

Abstract
The use of novel drugs and chemicals requires reliable data on their potential toxic effects on humans. Current test systems are mainly based on animals or in vitro–cultured animal-derived cells and do not or not sufficiently mirror the situation in humans. Therefore, in vitro models based on human pluripotent stem cells (hPSCs) have become an attractive alternative. The article summarizes the characteristics of pluripotent stem cells, including embryonic carcinoma and embryonic germ cells, and discusses the potential of pluripotent stem cells for safety pharmacology and toxicology. Special attention is directed to the potential application of embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs) for the assessment of developmental toxicology as well as cardio- and hepatotoxicology. With respect to embryotoxicology, recent achievements of the embryonic stem cell test (EST) are described and current limitations as well as prospects of embryotoxicity studies using pluripotent stem cells are discussed. Furthermore, recent efforts to establish hPSC-based cell models for testing cardio- and hepatotoxicity are presented. In this context, methods for differentiation and selection of cardiac and hepatic cells from hPSCs are summarized, requirements and implications with respect to the use of these cells in safety pharmacology and toxicology are presented, and future challenges and perspectives of using hPSCs are discussed.
Keywords: Embryonic stem (ES) cells, Embryonic carcinoma (EC) cells, Induced pluripotent stem (iPS) cells, Mouse, Human, Pluripotency, Differentiation, Embryonic stem cell test (EST), In vitro embryotoxicity, Cardiotoxicity, Hepatotoxicity
Introduction
During their whole life, human beings are exposed to xenobiotics, such as diverse chemical substances, pharmaceutical drugs and other potentially hazardous chemical and physical environmental factors. These expositions may hold serious risks for the individual and may lead to tissue-specific damages, but may also result in the development of tumours. In the case that early embryos or germ cells are affected, cytotoxic, mutagenic or teratogenic xenobiotics may induce embryotoxic effects or malformations of the offspring.
At present, a hierarchy of accepted test systems using in vitro and in vivo models are in use to analyse the potential toxic effects of pharmaceutical drugs and chemicals (OECD 2008; Stummann et al. 2009a; reviewed in Vojnits and Bremer 2010).
Many toxicological test systems are problematic, because toxicity tests using animals are not representative for human beings due to species-specific pharmaco-toxicological effects. Such inter-species differences were detected for example, in the cases of 13-cis retinoic acid (Anon 1987; Hendrickx 1998) and of thalidomide (Nau 1990; Nau 1993; Tzimas et al. 1994). Specifically, the dramatic consequences of the teratogenic effects of thalidomide in human beings urgently led to the need of new and optimized human-specific test systems.
Another aspect is the high number of animals that are required for toxicology testing. For example, the necessity to raise data on the potential toxicity of at least 30.000 chemical compounds under the REACH (Registration, Evaluation, Authorisation and Restriction of Chemicals) programme of the European Union is expected to require up to 10 million experimental animals (Gilbert 2010). In addition, animal tests are not adequately standardized. While human cellular test systems would overcome the problem of species specificity, in vitro–cultured human immortalized cell lines do not represent normal cell types, and human primary cells cultured in vitro usually loose their tissue-specific functions.
The recent applications of pluripotent stem cells and their derivatives in toxicology and drug research provide new alternatives to the standard routine tests performed by the industry and offer new strategies for chemical safety assessment (reviewed in Laustriat et al. 2010; Trosko and Chang 2010). Pluripotent stem cells (PSCs) are characterized by unique capacities of self-renewal and differentiation and have significant advantages in comparison with somatic cells. PSCs are available as undifferentiated embryonic stem cells (ESCs) and can be cultured in vitro as permanent lines. Specifically, human ESCs (hESCs) established from surplus embryos after in vitro fertilization are available with more than thousand individual cell lines (Löser et al. 2010).
The most important and relevant factor for toxicity testing is the functional capacity of the in vitro–cultured cells. ESCs do not need immortalization to maintain proliferation capacity and ESC derivatives are functional in vitro and after transplantation in vivo. ESCs recapitulate in vitro early processes of embryonic development and show tissue-specific expression profiles. ESCs are amenable to genetic modifications, and genetic markers for selection and cell enrichment can be introduced into the cells. In combination with three-dimensional (3D) culture supported by extracellular matrix (ECM) proteins, human stem cell-based systems can mimic the microenvironment of the in vivo niche, thus providing adequate human-specific models for pharmaco-toxicology (reviewed in Trosko and Chang 2010).
One of the major problems for the pharmaceutical industry is the late-stage attrition of novel compounds. For example, between 1991 and 2000 only 11% of compounds that entered a clinical trial were registered, and even after registration 23% of compounds failed. Safety issues accounted for 30% of late attrition and are, in addition to late manifestation of low efficacy, a major hurdle in the development and market introduction of novel drugs (Kola and Landis 2004). Consequently, there is an urgent need for improved test systems that have the capacity to predict toxicity in a human-specific manner and in an early phase of drug development.
