

Abstract
Heat shock protein 72 (Hsp72), a canonical intracellular molecular chaperone, may also function as an extracellular danger signal for the innate immune system. To further delineate the biological role of Hsp72 in the innate immune system, we generated two truncated versions of the full length human Hsp72 (N-terminus Hsp72, amino acids 1–430; and C-terminus Hsp72 amino acids 420–641) and directly compared their ability to activate cells from the macrophage/monocyte lineage. In RAW 264.7 macrophages transfected with a NF-κB-dependent luciferase reporter plasmid, C-terminus Hsp72 was a more potent inducer of NF-κB activity than N-terminus Hsp72, and this effect did not seem to be secondary to endotoxin contamination. C-terminus Hsp72-mediated activation of the NF-κB pathway was corroborated by increased activation of IκB kinase, degradation of IκBα, and increased NF-κB-DNA binding. C-terminus Hsp72 was a more potent inducer of tumor necrosis factor-α (TNFα) expression in RAW 264.7 macrophages and in primary murine peritoneal macrophages from wild-type mice. C-terminus Hsp72 did not induce TNFα expression in primary murine peritoneal macrophages from Toll-like receptor (TLR4) mutant mice, indicating a role for TLR4. In human THP-1 mononuclear cells, C-terminus Hsp72 induced tolerance to subsequent LPS stimulation, whereas N-terminus Hsp72 did not induce tolerance. Finally, control experiments using equimolar amounts of N-terminus or C-terminus Hsp72 demonstrated a higher biological potency for C-terminus Hsp72. These data demonstrate that the ability of human Hsp72 to serve as an activator for cells of the macrophage/monocyte lineage primarily lies in the C-terminus region spanning amino acids 420–641.
1. INTRODUCTION
Heat shock protein 72 (Hsp72) is well recognized as the major, intracellular, stress-inducible HSP allowing for cellular adaptation to severe biological stress [1]. It is now also known that Hsp72 exists in the extracellular compartment in the context of various clinical conditions [2,3]. For example, we have reported increased levels of Hsp72 in the serum of children with septic shock [4], and Lai and colleagues reported increased levels of Hsp72 in the cerebral spinal fluid of children with traumatic brain injury [5]. Clinical studies documenting increased levels of Hsp72 in the extracellular compartment have also reported correlations between measured Hsp72 levels and illness severity [4–6], thus raising the possibility that extracellular Hsp72 plays an important biological role in humans.
It has been proposed that a major biological role for extracellular Hsp72 is that of a signal for the innate immune system [7,8]. This hypothesis centers, in large part, on the ability of Hsp72 to induce NF-κB activation and pro-inflammatory gene expression in cells from the macrophage/monocyte lineage [9–11]. There is also evidence that Hsp72-mediated signaling in the innate immune system involves Toll-like receptors (TLRs) and CD14 [10–13]. Our own work has demonstrated that extracellular Hsp72 can reprogram macrophages/monocytes such that they have blunted responses to subsequent endotoxin simulation in an analogous manner to that of the phenomenon known as “endotoxin tolerance” [14,15]. Finally, we recently demonstrated that lung epithelial cells are also potential in vitro and in vivo targets for Hsp72 signaling via TLR4 [16].
Given the growing interest in Hsp72-mediated signaling in the innate immune system, we have been interested in further elucidating the biological properties of extracellular human Hsp72. In the current work we have generated two recombinant, truncated versions of the full length human Hsp72, and have used these two truncated proteins to begin testing the hypothesis that a specific region of Hsp72 accounts for its ability to activate cells of the macrophage/monocyte lineage.
2. MATERIALS AND METHODS
2.1. Cell culture
RAW 264.7 murine peritoneal macrophages (American Type Culture Collection, ATCC, Bethesda, MD) were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM, Gibco BRL, Grand Island, NY) containing 10% fetal bovine serum (FBS), 100 U/mL penicillin, 0.1 mg/mL streptomycin, 20 mM HEPES buffer, and 2.2 g/L sodium bicarbonate (Sigma, St. Louis, MO), at 37º C in a room air/5% CO2 tissue culture incubator. The human acute monocytic leukemia cell line, THP-1 (ATCC), was maintained in RPMI 1640 medium containing 10% FBS, 60 μg/ml kanamycin, 3.5 × 10−4% 2-β-mercaptoethanol, 2 mM L-glutamine, 10 mM HEPES, and 0.11 gm/L sodium pyruvate, at 37º C in a room air/5% CO2 tissue culture incubator.
2.2. Isolation of Murine Peritoneal Macrophages
Primary peritoneal macrophages were isolated from 6 to 8 week old male C3H/HeJ and C3H/HeOuJ mice (Jackson Laboratory, Bar Harbor, ME) via peritoneal lavage. Mice were housed in a laminar hood in a virus-free animal facility prior to isolation of macrophages. All experiments were conducted in accordance with the National Institutes of Health Guidelines for the Use of Laboratory Animals (National Institutes of Health Publication 85-23, revised 1996) and with approval of the Institutional Animal Care and Use Committee, Cincinnati Children’s Research Foundation. Animals were acclimatized for 7 days prior to surgical manipulation and maintained on 12-h light/dark cycles with access to food and water ad libitum. Briefly, mice were anesthetized with isoflurane and the peritoneal fascia was exposed by dissection. Three mL of sterile PBS was injected through the fascia into the peritoneal cavity using a sterile 22-G needle. Peritoneal fluid was then withdrawn through the fascia with an 18-G needle. The recovered peritoneal fluid was centrifuged at 1500 rpm × 10 min, and the pellet was isolated, resuspended in DMEM containing 10% FBS, and plated onto 24-well plates. Peritoneal macrophages were allowed to adhere for 1 hour at 37º C. This protocol typically results in >90% macrophage purity based on cytospin of the sample.
