

SUMMARY
Epigenetics involves molecular mechanisms related to gene expression independent of DNA sequence, mostly mediated by modification of chromatin histones. It has recently been suggested that these transcriptional changes may be implicated in the pathophysiology of mood disorders. In addition, histone deacetylase (HDAC) inhibitors have been shown to control epigenetic programming associated with the regulation of cognition and behavior, and may reverse dysfunctional epigenetic regulation associated with early life events in preclinical models. In this context, the active and continuous adaptation of chromatin, and the access of gene promoters to transcription factor mechanisms may represent a potential therapeutic target in the treatment of mood disorders such as bipolar disorder (BD) and major depressive disorder (MDD). Notably, the standard mood stabilizer valproate (VPA) has been shown to modulate the epigenome by inhibiting HDACs. However, several potential limitations are associated with this class of agents, including lack of selectivity for specific HDAC isoforms as well as risk of potentially serious side effects. Further studies regarding the potential role of chromatin remodeling in the mechanism of action of antidepressants and mood stabilizers are necessary to clarify the potential role of this class of agents as therapeutics for mood disorders.
Keywords: Bipolar disorder, Depression, Epigenetics, Histones, Pathophysiology, Treatment, Valproate
Introduction
Mood disorders such as major depressive disorder (MDD) and bipolar disorder (BD) affect the lives and functioning of millions of individuals worldwide. Currently available treatments for these disorders are inadequate for many. This lack of efficacy increases the prevalence of residual symptoms, functional impairment, episode relapses, suicide risk, and psychosocial disability [1].
An increasing number of studies have evaluated the potential therapeutic relevance of early intervention in BD, and this work includes continuous evaluation of prodromal symptoms and identification of biological markers [2]. The potential clinical relevance of neuroprotective agents as a suitable form of intervention during the early stages of the illness has also been described (reviewed in [3, 4]). It is possible that these early interventions may reverse dysfunctional epigenetic programming associated with the early traumatic events thought to trigger the onset of mood episodes (e.g., the high rates of comorbid BD and posttraumatic stress disorder [PTSD], or increased suicide risk in victims of childhood abuse) [5, 6].
In this article, we review the manner in which transcriptional epigenetic changes may be implicated in the pathophysiology of mood disorders, and how histone deacetylase (HDAC) inhibitors may regulate epigenetic programming. We also propose that dysfunctional epigenetic regulation associated with early life events may play an indirect role in the pathophysiology of mood disorders [7]. We further explore how the active and continuous adaptation of chromatin and the access of gene promoters to transcription factor mechanisms may represent a potential therapeutic target in mood disorders. Notably, the standard mood stabilizer valproate (VPA) modulates the epigenome by inhibiting HDACs, with potential implications in BD. Limitations of this class of agents are also reviewed.
Epigenetics: The Critical Balance between Histone Acetylation and Deacetylation
Epigenetic modifications are reversible chromatin rearrangements that control gene expression without modifying DNA sequence [8, 9]. Although DNA methylation and histone modifications are the most studied epigenetic mechanisms, other epigenetic processes have been shown to control gene function (e.g., the small noncoding RNA‐mediated regulation of gene expression and chromatin remodeling [10]. Histones are small proteins organized in complexes with DNA, which together belong to the nucleosome core. The histone octamer tails are composed of lysine aminoacids that interact with the negative charges on the DNA backbone [11]. Histone tails are involved in the dynamic regulation of chromatin structure and gene transcription [12].
Histones can be acetylated (OCH2CH3) or deacetylated at lysine residues, and these processes are controlled by histone acetyltransferases (HATs), and HDACs, respectively (Figure 1) [13]. More than 10 HDAC enzymes and multiple HATs participate in chromatin remodeling and gene expression. HDACs remove an acetyl group from the N‐terminal tails of histone proteins, leading to chromatin compaction and gene expression silencing [14]. Meanwhile, HATs regulate transcription and contact to genomic DNA by adding acetyl groups and increase acetylation, thus loosening the nucleosomes and providing access to DNA and docking sites for transcriptional factors [15]. In other words, when the DNA needs to interact with proteins or transcription factors, the histones weaken the interaction of their tails with other nucleosomes by inducing the acetylation of lysines, which removes the positive charge. Meanwhile, deacetylation of histones induces a tighter envelope of proteins around which the DNA winds, thereby blocking gene expression of diverse key proteins by physically limiting the access of transcription factors. DNA methylation generally directly reduces gene transcription in addition to histone deacetylation (see Figure 1).
Figure 1.
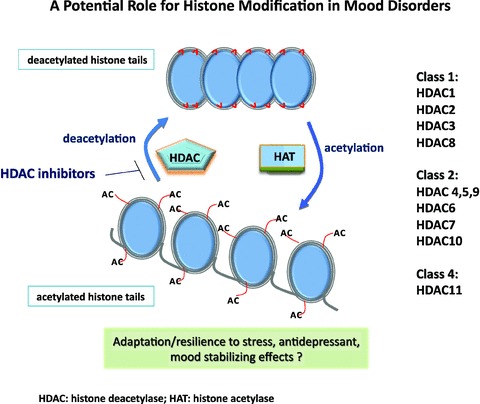
Histone deacetylation involves the removal of acetyl groups from lysine on N‐termini of histones by histone deacetylase (HDAC) enzymes. The consequent inactive and silent chromatin blocks gene transcription by limiting contact with promoter regions. In the opposite direction, the tails of histones can be acetylated at different sites generating an accessible form of chromatin. The deacetylated histone tails connect tightly to shape a compressed, unattainable form of chromatin. In addition to histone deacetylation, methylation of DNA may also be considered relevant in the context of epigenetics in mood disorders and involves the covalent binding of a methyl group by enzymes called DNA methyltransferases; following promoter methylation, reduced gene transcription generally occurs.