While initial hopes raised by hESCs were directed towards their potential use in replacement therapies, the focus of research has now shifted to the development of pluripotent cell-based models for drug research, toxicological test systems and disease modelling in vitro. Of the top 20 pharmaceutical companies, 14 (70%) have launched research activities involving stem cells and 64% of those are dealing with hESCs (Jensen et al. 2009). Collaborative research activities between large pharmaceutical companies and research institutions or small start-up companies have been initiated within the last years to develop hPSC-based in vitro systems for drug discovery and predictive toxicology (Baker 2010). The increasing interest of the pharmaceutical industry in using pluripotent human stem cells in safety pharmacology and toxicology is also illustrated by collaborative programmes between the industry and research institutions on the development of hPSC-based in vitro test systems, such as the consortium Stem Cells for Safer Medicine (SC4SM; Jha 2007).
The following article will discuss the unique properties of different types of pluripotent stem cells for toxicological studies with special consideration of hPSCs and their use in developmental as well as in cardiac and hepatic toxicology. Future developments may also result in the establishment of test systems using hPSC-derived keratinocytes (Green et al. 2003; Guenou et al. 2009; reviewed in Laustriat et al. 2010), fibroblasts (Cao et al. 2008) or neural cells (Zeng et al. 2006; Ylä-Outinen et al. 2010). Here, we concentrate on current efforts to establish hPSC-based models for studies of cardiotoxicity and hepatotoxicity: both cell types are being applied to many toxicology studies so far and are good examples to demonstrate the potential and the current problems of stem cell-based in vitro toxicological test systems. With respect to ethical and legal considerations of using hESCs for research and commercial applications we refer to national (e.g. German Stem Cell Act 2002; NIH Guidelines 2009) and international guidelines (e.g. ISSCR Guidelines 2006; Guidelines of the European Group on Ethics in Science 2007) and to published articles (Leist et al. 2008).
Characteristics of in vitro–cultured pluripotent stem cells
Embryonic carcinoma (EC) cells
The first pluripotent stem cells were detected as the stem cells of testicular germ cell tumours in specific strains (Sv129/ter) of mice, called teratocarcinomas (Stevens and Little 1954; Stevens 1964; Stevens 1970). The tumours contained embryonic carcinoma (EC) cells, which were able to multiply and generate differentiated cells, thus fulfilling the criteria of being a stem cell. Besides testicular teratomas, mouse strains that spontaneously develop ovarian teratomas as result of defective oocyte development also were described (Andrews 2002).
Teratocarcinoma-derived EC cells were transferred to in vitro culture and established as permanent cell lines, which proliferated indefinitely and retained the ability to differentiate in vitro into various cell types (Kahan and Ephrussi 1970; Evans 1972; Jakob et al. 1973). EC cells have been cultivated in the undifferentiated state on mouse embryonic fibroblasts (MEF) used as feeder layer (FL) cells and differentiated by treatment with substances, such as retinoic acid (RA) or dimethyl sulfoxide (DMSO) and by cultivation in “hanging drops” or in suspension to form cell aggregates called “embryoid bodies” (EBs). EBs are not embryo-derivatives, but are composed of progenitor cells of all three primary germ layers, which are irregularly arranged in the aggregates. After in vitro differentiation of EC cells with chemical inducers, tissue-specific genes are expressed and specialized cells including developing heart, skeletal muscle, cartilage, or nerve cells are formed (McBurney et al. 1982; Wobus et al. 1994b).
Using this cellular model, for the first time, embryonic development could be studied under in vitro conditions, but definitive pluripotency, that is in vivo development into both somatic and germ line cells, has not been shown. To test the developmental capacity, EC cells were transplanted into mouse blastocysts and after transfer of the blastocysts into the uterus of pseudo-pregnant foster mothers, resulted in chimeric mice, in which variable somatic tissues partially originated from EC cells (=“injection chimeras”; Brinster 1974). However, EC cells did not colonize the germ line and frequently gave rise to the formation of tumours in the offspring (Papaioannou et al. 1978), limiting their further use in developmental biology. Obviously, due to the formation of chromosome and gene mutations that appeared during the extra-uterine growth of embryonic cells as teratocarcinomas, the tissue participation of EC cells in chimeras was limited and tumours developed in the animals. However, EC cells are still used as cellular in vitro model. Specifically, human EC cells isolated from human testicular or ovarian teratocarcinomas have been applied to generate human-specific cell surface antigens (Andrews 2002).
