reports have shown an increase in microhemorrhages in different AD mouse models following passive intra-peritoneal immunization with different monoclonal antibodies with high affinity for Aβ plaques and CAA (50-52). Microhemorrhages following active immunization in animal models have also been reported but only in two studies, hence this appears to be less of a problem with this approach (53, 54). In transgenic mouse models, Aβ antibodies can both prevent the deposition of vascular amyloid, and remove it thus contribute to vascular repair. On the other hand, the autopsies from the AN1792 trial indicated no clearance of vascular amyloid and in one of these cases numerous cortical bleeds were found, which are typically rare in AD patients, (41). This is an important issue as CAA is present in virtually all AD cases, with approximately 20% of AD patients having “severe” CAA (55). Furthermore CAA is present in about 33% of cognitively normal elderly, control populations (56). The need for vascular repair and regeneration during Aβ immunotherapy represents another argument for early treatment as well as an argument favoring subtle clearance over a longer time period.
PHOSPHORYLATED TAU AS AN IMMUNE TARGET
Neurofibrillary tangles (NFTs) are a major pathologic hallmark of AD. NFTs are intraneuronal inclusion bodies that consist of an accumulation of paired helical filaments (PHFs), which biochemically are mainly composed of abnormally phosphorylated tau. Recently there is increasing focus on phosphorylated tau as an immunotherapeutic target (57-59). In the CNS, human tau is expressed in 6 isoforms arising from alternative mRNA splicing from a single gene on chromosome 17q21, containing 16 Exons (see Figure 1) (60, 61). The size range of the six isoforms is between 352 and 441 amino acids, which differ by the absence or presence of 29 (Exon 2) or 58 (Exon 2 + Exon 3) amino acids inserts in the amino-terminal. The carboxy-terminal half of tau contain three or four semi-homologous repeat of 31 or 32 amino acids, encoded by Exon 10. The repeats (3R, 4R) correspond to the microtubule binding region of protein tau. (see Figure1). Stabilization of microtubules by tau is essential for the maintenance of neuronal cell morphology and transport of organelles. In addition, tau has other roles such as interactions with kinesin -1 and the complex dynactin/dynein (62, 63). Tau plays also a crucial role in neuronal cell architecture by interacting with plasma membrane or cytoskeleton proteins such as actin, spectrin and neurofilament proteins. Several mutations have been detected in the tau gene in FTDP-17 and other tauopathies, however none have been linked to AD (64). Most of these mutations affect the binding of tau to microtubules or enhance the aggregation of tau into fibrils. Other intronic mutations that affect the splicing of Exon 10 induce an increase of isoforms with 4 repeats. In AD, tau is hyperphosphorylated at all phosphorylated sites with 9 phosphates per molecule in comparison to normal brain tau that has 2 to 3 phosphorylated residues (65). Other studies suggested that changes in tau splice forms are related to neurodegeneration. In some animal models expressing mutated Tau there is an increase of 4R versus 3R tau (66). The functional significance of a shift in the 3Rtau/4Rtau ratio remains unclear, but four -repeat tau binds microtubules with a higher affinity than three- repeat tau (67).
Figure 1.
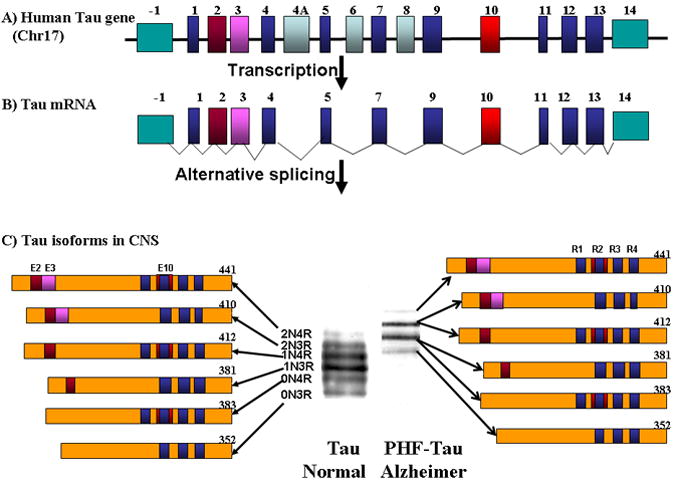
Shows a schematic representation of the human tau gene which is located on chromosome 17q21, and spans more than 130 kb. This gene is composed of 16 Exons. (A). Exons 1 and 14 are transcribed but not translated (turquoise color). The Exons 4A, 6 and 8 are not transcribed in human (light blue/charcoal color) (B). In the human brain, 6 tau isoforms ranging from 352 to 441 amino acids are generated by alternative splicing of Exons 2, 3, and 10 (shown in brown/red, pink and red respectively) from a single gene. Exons 1, 4, 5, 7, 9, 11, 12 and 13 (blue color) are included in all isoforms. Exon 3 is always included with Exon 2. The microtubule binding domains are indicated by R1, R2, R3 and R4, which correspond respectively to Exon 9, 10, 11 and 12, respectively. (C). Extraction of tau proteins and PHF-Tau from normal and Alzheimer brain respectively, shows by immunoblotting six bands between 45-68 kDa which correspond to different tau isoform in normal brain, while in PHF-tau 4 bands are detected between 60-74 KDa which corresponds to the aggregation of 6 hyperphosphorylated tau isoforms in the AD brain.
Normal tau and PHF tau differ in molecular weight and banding pattern as seen in Figure 1. Normal tau has 6 bands between 45 and 68 kDa, while PHF-Tau has 4 bands between 60 and 74 kDa (see Figure 1) (68, 69). The diversity of tau isoforms is related to various post-translational modifications such as phosphorylation, glycosylation, glycation, ubiquitination, nitration (70). The splicing regulation of the tau gene and the relative expression of isoforms is not significantly changed in sporadic AD (Figure 2) (71). Tau has multiple phosphorylation sites that were characterized using phospho-tau dependant antibodies (see Figure 3). 71 out of the 85 potential phosphorylated sites have been shown to be phosphorylated in physiological or pathological conditions (72, 73). More than 20 protein kinases have been implicated in the phosphorylation of tau proteins, with glycogen synthase kinase-3β (GSK-3β) and cyclin-independent kinase (cdk5) thought to play the most important role in phosphorylation under pathological condition (72-75).
Figure 2.
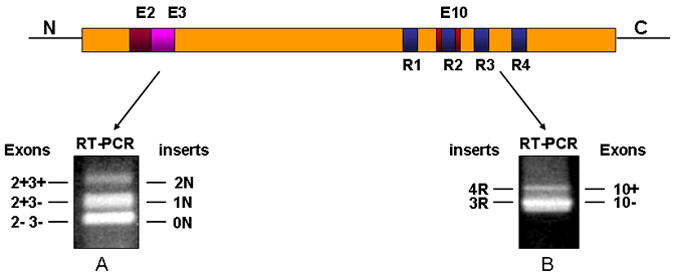
Shows the electrophoresis of RT-PCR amplification products of the 5’ domain of tau mRNAs (A), of the 3’ domain of tau mRNAs (B). Extraction of RNA was performed in from the cerebellar cortex of a sporadic Alzheimer’s disease patient. The expression of mRNA by RT-PCR shows different isoforms of human tau detected in the N-terminal (0N, 1N, 2N) and in the C-terminal (3R, 4R). Symbol (+) indicates with exon, while (-) indicates without exon.
Figure 3.
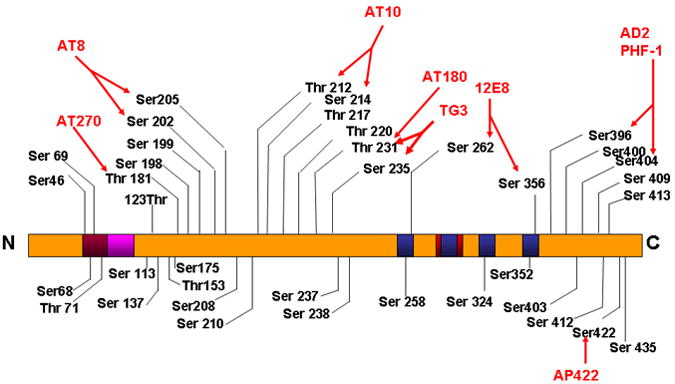
In the human Alzheimer brain, more than 40 phosphorylation sites on tau have been identified and localized in the proline-rich domain and in the C-terminal region. Phosphorylated sites are identified with 8 phospho-tau specific antibodies as indicated in figure.1C, with a red color. It has been suggested that the phosphorylation at Ser 262/356 is responsible for the detachment of tau from microtubules.
Several transgenic mice models that express human tau with FTDP-17 mutation have been produced (see Table 1). Some of these mice display neurofibrillary tangles, neuronal death and behavioral deficits (76-85) except a Tg mice model that expresses a mutated (N279K) tau that shows behavioral deficits without formation of NFTs or neuronal loss (86). In these models there is disruption of axon transport due to the tau expression that induces synaptic and neuronal loss. Another Tg tau mice model was developed expressing the mutated P301S tau which shows synaptic loss that precedes tangles formation (84). The distribution of neurofibrillary tangles in most of these tauopathies models are in contrast to Alzheimer’s disease, since NFT is localized in different brain regions such as the brain stem, spinal cord or in fronto-temporal cortex instead of the entorhinal region, hippocampus and neocortex as observed in AD (87). In order to generate a more ideal model for AD, other researchers have used a single wild-type human tau to generate a transgenic model; however, most of these models did not develop NFT, with the except of two models: One expressing ON3R wild-type tau with few NFTs in aged animals (80) and another with abundant NFT that expresses all 6 human tau isoforms on a knockout background for murine tau (88, 89). The absence of tangles in mice that expressed a single wild-type human tau is likely due to the endogenous tau inhibiting the formation of NFT-like pathology.