2.3. Generation of truncated Hsp72 proteins
The generation of full-length, recombinant human Hsp-72 was previously described in detail [15]. Using the full-length Hsp72 cDNA expression plasmid (puC-HSP-70, Stressgen, Victoria, British Columbia) as a template, we generated two truncated proteins:N-terminus Hsp72 (amino acids 1 to 430) and C-terminus Hsp72 (amino acids 420 to 641). The rationale for choosing these two particular sequences was simply based on the presence of convenient restriction sites in puC-HSP-70.
N-terminus Hsp72 was generated by digesting puC-HSP-70 with BamHI and SmaI and subcloning the resulting restriction digest product into the BamHI and PvuII cloning sites of the pRSET expression plasmid (Invitrogen, Carlsbad, CA). The nucleotide sequence of pRSET-N-terminus-Hsp72 was verified by the Cincinnati Children’s Research Foundation DNA Core facility. Subsequently, the E. coli strain BD21(DE3) (Novagen, Madison, WI) was transformed with pRSET-N-terminus-Hsp72 and grown for 16 h at 37°C in Luria-Bertani broth supplemented with 100 mg/ml ampicillin. The resulting cultures were diluted 100-fold with fresh Luria-Bertani medium and cultured at 37° C for 3 h while shaking at 250 rpm. Protein expression was induced by the addition of 1M isopropyl-β-D-thiogalactoside to a final concentration of 1.0 mM for 3 h while shaking at 37°C. The induced cells were lysed in BugBuster lysis buffer (Novagen) supplemented with 1:1000 benzonase nuclease. Cells were lysed for 30 minutes at room temperature with rocking. Cell debris was removed by centrifugation and the cell extracts were then loaded into a His-Bind Ni-NTA resin column (Novagen). The column was washed and the N-terminus-Hsp72 was eluted with elution buffer according to the manufacturer’s instructions. The protein was further purified using Endotrap Blue resin (Lonza-Walkersville, Atlanta, GA), according to the manufacturer’s instructions.
C-terminus Hsp72 was generated by digesting puC-HSP-70 with Bgl and HindIII, and subcloning the resulting restriction digest product into the corresponding cloning sites of the pRSET expression plasmid (Invitrogen). The nucleotide sequence of pRSET-C-terminus-Hsp72 was verified by the Cincinnati Children’s Research Foundation DNA Core facility. Subsequent bacterial transformation, induction of protein expression, protein isolation, and endotoxin removal procedures were conducted as described above.
The amino acid sequences of N-terminus and C-terminus Hsp72 were subsequently verified by the Cincinnati Children’s Research Foundation Proteomics Core facility, followed by 100% identity confirmation based on a NCBI Blast search.
Preparations of N-terminus and C-terminus Hsp72 were analyzed for endotoxin contamination by an independent laboratory (Charles River Laboratories, Wilmington, MA). The endotoxin concentration of the Hsp72 preparations was 20 ng/mg of protein. Thus, at the highest concentrations of Hsp72 used in our studies (1000 ng/ml, see Results section), the maximum concentration of endotoxin present in the Hsp72 was 20 picograms/ml.
2.4. Transient transfection and luciferase reporter assay
RAW 264.7 macrophages were transiently transfected with a 5xNF-κB luciferase reporter plasmid (Stratagene, La Jolla, CA) in duplicate, in six-well plates, at a density of 200,000 cells per well by incubation with FuGENE 6 (Roche Molecular Biochemicals, Indianapolis, IN) and serum free DMEM overnight. After transfection, cells were washed once with PBS and treated with the experimental conditions.
THP-1 cells were transiently transfected with the 5xNF-κB luciferase reporter plasmid using DEAE-dextran. Briefly, 1×106 cells per condition were pelleted and washed with S-TBS (25 mM Tris·Cl, pH 7.4, 137 mM NaCl, 5 mM KCl, 0.6 mM Na2HPO4, 0.7 mM CaCl2, and 0.5 mM MgCl2). Cells were resuspended at 1 ×106 cells/ml in S-TBS. One μg/ml of the NF-κB reporter plasmid and DEAE-dextran (100ng/ml of cells) were added to the cells. The cells were incubated at 37° C for 20min, washed twice with STBS, resuspended, and cultured in complete RPMI medium. Twenty-four hrs later cells were exposed to the experimental conditions.
All transfections were performed by co-transfection with a plasmid constitutively expressing Renilla luciferase (Promega, Madison, WI) in order to control for transfection efficiency. Cellular proteins were extracted and analyzed for dual-luciferase activity according to the manufacturer’s instructions (Promega) using a Glomax 96 microplate luminometer (Promega). NF-κB-dependent luciferase activity was normalized to the respective Renilla luciferase activity and reported as fold induction over control cells (cells that were transfected and treated with culture media alone).
2.5. Other assays
Electromobility shift assays (EMSA) for NF-κB-DNA binding, Western blot analyses for IκBα degradation, and in vitro IκB kinase assays were performed using standard procedures in our laboratories as previously published [14–17]. Enzyme-linked immunosorbent assay (ELISA) for murine tumor necrosis factor-α (TNFα) was performed using an ELISA kit and according the manufacturer’s instructions (Biosource, Camarillo, CA).
2.6. Statistical analyses
Continuous variables were analyzed using ANOVA and Bonferroni corrections for multiple comparisons (SigmaStat, Systat Software, San Jose, CA). Significance was set at a p value < 0.05.