Embryonic stem (ES) and embryonic germ (EG) cells
Instead of cultivating embryo-derived EC cells of mice, in 1981 early mouse embryos were directly transferred into tissue culture and established as permanent embryonic stem (ES) cells (Evans and Kaufman 1981; Martin 1981). ESCs have been cultured on MEF-FL cells, and after the identification of the differentiation-inhibiting activity (DIA) that represented the leukaemia-inhibitory factor (LIF; Smith et al. 1988; Williams et al. 1988), different murine (m) ESC lines were established, which were able to proliferate and differentiate into cell types of all three primary germ layers. The cells formed in vitro functional cells of the heart, skeletal muscle, nerve system, blood, vascular, liver, pancreas and other tissues, thereby recapitulating early developmental processes (reviewed in Wobus and Boheler 2005). Later on, ESCs were also induced to differentiate into female (Hübner et al. 2003) and male (Toyooka et al. 2003) germ-like cells.
The true pluripotency of mESCs was shown by injection into blastocysts (Bradley et al. 1984), a technique that was later modified by aggregating ESCs and blastomeres, called the “sandwich technique” (Nagy et al. 1993) or “tetraploid embryo complementation” (Eggan et al. 2001). These methods allowed the generation of offspring that completely originated from ESCs. Furthermore, techniques for the genetic manipulation of ESCs by introducing genes (gain-of-function) or selectively turning off genes (loss-of-function) were established (reviewed in Wobus and Boheler 2005). In gene-targeting (loss-of-function) experiments, ESCs served as vehicle for the selective inactivation of genes by homologous recombination (Thomas and Capecchi 1987), which so far resulted in the creation of more than thousand “knock-out mice” with specific genetic defects.
At that time, only a few groups analysed the in vitro differentiation of mESCs. This changed in 1998, when James Thompson succeeded in the establishment of the first human (h) ESC lines from the inner cell mass (ICM) of human blastocysts (Thomson et al. 1998). hESCs show indefinite proliferation on FL cells, a normal karyotype and high developmental capacity in vitro (Reubinoff et al. 2000; reviewed in Stojkovic et al. 2004; Wobus and Boheler 2005; Murry and Keller 2008). The pluripotency of hESCs is usually tested by teratoma formation after transplantation into immunodeficient mice. The generation of specialized cell types from hESCs opened the perspective of generating functional human cells for regenerative therapies.
At about the same time as the first hESC derivation, human embryonic germ (EG) cells were established from 5- to 7-week-old aborted human foetuses (Shamblott et al. 1998). Human EG cells showed multi-lineage differentiation potential, but limited proliferation and could be propagated only as EB derivatives. This is in contrast to murine EG cells, which were already generated in 1992 by in vitro culture of primordial germ cells from 9.5 to 11.5 d p.c. mouse embryos (Matsui et al. 1992; Resnick et al. 1992; Labosky et al. 1994). Murine EG cells showed properties similar to those of mESCs and were able to re-enter the germ line (Labosky et al. 1994; Stewart et al. 1994, see Table 1 ). When human EG cell-differentiated neural derivatives were transplanted into an animal model for neurorepair, they showed some regenerative potential (Kerr et al. 2003), suggesting that hEG cells might possibly be an alternative to hESCs for therapeutic use. However, the difficult isolation from human foetuses and the limited proliferative capacity restrict the applicability of hEG cells.
Table 1.
Properties of mouse and human pluripotent cell populations grown in vitro (modif. according to Pera and Tam 2010 and Durcova-Hills et al. 2008)
| Types of stem cells | Stem cell-associated genes | Cell surface markers | Response to factors | Developmental potential | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Oct4 | Nanog | Sox2 | Klf4 | Dppa3 | Rex1 | Gbx2 | Fgf5 | SSEA1 | SSEA3 SSEA4 | Alkaline phosphatase | LIF | Nodal and/or activin | FGF2 | Teratoma formation | Chimaera formation | |
| Mouse EC cells | + | +? | ? | ? | ? | +? | ? | ? | + | – | + | –? | –? | – | + | – (only somatic) |
| Mouse ES cells | + | + | + | + | + | + | + | – | + | – | + | + | –# | – | + | + |
| Mouse EG cells | + | + | + | + | + | + | ? | –? | + | – | + | + | –? | + | + | + |
| Mouse EPL cells | + | + | + | ND | – | – | – | + | + | – | + | +§ | ND | ND | + | – |
| Mouse EpiSCs | + | + | + | – | – | – | – | + | + | – | – | – | + | +& | + | – |
| Human EC cells | + | + | + | ? | ? | + | ? | ? | – | + | + | – | ? | ND! | ||
| Human ES cells | + | + | + | + | + | + | + | – | – | + | + | – | + | + | + | ND! |
+, The gene or cell surface marker is expressed, the growth factors are required for self-renewal or the cells form teratomas or chimaeras; –, the gene or cell surface marker is not expressed, the indicated factors are not required for self-renewal, or the cells do not form tumours or chimaeras, ND, not done; ND!, cannot to be done for obvious ethical reasons!