Table1.
Shows the different transgenic models used to study tau related pathology which express wild-type (WT) tau or mutated tau that has been identified in various FTDP-17 pedigrees.
| Tau Isoform | Tau Mutation | Presenilin cDNA | APP cDNA | Promotor | NFT | Citation |
|---|---|---|---|---|---|---|
| 2N4R | WT | — | — | Thy-1 | NO | (150) |
| 2N4R | WT | — | — | Thy-1.2 | NO | (151) |
| 2N4R | WT | — | — | Thy-1.2 | NO | (152) |
| 0N3R | WT | — | — | HMG-CoAR | NO | (153) |
| 0N3R | WT | — | — | PrP | NO | (154) |
| 3R | WT | — | — | Mouse tubulin Tα1 | NO | (155) |
| 0N3R | WT | PS1 | — | HMG-CoAR | NO | (156) |
| M146L | ||||||
| 0N3R | WT | PS1 | APP 751(SL) | HMG-CoAR and Thy-1 | NO | (157) |
| M146L | ||||||
| 2N4R | WT/KoKI | — | — | Thy-1 | No | (158) |
| 6 isoforms human | WT | — | — | Yes | (88) | |
| 0N4R | P301L | — | — | PrP | Yes | (159) |
| 0N4R | P301L | — | APPsw | PrP | Yes | (79) |
| 0N4R | P301S | — | Thy1.2 | Yes | (160) | |
| 0N4R | P301L | PS1 | APPsw | Thy1.2 | Yes | (10) |
| M146V | ||||||
| 2N4R | P301L | — | — | Thy-1.2 | Yes | (9) |
| 2N4R | G272V | — | — | PrP | Yes | (161) |
| 2N4R | M337V | — | — | PDGF | Yes | (81) |
| 2N4R | R406W | — | — | CaMKII | Yes | (162) |
| 2N4R | R406W | — | — | Thy-1 | Yes | (163) |
| P301L | ||||||
| G272V | ||||||
| 2N4R | P301L | — | — | Thy-1 | Yes | (158) |
| 1N2R | G272V | Thy1.2 | Yes | (164) | ||
| P301S | ||||||
Recently, it has been shown that active immunization of Tg mice P301L with a phospho-tau peptide (containing the phosphorylated PHF-1 epitopes Ser 396, Ser 404) for 2 to 5 months could prevent tau related pathology (90, 91).
These particular phosphorylation epitopes were chosen since these sites have been shown to increase the fibrillogenic nature of tau and contribute into paired helical filaments formation (92, 93). Histological and biochemical analyses showed a reduction of aggregated tau in the brain and improve performance on motor tasks(90). This study clearly documented that it is possible to reduce tau related pathology with active immunization.
At first examination it is difficult to understand how an antibody response to a protein which is accumulating intra-cellularly can have beneficial effects. However, such an outcome is supported by a study of immunization in a Parkinson’s disease transgenic mouse model with α-synuclein showing a reduction of intracellular α-synuclein aggregates (94). An additional study has shown that antibodies against Aβ can be internalized in AD neuronal culture models of Aβ accumulation and clear intra-neuronal Aβ aggregates via the endosomal–lysosomal pathway (95). Furthermore recent evidence has shown that extracellular tau aggregates can be internalized and promote the fibrillization of intracellular full length tau in a tissue culture model (96) and that injection of fibrillar tau brain extract into the brains of transgenic wild-type expressing mice can induce the formation of human tau into filaments, as well as the spread of pathology from the site of injection into neighboring brain regions (97). This type of “infectivity” of abnormal protein conformation from outside the cell has also been demonstrated for polyglutamine aggregates (98) and is well characterized in prion disease (99, 100). Hence if the spread of PHF pathology in AD occurs via a prion like mechanism, anti-phosphorylated tau antibodies would not need to enter cells in order to be effective.
QUESTIONS TO ADDRESS FOR A NEW GENERATION OF AD VACCINES
An initial question which needs to be addressed is when to begin a vaccination protocol. Extensive neuropathological data has established that by the time the earliest clinical signs of AD emerge, Aβ deposition may be close to reaching its peak and that NFT formation and neuronal loss are substantial but have not yet reached peak levels (101, 102). This would suggest that amyloid directed therapy would need to begin very early, perhaps even before the mild cognitive impairment stage, in order to have a maximal effect.
This is consistent with the albeit limited autopsy data from the initial AN1792 study that showed that despite evidence of very significant amyloid plaque removal in 6 out of the 8 autopsy subjects, which correlated with the anti-Aβ titer, in the overall group there was no evidence of improved survival or an improvement in the time to severe dementia (22). Hence there is a need for identification of markers predicting the conversion from normal aging to very mild dementia/mild cognitive impairment. These include CSF biomarkers such as increased p-tau/Aβ42 ratios in cognitively normal individuals which enhance the probability of conversion to MCI (103). Early FDG-PET changes in hippocampal glucose metabolism can predict the conversion of normal cognition to pathologically verified AD (104). Studies in AD Tg models suggest that paramagnetic amyloid binding ligands utilizing magnetic resonance imaging have potential for early amyloid detection and following treatment effects (105, 106). However currently, direct imaging of amyloid depositions with agents such as PIB, using PET imaging, is the most promising method for identification of early amyloid deposits and identifying patients who will likely convert to MCI from normal aging and MCI to early AD (107, 108). An alternative approach is to immunize targeting both Aβ deposition and tau related pathology.
Such an approach has a higher probability of having a more clear effect on the clinical course, even if started when clinical symptoms are evident. Furthermore if, as discussed above, tau pathology is not downstream from amyloid deposit but represents a parallel pathology related to a common upstream cause, it will be essential to target tau related pathology regardless of how early vaccination treatment is initiated.
Another significant issue which needs to be addressed in future studies is the development of better models for pre-clinical testing of vaccination approaches. There are many shortcomings to current Tg models of AD pathology. These include the fact that Tg amyloid deposits typically lack the extensive post-translational modifications of AD amyloid and thus are much more soluble, presumably allowing them to be cleared more easily (109). The rodent immune system is quite different from the human immune system, leading to significant differences in the toxic responses to amyloid deposition (110). Most current tau Tg models reflect FTDP related pathology in contrast to AD tau pathology, as discussed above (87). Relatively few of the vaccination approaches being developed have been tested in non-human primates or other non-transgenic models of AD which may provide more accurate models of the type of immune response that might be elicited in aged humans as well as better reflecting the combination of true human Aβ and tau related pathology(111-113). These models include the rhesus monkey, the vervet monkey, mouse lemurs and aged beagles (113-118). It is striking that in a recent 22 month active immunization trial in aged beagles, despite an ~80% reduction in cortical Aβ immunoreactivity, little cognitive improvement on multiple measures of learning and memory could be detected (119). However, improvements in some executive functions were found, mirroring the modest improvements seen only in the z-score of the Neurological Test Battery among patients in the AN1782 trial. These results reinforce the need to begin immunomodulation very early in the disease progression focusing on preventing Aβ deposition rather than clearance of preexisting lesions, as well as the likely need to target tau related pathology concurrently.
Active vaccination approaches under development are aiming to avoid the excessive Th1 stimulation that resulted in encephalitis in a proportion of the AN1792 patients. Concurrently the formulation of any active vaccine also has to overcome the problem of immunosenescence in the target patient population. One promising approach taken by several investigators is to alter the sequence of the Aβ peptide immunogen in order to remove or alter the major Th1 stimulator sites in the carboxyl terminus and the middle portion of Aβ, while focusing on the major Th2 stimulator site in the amino terminus (28, 33, 120-122). These Aβ homologous peptide immunogens can be combined with various co-stimulator epitopes. An example of this approach is a combination with a synthetic, non-natural Pan HLA DR-binding epitope PADRE (122) or linkage to viral-like particles (VLP) (123-125) to induce a primarily humoral immune response. These can be further combined with other immunostimulator carriers. For example the Aβ Th2 amino terminal epitope can be combined with PADRE and macrophage derived chemokine (MDC) in a DNA epitope vaccine to drive robust Th2 responses (126). The choice of adjuvant is also an important consideration. The use of polysorbate 80, a strong Th1 stimulate adjuvant, in the AN1792 trial is one likely contributing factor to the encephalitis in a minority of patients. Use of adjuvants such as alum which drive primarily a Th2 response is preferable (29, 119). The route of immunization also plays an important role. Stimulating mucosal immunity by vaccinating nasally, via the gut or transcutaneously has been shown to drive strong Th2 responses (127-129). An alternative, non-mutually exclusive approach to enhance vaccine design is to stimulate innate immunity and enable microglia/macrophages to phagocytose amyloid deposits. Over 20 yrs ago, H.Wisniewski noted that while brain-resident macrophages were unable to phagocytose amyloid, brain-infilrating macrophages are plaque competent (130). A number of recent studies suggest that only a small percentage of plaques are associated with peripheral origin macrophages and that these are required for plaque clearance (131-133). Vaccination approaches based on this knowledge are now being developed. Stimulation of peripheral macrophages to enter the CNS and phagocytose amyloid has been achieved by stimulation of the Toll-like receptor 9 using CpG (134, 135), via blockade of the CD40/CD40L interaction (136) and by blockade of TGFβ-Smad2/3 innate signaling pathway (137). These innate immunity stimulatory approaches can be used alone or in combination with adaptive immunity stimulation. Stimulating the innate immune system has the added potential advantage that it could be effective against both Aβ and tau related pathologies.