It is important to emphasize that dysregulation in the delicate interplay between HAT and HDAC function may lead to cellular dysfunction, including uncontrolled cell growth of abnormal cells and/or apoptosis. Enhanced acetylation of histones and decreased methylation of DNA (CH3) change chromatin structure and facilitate access to transcription factors and gene expression, but the balance between these effects is critical to maintaining cellular homeostasis. For instance, abundant deacetylated histones are usually present in pathological conditions, particularly those associated with DNA hypermethylation, chromatin condensation, and gene silencing [7]. Meanwhile, excessive DNA acetylation may also lead to the overproduction of certain proteins (e.g., proapoptotic), thus inducing deleterious effects.
A Potential Role for Histones in the Pathophysiology of Mood Disorders
Recently, there has been increasing appreciation of gene‐environment interactions and early life events in mood disorders, which have not traditionally been considered developmental disorders. Epigenetics has been proposed to mediate diverse environmental aspects involved in the pathophysiology of major psychotic disorders [7]. Within a translational framework in psychiatry, epigenetic changes may involve differential gene expression induced by various early life events, as well as developmental and environmental aspects affecting critical limbic circuits. These modifications intrinsically entail phenotypic and functional differences that affect cognition and behavior in a temporally long‐lasting manner. Thus, epigenetic regulation can be considered a key molecular mechanism for “cellular memory.” For instance, recent preclinical findings from models of gene‐environment interactions suggest that maternal behavior produces stable alterations of DNA methylation and chromatin structure, providing a mechanism for the long‐term effects of maternal care on gene expression in the offspring [16]. Decreased levels of glucocorticoid receptor mRNA, as well as mRNA transcripts bearing the glucocorticoid receptor 1F splice variant have recently been observed in postmortem hippocampus obtained from suicide victims with a history of childhood abuse as compared to those from either suicide victims with no childhood abuse or controls; increased cytosine methylation of an NR3C1 promoter was also found in these individuals [17]. These findings suggest a common effect of parental care (adverse or positive experience dependent) on the epigenetic regulation of hippocampal glucocorticoid receptor expression in humans and animals [17].
HDAC inhibition directly or indirectly affects 2–5% of all genes [18]. In humans, HDAC enzymes can be divided into four major classes [19]. Class I HDACs include HDAC1, 2, 3, and 8. Class II HDACs include HDAC4, 5, 6, 7, 9, and 10; these are further divided into two subclasses—IIa (HDAC4, 5, 7, and 9) and IIb (HDAC6 and 10)—according to their structural similarities. Class I and II HDACs have been most extensively investigated in the central nervous system (CNS). HDAC inhibitors prevent histone deacetylation by selectively inactivating Class I or II HDACs, thereby enhancing levels of histone acetylation in the brain. Meanwhile, Class III HDACs (also known as sirtuins) differ structurally and functionally from other HDACs; in humans, the class III HDACs include seven members. The deacetylation reaction mediated by sirtuins is coupled to the cleavage of nicotinamide adenine dinucleotide (NAD+), yielding nicotinamide and 2′‐O‐acetyl ADP‐ribose, along with the deacetylated lysine residue within the protein substrate [20].
Recent studies suggest the involvement of HDAC dysfunction in the pathophysiology of mood disorders, although it is important to mention that most of these studies describe preliminary data. For instance, decreased HDAC2 protein expression was identified in the nucleus accumbens (NAc) of individuals with MDD [21]. In another recent postmortem investigation, individuals with BD had higher baseline levels of total acetylated histone 3 levels compared to subjects with schizophrenia [22]. Another recent study evaluated 11 HDACs in the peripheral leukocytes of subjects with MDD and BD during euthymia or depressive episodes and found increased expression of HDAC2 and HDAC 5 mRNA during depressive episodes compared to controls and patients in remission, suggesting a state‐dependent alteration [23]. In individuals with BD, HDAC4 mRNA also showed a state‐dependent increase (during depressive episodes), while HDAC6 and HDAC 8 were decreased in both symptomatic and euthymic subjects with BD compared to controls [23]. Repressor element‐1 silencing transcription factor (REST), which is also associated with changes in transcriptional regulation, showed a state‐dependent decrease in subjects with MDD during a depressive episode [24]. REST negatively regulates genes that contain the repressor element‐1 (RE‐1) binding site and has many well‐defined target genes associated with the pathophysiology of MDD, such as corticotropin releasing hormone (CRH), brain‐derived neurotrophic factor (BDNF), and the serotonin 1A receptor. Repression of RE‐1‐containing genes by REST is sensitive to the HDAC inhibitor Trichostatin A (TSA), indicating that histone deacetylation may play a role in repression by REST through RE‐1 [25].