3. RESULTS
3.1. NF-κB activation in response to truncated Hsp72
In these initial experiments we evaluated the ability of truncated Hsp72 proteins to activate NF-κB. RAW 264.7 macrophages were co-transfected with an NF-κB-dependent luciferase reporter plasmid and a Renilla luciferase plasmid (control for transfection efficiency), then subsequently treated with increasing log-fold concentrations of either N-terminus Hsp72 or C-terminus Hsp72, or a panel of control conditions. As shown in Figure 1A, treatment with N-terminus Hsp72 led to NF-κB activation (increased luciferase activity) only at the highest concentration tested (1000 ng/ml). In contrast, treatment with C-terminus Hsp72 increased activation of NF-κB in a dose response manner. At 100 ng/ml of C-terminus Hsp72 the degree of NF-κB activation was similar to that of N-terminus Hsp72 at 1000 ng/ml. Increasing the concentration of C-terminus Hsp72 to 1000 ng/ml caused a further increase of NF-κB activation that was equivalent to that of the lipopolysaccharide (LPS, E. coli O55:B5)positive control (100 ng/ml). Furthermore, the amount of C-terminus Hsp72-mediated NF-κB activity was similar to that of an equimolar concentration of full length Hsp72 (Figure 1B).
Figure 1.
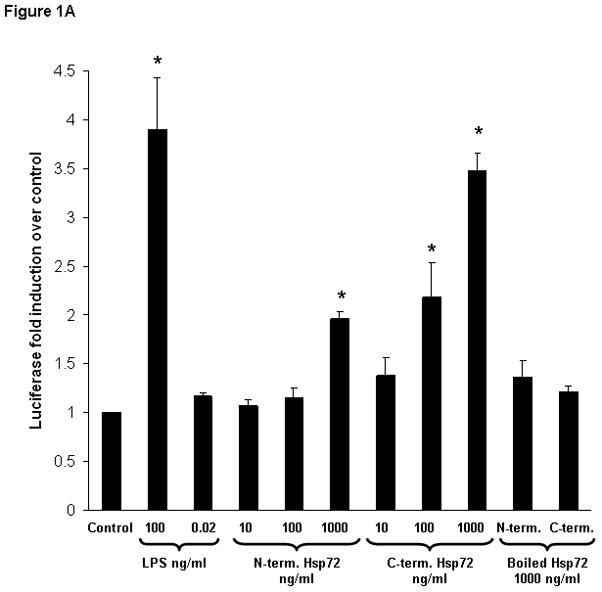
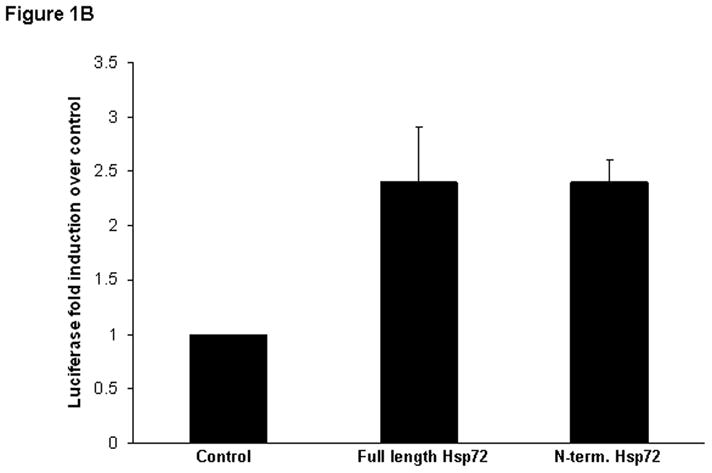
Figure 1A. Luciferase assay demonstrating the differential effects of N-terminus Hsp72 and C-terminus Hsp72 on NF-κB activation. RAW 264.7 macrophages were transiently transfected with a NF-κB-dependent luciferase reporter plasmid and treated with the indicated conditions for 4 hours. As a control for transfection efficiency, cells were co-transfected with a Renilla luciferase reporter plasmid. Control cells were treated with media. As a positive control one group of cells was treated with LPS (100 ng/ml). To control for LPS contamination of the Hsp72 preparations, a separate group of cells was treated with either an equivalent amount of LPS (0.02 ng/ml) as that found in the highest concentration of Hsp72, or Hsp72 preparations subjected to boiling for 1 hour prior to addition to the cells.
Figure 1B. Luciferase assay demonstrating equivalent luciferase induction in RAW 264.7 macrophages transiently transfected with a NF-κB-dependent luciferase reporter plasmid and treated with equimolar concentrations of full length Hsp72 or C-terminus Hsp72 for 4 hours.
In both Figures, data are expressed as fold induction (± S.E.M.) of luciferase activity over control cells and are normalized for the respective Renilla luciferase activity. Data represent 5 individual experiments, with each experimental condition performed in duplicate within each experiment, and reported as the mean fold induction over control ± S.E.M. *p < 0.05 versus control (ANOVA with Bonferroni correction for multiple comparisons).
The potential effect of LPS contamination of the Hsp72 preparations was evaluated by treating a separate group of cells with an equivalent amount of LPS (0.02 ng/ml) as that found in the 1000 ng/ml concentration of the Hsp72 preparations (heretofore called the “LPS contamination control”). Treatment with the LPS contamination control did not significantly increase NF-κB activation (Figure 1A). Wefurther evaluated the effects of LPS contamination by boiling the two truncated Hsp72 preparations (1000 ng/ml) for 1 hour prior to treating the cells. Treatment with the boiled (and presumably denatured) Hsp72 truncated proteins abolished the ability to induce NF-κB (Figure 1A).