Another important issue for future vaccination approaches is what is the best target for either active or passive immunization? Abundant evidence both in vivo and in vitro suggests that the most toxic species of Aβ are oligomers or Aβ derived diffusible ligands (ADDLs) (138, 139) with a similar line of evidence suggesting that tau oligomers are the most toxic form of phosphorylated tau (59, 84). Active vaccination or use of monoclonal antibodies that specifically target Aβ oligomers, tau oligomers or preferably both would be an ideal way to block AD related toxicity.
A small number of pre-clinical studies targeting Aβ oligomers suggest that this methodology is potentially powerful and in the need of further development (140-144). An additional important factor to consider is that emerging evidence suggests that monomeric Aβ peptides have normal physiological functions in the brain such as neuroprotection and modulating LTP (145, 146), with phosphorylated tau also having a normal role (58). Targeting only oligomeric Aβ or tau would avoid the potential of interfering with these physiological functions. A novel immunotherapeutic approach is to target the shared abnormal β-sheet conformation of amyloid proteins using conformationally specific antibodies or active immunization that favors such a conformational response (140, 141, 147). Such an approach has the advantage that both Aβ and tau related pathologies would be addressed concurrently.
CONCLUSION
Numerous therapeutic approaches are under development for AD; however, harnessing the immune system to clear both Aβ and tau related pathology is perhaps the most promising and advanced approach. Abnormal protein conformation is thought to be not only the underlying pathogenesis of AD but also of a long list of neurodegenerative conditions, such as prion disease, Parkinson’s disease and Huntington’s chorea, with immunomodulation having the potential to be a disease altering therapeutic approach for all these disorders. For example it has been shown that prion directed mucosal vaccination can prevent infection from an exogenous source (148, 149). Ultimately, directing the immune system to clear the highly toxic abnormal oligomeric conformers that characterize multiple neurodegenerative diseases has the potential to dramatically alter the course of a wide spectrum of age associated diseases.
Acknowledgments
This manuscript was supported by NIH grants AG20245 and AG15408, as well as Alzheimer’s Association grant IIRG-06-26434.
References
- 1.Brookmeyer R, Johnson E, Ziegler-Graham K, Arrighi HM. Forecasting the global burden of Alzheimer’s disease. Alz Dementia. 2007;3:186–191. doi: 10.1016/j.jalz.2007.04.381. [DOI] [PubMed] [Google Scholar]
- 2.Rafii MS, Aisen PS. Recent developments in Alzheimer’s disease therapeutics. BMC Med. 2009;7:7. doi: 10.1186/1741-7015-7-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brody DL, Holtzman DM. Active and passive immunotherapy for neurodegenerative diseass. Annual Review of Neuroscience. 2008;31:175–193. doi: 10.1146/annurev.neuro.31.060407.125529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wisniewski T, Konietzko U. Amyloid-β immunization for Alzheimer’s disease. Lancet Neurol. 2008;7:805–811. doi: 10.1016/S1474-4422(08)70170-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 6.Tanzi RE, Bertram L. Twenty years of the Alzheimer’s disease amyloid hypothesis: a genetic perspective. Cell. 2005;120:545–555. doi: 10.1016/j.cell.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 7.Hardy J. A hundred years of Alzheimer’s disease research. Neuron. 2006;52:3–13. doi: 10.1016/j.neuron.2006.09.016. [DOI] [PubMed] [Google Scholar]
- 8.Lemere CA, Blusztajn JK, Yamaguchi H, Wisniewski T, Saido TC, Selkoe DJ. Sequence of deposition of heterogeneous amyloid β-peptides and APO E in Down syndrome: implications for initial events in amyloid plaque formation. Neurobiology of Disease. 1996;3:16–32. doi: 10.1006/nbdi.1996.0003. [DOI] [PubMed] [Google Scholar]
- 9.Gotz J, Chen F, van DJ, Nitsch RM. Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Abeta 42 fibrils. Science. 2001;293:1491–1495. doi: 10.1126/science.1062097. [DOI] [PubMed] [Google Scholar]
- 10.Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 11.King ME, Kan HM, Baas PW, Erisir A, Glabe CG, Bloom GS. Tau-dependent microtubule disassembly initiated by prefibrillar beta-amyloid. J Cell Biol. 2006;175:541–546. doi: 10.1083/jcb.200605187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Liao Z, Lieberburg I, Motter R, Mutter L, Soriano F, Shopp G, Vasquez N, Vandevert C, Walker S, Wogulis M, Yednock T, Games D, Seubert P. Immunization with amyloid-β attenuates Alzheimer disease-like pathology in the PDAPP mice. Nature. 1999;400:173–177. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- 13.Sigurdsson EM, Scholtzova H, Mehta P, Frangione B, Wisniewski T. Immunization with a non-toxic/non-fibrillar amyloid-β homologous peptide reduces Alzheimer’s disease associated pathology in transgenic mice. American Journal of Pathology. 2001;159:439–447. doi: 10.1016/s0002-9440(10)61715-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morgan D, Diamond DM, Gottschall PE, Ugen KE, Dickey C, Hardy J, Duff K, Jantzen P, DiCarlo G, Wilcock D, Connor K, Hatcher J, Hope C, Gordon M, Arendash GW. Aβ peptide vaccination prevents memory loss in an animal model of Alzheimer’s disease. Nature. 2001;408:982–985. doi: 10.1038/35050116. [DOI] [PubMed] [Google Scholar]
- 15.Janus C, Pearson J, McLaurin J, Mathews PM, Jiang Y, Schmidt SD, Chishti MA, Horne P, Heslin D, French J, Mount HT, Nixon RA, Mercken M, Bergeron C, Fraser PE, George-Hyslop P, Westaway D. Aβ peptide immunization reduces behavioural impairment and plaques in a model of Alzheimer’s disease. Nature. 2000;408:979–982. doi: 10.1038/35050110. [DOI] [PubMed] [Google Scholar]
- 16.Oddo S, Caccamo A, Tran L, Lambert MP, Glabe CG, Klein WL, LaFerla FM. Temporal profile of amyloid-beta (Abeta) oligomerization in an in vivo model of Alzheimer disease. A link between Abeta and tau pathology. Journal of Biological Chemistry. 2006;281:1599–1604. doi: 10.1074/jbc.M507892200. [DOI] [PubMed] [Google Scholar]
- 17.Blurton-Jones M, LaFerla FM. Pathways by which Abeta facilitates tau pathology. Curr Alzheimer Res. 2006;3:437–448. doi: 10.2174/156720506779025242. [DOI] [PubMed] [Google Scholar]
- 18.McKee AC, Carreras I, Hossain L, Ryu H, Klein WL, Oddo S, LaFerla FM, Jenkins BG, Kowall NW, Dedeoglu A. Ibuprofen reduces Abeta, hyperphosphorylated tau and memory deficits in Alzheimer mice. Brain Research. 2008;1207:225–236. doi: 10.1016/j.brainres.2008.01.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Naslund J, Haroutunian V, Mohs R, Davis KL, Davies P, Greengard P, Buxbaum JD. Correlation between elevated levels of amyloid beta-peptide in the brain and cognitive decline. Journal of the American Medical Association. 2000;283:1571–1577. doi: 10.1001/jama.283.12.1571. [DOI] [PubMed] [Google Scholar]
- 20.Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med. 2008 doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meyer-Luehmann M, Coomaraswamy J, Bolmont T, Kaeser S, Schaefer C, Kilger E, Neuenschwander A, Abramowski D, Frey P, Jaton AL, Vigouret JM, Paganetti P, Walsh DM, Mathews PM, Ghiso J, Staufenbiel M, Walker LC, Jucker M. Exogenous induction of cerebral beta-amyloidogenesis is governed by agent and host. Science. 2006;313:1781–1784. doi: 10.1126/science.1131864. [DOI] [PubMed] [Google Scholar]
- 22.Holmes C, Boche D, Wilkinson D, Yadegarfar G, Hopkins V, Bayer A, Jones RW, Bullock R, Love S, Neal JW, Zotova E, Nicoll JAR. Long term effects of Aβ42 immunization in Alzheimer’s disease: immune response, plaque removal and clinical function. Lancet. 2008;372:216–223. doi: 10.1016/S0140-6736(08)61075-2. [DOI] [PubMed] [Google Scholar]