Preliminary data also suggest that HDACs regulate brain histone‐RELN and histone‐GAD67 promoter interactions [26]. The Reelin and GAD67 promoters are activated by epigenetic drugs that facilitate the disruption of local repressor complexes [27]. In psychiatric disorders, GAD67 expression was strongly and negatively correlated with mRNA expression levels of HDAC1, HDAC3, and HDAC4 [28]. Interestingly, these effects at GAD67 and RELN were described as consistent vulnerability factors for psychoses [29]. Specifically, prefrontal cortex GAD67 and RELN expression were significantly decreased in individuals with BD and schizophrenia who had a history of psychotic symptoms compared to controls and individuals with MDD without psychosis [29]. Similarly, a significant downregulation of GAD and RELN expression in cortical gamma aminobutyric acid (GABA) interneurons was described in individuals with BD, and this was potentially associated with epigenetic hypermethylation [29]. These effects at RELN and GAD67 promoters involve dysfunctions in chromatin remodeling controlled by GABAergic neurotransmission, which has been directly implicated in the pathophysiology of BD [30]. With regard to potential rescue effects at these targets using HDAC inhibitors (see below), diverse models noted that significantly increased expression of these proteins seem to involve epigenetic mechanisms [31, 32].
Also, chronic social defeat stress enhanced acetylated histone H3, thus decreasing HDAC2 levels in the NAc [21]. These effects have been described as mediating long‐lasting positive neuronal adaptive effects. HDACs also directly interact and deacetylate nonhistone proteins such as cAMP response‐element‐binding protein (CREB) [33], which has been directly and indirectly implicated in the pathophysiology of BD [34, 35]. Other transcription factors and cofactors controlling chromatin remodeling are similarly believed to regulate memory formation and consolidation [36].
HDACs and HATs are also believed to play a pivotal role in regulating synaptic plasticity [37, 38, 39, 40, 41], a mechanism that has been proposed to be involved in the pathophysiology and therapeutics of mood disorders such as BD [42].
The Potential Therapeutic Role of HDAC Inhibitors in Mood Disorders
Class I and II HDAC inhibitors include small‐molecule hydroxamates (the most potent HDAC inhibitors) such as TSA, suberoylanilide hydroxamic acid (SAHA)/vorinostat and scriptaid, and derivatives of aliphatic acid such as sodium butyrate (SB), sodium phenylbutyrate (SPB), and VPA. Cyclic tetrapeptides such as apicidin, trapoxin, depsipeptide (FK‐228)/romidepsin, and benzamides such as MS‐275/SNDX‐275 and Cl‐994, are different classes of HDAC inhibitors [43]. Benzamides and cyclic peptides have been evaluated in diverse clinical trials. VPA (described elsewhere) is a small chain fatty acid and does not have a strong HDAC inhibition constant [9]. HDAC inhibitors such as SB, phenylbutyrate (PB), and TSA induce histone hyperacetylation and cause chromatin remodeling, also increasing the expression of proteins that directly modulate the expression of neuroprotective/neurotrophic proteins [44].
Because data suggest that deficits in molecular brain adaptations, including chromatin remodeling and other epigenetic changes, may play a potential role in the pathophysiology of mood disorders, it is possible that therapeutics for mood disorders could work by strengthening the synaptic connections necessary for long‐term behavioral changes. Within this paradigm, it has been suggested that early life stressors may increase vulnerability to the first episode for individuals susceptible to BD [45]; these events may be consistent with subsequent deficits in the ability to produce more “plastic” chromatin, also indirectly supporting a putative role for HDAC inhibitors as therapeutics for BD. However, it is important to note that there is no specific isoform or molecular target able to explain the potential therapeutic effects of HDAC inhibitors in neuropsychiatric disorders.
Due to their potential ability to reverse dysfunctional epigenetic regulation, diverse CNS‐penetrant HDAC inhibitors have been suggested as potential therapeutics for the treatment of mood disorders. Preclinical studies found that central infusion of an HDAC inhibitor reversed/blocked increased histone deacetylation, DNA methylation, and hypothalamic–pituitary–adrenal (HPA) stress responses in rodent models of depression associated with early life events [16]. Similarly, two preclinical studies evaluating SB, a nonspecific class I and II HDAC inhibitor, found that it induced antidepressant‐like effects [46, 47]. Infusion of two selective class I and II HDAC inhibitors into the mouse NAc also induced significant antidepressant‐like effects [21].
With regard to mechanisms of action, diverse class I or class II HDAC inhibitors have been shown to protect neurons against oxidative stress‐induced neuronal death [48]. Notably, pulse treatment for 2 h with the HDAC inhibitor TSA protected against oxidative stress‐induced neuronal injury [49]. However, repeated exposure to this agent in cultured cortical neurons induced toxicity [49]. In mood disorders research, increased oxidative stress has been consistently described in subjects with BD, and the mood stabilizer lithium has been shown to exert potent antioxidant effects [50, 51]. Also, Leng and colleagues recently showed that treatment with lithium, HDAC1 siRNA, or other HDAC inhibitors induced synergistic neuroprotective effects in vitro[52]; these effects were further corroborated by lithium‐induced phospho‐acetylation of histone H3 observed in the amygdala [53].