#One study shows long-term self-renewal; § EPL cells grown in LIF revert to an ES-like state; & EpiSCs are derived and maintained in the presence of these factors, but dependence on the factors for self-renewal has not been rigorously examined
It should be also mentioned that murine germ line stem cells were also isolated from adult testes and cultivated as spermatogonial stem cells (SSCs, Guan et al. 2006). These cells showed multi-lineage potential in vitro, but no germ line participation, whereas another germ cell-derived murine stem cell line showed true pluripotency (Ko et al. 2009). However, the derivation of pluripotent stem cell lines from adult human testis (Conrad et al. 2008) has not been repeated so far and is controversial (Ko et al. 2010).
Because of intensive public and scientific discussion on ethical implications of destroying early embryos in the process of hESC derivation, alternative sources for human pluripotent stem cell lines have been exploited. For example, hESC lines have also been obtained from single blastomeres of human embryos without embryo destruction (Chung et al. 2008). In addition, developmentally arrested embryos or clinically unsuitable embryos found to be aneuploid on preimplantation genetic screening (PGS) were used to produce hESCs (Zhang et al. 2006; Peura et al. 2007; Peura et al. 2008; Narwani et al. 2010). Interestingly, the majority of embryos with chromosomal aneuploidies resulted in genetically normal hESC lines, suggesting mosaicism of preimplantation human embryonic cells (Peura et al. 2008; Narwani et al. 2010). At present, there are more than 100 hESC lines derived from clinically unsuitable embryos analysed by preimplantation genetic diagnosis (PGD) for genetic alterations linked to heritable diseases. These “disease-specific” hESC lines (overview in Löser et al. 2010) are considered as valuable cellular models for investigating the pathogenesis of hereditary diseases and for establishing novel diagnostic and therapeutic strategies (reviewed in Colman and Dreesen 2009).
As mentioned earlier, the ability of pluripotent cells to grow in vitro for a prolonged time allowed the introduction of genes into the genome (gain-of-function) or the inactivation of preselected genes by gene targeting via homologous recombination (loss-of-function). Both techniques are routine strategies for mouse ESCs and allowed to analyse the effects of constitutively expressed genes (e.g. Rohwedel et al. 1995; Blyszczuk et al. 2003) or of gene inactivation on in vitro differentiation (e.g. Fassler et al. 1996; reviewed in Prelle et al. 2002; Wobus and Boheler 2005). The in vitro loss-of-function strategy was extremely helpful to analyse the molecular and cellular properties of knock-out murine ESCs leading to embryonic lethality in vivo. For example, β1 integrin-deficient embryos are dying around the time of implantation, but the role of β1 integrin deficiency on early development could only be analysed by in vitro differentiation into cardiac, skeletal muscle and neural cells (Fassler et al. 1996; Rohwedel et al. 1998).
Further advances in genetic modifications of ESCs were integration-independent extra-chromosomal expression, recombineering and RNA interference (reviewed in Wobus and Boheler 2005; Zeng and Rao 2008). These advanced genetic modification methods require the establishment of further strategies, including vector design (DNA, BACs, YACs), gene expression from constitutive or regulated promoters, positive or negative selection, and the establishment of specific transfection routes.
Whereas the technique of homologous recombination in mESCs resulted in thousands of knock-out animals, the adaptation of this technique to hESCs is difficult, partially due to low homologous recombination efficiencies and targeting to non-expressed genes. Homologous recombination in hESCs was first demonstrated by Zwaka and Thomson (2003) using both knock-out and knock-in strategies. Inactivation was successful for the hypoxanthin phosphoribosyl transferase (hprt) 1 gene since it is located on the X chromosome, and inactivation of the hprt1 gene was achieved in the male (XY) hESC line H1.1. For the knock-in strategy, oct4/gfp reporter hESC lines were created, in which the GFP reporter gene was inserted into one allele of the pluripotency gene Oct4. The homologous recombination efficiency for both genes was 10−5 to 10−6, which is in the range of those observed in mESCs (Zwaka and Thomson 2003). There are only few other examples of successful gene targeting in hESCs until now (e.g. Urbach et al. 2004; Irion et al. 2007; Table 2), and the low efficiency of homologous recombination is still limiting studies on genetically modified hESCs (reviewed in Nakayama 2010; Tenzen et al. 2010). More recently, zinc-finger nucleases enhanced homologous recombination was shown to work efficiently for introducing genetic modifications into specific sites of hESCs (Lombardo et al. 2007; Hockemeyer et al. 2009; Zou et al. 2009). In addition, RNAi technology and gene trapping were successfully applied to hESCs to silence gene function (Zaehres et al. 2005; reviewed in Ben-Nun and Benvenisty 2006; see also Zeng and Rao 2008).
Table 2.