- 23.Small SA, Duff K. Linking Abeta and tau in late-onset Alzheimer’s disease: a dual pathway hypothesis. Neuron. 2008;60:534–542. doi: 10.1016/j.neuron.2008.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Castellani RJ, Lee HG, Zhu X, Perry G, Smith MA. Alzheimer disease pathology as a host response. J Neuropathol Exp Neurol. 2008;67:523–531. doi: 10.1097/NEN.0b013e318177eaf4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shioi J, Georgakopoulos A, Mehta P, Kouchi Z, Litterst CM, Baki L, Robakis NK. FAD mutants unable to increase neurotoxic Abeta 42 suggest that mutation effects on neurodegeneration may be independent of effects on Abeta. Journal of Neurochemistry. 2007;101:674–681. doi: 10.1111/j.1471-4159.2006.04391.x. [DOI] [PubMed] [Google Scholar]
- 26.Solomon B, Koppel R, Frankel D, Hanan-Aharon E. Disaggregation of Alzheimer β-amyloid by site-directed mAb. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:4109–4112. doi: 10.1073/pnas.94.8.4109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Solomon B. Antibody-mediated immunotherapy for Alzheimer’s disease. Curr Opin Investig Drugs. 2007;8:519–524. [PubMed] [Google Scholar]
- 28.Sigurdsson EM, Knudsen EL, Asuni A, Sage D, Goni F, Quartermain D, Frangione B, Wisniewski T. An attenuated immune response is sufficient to enhance cognition in an Alzheimer’s disease mouse model immunized with amyloid-β derivatives. Journal of Neuroscience. 2004;24:6277–6282. doi: 10.1523/JNEUROSCI.1344-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Asuni A, Boutajangout A, Scholtzova H, Knudsen E, Li Y, Quartermain D, Frangione B, Wisniewski T, Sigurdsson EM. Aβ derivative vaccination in alum adjuvant prevents amyloid deposition and does not cause brain microhemorrhages in Alzheimer’s model mice. Eur J Neurosci. 2006;24:2530–2542. doi: 10.1111/j.1460-9568.2006.05149.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Lieberburg I, Motter R, Nguyen M, Soriano F, Vasquez N, Weiss K, Welch B, Seubert P, Schenk D, Yednock T. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of alzheimer disease. Nat Med. 2000;6:916–919. doi: 10.1038/78682. [DOI] [PubMed] [Google Scholar]
- 31.DeMattos RB, Bales KR, Cummins DJ, Dodart JC, Paul SM, Holtzman DM. Peripheral anti-A beta antibody alters CNS and plasma A beta clearance and decreases brain A beta burden in a mouse model of Alzheimer’s disease. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:8850–8855. doi: 10.1073/pnas.151261398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lemere CA, Maron R, Selkoe DJ, Weiner HL. Nasal vaccination with beta-amyloid peptide for the treatment of Alzheimer’s disease. DNA and Cell Biology. 2001;20:705–711. doi: 10.1089/10445490152717569. [DOI] [PubMed] [Google Scholar]
- 33.Sigurdsson EM, Scholtzova H, Mehta P, Frangione B, Wisniewski T. Immunization with a nontoxic/nonfibrillar amyloid-β homologous peptide reduces Alzheimer’s disease associated pathology in transgenic mice. American Journal of Pathology. 2001;159:439–447. doi: 10.1016/s0002-9440(10)61715-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sigurdsson EM, Frangione B, Wisniewski T. Immunization for Alzheimer’s disease. Drug Development Research. 2002;56:135–142. [Google Scholar]
- 35.Wisniewski T, Frangione B. Immunological and anti-chaperone therapeutic approaches for Alzheimer’s disease. Brain Pathology. 2005;15:72–77. doi: 10.1111/j.1750-3639.2005.tb00102.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bayer AJ, Bullock R, Jones RW, Wilkinson D, Paterson KR, Jenkins L, Millais SB, Donoghue S. Evaluation of the safety and immunogenicity of synthetic Aβ42 (AN1792 in patients with AD. Neurology. 2005;64:94–101. doi: 10.1212/01.WNL.0000148604.77591.67. [DOI] [PubMed] [Google Scholar]
- 37.Pride M, Seubert P, Grundman M, Hagen M, Eldridge J, Black RS. Progress in the active immunotherapeutic approach to Alzheimer’s disease: clinical investigations into AN1792-associated meningoencephalitis. Neurodegener Dis. 2008;5:194–196. doi: 10.1159/000113700. [DOI] [PubMed] [Google Scholar]
- 38.Wisniewski T. Practice point commentary on “Clinical effects of Aβ immunization (AN1792 in patients with AD in an interupted trial”. Nature Clinical Practice Neurology. 2005;1:84–85. [Google Scholar]
- 39.Boche D, Nicoll JA. The role of the immune system in clearance of Abeta from the brain. Brain Pathology. 2008;18:267–278. doi: 10.1111/j.1750-3639.2008.00134.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bombois S, Maurage CA, Gompel M, Deramecourt V, kowiak-Cordoliani MA, Black RS, Lavielle R, Delacourte A, Pasquier F. Absence of beta-amyloid deposits after immunization in Alzheimer disease with Lewy body dementia. Arch Neurol. 2007;64:583–587. doi: 10.1001/archneur.64.4.583. [DOI] [PubMed] [Google Scholar]
- 41.Ferrer I, Boada RM, Sanchez Guerra ML, Rey MJ, Costa-Jussa F. Neuropathology and pathogenesis of encephalitis following amyloid-beta immunization in Alzheimer’s disease. Brain Pathology. 2004;14:11–20. doi: 10.1111/j.1750-3639.2004.tb00493.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Masliah E, Hansen L, Adame A, Crews L, Bard F, Lee C, Seubert P, Games D, Kirby L, Schenk D. Aβ vaccination effects on plaque pathology in the absence of encephalitis in Alzheimer disease. Neurology. 2005;64:129–131. doi: 10.1212/01.WNL.0000148590.39911.DF. [DOI] [PubMed] [Google Scholar]
- 43.Nicoll JA, Wilkinson D, Holmes C, Steart P, Markham H, Weller RO. Neuropathology of human Alzheimer disease after immunization with amyloid-beta peptide: a case report. Nat Med. 2005;9:448–452. doi: 10.1038/nm840. [DOI] [PubMed] [Google Scholar]
- 44.Nicoll JA, Barton E, Boche D, Neal JW, Ferrer I, Thompson P, Vlachouli C, Wilkinson D, Bayer A, Games D, Seubert P, Schenk D, Holmes C. Abeta species removal after abeta42 immunization. J Neuropathol Exp Neurol. 2006;65:1040–1048. doi: 10.1097/01.jnen.0000240466.10758.ce. [DOI] [PubMed] [Google Scholar]
- 45.Sadowski M, Wisniewski T. Disease modifying approaches for Alzheimer’s pathology. Current Pharmaceutic Design. 2007;13:1943–1954. doi: 10.2174/138161207781039788. [DOI] [PubMed] [Google Scholar]
- 46.Hock C, Konietzko U, Straffer JR, Tracy J, Signorell A, Muller-Tillmanns B, Lemke U, Henke K, Moritz E, Garcia E, Axel Wollmar M, Umbricht D, de Quervain DJF, Hofmann M, Maddalena A, Papassotiropoulos A, Nitsch RM. Antibodies against β–amyloid slow cognitive decline in Alzheimer’s disease. Neuron. 2003;38:547–554. doi: 10.1016/s0896-6273(03)00294-0. [DOI] [PubMed] [Google Scholar]
- 47.Hock C, Konietzko U, Paspassotiropoulos A, Wollmer A, Streffer J, von Rotz RC, Davey G, Moritz E, Nitsch RM. Generation of antibodies specific for β-amyloid by vaccination of patients with Alzheimer disease. Nat Med. 2002;8:1270–1276. doi: 10.1038/nm783. [DOI] [PubMed] [Google Scholar]
- 48.Gilman S, Koller M, Black RS, Jenkins L, Griffith SG, Fox NC, Eisner L, Kirby L, Boada Rovira M, Forette F, Orgogozo JM. Clinical effects of Aβ immunization (AN1792 in patients with AD in an interupted trial. Neurology. 2005;64:1553–1562. doi: 10.1212/01.WNL.0000159740.16984.3C. [DOI] [PubMed] [Google Scholar]
- 49.Relkin NR, Szabo P, Adamiak B, Burgut T, Monthe C, Lent RW, Younkin S, Younkin L, Schiff R, Weksler ME. 18-Month study of intravenous immunoglobulin for treatment of mild Alzheimer disease. Neurobiol Aging. 2008 Feb 20; doi: 10.1016/j.neurobiolaging.2007.12.021. [DOI] [PubMed] [Google Scholar]
- 50.Pfeifer M, Boncristiano S, Bondolfi L, Stalder A, Deller T, Staufenbiel M, Mathews PM, Jucker M. Cerebral hemorrhage after passive anti-Aβ immunotherapy. Science. 2002;298:1379. doi: 10.1126/science.1078259. [DOI] [PubMed] [Google Scholar]