Besides the direct effects seen at neurons, HDAC inhibitors also appear to be neuroprotective in nonneuronal cells not intrinsically associated with histone acetylation. For instance, HDAC inhibition in astrocytes protected neurons by inducing the release of BDNF and glial‐derived neurotrophic factor (GDNF) [54, 55]. Anxiety is often present in mood disorders. In models of anxiety such as extinction of conditioned fear, a significant increase in histone H4 acetylation around the BDNF P4 gene promoter was noted; this effect also upregulated BDNF exon I and IV mRNA expression in the prefrontal cortex after treatment with VPA (described in the next section) [56]. Other studies found that HDAC inhibitors enhanced initial learning in contextual fear conditioning [39, 57], potentially involving the extracellular signal‐regulated kinase‐activated protein kinase (ERK/MAPK) pathway, which has also been associated with the pathophysiology of mood disorders [58].
Enzymes and transcription factors able to generate more “plastic” chromatin via histone acetylation may be involved in memory formation and may also be relevant as therapeutics for mood disorders. Indeed, HDAC inhibitors enhance extinction for contextual fear, perhaps by modulating transcription in the hippocampus [59].
VPA and other HDAC Inhibitors in Mood Disorders and Beyond
VPA can be considered the prototypical HDAC inhibitor in mood disorders research. Indeed, the fact that VPA acts as an HDAC inhibitor underscores the possibility that this mechanism may be involved in its mood stabilizing properties. At therapeutic levels, VPA relieves HDAC‐dependent transcriptional repression and causes hyperacetylation of histones in cells and in vivo[60]. VPA also inhibits HDAC activity in vitro, most probably by binding to the catalytic center of HDACs, as well as indirectly through transcriptional activation of several promoters [60].
In preclinical models, VPA blocked methionine‐induced RELN promoter hypermethylation and RELN mRNA downregulation, which resulted in improved social interaction and prepulse inhibition [61]. Similarly, VPA induced a greater than 5‐fold increase in GAD67 mRNA and enhanced total acetylated histone 3 levels [61]. VPA also inhibited the release of proinflammatory factors from microglia [62]. It is important to note that in preclinical models, VPA's ability to inhibit HDAC may also mediate several severe side effects, such as teratogenicity or polycystic ovarian syndrome. For instance, Gurvich and colleagues found that VPA and other HDAC inhibitors caused defects similar to spina bifida in Xenopus and zebrafish [63]. Further studies with VPA are necessary to clarify this issue. VPA also induces hyperacetylation of histone H3 in the α‐syn promoter region in neuroblastoma SH‐SY5Y cells [64], with a potential role in plasticity. Overall, VPA has been shown to inhibit HDAC activity in vitro in several models, thus suggesting it may play a relevant role in neuroprotection (for a review see Ref. 65).
Clinical investigations noted that individuals with BD had decreased acetylated histone 3 (H3K9, K14ac) and acetylated histone 4 protein levels that were upregulated after 4 weeks of treatment with VPA [66]; similar—but less pronounced—results were noted in subjects with schizophrenia. HDAC activity has also been shown to be dysfunctional in neurological disease [48, 67, 68], and overactivation of HDACs has been associated with misregulation, overexpression, mutation, and amplification of diverse proteins involved in oncogenesis [69].
Diverse HDAC inhibitors are neuroprotective, and this effect is believed to be due to their ability to target epigenetic functions [44]. These agents have been tested in both cell culture models and rodent models of diverse neurodegenerative diseases. For instance, the HDAC inhibitor TSA protected cortical neurons against oxygen and glucose deprivation secondary to ischemia [70]. As noted earlier, HDAC inhibitors such as TSA and SB enhance the expression of GDNF and BDNF in astrocytes, thus protecting dopaminergic neurons [55]. Diverse HDAC inhibitors, such as HDACi 4b, SB, and SAHA improved motor performance in animal models of Huntington's Disease, and these improvements were associated with increased brain histone acetylation [71, 72, 73]. Other agents—such as VPA, SB, and LBH589—are being evaluated in clinical studies to treat spinal muscular atrophy (SMA), Alzheimer's Disease, and amyotrophic lateral sclerosis (ALS) [43]. Indeed, treatment with HDAC inhibitors improved the ALS phenotype in animal models [74]. Studies also suggest that HDAC inhibitors may play a potential therapeutic role in treating Friedreich's ataxia (FRDA), a disorder caused by a mutation within intron 1 of the frataxin (FXN) gene that induces hypoacetylation of histones H3 and H4 [75, 76]. Notably, two studies have shown that a new class of HDAC inhibitors reversed FXN gene silencing in human and preclinical models. This effect was observed in primary lymphocytes from individuals with FRDA as well as a murine model of the disease [76, 77].
Conclusions and Perspectives
Protein acetylation has been widely described as a key posttranslational modification that controls diverse cellular functions. Transcriptional dysregulation may contribute to the molecular pathophysiology of mood disorders. The active and continuous adaptations of chromatin and the access of gene promoters to transcription factor mechanisms offers solid rationale for the potential therapeutic use of HDAC inhibitors in mood disorders. The study of HDAC inhibitors in several behavioral models and at the level of gene expression in mood disorders is promising; furthermore, these agents may also directly target synaptic plasticity and the expression of neurotrophic factors necessary for long‐term behavior and cognitive changes observed in neurons and glial cells.