3.2. Confirmation of NF-κB activation
The major steps leading to NF-κB activation include activation of IκB kinase (IKK), leading to phosphorylation of the NF-κB inhibitory protein, IκBα, leading to degradation of IκBα, and subsequent liberation of NF-κB to translocate to the nucleus and bind to DNA target sequences. To confirm the differential effects of N-terminus Hsp72 and C-terminus Hsp72 on NF-κB activation in RAW 264.7 macrophages, we measured these three key steps in the NF-κB activation pathway.
Using EMSA we confirmed that C-terminus Hsp72 had a greater ability to induce NF-κB-DNA binding than N-terminus Hsp72 (Figure 2, lanes 4 through 9). The specificity of this binding was indicated by cold competitor and supershift assays (Figure 2, lanes 10 through 12), and by the lack of NF-κB-DNA binding in cells treated with the LPS contamination control (Figure 2, lane 3). Using Western blot analysis we confirmed that C-terminus Hsp72 had a greater ability to induce degradation of IκBα than N-terminus Hsp72 (Figure 3, lanes 4 through 9). The specificity of this effect was again indicated by the lack of IκBα degradation in cells treated with the LPS contamination control (Figure 3, lane 3). Using an in vitro kinase assay we confirmed that C-terminus Hsp72 had a greater ability to induce IKK activity than the N-terminus Hsp72 (Figure 4, lanes 6 through 13).
Figure 2.

Representative EMSA demonstrating the differential effects of N-terminus Hsp72 and C-terminus Hsp72 on NF-κB-DNA binding. RAW 264.7 macrophages weretreated with the indicated conditions for 1 hour and then cells were harvested for isolation of nuclear proteins. Control cells were treated with media. As a positive control one group of cells was treated with LPS (100 ng/ml). To control for LPS contamination of the Hsp72 preparations a separate group of cells was treated with an equivalent amount of LPS (0.02 ng/ml) as that found in the highest concentration of Hsp72. Cold competitor and supershift assays were performed as described in the Methods section. The gel is representative of 3 different experiments with similar results.
Figure 3.
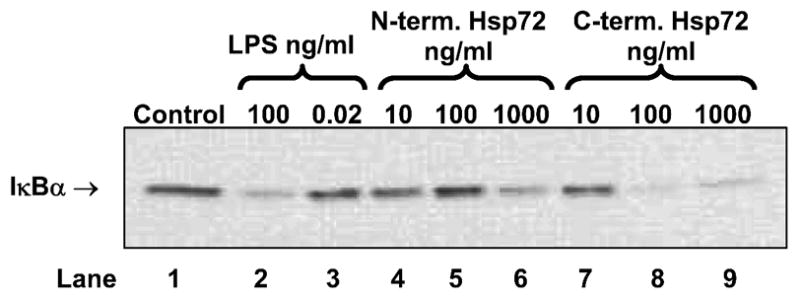
Representative Western blot demonstrating the differential effects of N-terminus Hsp72 and C-terminus Hsp72 on IκBα degradation. RAW 264.7 macrophages were treated with the indicated conditions for 30 minutes and then cells were harvested for cellular protein isolation. Control cells were treated with media. As a positive control one group of cells was treated with LPS (100 ng/ml). To control for LPS contamination of the Hsp72 preparations, a separate group of cells was treated with an equivalent amount of LPS (0.02 ng/ml) as that found in the highest concentration of Hsp72. The gel is representative of 3 different experiments with similar results.
Figure 4.
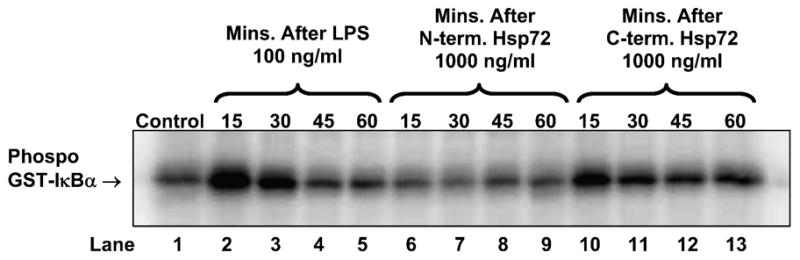
Representative in vitro IKK assay demonstrating the differential effects of N-terminus Hsp72 and C-terminus Hsp72 on IKK activity. RAW 264.7 macrophages were treated with the indicated conditions and for the indicated time periods, and then cells were harvested for cellular protein isolation. Control cells were treated with media. As a positive control one group of cells was treated with LPS (100 ng/ml). The gel is representative of 3 different experiments with similar results.
Collectively, these data demonstrate that the C-terminus of Hsp72 has a greater ability to induce NF-κB activation in RAW 264.7 macrophages than the N-terminus of Hsp72. This effect is evident from activation of IKK, through degradation of IκBα, through induction NF-κB-DNA binding, and finally through induction of NF-κB dependent gene expression (luciferase).
3.3. Proinflammatory gene expression in response to truncated Hsp72
In these experiments we further evaluated the ability of truncated Hsp72 to induce a proinflammatory phenotype in RAW 264.7 macrophages by measuring TNFα expression in the media of treated cells. Cells were treated with increasing log-fold concentrations of either N-terminus Hsp72 or C-terminus Hsp72, or a panel of control conditions. As shown in Figure 5, treatment with N-terminus Hsp72 led to TNFα expression only at the highest concentration tested (1000 ng/ml). In contrast, treatment with C-terminus Hsp72 increased TNFα expression in a dose response manner. At 100 ng/ml of C-terminus Hsp72 the degree of TNFα expression was similar to that of N-terminus Hsp72 at 1000 ng/ml. Increasing the concentration of C-terminus Hsp72 to 1000 ng/ml caused a further increase of TNFα expression that was similar to that of the LPS positive control (100 ng/ml). Treatment with the LPS contamination control did not significantly induce TNFα expression above baseline (control) levels.