- 51.Wilcock DM, Rojiani A, Rosenthal A, Subbarao S, Freeman MJ, Gordon MN, Morgan D. Passive immunization against Abeta in aged APP-transgenic mice reverses cognitive deficits and depletes parenchymal amyloid deposits in spite of increased vascular amyloid and microhemorrhage. J Neuroinflammation. 2004;1:24. doi: 10.1186/1742-2094-1-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Racke MM, Boone LI, Hepburn DL, Parsadanian M, Bryan MT, Ness DK, Piroozi KS, Jordan WH, Brown DD, Hoffman WP, Holtzman DM, Bales KR, Gitter BD, May PC, Paul SM, DeMattos RB. Exacerbation of cerebral amyloid angiopathy-associated microhemorrhages in amyloid precursor protein transgenic mice by immunotherapy is dependent on antibody recognition of deposited forms of amyoid beta. Journal of Neuroscience. 2005;25:629–636. doi: 10.1523/JNEUROSCI.4337-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wilcock DM, Jantzen PT, Li Q, Morgan D, Gordon MN. Amyloid-beta vaccination, but not nitro-nonsteroidal anti-inflammatory drug treatment, increases vascular amyloid and microhemorrhage while both reduce parenchymal amyloid. Neuroscience. 2007;144:950–960. doi: 10.1016/j.neuroscience.2006.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Petrushina I, Ghochikyan A, Mkrtichyan M, Mamikonyan G, Movsesyan N, Ajdari R, Vasilevko V, Karapetyan A, Lees A, Agadjanyan MG, Cribbs DH. Mannan-Abeta28 conjugate prevents Abeta-plaque deposition, but increases microhemorrhages in the brains of vaccinated Tg2576 (APPsw) mice. J Neuroinflammation. 2008;5:42. doi: 10.1186/1742-2094-5-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jellinger KA. Alzheimer disease and cerebrovascular pathology: an update. J Neural Transm. 2002;109:813–836. doi: 10.1007/s007020200068. [DOI] [PubMed] [Google Scholar]
- 56.Zhang-Nunes SX, Maat-Schieman ML, Van Duinen SG, Roos RA, Frosch MP, Greenberg SM. The cerebral beta-amyloid angiopathies: hereditary and sporadic. Brain Pathology. 2006;16:30–39. doi: 10.1111/j.1750-3639.2006.tb00559.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sigurdsson EM. Immunotherapy targeting pathological tau protein in Alzheimer’s disease and related tauopathies. J Alzheimers Dis. 2008;15:157–168. doi: 10.3233/jad-2008-15202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Noble W, Garwood CJ, Hanger DP. Minocycline as a potential therapeutic agent in neurodegenerative disorders characterised by protein misfolding. Prion. 2009;3 doi: 10.4161/pri.3.2.8820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kayed R, Jackson GR. Prefilament tau species as potential targets for immunotherapy for Alzheimer disease and related disorders. Curr Opin Immunol. 2009;21:359–363. doi: 10.1016/j.coi.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 60.Goedert M, Wischik CM, Crowther RA, Walker JE, Klug A. Cloning and sequencing of the cDNA encoding a core protein of the paired helical filament of Alzheimer disease: identification as the microtubule-associated protein tau. Proc Natl Acad Sci U S A. 1988;85:4051–4055. doi: 10.1073/pnas.85.11.4051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Goedert M, Spillantini MG, Jakes R, Rutherford D, Crowther RA. Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron. 1989;3:519–526. doi: 10.1016/0896-6273(89)90210-9. [DOI] [PubMed] [Google Scholar]
- 62.Utton MA, Noble WJ, Hill JE, Anderton BH, Hanger DP. Molecular motors implicated in the axonal transport of tau and alpha-synuclein. J Cell Sci. 2005;118:4645–4654. doi: 10.1242/jcs.02558. [DOI] [PubMed] [Google Scholar]
- 63.Magnani E, Fan J, Gasparini L, Golding M, Williams M, Schiavo G, Goedert M, Amos LA, Spillantini MG. Interaction of tau protein with the dynactin complex. EMBO J. 2007;26:4546–4554. doi: 10.1038/sj.emboj.7601878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gendron TF, Petrucelli L. The role of tau in neurodegeneration. Mol Neurodegener. 2009;4:13. doi: 10.1186/1750-1326-4-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Morishima-Kawashima M, Hasegawa M, Takio K, Suzuki M, Yoshida H, Watanabe A, Titani K, Ihara Y. Hyperphosphorylation of tau in PHF. Neurobiol Aging. 1995;16:365–371. doi: 10.1016/0197-4580(95)00027-c. [DOI] [PubMed] [Google Scholar]
- 66.Sergeant N, David JP, Lefranc D, Vermersch P, Wattez A, Delacourte A. Different distribution of phosphorylated tau protein isoforms in Alzheimer’s and Pick’s diseases. FEBS Letters. 1997;412:578–582. doi: 10.1016/s0014-5793(97)00859-4. [DOI] [PubMed] [Google Scholar]
- 67.Butner KA, Kirschner MW. Tau protein binds to microtubules through a flexible array of distributed weak sites. J Cell Biol. 1991;115:717–730. doi: 10.1083/jcb.115.3.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Brion JP, Flament-Durand J, Dustin P. Alzheimer’s disease and tau proteins. Lancet. 1986;2:1098. doi: 10.1016/s0140-6736(86)90495-2. [DOI] [PubMed] [Google Scholar]
- 69.Brion JP. Immunological demonstration of tau protein in neurofibrillary tangles of Alzheimer’s disease. J Alzheimers Dis. 2006;9:177–185. doi: 10.3233/jad-2006-9s321. [DOI] [PubMed] [Google Scholar]
- 70.Wang JZ, Liu F. Microtubule-associated protein tau in development, degeneration and protection of neurons. Prog Neurobiol. 2008;85:148–175. doi: 10.1016/j.pneurobio.2008.03.002. [DOI] [PubMed] [Google Scholar]
- 71.Boutajangout A, Boom A, Leroy K, Brion JP. Expression of tau mRNA and soluble tau isoforms in affected and non-affected brain areas in Alzheimer’s disease. FEBS Letters. 2004;576:183–189. doi: 10.1016/j.febslet.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 72.Buee L, Bussiere T, Buee-Scherrer V, Delacourte A, Hof PR. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res Brain Res Rev. 2000;33:95–130. doi: 10.1016/s0165-0173(00)00019-9. [DOI] [PubMed] [Google Scholar]
- 73.Sergeant N, Bretteville A, Hamdane M, Caillet-Boudin ML, Grognet P, Bombois S, Blum D, Delacourte A, Pasquier F, Vanmechelen E, Schraen-Maschke S, Buee L. Biochemistry of Tau in Alzheimer’s disease and related neurological disorders. Expert Rev Proteomics. 2008;5:207–224. doi: 10.1586/14789450.5.2.207. [DOI] [PubMed] [Google Scholar]
- 74.Baumann K, Mandelkow EM, Biernat J, Piwnica-Worms H, Mandelkow E. Abnormal Alzheimer-like phosphorylation of tau-protein by cyclin-dependent kinases cdk2 and cdk5. FEBS Letters. 1993;336:417–424. doi: 10.1016/0014-5793(93)80849-p. [DOI] [PubMed] [Google Scholar]
- 75.Hamdane M, Sambo AV, Delobel P, Begard S, Violleau A, Delacourte A, Bertrand P, Benavides J, Buee L. Mitotic-like tau phosphorylation by p25-Cdk5 kinase complex. J Biol Chem. 2003;278:34026–34034. doi: 10.1074/jbc.M302872200. [DOI] [PubMed] [Google Scholar]
- 76.Gotz J, Barmettler R, Ferrari A, Goedert M, Probst A, Nitsch RM. In vivo analysis of wild-type and FTDP-17 tau transgenic mice. Annals of the New York Academy of Science. 2000;920:126–133. doi: 10.1111/j.1749-6632.2000.tb06914.x. [DOI] [PubMed] [Google Scholar]
- 77.Gotz J, Chen F, Barmettler R, Nitsch RM. Tau filament formation in transgenic mice expressing P301L tau. J Biol Chem. 2001;276:529–534. doi: 10.1074/jbc.M006531200. [DOI] [PubMed] [Google Scholar]
- 78.Gotz J, Ittner LM. Animal models of Alzheimer’s disease and frontotemporal dementia. Nature Reviews in Neuroscience. 2008;9:532–544. doi: 10.1038/nrn2420. [DOI] [PubMed] [Google Scholar]
- 79.Lewis J, Dickson D, Lin WL, Chisholm L, Corral A, Jones G, Yen SH, Sahara N, Skipper L, Yager D, Eckman C, Hardy J, Hutton M, McGowan E. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science. 2001;293:1487–1491. doi: 10.1126/science.1058189. [DOI] [PubMed] [Google Scholar]
- 80.Ishihara T, Zhang B, Higuchi K, Yoshiyama Y, Trojanowski JQ, Lee VM. Age-dependent induction of congophilic neurofibrillary tau inclusions in tau transgenic mice. American Journal of Pathology. 2001;158:555–562. doi: 10.1016/S0002-9440(10)63997-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tanemura K, Akagi T, Murayama M, Kikuchi N, Murayama O, Hashikawa T, Yoshiike Y, Park JM, Matsuda K, Nakao S, Sun X, Sato S, Yamaguchi H, Takashima A. Formation of filamentous tau aggregations in transgenic mice expressing V337M human tau. Neurobiol Dis. 2001;8:1036–1045. doi: 10.1006/nbdi.2001.0439. [DOI] [PubMed] [Google Scholar]
- 82.Egashira N, Iwasaki K, Takashima A, Watanabe T, Kawabe H, Matsuda T, Mishima K, Chidori S, Nishimura R, Fujiwara M. Altered depression-related behavior and neurochemical changes in serotonergic neurons in mutant R406W human tau transgenic mice. Brain Research. 2005;1059:7–12. doi: 10.1016/j.brainres.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 83.Lee VM, Kenyon TK, Trojanowski JQ. Transgenic animal models of tauopathies. Biochim Biophys Acta. 2005;1739:251–259. doi: 10.1016/j.bbadis.2004.06.014. [DOI] [PubMed] [Google Scholar]