However, one concern about this class of agents is their risk of nonspecific DNA effects, which could lead to severe side effects and limits their potential clinical usefulness. Thus, this lack of selectivity and consequent potential cytotoxicity must be carefully evaluated (e.g., cardiovascular side effects were described with HDAC inhibitors in clinical trials for cancer [43]). The half‐life and affinity of any HDAC inhibitor may also define its potential risk for toxicity and undesirable side effects in vivo. The synthesis of more specific HDAC inhibitors is still in its infancy, as is the identification of nonhistone targets of these agents.
New agents able to target specific HDAC inhibitors and induce selective chromatin remodeling and gene expression effects are necessary. Key to the study of these agents in psychiatric research is our ability to define an appropriate dose, as well as identify HDAC inhibitors able to cross the blood–brain barrier. Currently, only SAHA, SB, and VPA have been consistently shown to cross the blood–brain barrier, although it appears that the structural properties of identified benzamides may allow greater brain penetration. Overall, further studies are necessary to clarify the promising role of HDACs in the pathophysiology and therapeutics of mood disorders.
Author Contributions
RMV conceptualized and conducted the review, wrote the manuscript. LI conducted the review, wrote the manuscript. CAZ conceptualized and conducted the review, wrote the manuscript.
Conflict of Interest
The authors report no conflicts of interest, financial or otherwise.
Acknowledgments
Ioline Henter provided invaluable editorial assistance. Funding for this work was supported by the Intramural Research Program of the National Institute of Mental Health, National Institutes of Health, and Department of Health and Human Services (IRP‐NIMH‐NIH‐DHHS).
References
- 1. Judd LL, Schettler PJ, Akiskal HS, Coryell W, Leon AC, Maser JD, Solomon DA. Residual symptom recovery from major affective episodes in bipolar disorders and rapid episode relapse/recurrence. Arch Gen Psychiatry 2008;65:386–394. [DOI] [PubMed] [Google Scholar]
- 2. Berk M, Hallam K, Lucas N, et al Early intervention in bipolar disorder: Opportunities and pitfalls. Med J Aust 2007;187(Suppl 7):S11–S14. [DOI] [PubMed] [Google Scholar]
- 3. McGorry PD, Nelson B, Amminger GP, et al Intervention in individuals at ultra high risk for psychosis: A review and future directions. J Clin Psychiatry 2009;70:1206–1212. [DOI] [PubMed] [Google Scholar]
- 4. Salvadore G, Drevets WC, Henter ID, Zarate CA, Manji HK. Early intervention in bipolar disorder, part I: Clinical and imaging findings. Early Interv Psychiatry 2008;2:122–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Curtis C. Sexual abuse and subsequent suicidal behaviour. Exacerbating factors and implications for recovery. J Child Sex Abuse 2006;15:1–21. [DOI] [PubMed] [Google Scholar]
- 6. Otto MW, Perlman CA, Wernicke R, Reese HE, Bauer MS, Pollack MH. Posttraumatic stress disorder in patients with bipolar disorder: A review of prevalence, correlates, and treatment strategies. Bipolar Disord 2004;6:470–479. [DOI] [PubMed] [Google Scholar]
- 7. Rutten BP, Mill J. Epigenetic mediation of environmental influences in major psychotic disorders. Schizophr Bull 2009;35:1045–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kato T, Iwamoto K, Kakiuchi C, Kuratomi G, Okazaki Y. Genetic or epigenetic difference causing discordance between monozygotic twins as a clue to molecular basis of mental disorders. Mol Psychiatry 2005;10:622–630. [DOI] [PubMed] [Google Scholar]
- 9. Santini V, Gozzini A, Ferrari G. Histone deacetylase inhibitors: Molecular and biological activity as a premise to clinical application. Curr Drug Metab 2007;8:383–393. [DOI] [PubMed] [Google Scholar]
- 10. Allis CD, Jenuwein T, Reinberg D. Overview and concepts In: Allis CD, Jenuwein T, Reinberg D, et al, editors. Epigenetics. Cold Spring Harbor , NY : Cold Spring Harbor Laboratory Press, 2007;23–61. [Google Scholar]
- 11. Monneret C. Histone deacetylase inhibitors. Eur J Med Chem 2005;40:1–13. [DOI] [PubMed] [Google Scholar]
- 12. Wolffe AP. Nucleosome positioning and modification: Chromatin structures that potentiate transcription. Trends Biochem Sci 1994;19:240–244. [DOI] [PubMed] [Google Scholar]
- 13. Shahbazian MD, Grunstein M. Functions of site‐specific histone acetylation and deacetylation. Annu Rev Biochem 2007;76:75–100. [DOI] [PubMed] [Google Scholar]
- 14. Kurdistani SK, Tavazoie S, Grunstein M. Mapping global histone acetylation patterns to gene expression. Cell 2004;117:721–733. [DOI] [PubMed] [Google Scholar]
- 15. Peterson CL, Laniel MA. Histones and histone modifications. Curr Biol 2004;14:R546–R551. [DOI] [PubMed] [Google Scholar]