Figure 5.

ELISA demonstrating the differential effects of N-terminus Hsp72 and C-terminus Hsp72 on TNFα production. RAW 264.7 macrophages were treated with the indicated conditions for 24 hours. Control cells were treated with media. As a positive control one group of cells was treated with LPS (100 ng/ml). To control for LPS contamination of the Hsp72 preparations a separate group of cells was treated with an equivalent amount of LPS (0.02 ng/ml) as that found in the highest concentration of Hsp72. Data are expressed as the mean nanograms/ml (± S.E.M.) of TNFα and represent 5 individual experiments, with each experimental condition performed in duplicate within each experiment. *p < 0.05 versus control (ANOVA with Bonferroni correction for multiple comparisons).
These data involving TNFα expression closely parallel the above data related to activation of the NF-κB pathway, and therefore provide further evidence of the differential effects of N-terminus and C-terminus Hsp72 on inducing a proinflammatory phenotype in RAW 264.7 macrophages.
3.4. TNFα expression in primary murine macrophages treated with truncated Hsp72
To ensure that our observations are not restricted to a particular transformed cell line, in these experiments we tested the ability of truncated Hsp72 to induce a proinflammatory phenotype (i.e. TNFα expression) in primary peritoneal macrophages derived from wild-type mice (C3H/HeOuJ mice). As shown in Figure 6 (black bars), treatment with 1000 ng/ml of N-terminus Hsp72 did not significantly induce TNFα expression in wild-type peritoneal macrophages. In contrast, treatment with 1000 ng/ml of C-terminus Hsp72 significantly increased TNFα expression in wild-type peritoneal macrophages and almost to the same level as that of the LPS positive control (100 ng/ml). Treatment with the LPS contamination control or boiled C-terminus Hsp72 did not induce TNFα expression.
Figure 6.

ELISA demonstrating the differential effects of N-terminus Hsp72 and C-terminus Hsp72 on TNFα production using primary peritoneal macrophages from wild-type (C3H/HeOuJ) and TLR4 mutant (C3H/HeJ) mice. Primary peritoneal macrophages were treated with the indicated conditions for 24 hours. Control cells were treated with media. As a positive control one group of cells was treated with LPS (100 ng/ml). To control for LPS contamination of the Hsp72 preparations a separate group of cells was treated with an equivalent amount of LPS (0.02 ng/ml) as that found in the highest concentration of Hsp72. Data are expressed as the mean picograms/ml (± S.E.M.) of TNFα and represent 3 individual experiments, with each experimental condition performed in duplicate within each experiment. *p < 0.05 versus respective control (ANOVA with Bonferroni correction for multiple comparisons).
3.5. Role of TLR4
To evaluate the role of TLR4 in C-terminus Hsp72-mediated activation of primary peritoneal macrophages, we conducted identical experiments using peritoneal macrophages from TLR4 mutant mice (C3H/HeJ mice). As shown in Figure 6 (gray bars) C-terminus Hsp72 was not able to induce TNFα expression in primary peritoneal macrophages derived from TLR4 mutant mice. In summary, Figure 6 demonstrates that the observed differential biological effects of N-terminus and C-terminus Hsp72 are not simply an epiphenomenon of a transformed cell line (i.e. RAW 264.7 macrophages). Rather, this differential effect is also operative in primary murine peritoneal macrophages. Furthermore, C-terminus-mediated macrophage activation appears to be TLR4-dependent.
3.6. Induction of endotoxin tolerance
We previously reported that preconditioning human mononuclear cells with full length recombinant Hsp72, at concentrations below the threshold for NF-κB induction, induced tolerance to subsequent LPS stimulation [15]. Accordingly, in these subsequent experiments we tested the ability of truncated Hsp72 to induce tolerance in a human mononuclear cell line. THP-1 cells were co-transfected with a NF-κB-dependent luciferase reporter plasmid and a Renilla luciferase plasmid (control for transfection efficiency), then subsequently preconditioned with 30 ng/ml of either N-terminus or C-terminus Hsp72 for 18 hours. After the 18 hour preconditioning period, cells were washed and treated with LPS at a concentration of 1 μg/ml for 4 hours. As shown in Figure 7, LPS significantly induced luciferase activity in cells that were not preconditioned, and the level of induction was not significantly affected by preconditioning with N-terminus Hsp72. In contrast, preconditioning with C-terminus Hsp72 significantly attenuated LPS-mediated luciferase induction. These data demonstrate that C-terminus Hsp72 has a more biologically potent ability to induce endotoxin tolerance in THP-1 cells compared to N-terminus Hsp72.
Figure 7.