Methods for targeted gene modifications in human pluripotent stem cells
| Method | References | Cell type(s) and cell line(s) used | Targeted gene(s) | Purpose of research; comments |
|---|---|---|---|---|
| Common Homologous Recombination (HR) | Zwaka and Thomson (2003) | hES (H1.1) | HPRT1; OCT4 | Proof-of-concept for homologous recombination in hES cells |
| Urbach et al. (2004) | hES (H13) | HPRT1 | Modelling of Lesch-Nyhan syndrome in hES cells | |
| Irion et al. (2007) | hES (HES-2) | Rosa 26 human equivalent | Definition of a site suitable for gene transfer in hES cells | |
| Davis et al. (2008) | hES (HES-3) | MIXL1 | Establishment of cell lines for lineage selection in hematopoietic differentiation; Cre-lox-mediated excision of marker genes | |
| Suzuki et al. (2008) | hES (KhES1, KhES3) | HPRT1 | Proof-of-concept of adeno-associated virus (AAV)-based gene transfer for HR in hES cells | |
| Mitsui et al. (2009) | hES (KhES1; KhES3) hiPS (243H7; 246H1) | HPRT1; NANOG | Proof of concept for efficiency of HR in hES cells using adeno-associated virus-based gene transfer vectors | |
| Ruby and Zheng (2009) | hES (HUES9) | FEZF2 | Proof-of-concept for effective HR in HUES9 cells, establishment of cell lines for lineage selection of subcortical projection neurons | |
| Xue et al. (2009) | hES (BG01) | OLIG2 | Establishment of cell lines for lineage selection in neural differentiation | |
| Khan et al. (2010) | hES (H1; BG01; BG02) hiPS (several clones) | HPRT1; HMGA1; Fibroblasts subsequently used for hiPS cell generation | Proof-of-concept of AAV-based gene transfer for HR in hES and hiPS cells; hiPS cell generation from human cells subsequent to gene targeting | |
| Song et al. (2010) | hES (HUES3, HUES8, HUES9) | P53, ATM1; HPRT1 | Establishment of p53−/− and ATM−/− hES cell lines; use of bacterial artificial chromosome (BAC)-based targeting vectors | |
| Zinc finger nuclease (ZFN)—enhanced homologous recombination | Lombardo et al. (2007) | hES (HUES1, HUES3) | CCR5 | Proof-of-concept of zinc-finger nuclease (ZFN)–enhanced HR in multiple human cell types including hES cells; use of an integrase-defective lentiviral vector for gene transfer |
| Hockemeyer et al. (2009) | hES (BG01); hiPS (several clones) | Oct4; PP1R12C (AAVS1 locus); PITX3 | Proof-of-concept for zinc-finger nuclease (ZFN)–enhanced HR into expressed and non-expressed sites in hES cells | |
| Zou et al. (2009) | hES (H9, H1) hiPS (two lines) | Lentivirally transduced mutated eGFP gene; PIG-A | Proof-of-concept for zinc-finger nuclease (ZFN)–enhanced HR in hES cells; targeting of a disease-related gene in hES cells | |
| Site-specific (phage-derived) recombination systems | Di Dominico et al. (2008) | hES (H1) | HPRT1 | Establishment of a Cre/Lox P-based gene replacement system; proof-of-concept for effective recombinase-mediated cassette exchange |
| Thyagarajan et al. (2008) | hES (BG01v) | Pseudo attP sites in the human genome | Proof-of-concept for functionality of the Streptomyces phage phiC31-based recombination system in hES cells | |
| Sakurai et al. (2010) | hES (KhES1) | HPRT1 | Establishment of a Cre/Lox P-based gene replacement system |
The comparative investigation of mouse and human ESCs revealed significant differences between both species. hESCs grow independent of LIF (but need FL cells or FL-conditioned medium) but are dependent on FGF2- and Activin/Nodal-controlled signalling pathways (reviewed in Schnerch et al. 2010). hESCs partially express different cell surface molecules than mESCs (e.g. SSEA-3/4, instead of SSEA-1). Comparative gene expression analyses indicated a higher similarity of hESCs with murine postimplantation-derived epiblast stem cells (mEpiSC) than with blastocyst-derived mESCs (Brons et al. 2007; Tesar et al. 2007).
In summary, both murine and human ESCs resemble pluripotent cells of the ICM, multiply almost indefinitely and differentiate into derivatives of all three primary germ layers. Thus, ESCs fulfil the criteria of “true” stem cells and represent an “immortal” cell population, which in contrast to somatic cells of the body (under adequate culture conditions) may be maintained without signs of ageing. However, due to their unlimited proliferation in vitro and a high telomerase activity, ESCs may also behave as tumour cells (reviewed in Wobus 2010). Specifically, for hESCs the only test for pluripotency includes the transplantation of cells into immunodeficient mice resulting in teratoma (or, in rare cases, teratocarcinoma) formation. On the other hand, in vitro differentiation into functional cell derivatives of the three germ layers opens the unique potential for applications of hESCs as cellular model in pharmaco-toxicology and developmental toxicology.