- 16. Weaver IC, Cervoni N, Champagne FA, et al Epigenetic programming by maternal behavior. Nat Neurosci 2004;7:847–854. [DOI] [PubMed] [Google Scholar]
- 17. McGowan PO, Sasaki A, D’Alessio AC, et al Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nat Neurosci 2009;12:342–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Van Lint C, Emiliani S, Verdin E. The expression of a small fraction of cellular genes is changed in response to histone hyperacetylation. Gene Expr 1996;5:245–253. [PMC free article] [PubMed] [Google Scholar]
- 19. Carey N, La Thangue NB. Histone deacetylase inhibitors: Gathering pace. Curr Opin Pharmacol 2006;6:369–375. [DOI] [PubMed] [Google Scholar]
- 20. Kazantsev AG, Thompson LM. Therapeutic application of histone deacetylase inhibitors for central nervous system disorders. Nat Rev Drug Discov 2008;7:854–868. [DOI] [PubMed] [Google Scholar]
- 21. Covington HE 3rd, Maze I, LaPlant QC, et al Antidepressant actions of histone deacetylase inhibitors. J Neurosci 2009;29:11451–11460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gavin DP, Kartan S, Chase K, Jayaraman S, Sharma SP. Histone deacetylase inhibitors and candidate gene expression: An in vivo and in vitro approach to studying chromatin remodeling in a clinical population. J Psychiatr Res 2009;43:870–876. [DOI] [PubMed] [Google Scholar]
- 23. Hobara T, Uchida S, Otsuki K, et al Altered gene expression of histone deacetylases in mood disorder patients. J Psychiatr Res 2010;44:263–270. [DOI] [PubMed] [Google Scholar]
- 24. Otsuki K, Uchida S, Wakabayashi Y, Matusbara T, Hobara T, Funato H, Watanabe Y. Aberrant REST‐mediated transcriptional regulation in major depressive disorder. J Psychiatr Res 2010;44:378–384. [DOI] [PubMed] [Google Scholar]
- 25. Ng HH, Bird A. Histone deacetylases: Silencers for hire. Trends Biochem Sci 2000;25:121–126. [DOI] [PubMed] [Google Scholar]
- 26. Simonini MV, Camargo LM, Dong E, Maloku E, Veldic M, Costa E, Guidotti A. The benzamide MS‐275 is a potent, long‐lasting brain region‐selective inhibitor of histone deacetylases. Proc Natl Acad Sci USA 2006;103:1587–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kundakovic M, Chen Y, Guidotti A, Grayson DR. The Reelin and GAD67 promotors are activated by epigenetic drugs that facilitate the disruption of local repressor complexes. Mol Pharmacol 2009;75:342–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sharma R, Ottenhof T, Rzeczkowska PA, Niles NP. Epigenetic targets for melatonin: Induction of histone H3 hyperacetylation and gene expression in C17.2 neural stem cells. J Pineal Res 2008;45:277–284. [DOI] [PubMed] [Google Scholar]
- 29. Guidotti A, Auta J, Davis JM, et al Decrease in reelin and glutamic acid decarboxylase67 (GAD67) expression in schizophrenia and bipolar disorder: A postmortem brain study. Arch Gen Psychiatry 2000;57:1061–1069. [DOI] [PubMed] [Google Scholar]
- 30. Benes FM, Berretta S. GABAergic interneurons: Implications for understanding schizophrenia and bipolar disorder. Neuropsychopharmacology 2001;25:1–27. [DOI] [PubMed] [Google Scholar]
- 31. Dong E, Guidotti A, Grayson DR, Costa E. Histone hyperacetylation induces demethylation of reelin and 67‐kDa glutamic acid decarboxylase promoters. Proc Natl Acad Sci USA 2007;104:4676–4681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kundakovic M, Chen Y, Costa E, Grayson DR. DNA methyltransferase inhibitors coordinately induce expression of the human reelin and glutamic acid decarboxylase 67 genes. Mol Pharmacol 2007;71:644–653. [DOI] [PubMed] [Google Scholar]
- 33. Lu Q, Hutchins AE, Doyle CM, Lundblad JR, Kwok RP. Acetylation of cAMP‐responsive element‐binding protein (CREB) by CREB‐binding protein enhances CREB‐dependent transcription. J Biol Chem 2003;278:15727–15734. [DOI] [PubMed] [Google Scholar]
- 34. Mamdani F, Alda M, Grof P, Young LT, Rouleau G, Turecki G. Lithium response and genetic variation in the CREB family of genes. Am J Med Genet B Neuropsychiatr Genet 2008;147B:500–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Manji HK, Quiroz JA, Payne JL, Singh J, Lopes BP, Viegas JS, Zarate CA. The underlying neurobiology of bipolar disorder. World Psychiatry 2003;2:136–146. [PMC free article] [PubMed] [Google Scholar]
- 36. Wood MA, Hawk JD, Abel T. Combinatorial chromatin modifications and memory storage: A code for memory? Learn Mem 2006;13:241–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Alarcon JM, Malleret G, Touzani K, Vronskaya S, Ishii S, Kandel ER, Barco A. Chromatin acetylation, memory, and LTP are impaired in CBP+/– mice: A model for the cognitive deficit in Rubinstein‐Taybi syndrome and its amelioration. Neuron 2004;42:947–959. [DOI] [PubMed] [Google Scholar]