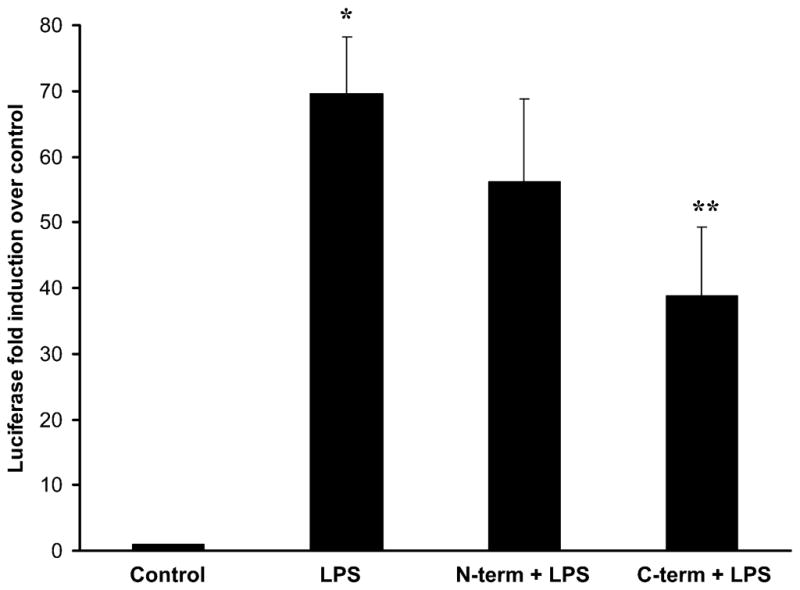
Luciferase assay demonstrating the differential ability of N-terminus Hsp72 and C-terminus Hsp72 to induce tolerance to subsequent LPS stimulation. THP-1 human mononuclear cells were transiently transfected with a NF-κB-dependent luciferase reporter plasmid. As a control for transfection efficiency, cells were co-transfected with a Renilla luciferase reporter plasmid. Control cells were maintained in basal growth media throughout the experiment. LPS treated cells were maintained in basal growth media then treated with LPS (1 μg/ml) for 4 hours. One group of cells was preconditioned with N-terminus Hsp72 (30 ng/ml) for 18 hours, then subsequently treated with LPS (1 μg/ml) for 4 hours. The final group of cells was preconditioned with C-terminus Hsp72 (30 ng/ml) for 18 hours, then subsequently treated with LPS (1 μg/ml) for 4 hours. Data are expressed as mean fold induction over control (± S.E.M.) of luciferase activity over control cells and are normalized for the respective Renilla luciferase activity. Data represent 6 individual experiments, with each experimental condition performed in duplicate within each experiment. *p < 0.05 versus control and ** p < 0.05 versus LPS alone (ANOVA with Bonferroni correction for multiple comparisons).
3.7. Biological activity of equimolar concentrations of truncated Hsp72
The data presented above support the assertion that C-terminus Hsp72 has more biological activity than N-terminus Hsp72. A potential flaw in these experiments, however, is the fact that all comparisons of N-terminus and C-terminus Hsp72 were made on the basis of equivalent weight per volume. The C-terminus Hsp72 (amino acids 420 to 641) has 209 less amino acids than the N-terminus Hsp72 (amino acids 1to 430). Thus, treating cells with equal, weight per volume concentrations would lead to ~2 fold greater molar amount of C-terminus Hsp72 than that of N-terminus Hsp72. This discrepancy could have the potential to artificially generate the differential biological activities described above. Accordingly, we conducted additional control experiments in which we transfected RAW 264.7 macrophages with the NF-κB-dependent luciferase reporter plasmid and treated them with equimolar amounts of truncated Hsp72. As shown in Figure 8, treatment with 4.2 pmol of C-terminus Hsp72 (equivalent to 100 ng/ml) significantly increased luciferase activity relative to control cells. In contrast, treatment with 4.2 pmol of N-terminus Hsp72 did not significantly increase luciferase activity relative to control cells. These data indicate that our observations regarding differential biological activity of N-terminus and C-terminus Hsp72 are not artifacts of comparisons based on equivalent weight per volume concentrations.
Figure 8.

Luciferase assay demonstrating the effect of dosing with equimolar concentrations of N-terminus and C-terminus Hsp72. RAW 264.7 macrophages were transiently transfected with a NF-κB-dependent luciferase reporter plasmid and treated with the indicated conditions for 4 hours. As a control for transfection efficiency, cells were co-transfected with a Renilla luciferase control reporter plasmid. Control cells were treated with media. The other groups were treated with 4.2 pmol N-terminus or C-terminus Hsp72 as indicated. Data are expressed as mean fold induction over control (± S.E.M.) of luciferase activity over control cells and are normalized for the respective Renilla luciferase activity. Data represent 3 individual experiments, with each experimental condition performed in duplicate within each experiment. *p < 0.05 versus control (ANOVA with Bonferroni correction for multiple comparisons).
4. DISCUSSION
The biology of Hsp72-mediated signaling in the innate immune system is an evolving field [7-9,12,18]. Our current work adds to the field by demonstrating that the C-terminus portion of human Hsp72, spanning amino acids 420 to 641, possesses substantially higher biological activity, in cells of the monocyte/macrophage lineage, than the N-terminus portion spanning amino acids 1 to 430. This assertion is supported at multiple levels, including the demonstration of biological activity in three different experimental models:a murine macrophage cell line (RAW 264.7 macrophages), primary murine peritoneal macrophages, and a human mononuclear cell line (THP-1 cells). In addition, the degree of biological activity demonstrated by C-terminus Hsp72 iscomparable to what we observe when using equimolar full length Hsp72 in these cell models [15]. Importantly, the differential biological activity between N-terminus and C-terminus Hsp72 remained evident when the two truncated proteins were compared using an equimolar dosing strategy.
We have demonstrated that C-terminus Hsp72 activates the NF-κB pathway as measured by increased NF-κB-dependent luciferase activity, increased activation of IκB kinase, degradation of IκBα, and increased NF-κB-DNA binding. Using TNFα as readout for pro-inflammatory gene expression, we have demonstrated that C-terminus Hsp72 also induces pro-inflammatory gene expression, and that this effect is dependent on TLR4. These data are consistent with the work of Asea et al, which first reported that full length Hsp72 induced NF-κB activation, in a TLR-dependent manner, in cells of the innate immune system [10,11], and our own work demonstrating full length Hsp72-mediated and TLR-dependent activation of NF-κB in respiratory epithelium [16].