- 84.Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, Maeda J, Suhara T, Trojanowski JQ, Lee VM. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007;53:337–351. doi: 10.1016/j.neuron.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 85.Murakami T, Paitel E, Kawarabayashi T, Ikeda M, Chishti MA, Janus C, Matsubara E, Sasaki A, Kawarai T, Phinney AL, Harigaya Y, Horne P, Egashira N, Mishima K, Hanna A, Yang J, Iwasaki K, Takahashi M, Fujiwara M, Ishiguro K, Bergeron C, Carlson GA, Abe K, Westaway D, St George-Hyslop P, Shoji M. Cortical neuronal and glial pathology in TgTauP301L transgenic mice: neuronal degeneration, memory disturbance, and phenotypic variation. Am J Pathol. 2006;169:1365–1375. doi: 10.2353/ajpath.2006.051250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Taniguchi T, Doe N, Matsuyama S, Kitamura Y, Mori H, Saito N, Tanaka C. Transgenic mice expressing mutant (N279K) human tau show mutation dependent cognitive deficits without neurofibrillary tangle formation. FEBS Letters. 2005;579:5704–5712. doi: 10.1016/j.febslet.2005.09.047. [DOI] [PubMed] [Google Scholar]
- 87.Zilka N, Korenova M, Novak M. Misfolded tau protein and disease modifying pathways in transgenic rodent models of human tauopathies. Acta Neuropathologica. 2009;118:71–86. doi: 10.1007/s00401-009-0499-y. [DOI] [PubMed] [Google Scholar]
- 88.Andorfer C, Kress Y, Espinoza M, de SR, Tucker KL, Barde YA, Duff K, Davies P. Hyperphosphorylation and aggregation of tau in mice expressing normal human tau isoforms. J Neurochem. 2003;86:582–590. doi: 10.1046/j.1471-4159.2003.01879.x. [DOI] [PubMed] [Google Scholar]
- 89.Andorfer C, Acker CM, Kress Y, Hof PR, Duff K, Davies P. Cell-cycle reentry and cell death in transgenic mice expressing nonmutant human tau isoforms. J Neurosci. 2005;25:5446–5454. doi: 10.1523/JNEUROSCI.4637-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Asuni AA, Boutajangout A, Quartermain D, Sigurdsson EM. Immunotherapy targeting pathological tau conformers in a tangle mouse model reduces brain pathology with associated functional improvements. J Neurosci. 2007;27:9115–9129. doi: 10.1523/JNEUROSCI.2361-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sigurdsson EM. Tau-Focused Immunotherapy for Alzheimer’s Disease and Related Tauopathies. Curr Alzheimer Res. 2009 doi: 10.2174/156720509789207930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Eidenmuller J, Fath T, Maas T, Pool M, Sontag E, Brandt R. Phosphorylation-mimicking glutamate clusters in the proline-rich region are sufficient to simulate the functional deficiencies of hyperphosphorylated tau protein. Biochem J. 2001;357:759–767. doi: 10.1042/0264-6021:3570759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Fath T, Eidenmuller J, Brandt R. Tau-mediated cytotoxicity in a pseudohyperphosphorylation model of Alzheimer’s disease. J Neurosci. 2002;22:9733–9741. doi: 10.1523/JNEUROSCI.22-22-09733.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Masliah E, Rockenstein E, Adame A, Alford M, Crews L, Hashimoto M, Seubert P, Lee M, Goldstein J, Chilcote T, Games D, Schenk D. Effects of alpha-synuclein immunization in a mouse model of Parkinson’s disease. Neuron. 2005;46:857–868. doi: 10.1016/j.neuron.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 95.Tampellini D, Magrane J, Takahashi RH, Li F, Lin MT, Almeida CG, Gouras GK. Internalized antibodies to the Abeta domain of APP reduce neuronal Abeta and protect against synaptic alterations. J Biol Chem. 2007;282:18895–18906. doi: 10.1074/jbc.M700373200. [DOI] [PubMed] [Google Scholar]
- 96.Frost B, Jacks RL, Diamond MI. Propagation of tau misfolding from the outside to the inside of a cell. J Biol Chem. 2009;284:12845–12852. doi: 10.1074/jbc.M808759200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Clavaguera F, Bolmont T, Crowther RA, Abramowski D, Frank S, Probst A, Fraser G, Stalder AK, Beibel M, Staufenbiel M, Jucker M, Goedert M, Tolnay M. Transmission and spreading of tauopathy in transgenic mouse brain. Nature Cell Biology. 2009 doi: 10.1038/ncb1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ren PH, Lauckner JE, Kachirskaia I, Heuser JE, Melki R, Kopito RR. Cytoplasmic penetration and persistent infection of mammalian cells by polyglutamine aggregates. Nature Cell Biology. 2009;11:219–225. doi: 10.1038/ncb1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Aguzzi A. Cell biology: Beyond the prion principle. Nature. 2009;459:924–925. doi: 10.1038/459924a. [DOI] [PubMed] [Google Scholar]
- 100.Sadowski M, Verma A, Wisniewski T. Infectious Disease of the Nervous System: Prion Diseases. In: Bradley WG, Daroff RB, Fenichel GM, Jankovic J, editors. Neurology in Clinical Practice. Philadelphia: Elsevier; 2008. pp. 1567–1581. [Google Scholar]
- 101.Price JL, Morris JC. Tangles and plaques in nondemented aging and “preclinical” Alzheimer’s disease. Annals of Neurology. 1999;45:358–368. doi: 10.1002/1531-8249(199903)45:3<358::aid-ana12>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 102.Tarawneh R, Holtzman DM. Critical issues for successful immunotherapy in Alzheimer’s disease: development of biomarkers and methods for early detection and intervention. CNS Neurol Disord Drug Targets. 2009;8:144–159. doi: 10.2174/187152709787847324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Fagan AM, Roe CM, Xiong C, Mintun MA, Morris JC, Holtzman DM. Cerebrospinal fluid tau/beta-amyloid(42) ratio as a prediction of cognitive decline in nondemented older adults. Archives of Neurology. 2007;64:343–349. doi: 10.1001/archneur.64.3.noc60123. [DOI] [PubMed] [Google Scholar]
- 104.Mosconi L, Mistur R, Switalski R, Tsui WH, Glodzik L, Li Y, Pirraglia E, De SS, Reisberg B, Wisniewski T, De Leon MJ. FDG-PET changes in brain glucose metabolism from normal cognition to pathologically verified Alzheimer’s disease. Eur J Nucl Med Mol Imaging. 2009;36:811–822. doi: 10.1007/s00259-008-1039-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sigurdsson EM, Wadghiri YZ, Mosconi L, Blind JA, Knudsen E, Asuni A, Tsui WH, Sadowski M, Turnbull D, de Leon M, Wisniewski T. A non-toxic ligand for voxel-based MRI analysis of plaques in AD transgenic mice. Neurobiol Aging. 2008;29:836–877. doi: 10.1016/j.neurobiolaging.2006.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Scholtzova H, Wadghiri YZ, Douadi M, Sigurdsson EM, Li Y, Quartermain D, Banerjee P, Wisniewski T. A NMDA receptor antagonist leads to behavioral improvement and amyloid reduction in Alzheimer’s disease model transgenic mice shown by micro-magnetic resonance imaging. Journal of Neuroscience Research. 2008;86:2784–2791. doi: 10.1002/jnr.21713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Rowe CC, Ng S, Ackermann U, Gong SJ, Pike K, Savage G, Cowie TF, Dickinson KL, Maruff P, Darby D, Smith C, Woodward M, Merory J, Tochon-Danguy H, O’Keefe G, Klunk WE, Mathis CA, Price JC, Masters CL, Villemagne VL. Imaging beta-amyloid burden in aging and dementia. Neurology. 2007;68:1718–1725. doi: 10.1212/01.wnl.0000261919.22630.ea. [DOI] [PubMed] [Google Scholar]
- 108.Forsberg A, Engler H, Almkvist O, Blomquist G, Hagman G, Wall A, Ringheim A, Langstrom B, Nordberg A. PET imaging of amyloid deposition in patients with mild cognitive impairment. Neurobiol Aging. 2008;29:1456–1465. doi: 10.1016/j.neurobiolaging.2007.03.029. [DOI] [PubMed] [Google Scholar]
- 109.Roher AE, Kokjohn TA. Of mice and men: The relevance of transgenic mice Abeta immunizations to Alzheimer’s disease. J Alzheimers Dis. 2002;4:431–434. doi: 10.3233/jad-2002-4509. [DOI] [PubMed] [Google Scholar]
- 110.Tenner AJ, Fonseca MI. The double-edged flower: roles of complement protein C1q in neurodegenerative diseases. Advances in Experimental Medicine and Biology. 2006;586:153–176. doi: 10.1007/0-387-34134-X_11. [DOI] [PubMed] [Google Scholar]
- 111.Gandy S, DeMattos RB, Lemere CA, Heppner FL, Leverone J, Aguzzi A, Ershler WB, Dai J, Fraser P, St George Hyslop P, Holtzman DM, Walker LC, Keller ET. Alzheimer’s Aβ vaccination of rhesus monkeys (Macaca mulatta) Alzheimer’s Disease and Associated Diseases. 2004;18:44–46. doi: 10.1097/00002093-200401000-00009. [DOI] [PubMed] [Google Scholar]