Induced pluripotent stem (iPS) cells
An alternative pluripotent stem cell type of significance for toxicological studies has been introduced only a few years ago: In 2006, Shinja Yamanaka established so-called induced pluripotent stem cells (iPSCs) by reprogramming murine fibroblasts by viral transfer of four genes associated with pluripotency, Oct4, Sox2, Klf4 and c-Myc (Takahashi and Yamanaka 2006). These data demonstrated that the specialized somatic cells can be reversed into a pluripotent state in vitro. Reprogramming of in vitro–cultured mammalian cells to pluripotency occurred solely by expression of virally transferred pluripotency-associated genes. The robustness of the method was independently confirmed by three groups (Maherali et al. 2007; Okita et al. 2007; Wernig et al. 2007) and only 1 year later, reprogramming of human fibroblasts into human iPS cells was achieved (Takahashi et al. 2007; Yu et al. 2007). Meanwhile, reprogramming of diverse somatic cell types of various mammalian species into iPS cells with proliferation, morphology, gene expression, imprinting and chromatin profiles similar to ESCs has been shown (reviewed in Nishikawa et al. 2008; Amabile and Meissner 2009). Pluripotency of murine iPS cells was confirmed by blastocyst injection or “tetraploid embryo complementation”, which resulted in the contribution of the iPS cells to embryo formation (Maherali et al. 2007; Wernig et al. 2007) and, only 2 years later, in the birth of living mice completely derived from somatic donor cells (Boland et al. 2009; Kang et al. 2009; Zhao et al. 2009b).
Progress in iPS cell generation was achieved by establishing techniques that avoid stable integration of foreign genetic material into the host cell genome in the process of reprogramming, for example, by transgene removal following viral transduction of the pluripotency genes in iPS cells (Kaji et al. 2009; Soldner et al. 2009; Woltjen et al. 2009), the generation of iPS cells using non-integrating viral vectors or plasmids to provide the reprogramming factors (Stadtfeld et al. 2008; Yu et al. 2009; Zhou and Freed 2009) and the transduction of recombinant cell-penetrating proteins (Oct4, Sox2, Klf4, c-Myc) into mouse (Zhou et al. 2009) or human (Kim et al. 2009) cells. In addition, reprogramming with small molecules acting as chromatin-modifying agents to replace the potentially oncogenic reprogramming factors increased the reprogramming efficiency (Huangfu et al. 2008; Shi et al. 2008; reviewed in Seifinejad et al. 2010).
These new reprogramming technologies would enable not only the generation of pluripotent cells for autologous cell therapies, but would also allow for the application of human iPS cells to specific issues in drug research and in safety pharmacology or toxicology studies (reviewed in Ebert and Svendsen 2010; Phillips and Crook 2010; Rowntree and McNeish 2010), as well as for the analysis of pathomechanisms of heritable diseases with “disease-specific” iPS cells (e.g. Dimos et al. 2008; Park et al. 2008; Ebert et al. 2009; reviewed in Saha and Jaenisch 2009; Lee and Studer 2010).
To overcome the limitations with respect to transgenesis and gene targeting in hESCs (see “Embryonic stem (ES) and embryonic germ (EG) cells”), an alternative strategy was proposed: reprogramming of human somatic cells using five reprogramming factors (Oct4, Sox2, Klf4, c-Myc, Nanog) in the presence of LIF yielded cells of a human pluripotent state comparable to that of mouse ESCs (Buecker et al. 2010). This state was found to facilitate homologous recombination-mediated gene targeting also in human pluripotent stem cells, but required the continued expression of the five reprogramming factors. A pluripotency stage of hiPSCs comparable to that of mESCs was also found by Hanna et al. (2010) when reprogrammed cells were cultured in the presence of a combination of the ERK1/2 inhibitor PD0325901, the GSK3 inhibitor CHIR99021 and LIF (Hanna et al. 2010).
However, there is a strong need to further analyse and improve the reprogramming process. Several critical parameters such as low reprogramming efficiencies (usually between 0.0001 and 0.1%; Maherali et al. 2007; Wernig et al. 2007), only partial reprogramming (see Nagy and Nagy 2010) and genetic instability of the generated cells may be obstacles to the routine application of hiPSCs. Specifically, the “epigenetic memory” of hiPSCs (Kim et al. 2010b; Polo et al. 2010), the cell cycle status and the genetic and epigenetic background of the somatic cells may affect the reprogramming process (Jalving and Schepers 2009).