We previously reported that preconditioning THP-1 cells or human peripheral blood mononuclear cells with full length Hsp72, at a low concentration that does not measurably activate the NF-κB pathway, reprograms the mononuclear cell response to subsequent LPS stimulation [15]. This observation is analogous to the well described phenomenon known as “endotoxin tolerance” in which preconditioning with low concentrations of endotoxin leads to resistance to subsequent endotoxin-mediated pro-inflammatory responses [19]. In another report, we have corroborated the ability of Hsp72 to induce endotoxin tolerance using conditioned media from macrophages subjected to heat shock [14]. This conditioned media, containing endogenously produced Hsp72, was demonstrated to also induce endotoxin tolerance when applied tonaïve, non heat shock-exposed RAW 264.7 macrophages. The specificity of Hsp72 in this process was demonstrated by experiments directed at depletion of Hsp72 from the conditioned media, which subsequently lost its ability to induce endotoxin tolerance. The current data add to these previous observations by demonstrating that C-terminus Hsp72, at an identical concentration to that of our previous studies [15], was also able to induce endotoxin tolerance in THP-1 cells. Thus, the 420 to 641 amino acid C-terminus sequence of human Hsp72 is sufficient to induce activation of NF-κB-related pro-inflammatory responses and to induce endotoxin tolerance.
Gastpar and colleagues previously reported that a 14-mer peptide sequence of human Hsp72 (TKDNNLLGREFELSG) has the ability to induce migration and cytolytic activity in human natural killer cells [20]. This 14-mer peptide spans amino acids 450 to 463 of human Hsp72. The C-terminus Hsp72 that we describe in the current manuscript contains this entire 14 amino acid sequence, thus adding to the biological plausibility of our current findings. However, using the identical experimental models and protocols that yielded our current findings, a synthetic 14-mer sequence identical to that described by Gastpar et al has not consistently induced NF-κB activation or endotoxin tolerance when compared to a scrambled control 14-mer peptide (unpublished data). Thus, it is possible that an epitope, different from the epitope reported by Gastpar and colleagues, exists within the C-terminus of human Hsp72 having relative specificity for cells of the macrophage/monocyte lineage. In this regard, we have tested a battery of synthetic 15-mer sequences spanning all portions of the C-terminus of human Hsp72, and to date, we have not been able to identify a specific sequence within the C-terminus to accountfor the observed biological activity. Thus, the exact portion of the C-terminus of human Hsp72 responsible for our observations remains to be determined.
Wang and colleagues previously reported that the C-terminus portion of Mycobacterium tuberculosis Hsp70 (amino acids 359–610) was capable of inducing cytokine production in human monocytes, whereas the N-terminus portion of Mycobacterium tuberculosis Hsp70 did not have this capacity [21]. This same group of investigators subsequently found a 20-mer epitope within the C-terminus portion of Mycobacterium tuberculosis Hsp70 (amino acids 407 to 426; sequence QPSVQIQVYQGEREIAAHNK) having the ability to modulate cytokine production and maturation of dendritic cells [22]. Based on a NCBI Blast search, we found that the first 13 amino acids of this 20-mer sequence derived from Mycobacterium tuberculosis Hsp70 has 76% homology with amino acids 435 to 447 of human Hsp72. In the same publication, Wang and colleagues identified a 40-mer epitope within the C-terminus portion of Mycobacterium tuberculosis Hsp70 (amino acidsto 457 to 496) having the ability to inhibit cytokine production and maturation of dendritic cells [22]. Based on a NCBI Blast search, we found that this 40-mer inhibitory sequence derived from Mycobacterium tuberculosis Hsp70 has 53% homology with amino acids 485 to 525 of human Hsp72. Collectively, these data by Wang and colleagues involving the C-terminus of Mycobacterium tuberculosis Hsp70 further add to the biological plausibility of our current findings and will serve to direct our future studies focused on the C-terminus of human Hsp72.
An important issue surrounding our current studies, as well as any study involving recombinant proteins that are applied to cells of the innate immune system, isthat of endotoxin contamination functioning as a confounding variable. Indeed, previous studies have suggested that the putative biological signaling properties of recombinant HSPs are merely artifacts of endotoxin contamination [23,24]. Our current studies have taken a number of steps to address this highly important issue of endotoxin contamination. These include a protocol for endotoxin removal, independent verification of low endotoxin contamination in the protein preparations, and experimental controls directly addressing endotoxin contamination. Two of our previous studies also support direct biological activity of Hsp72, rather than artifactual endotoxin contamination. First, we have demonstrated that media from cells exposed to heat shock, containing endogenously produced Hsp72 and thus no external endotoxin source, is capable of inducing endotoxin tolerance [14]. Second, we have demonstrated that a human respiratory epithelial cell line, which is typically highly resistant to endotoxin stimulation, readily responds to Hsp72 stimulation at concentration ranges similar to the ones used in the current studies [16]. Thus, although we cannot completely rule out the confounding effects of minute amounts of endotoxin contamination, we contend that the data reported in the current study reflect, at least in part, direct biological activity of Hsp72.
In summary, we have demonstrated that the C-terminus of human Hsp72, which represents the conserved substrate binding domain of full length Hsp72 [25,26], seems to be primarily responsible for the biological activity of Hsp72 in cells of the macrophage/monocyte lineage. Our data are consistent with previous data involving other cells of the innate immune system and Hsp72 of microbial origin [20–22]. Finally, the data provide the foundation for identifying a specific amino acid sequence within Hsp72 conferring the ability to serve as an activator for monocytes and macrophages.