- 112.Lemere CA, Beierschmitt A, Iglesias M, Spooner ET, Bloom JK, Leverone JF, Zheng JB, Seabrook TJ, Louard D, Li D, Selkoe DJ, Palmour RM, Ervin FR. Alzheimer’s disease abeta vaccine reduces central nervous system abeta levels in a non-human primate, the Caribbean vervet. American Journal of Pathology. 2004;165:283–297. doi: 10.1016/s0002-9440(10)63296-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Trouche SG, Asuni A, Rouland S, Wisniewski T, Frangione B, Verdier JM, Sigurdsson EM, Mestre-Frances N. Antibody response and plasma Aβ1-40 in young Microcebus Murinus primates immunized with Aβ1-42 and its derivatives. Vaccine. 2009;27:957–964. doi: 10.1016/j.vaccine.2008.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Gandy S, DeMattos RB, Lemere CA, Heppner FL, Leverone J, Aguzzi A, Ershler WB, Dai J, Fraser P, St George Hyslop P, Holtzman DM, Walker LC, Keller ET. Alzheimer’s Aβ vaccination of rhesus monkeys (Macaca mulatta) Mechanisms Ageing and Develop. 2004;125:149–151. doi: 10.1016/j.mad.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 115.Lemere CA, Iglesias M, Spooner ET, Bloom JK, Leverone JF, Li D, Zheng JB, Seabrook TJ, Selkoe D, Ervin FR, Palmour RM, Beiershcmitt A. Aβ immunization in aged vervet monkeys reduces Aβ levels in brain and CSF. Soc Neurosci Abst. 2003;133(5):5. [Google Scholar]
- 116.Lemere CA, Maier M, Jiang L, Peng Y, Seabrook TJ. Amyloid-beta immunotherapy for the prevention and treatment of Alzheimer disease: lessons from mice, monkeys, and humans. Rejuvenation Res. 2006;9:77–84. doi: 10.1089/rej.2006.9.77. [DOI] [PubMed] [Google Scholar]
- 117.Vasilevko V, Head E. Immunotherapy in a natural model of Abeta pathogenesis: the aging beagle. CNS Neurol Disord Drug Targets. 2009;8:98–113. doi: 10.2174/187152709787847333. [DOI] [PubMed] [Google Scholar]
- 118.Cotman CW, Head E. The canine (dog) model of human aging and disease: dietary, environmental and immunotherapy approaches. J Alzheimers Dis. 2008;15:685–707. doi: 10.3233/jad-2008-15413. [DOI] [PubMed] [Google Scholar]
- 119.Head E, Pop V, Vasilevko V, Hill M, Saing T, Sarsoza F, Nistor M, Christie LA, Milton S, Glabe C, Barrett E, Cribbs D. A two-year study with fibrillar beta-amyloid (Abeta) immunization in aged canines: effects on cognitive function and brain Abeta. J Neurosci. 2008;28:3555–3566. doi: 10.1523/JNEUROSCI.0208-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Cribbs DH, Ghochikyan A, Vasilevko V, Tran M, Petrushina I, Sadzikava N, Babikyan D, Kesslak P, Kieber-Emmons T, Cotman CW, Agadjanyan MG. Adjuvant-dependent modulation of Th1 and Th2 responses to immunization with beta-amyloid. Int Immunol. 2003;15:505–514. doi: 10.1093/intimm/dxg049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Maier M, Seabrook TJ, Lazo ND, Jiang L, Das P, Janus C, Lemere CA. Short amyloid-beta (Abeta) immunogens reduce cerebral Abeta load and learning deficits in an Alzheimer’s disease mouse model in the absence of an Abeta-specific cellular immune response. Journal of Neuroscience. 2006;26:4717–4728. doi: 10.1523/JNEUROSCI.0381-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Agadjanyan MG, Ghochikyan A, Petrushina I, Vasilevko V, Movsesyan N, Mkrtichyan M, Saing T, Cribbs DH. Prototype Alzheimer’s disease vaccine using the immunodominant B cell epitope from beta-amyloid and promiscuous T cell epitope pan HLA DR-binding peptide. J Immunol. 2005;174:1580–1586. doi: 10.4049/jimmunol.174.3.1580. [DOI] [PubMed] [Google Scholar]
- 123.Zamora E, Handisurya A, Shafti-Keramat S, Borchelt D, Rudow G, Conant K, Cox C, Troncoso JC, Kirnbauer R. Papillomavirus-like particles are an effective platform for amyloid-beta immunization in rabbits and transgenic mice. J Immunol. 2006;177:2662–2670. doi: 10.4049/jimmunol.177.4.2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Chackerian B, Rangel M, Hunter Z, Peabody DS. Virus and virus-like particle-based immunogens for Alzheimer’s disease induce antibody responses against amyloid-beta without concomitant T cell responses. Vaccine. 2006;24:6321–6331. doi: 10.1016/j.vaccine.2006.05.059. [DOI] [PubMed] [Google Scholar]
- 125.Jennings GT, Bachmann MF. The coming of age of virus-like particle vaccines. Biol Chem. 2008;389:521–536. doi: 10.1515/bc.2008.064. [DOI] [PubMed] [Google Scholar]
- 126.Movsesyan N, Ghochikyan A, Mkrtichyan M, Petrushina I, Davtyan H, Olkhanud PB, Head E, Biragyn A, Cribbs DH, Agadjanyan MG. Reducing AD-like pathology in 3xTg-AD mouse model by DNA epitope vaccine - a novel immunotherapeutic strategy. PloS ONE. 2008;3:e2124. doi: 10.1371/journal.pone.0002124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Lemere CA, Spooner ET, Leverone JF, Mori C, Clements JD. Intranasal immunotherapy for the treatment of Alzheimer’s disease: Escherichia coli LT and LT(R192G) as mucosal adjuvants. Neurobiol Aging. 2002;23:991–1000. doi: 10.1016/s0197-4580(02)00127-6. [DOI] [PubMed] [Google Scholar]
- 128.Seabrook TJ, Thomas K, Jiang L, Bloom J, Spooner E, Maier M, Bitan G, Lemere CA. Dendrimeric Abeta1-15 is an effective immunogen in wildtype and APP-tg mice. Neurobiol Aging. 2006 doi: 10.1016/j.neurobiolaging.2006.04.007. [DOI] [PubMed] [Google Scholar]
- 129.Nikolic WV, Bai Y, Obregon D, Hou H, Mori T, Zeng J, Ehrhart J, Shytle RD, Giunta B, Morgan D, Town T, Tan J. Transcutaneous beta-amyloid immunization reduces cerebral beta-amyloid deposits without T cell infiltration and microhemorrhage. Proc Natl Acad Sci U S A. 2007;104:2507–2512. doi: 10.1073/pnas.0609377104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Frackowiak J, Wisniewski HM, Wegiel J, Merz GS, Iqbal K, Wang KC. Ultrastructure of the microglia that phagocytose amyloid and the microglia that produce beta-amyloid fibrils. Acta Neuropathologica. 1992;84:225–233. doi: 10.1007/BF00227813. [DOI] [PubMed] [Google Scholar]
- 131.Jucker M, Heppner FL. Cerebral and peripheral amyloid phagocytes--an old liaison with a new twist. Neuron. 2008;59:8–10. doi: 10.1016/j.neuron.2008.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Butovsky O, Kunis G, Koronyo-Hamaoui M, Schwartz M. Selective ablation of bone marrow-derived dendritic cells increases amyloid plaques in a mouse Alzheimer’s disease model. Eur J Neurosci. 2007;26:413–416. doi: 10.1111/j.1460-9568.2007.05652.x. [DOI] [PubMed] [Google Scholar]
- 133.El KJ, Toft M, Hickman SE, Means TK, Terada K, Geula C, Luster AD. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat Med. 2007;13:432–438. doi: 10.1038/nm1555. [DOI] [PubMed] [Google Scholar]
- 134.Scholtzova H, Kascsak RJ, Bates KA, Boutajangout A, Kerr DJ, Meeker HC, Mehta PD, Spinner DS, Wisniewski T. Induction of Toll-like receptor 9 signaling as a method for ameliorating Alzheimer’s disease related pathology. Journal of Neuroscience. 2009;29:1846–1854. doi: 10.1523/JNEUROSCI.5715-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Tahara K, Kim HD, Jin JJ, Maxwell JA, Li L, Fukuchi K. Role of toll-like receptor signalling in Abeta uptake and clearance. Brain. 2006;129:3006–3019. doi: 10.1093/brain/awl249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Obregon D, Hou H, Bai Y, Nikolic WV, Mori T, Luo D, Zeng J, Ehrhart J, Fernandez F, Morgan D, Giunta B, Town T, Tan J. CD40L disruption enhances Abeta vaccine-mediated reduction of cerebral amyloidosis while minimizing cerebral amyloid angiopathy and inflammation. Neurobiol Dis. 2008;29:336–353. doi: 10.1016/j.nbd.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Town T, Laouar Y, Pittenger C, Mori T, Szekely CA, Tan J, Duman RS, Flavell RA. Blocking TGF-beta-Smad2/3 innate immune signaling mitigates Alzheimer-like pathology. Nat Med. 2008;14:681–687. doi: 10.1038/nm1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Glabe CG. Structural classification of toxic amyloid oligomers. J Biol Chem. 2008;283:29639–29643. doi: 10.1074/jbc.R800016200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Klybin I, Betts V, Blennow K, Zetterberg H, Wallin A, Lemere CA, Cullen WK, Welzel A, Peng Y, Wisniewski T, Selkoe DJ, Anwyl R, Walsh DM, Rowan MJ. Aβ dimer-containing human cerebrospinal fluid disrupts synaptic plasticity: prevention by systemic passive immunization. Journal of Neuroscience. 2008;28:4231–4237. doi: 10.1523/JNEUROSCI.5161-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Lee EB, Leng LZ, Zhang B, Kwong L, Trojanowski JQ, Abel T, Lee VM. Targeting amyloid-beta peptide (Abeta) oligomers by passive immunization with a conformation-selective monoclonal antibody improves learning and memory in Abeta precursor protein (APP) transgenic mice. J Biol Chem. 2006;281:4292–4299. doi: 10.1074/jbc.M511018200. [DOI] [PubMed] [Google Scholar]