It was further shown that the cell type of origin and the methods used for iPS cell generation can affect the functional properties of iPS cells (Polo et al. 2010) and the molecular characteristics defining their pluripotent state (Soldner et al. 2009). For example, viral integration of transgenes modified the epigenetic state of the cells. Accordingly, a higher similarity in the expression profiles was found between ‘factor-free’ hiPS and hESCs, whereas different hiPSC lines that had integrated DNA of the viral vector used for reprogramming showed different molecular patterns (Soldner et al. 2009). A first study on gene expression signatures of hESCs and hiPSCs disclosed functional and developmental differences between ES- and iPS-derived cells (Chin et al. 2009). However, a recent detailed comparison of a panel of different hESC and hiPSC lines revealed only subtle variations in chromatin structure or gene expression patterns among individual lines, but no consistent signatures that distinguish hiPS from hESC lines when examined after extended culture time (Guenther et al. 2010). Another study detected more differences in gene expression and histone modification profiles between hES and hiPSCs generated by different laboratories, suggesting that obviously methodical differences between separate studies rather than differences between both pluripotent cell types have to be considered (Newman and Cooper 2010).
However, other data may be also important to answer the question, whether or not hESCs and hiPSCs resemble similar pluripotent states. Murine iPS cells revealed a lower efficiency, higher senescence and greater variability with respect to their expansion and differentiation characteristics than mESCs (Feng et al. 2010; Hu et al. 2010). It was shown that a defined maternally imprinted region on chromosome 12 of miPS cells was important to define pluripotency. Only iPS cell lines with normally activated gene clusters were able to generate chimeras almost as efficiently as mESCs, whereas cells with silenced clusters were unable to give rise to chimeric animals (Stadtfeld et al. 2010). In the future, the epigenetic status of the chromosome 12 gene cluster eventually will allow the identification of iPS cell lines that have the full developmental potential known from ESCs. It remains to be shown whether similar “epigenetic hot spots” also exist in human iPS cells (Dolgin 2010).
Moreover, a close correlation between pluripotency and tumourigenicity as well as remarkable similarities between the reprogramming process leading to induced pluripotency and oncogenic transformation may have also a critical impact on toxicity studies (reviewed in Knoepfler 2009; Krizhanovsky and Lowe 2009; Wobus 2010). The following aspects may be important with respect to potential tumourigenesis of reprogrammed iPS cells: (1) the reprogramming factors are involved in tumour initiation (Jalving and Schepers 2009), (2) the suppression of the p53 pathway in pluripotent stem cells necessary for reprogramming may result in genomic instability (Krizhanovsky and Lowe 2009), (3) high telomerase activity required for unlimited proliferation is a property of both pluripotent stem and tumour cells (Marion et al. 2009; Vaziri et al. 2010; Zalzman et al. 2010) and (4) the in vitro culture required to establish iPS cells may induce chromosomal aberrations (Mayshar et al. 2010) and, possibly, epigenetic changes.
Nevertheless, reprogramming by induced pluripotency offers exciting perspectives for regenerative medicine and pathogenesis research, and also for toxicity testing and drug research (see Nishikawa et al. 2008). Specifically, both human ES and iPS cells have been proposed as in vitro test systems for pharmaco-toxicological studies (Sartipy et al. 2006; Caspi et al. 2009; Ebert and Svendsen 2010; Trosko and Chang 2010) and companies have started to establish new strategies based on hPSCs to provide new models for drug safety testing (Baker 2010). Examples of these applications and first results will be presented in the next sections.
Mutagenicity/genotoxicity studies using EC, EG and ES cells
Mutagenic substances induce gene mutations, but also structural and numerical chromosomal aberrations. Genotoxic factors are capable of damaging DNA, but they can also affect cellular components that regulate the fidelity of the genome, such as the spindle apparatus, topoisomerases, DNA repair systems and DNA polymerases. These effects may directly or indirectly involve the genetic material and result in developmental perturbations, abnormal embryogenesis and malformations. A combination of mutagenicity tests has been designed to detect potential consequences of exposure to chemicals. These include analyses of major endpoints of genetic damage associated with human diseases, such as gene mutations as well as structural and numerical chromosomal aberrations. Studies in the past clearly documented that no single assay can detect all genotoxic or mutagenic substances. Therefore, a battery of appropriate in vitro and in vivo tests to detect mutagenicity was established (reviewed in Eastmond et al. 2009).
Genotoxic effects on germ cells are of crucial importance because gene mutations or chromosome damage may be transferred to the offspring and future generations. Strategies for in vivo germ cell testing include the mammalian spermatogonial, chromosome aberration, dominant lethal (Russell and Russell 1954; Ehling 1974), specific locus (Ehling et al. 1978; Russell et al. 1981) and mouse heritable translocation assays (see Eastmond et al. 2009). These tests usually require large numbers of several generations of animals, which are time-consuming and expensive. Therefore, researchers have long been interested in developing alternative in vitro methods. Most of these assays were based on bacterial reverse mutation tests or on somatic cells, such as primary and permanent cell lines including transgenic reporter cell lines, but did not rely on embryonic or germ cells (Brown et al. 1995; reviewed in Spielmann 1998; Spielmann 2005).