Acknowledgments
Supported by NIH grants R01-GM061723 (H.R.W.), K12-HD047349 (S.E.P.), and K08-GM077432 (D.S.W.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wheeler DS, Wong HR. Free Radic Biol Med. 2007;42:1–14. doi: 10.1016/j.freeradbiomed.2006.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Calderwood SK, Mambula SS, Gray PJ, Jr, Theriault JR. FEBS Lett. 2007;581:3689–94. doi: 10.1016/j.febslet.2007.04.044. [DOI] [PubMed] [Google Scholar]
- 3.Johnson JD, Fleshner M. J Leukoc Biol. 2006;79:425–34. doi: 10.1189/jlb.0905523. [DOI] [PubMed] [Google Scholar]
- 4.Wheeler DS, Fisher LE, Jr, Catravas JD, Jacobs BR, Carcillo JA, Wong HR. Pediatr Crit Care Med. 2005;6:308–11. doi: 10.1097/01.PCC.0000161075.97355.2E. [DOI] [PubMed] [Google Scholar]
- 5.Lai Y, Kochanek PM, Adelson PD, Janesko K, Ruppel RA, Clark RS. J Neurotrauma. 2004;21:229–37. doi: 10.1089/089771504322972022. [DOI] [PubMed] [Google Scholar]
- 6.Pittet JF, Lee H, Morabito D, Howard MB, Welch WJ, Mackersie RC. J Trauma. 2002;52:611–7. doi: 10.1097/00005373-200204000-00001. discussion 617. [DOI] [PubMed] [Google Scholar]
- 7.Fleshner M, Johnson JD. Int J Hyperthermia. 2005;21:457–71. doi: 10.1080/02656730500088211. [DOI] [PubMed] [Google Scholar]
- 8.Moseley P. Immunopharmacology. 2000;48:299–302. doi: 10.1016/s0162-3109(00)00227-7. [DOI] [PubMed] [Google Scholar]
- 9.Asea A. Curr Immunol Rev. 2006;2:209–215. doi: 10.2174/157339506778018514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Asea A, Rehli M, Kabingu E, Boch JA, Bare O, Auron PE, Stevenson MA, Calderwood SK. J Biol Chem. 2002;277:15028–34. doi: 10.1074/jbc.M200497200. [DOI] [PubMed] [Google Scholar]
- 11.Asea A, Kraeft SK, Kurt-Jones EA, Stevenson MA, Chen LB, Finberg RW, Koo GC, Calderwood SK. Nat Med. 2000;6:435–42. doi: 10.1038/74697. [DOI] [PubMed] [Google Scholar]
- 12.Asea A. Handb Exp Pharmacol. 2008:111–27. doi: 10.1007/978-3-540-72167-3_6. [DOI] [PubMed] [Google Scholar]
- 13.Vabulas RM, Ahmad-Nejad P, Ghose S, Kirschning CJ, Issels RD, Wagner H. J Biol Chem. 2002;277:15107–12. doi: 10.1074/jbc.M111204200. [DOI] [PubMed] [Google Scholar]
- 14.Abboud PA, Lahni PM, Page K, Giuliano JS, Jr, Harmon K, Dunsmore KE, Wong HR, Wheeler DS. Shock. 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aneja R, Odoms K, Dunsmore K, Shanley TP, Wong HR. J Immunol. 2006;177:7184–92. doi: 10.4049/jimmunol.177.10.7184. [DOI] [PubMed] [Google Scholar]
- 16.Chase MA, Wheeler DS, Lierl KM, Hughes VS, Wong HR, Page K. J Immunol. 2007;179:6318–24. doi: 10.4049/jimmunol.179.9.6318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grossman BJ, Shanley TP, Odoms K, Dunsmore KE, Denenberg AG, Wong HR. Inflammation. 2002;26:129–37. doi: 10.1023/a:1015552515183. [DOI] [PubMed] [Google Scholar]
- 18.Kaczorowski DJ, Mollen KP, Edmonds R, Billiar TR. J Leukoc Biol. 2008;83:546–52. doi: 10.1189/jlb.0607374. [DOI] [PubMed] [Google Scholar]
- 19.Cavaillon JM, Adib-Conquy M. Crit Care. 2006;10:233. doi: 10.1186/cc5055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gastpar R, Gross C, Rossbacher L, Ellwart J, Riegger J, Multhoff G. J Immunol. 2004;172:972–80. doi: 10.4049/jimmunol.172.2.972. [DOI] [PubMed] [Google Scholar]
- 21.Wang Y, Kelly CG, Singh M, McGowan EG, Carrara AS, Bergmeier LA, Lehner T. J Immunol. 2002;169:2422–9. doi: 10.4049/jimmunol.169.5.2422. [DOI] [PubMed] [Google Scholar]
- 22.Wang Y, Whittall T, McGowan E, Younson J, Kelly C, Bergmeier LA, Singh M, Lehner T. J Immunol. 2005;174:3306–16. doi: 10.4049/jimmunol.174.6.3306. [DOI] [PubMed] [Google Scholar]
- 23.Gao B, Tsan MF. J Biol Chem. 2003;278:174–9. doi: 10.1074/jbc.M208742200. [DOI] [PubMed] [Google Scholar]
- 24.Gao B, Tsan MF. J Biol Chem. 2003;278:22523–9. doi: 10.1074/jbc.M303161200. [DOI] [PubMed] [Google Scholar]
- 25.Daugaard M, Rohde M, Jaattela M. FEBS Lett. 2007;581:3702–10. doi: 10.1016/j.febslet.2007.05.039. [DOI] [PubMed] [Google Scholar]
- 26.Bukau B, Weissman J, Horwich A. Cell. 2006;125:443–51. doi: 10.1016/j.cell.2006.04.014. [DOI] [PubMed] [Google Scholar]