- 141.Moretto N, Bolchi A, Rivetti C, Imbimbo BP, Villetti G, Pietrini V, Polonelli L, Del SS, Smith KM, Ferrante RJ, Ottonello S. Conformation-sensitive antibodies against alzheimer amyloid-beta by immunization with a thioredoxin-constrained B-cell epitope peptide. J Biol Chem. 2007;282:11436–11445. doi: 10.1074/jbc.M609690200. [DOI] [PubMed] [Google Scholar]
- 142.Mamikonyan G, Necula M, Mkrtichyan M, Ghochikyan A, Petrushina I, Movsesyan N, Mina E, Kiyatkin A, Glabe CG, Cribbs DH, Agadjanyan MG. Anti-A beta 1-11 antibody binds to different beta-amyloid species, inhibits fibril formation, and disaggregates preformed fibrils but not the most toxic oligomers. J Biol Chem. 2007;282:22376–22386. doi: 10.1074/jbc.M700088200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Lambert MP, Velasco PT, Chang L, Viola KL, Fernandez S, Lacor PN, Khuon D, Gong Y, Bigio EH, Shaw P, De Felice FG, Krafft GA, Klein WL. Monoclonal antibodies that target pathological assemblies of Abeta. J Neurochem. 2007;100:23–35. doi: 10.1111/j.1471-4159.2006.04157.x. [DOI] [PubMed] [Google Scholar]
- 144.Lambert MP, Velasco PT, Viola KL, Klein WL. Targeting generation of antibodies specific to conformational epitopes of amyloid beta-derived neurotoxins. CNS Neurol Disord Drug Targets. 2009;8:65–81. doi: 10.2174/187152709787601876. [DOI] [PubMed] [Google Scholar]
- 145.Puzzo D, Privitera L, Leznik E, Fa M, Staniszewski A, Palmeri A, Arancio O. Picomolar amyloid-beta positively modulates synaptic plasticity and memory in hippocampus. J Neurosci. 2008;28:14537–14545. doi: 10.1523/JNEUROSCI.2692-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Giuffrida ML, Caraci F, Pignataro B, Cataldo S, De BP, Bruno V, Molinaro G, Pappalardo G, Messina A, Palmigiano A, Garozzo D, Nicoletti F, Rizzarelli E, Copani A. Beta-amyloid monomers are neuroprotective. J Neurosci. 2009;29:10582–10587. doi: 10.1523/JNEUROSCI.1736-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Wisniewski T, Prelli F, Scholtzova H, Chung E, Mehta PD, Kascsak R, Kascsak R, Goni F. Immunotherapy targeting abnormal protein conformation. Alz Dementia. 2009;4:113. [Google Scholar]
- 148.Wisniewski T, Sigurdsson EM. Therapeutic approaches for prion and Alzheimer’s diseases. FEBS J. 2007;274:3784–3798. doi: 10.1111/j.1742-4658.2007.05919.x. [DOI] [PubMed] [Google Scholar]
- 149.Goni F, Prelli F, Schreiber F, Scholtzova H, Chung E, Kascsak R, Brown DR, Sigurdsson EM, Chabalgoity JA, Wisniewski T. High titers of mucosal and systemic anti-PrP antibodies abrogates oral prion infection in mucosal vaccinated mice. Neurosci. 2008;153:679–686. doi: 10.1016/j.neuroscience.2008.02.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Gotz J, Probst A, Spillantini MG, Schafer T, Jakes R, Burki K, Goedert M. Somatodendritic localization and hyperphosphorylation of tau protein in transgenic mice expressing the longest human brain tau isoform. EMBO J. 1995;14:1304–1313. doi: 10.1002/j.1460-2075.1995.tb07116.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Probst A, Gotz J, Wiederhold KH, Tolnay M, Mistl C, Jaton AL, Hong M, Ishihara T, Lee VM, Trojanowski JQ, Jakes R, Crowther RA, Spillantini MG, Burki K, Goedert M. Axonopathy and amyotrophy in mice transgenic for human four-repeat tau protein. Acta Neuropathologica. 2000;99:469–481. doi: 10.1007/s004010051148. [DOI] [PubMed] [Google Scholar]
- 152.Spittaels K, Van den HC, van DJ, Bruynseels K, Vandezande K, Laenen I, Geerts H, Mercken M, Sciot R, Van LA, Loos R, Van LF. Prominent axonopathy in the brain and spinal cord of transgenic mice overexpressing four-repeat human tau protein. Am J Pathol. 1999;155:2153–2165. doi: 10.1016/S0002-9440(10)65533-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Brion JP, Tremp G, Octave JN. Transgenic expression of the shortest human tau affects its compartmentalization and its phosphorylation as in the pretangle stage of Alzheimer’s disease. Am J Pathol. 1999;154:255–270. doi: 10.1016/S0002-9440(10)65272-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Ishihara T, Hong M, Zhang B, Nakagawa Y, Lee MK, Trojanowski JQ, Lee VM. Age-dependent emergence and progression of a tauopathy in transgenic mice overexpressing the shortest human tau isoform. Neuron. 1999;24:751–762. doi: 10.1016/s0896-6273(00)81127-7. [DOI] [PubMed] [Google Scholar]
- 155.Higuchi M, Ishihara T, Zhang B, Hong M, Andreadis A, Trojanowski J, Lee VM. Transgenic mouse model of tauopathies with glial pathology and nervous system degeneration. Neuron. 2002;35:433–446. doi: 10.1016/s0896-6273(02)00789-4. [DOI] [PubMed] [Google Scholar]
- 156.Boutajangout A, Leroy K, Touchet N, Authelet M, Blanchard V, Tremp G, Pradier L, Brion JP. Increased tau phosphorylation but absence of formation of neurofibrillary tangles in mice double transgenic for human tau and Alzheimer mutant (M146L) presenilin-1. Neuroscience Letters. 2002;318:29–33. doi: 10.1016/s0304-3940(01)02461-2. [DOI] [PubMed] [Google Scholar]
- 157.Boutajangout A, Authelet M, Blanchard V, Touchet N, Tremp G, Pradier L, Brion JP. Characterisation of cytoskeletal abnormalities in mice transgenic for wild-type human tau and familial Alzheimer’s disease mutants of APP and presenilin-1. Neurobiol Dis. 2004;15:47–60. doi: 10.1016/j.nbd.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 158.Terwel D, Lasrado R, Snauwaert J, Vandeweert E, Van HC, Borghgraef P, Van LF. Changed conformation of mutant Tau-P301L underlies the moribund tauopathy, absent in progressive, nonlethal axonopathy of Tau-4R/2N transgenic mice. J Biol Chem. 2005;280:3963–3973. doi: 10.1074/jbc.M409876200. [DOI] [PubMed] [Google Scholar]
- 159.Lewis J, McGowan E, Rockwood J, Melrose H, Nacharaju P, Van Slegtenhorst M, Gwinn-Hardy K, Murphy MP, Baker M, Yu X, Duff K, Hardy J, Corral A, Lin WL, Yen SH, Dickson D, Davies P, Hutton M. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat Genet. 2000;25:402–405. doi: 10.1038/78078. [DOI] [PubMed] [Google Scholar]
- 160.Allen B, Ingram E, Takao M, Smith MJ, Jakes R, Virdee K, Yoshida H, Holzer M, Craxton M, Emson PC, Atzori C, Migheli A, Crowther RA, Ghetti B, Spillantini MG, Goedert M. Abundant tau filaments and nonapoptotic neurodegeneration in transgenic mice expressing human P301S tau protein. J Neurosci. 2002;22:9340–9351. doi: 10.1523/JNEUROSCI.22-21-09340.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Gotz J, Tolnay M, Barmettler R, Chen F, Probst A, Nitsch RM. Oligodendroglial tau filament formation in transgenic mice expressing G272V tau. Eur J Neurosci. 2001;13:2131–2140. doi: 10.1046/j.0953-816x.2001.01604.x. [DOI] [PubMed] [Google Scholar]
- 162.Tatebayashi Y, Miyasaka T, Chui DH, Akagi T, Mishima K, Iwasaki K, Fujiwara M, Tanemura K, Murayama M, Ishiguro K, Planel E, Sato S, Hashikawa T, Takashima A. Tau filament formation and associative memory deficit in aged mice expressing mutant (R406W) human tau. Proc Natl Acad Sci U S A. 2002;99:13896–13901. doi: 10.1073/pnas.202205599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Lim F, Hernandez F, Lucas JJ, Gomez-Ramos P, Moran MA, Avila J. FTDP-17 mutations in tau transgenic mice provoke lysosomal abnormalities and Tau filaments in forebrain. Mol Cell Neurosci. 2001;18:702–714. doi: 10.1006/mcne.2001.1051. [DOI] [PubMed] [Google Scholar]
- 164.Schindowski K, Bretteville A, Leroy K, Begard S, Brion JP, Hamdane M, Buee L. Alzheimer’s disease-like tau neuropathology leads to memory deficits and loss of functional synapses in a novel mutated tau transgenic mouse without any motor deficits. Am J Pathol. 2006;169:599–616. doi: 10.2353/ajpath.2006.060002. [DOI] [PMC free article] [PubMed] [Google Scholar]