- 38. Guan Z, Giustetto M, Lomvardas S, et al Integration of long‐term‐memory‐related synaptic plasticity involves bidirectional regulation of gene expression and chromatin structure. Cell 2002;111:483–493. [DOI] [PubMed] [Google Scholar]
- 39. Levenson JM, O’Riordan KJ, Brown KD, Trinh MA, Molfese DL, Sweatt JD. Regulation of histone acetylation during memory formation in the hippocampus. J Biol Chem 2004;279:40545–40559. [DOI] [PubMed] [Google Scholar]
- 40. Vecsey CG, Hawk JD, Lattal KM, et al Histone deacetylase inhibitors enhance memory and synaptic plasticity via CREB:CBP‐dependent transcriptional activation. J Neurosci 2007;27:6128–6140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yeh SH, Lin CH, Gean PW. Acetylation of nuclear factor‐kappaB in rat amygdala improves long‐term but not short‐term retention of fear memory. Mol Pharmacol 2004;65:1286–1292. [DOI] [PubMed] [Google Scholar]
- 42. Machado‐Vieira R, Manji HK, Zarate CA Jr. The role of lithium in the treatment of bipolar disorder: Convergent evidence for neurotrophic effects as a unifying hypothesis. Bipolar Disord 2009;11(Suppl 2):92–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sleiman SF, Basso M, Mahishi L, Kozikowski AP, Donohoe ME, Langley B, Ratan RR. Putting the ‘HAT’ back on survival signalling: The promises and challenges of HDAC inhibition in the treatment of neurological conditions. Expert Opin Investig Drugs 2009;18:573–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Langley B, Gensert JM, Beal MF, Ratan RR. Remodeling chromatin and stress resistance in the central nervous system: Histone deacetylase inhibitors as novel and broadly effective neuroprotective agents. Curr Drug Targets CNS Neurol Disord 2005;4:41–50. [DOI] [PubMed] [Google Scholar]
- 45. Leverich GS, Altshuler LL, Frye MA, et al Factors associated with suicide attempts in 648 patients with bipolar disorder in the Stanley Foundation Bipolar Network. J Clin Psychiatry 2003;64:506–515. [DOI] [PubMed] [Google Scholar]
- 46. Schroeder FA, Lin CL, Crusio WE, Akbarian S. Antidepressant‐like effects of the histone deacetylase inhibitor, sodium butyrate, in the mouse. Biol Psychiatry 2007;62:55–64. [DOI] [PubMed] [Google Scholar]
- 47. Tsankova NM, Berton O, Renthal W, Kumar A, Neve RL, Nestler EJ. Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nat Neurosci 2006;9:519–525. [DOI] [PubMed] [Google Scholar]
- 48. Ryu H, Lee J, Olofsson BA, et al Histone deacetylase inhibitors prevent oxidative neuronal death independent of expanded polyglutamine repeats via an Sp1‐dependent pathway. Proc Natl Acad Sci USA 2003;100:4281–4286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Langley B, D’Annibale MA, Suh K, et al Pulse inhibition of histone deacetylases induces complete resistance to oxidative death in cortical neurons without toxicity and reveals a role for cytoplasmic p21(waf1/cip1) in cell cycle‐independent neuroprotection. J Neurosci 2008;28:163–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Andreazza AC, Kauer‐Sant’anna M, Frey BN, Bond DJ, Kapczinski F, Young LT, Yatham LN. Oxidative stress markers in bipolar disorder: A meta‐analysis. J Affect Disord 2008;111:135–144. [DOI] [PubMed] [Google Scholar]
- 51. Machado‐Vieira R, Andreazza AC, Viale CI, et al Oxidative stress parameters in unmedicated and treated bipolar subjects during initial manic episode: A possible role for lithium antioxidant effects. Neurosci Lett 2007;421:33–36. [DOI] [PubMed] [Google Scholar]
- 52. Leng Y, Liang MH, Ren M, Marinova Z, Leeds P, Chuang DM. Synergistic neuroprotective effects of lithium and valproic acid or other histone deacetylase inhibitors in neurons: Roles of glycogen synthase kinase‐3 inhibition. J Neurosci 2008;28:2576–2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kwon B, Houpt TA. Phospho‐acetylation of histone H3 in the amygdala after acute lithium chloride. Brain Res 2010;1333:36–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chen PS, Peng GS, Li G, et al Valproate protects dopaminergic neurons in midbrain neuron/glia cultures by stimulating the release of neurotrophic factors from astrocytes. Mol Psychiatry 2006;11:1116–1125. [DOI] [PubMed] [Google Scholar]
- 55. Wu X, Chen PS, Dallas S, et al Histone deacetylase inhibitors up‐regulate astrocyte GDNF and BDNF gene transcription and protect dopaminergic neurons. Int J Neuropsychopharmacol 2008;11:1123–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Bredy TW, Wu H, Crego C, Zellhoefer J, Sun YE, Barad M. Histone modifications around individual BDNF gene promoters in prefrontal cortex are associated with extinction of conditioned fear. Learn Mem 2007;14:268–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Chwang WB, O’Riordan KJ, Levenson JM, Sweatt JD. ERK/MAPK regulates hippocampal histone phosphorylation following contextual fear conditioning. Learn Mem 2006;13:322–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Yuan P, Zhou R, Wang Y, et al Altered levels of extracellular signal‐regulated kinase signaling proteins in postmortem frontal cortex of individuals with mood disorders and schizophrenia. J Affect Disord 2010;124:164–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lattal KM, Barrett RM, Wood MA. Systemic or intrahippocampal delivery of histone deacetylase inhibitors facilitates fear extinction. Behav Neurosci 2007;121:1125–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Gottlicher M, Minucci S, Zhu P, et al Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J 2001;20:6969–6978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Tremolizzo L, Rodriguez‐Menendez V, Sala G, Di Francesco JC, Ferrarese C. Valproate and HDAC inhibition: A new epigenetic strategy to mitigate phenotypic severity in ALS? Amyotroph Lateral Scler Other Motor Neuron Disord 2005;6:185–186. [DOI] [PubMed] [Google Scholar]
- 62. Chen PS, Wang CC, Bortner CD, et al Valproic acid and other histone deacetylase inhibitors induce microglial apoptosis and attenuate lipopolysaccharide‐induced dopaminergic neurotoxicity. Neuroscience 2007;149:203–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Gurvich N, Berman MG, Wittner BS, Gentleman RC, Klein PS, Green JB. Association of valproate‐induced teratogenesis with histone deacetylase inhibition in vivo. FASEB J 2005;19:1166–1168. [DOI] [PubMed] [Google Scholar]
- 64. Leng Y, Chuang DM. Endogenous alpha‐synuclein is induced by valproic acid through histone deacetylase inhibition and participates in neuroprotection against glutamate‐induced excitotoxicity. J Neurosci 2006;26:7502–7512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Chuang DM. The antiapoptotic actions of mood stabilizers: Molecular mechanisms and therapeutic potentials. Ann N Y Acad Sci 2005;1053:195–204. [DOI] [PubMed] [Google Scholar]
- 66. Sharma RP, Rosen C, Kartan S, Guidotti A, Costa E, Grayson DR, Chase K. Valproic acid and chromatin remodeling in schizophrenia and bipolar disorder: Preliminary results from a clinical population. Schizophr Res 2006;88:227–231. [DOI] [PubMed] [Google Scholar]
- 67. Nakajima T, Fukamizu A, Takahashi J, Gage FH, Fisher T, Blenis J, Montminy MR. The signal‐dependent coactivator CBP is a nuclear target for pp90RSK. Cell 1996;86:465–474. [DOI] [PubMed] [Google Scholar]
- 68. Steffan JS, Bodai L, Pallos J, et al Histone deacetylase inhibitors arrest polyglutamine‐dependent neurodegeneration in Drosophila. Nature 2001;413:739–743. [DOI] [PubMed] [Google Scholar]
- 69. Drummond DC, Noble CO, Kirpotin DB, Guo Z, Scott GK, Benz CC. Clinical development of histone deacetylase inhibitors as anticancer agents. Annu Rev Pharmacol Toxicol 2005;45:495–528. [DOI] [PubMed] [Google Scholar]
- 70. Meisel A, Harms C, Yildirim F, et al Inhibition of histone deacetylation protects wild‐type but not gelsolin‐deficient neurons from oxygen/glucose deprivation. J Neurochem 2006;98:1019–1031. [DOI] [PubMed] [Google Scholar]
- 71. Ferrante RJ, Kubilus JK, Lee J, et al Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington's disease mice. J Neurosci 2003;23:9418–9427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Hockly E, Richon VM, Woodman B, et al Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, ameliorates motor deficits in a mouse model of Huntington's disease. Proc Natl Acad Sci USA 2003;100:2041–2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Thomas EA, Coppola G, Desplats PA, et al The HDAC inhibitor 4b ameliorates the disease phenotype and transcriptional abnormalities in Huntington's disease transgenic mice. Proc Natl Acad Sci USA 2008;105:15564–15569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Petri S, Kiaei M, Kipiani K, Chen J, Calingasan NY, Crow JP, Beal MF. Additive neuroprotective effects of a histone deacetylase inhibitor and a catalytic antioxidant in a transgenic mouse model of amyotrophic lateral sclerosis. Neurobiol Dis 2006;22:40–49. [DOI] [PubMed] [Google Scholar]
- 75. Greene E, Mahishi L, Entezam A, Kumari D, Usdin K. Repeat‐induced epigenetic changes in intron 1 of the frataxin gene and its consequences in Friedreich ataxia. Nucleic Acids Res 2007;35:3383–3390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Herman D, Jenssen K, Burnett R, Soragni E, Perlman SL, Gottesfeld JM. Histone deacetylase inhibitors reverse gene silencing in Friedreich's ataxia. Nat Chem Biol 2006;2:551–558. [DOI] [PubMed] [Google Scholar]
- 77. Rai M, Soragni E, Jenssen K, et al HDAC inhibitors correct frataxin deficiency in a Friedreich ataxia mouse model. PLoS One 2008;3:e1958. [DOI] [PMC free article] [PubMed] [Google Scholar]