In the 1990s, pluripotent embryonic carcinoma (EC) and embryonic stem (ES) cells offered for the first time the opportunity to analyse the mutagenic or genotoxic and cytotoxic effects of chemical mutagens on embryonic cells in vitro (reviewed in Rohwedel et al. 2001). First studies that aimed at determining the mutagenic effects of the alkylating substances ENU (N-ethyl-N-nitrosourea), MNNG (N-methyl-N’-nitro-N-nitrosoguanidine) and the DNA crosslinking and alkylating agent mitomycin C(MMC) were performed on undifferentiated P19 EC cells. When compared to EPI-7 cells, an epithelial derivative of P19 cells, the undifferentiated P19 EC cells were found to be less sensitive with respect to the induction of 6-thioguanidine resistance (6-TGr), suggesting a higher or more efficient DNA repair capacity of undifferentiated in comparison with differentiated cells. But it has been also discussed that EC cells may be less sensitive because of their malignant status, which makes them more resistant to anticancer drugs (Sehlmeyer and Wobus 1994; Sehlmeyer and Wobus 1995; Sehlmeyer et al. 1996a).
Similarly, when the sister chromatid exchange (SCE) frequency was investigated as an endpoint of mutagenic effects (Vogel 1993), it was found that EC and ES cells reacted with the same or even lower sensitivity to ENU than differentiated cells (Bremer and Vogel 1999). A comparison of SCE frequencies of mouse EG cells and fibroblasts revealed a higher sensitivity of EG cells after treatment with MMS (methylmethane sulphonate), HU (hydroxyurea), MNU (methyl-nitrosourea), ENU and MMC (Klemm et al. 2001a; Klemm et al. 2001b). Based on these data, a biostatistical prediction model for genotoxicity on EG cells in vitro was established, which showed a correct classification for 5 genotoxic and three non-genotoxic test chemicals (Klemm et al. 2001a). In addition, numerous in vivo studies demonstrated that germ cells of different stages reacted differently to mutagenic compounds depending on the specific substance tested (Ehling 1974; Favor et al. 1990). But, it should be also considered that SCEs, chromosome aberrations and point mutations are different endpoints of mutagenesis that may considerably vary for specific mutagens, and different primary lesions may be involved (Sonoda et al. 1999).
Later on, transgenic ESC lines containing specific ‘loss-of-function’ mutations in genes regulating DNA repair or drug resistance metabolism have been used to analyse mechanisms of mutagenicity and genotoxicity. Such ESC lines were deficient for ERCC1 (‘excision repair complementation defective repair in Chinese hamster cells’), a gene involved in nucleotide excision repair (Van Sloun et al. 1999), for Msh2 and Msh3 involved in DNA mismatch repair (Abuin et al. 2000), for the gene encoding the ‘multidrug resistance-associated protein’ (MRP, Lorico et al. 1996) or the tumour suppressor p53 (Donehower et al. 1992). Using p53-deficient ESCs, effects of electromagnetic fields (EMF) have been analysed. Exposure of p53-deficient ESCs to EMF resulted in altered transcript levels, whereas wild-type ESCs that were treated in a similar way did not show these responses (Czyz et al. 2004a; Czyz et al. 2004b).
Also, ‘gain-of-function’ approaches have been applied to cytotoxicity studies with ESCs. Over-expression of the heat shock protein HSP27 in ESCs demonstrated that HSP27 is involved in protection against metal toxicity (Wu and Welsh 1996).
In summary, pluripotent stem cells including genetically altered EC or ES cell lines were found to be useful models to analyse genotoxic/mutagenic effects of drugs and xenobiotics. These cellular systems may be more relevant for germ cells or early embryos than for somatic cells, because ES and somatic cells differ in their mutation frequencies. Mutations were induced less frequently in ESCs than in somatic cells; however, it has to be noted that extended culture of mESCs may result in the accumulation of mutations (uniparental deficiency rather than loss of heterozygosity; Cervantes et al. 2002).
Embryotoxicity tests using pluripotent stem cells
The need to establish reliable in vitro tests for embryotoxicity of chemicals
The effects of chemicals or drugs on germ cells or early embryos may lead to infertility or impaired development of pre-implantation embryos and result in embryotoxic or teratogenic effects in the progeny (for review see Spielmann 2005; Krtolica et al. 2009). A system of versatile tests for reproductive toxicity had been established to systematically screen for such hazards present in the daily life, including chemical compounds that are components of drugs and pharmaceuticals. Such test systems are required for reproductive toxicological analyses and are part of governmental regulations. The OECD Guidelines for testing industrial and agrochemicals stipulate extensive screening and multi-generation studies including tests for teratological, reproductive and developmental effects, neurotoxicity, oestrogenic and androgenic properties and include extensive one- and two-generation reproductive toxicity studies (reviewed in Spielmann 2009). Chemicals that are parts of drugs have to be investigated in segment studies covering combined fertility and embryotoxicity (Segment 1), teratology (Segment 2) and pre- and postnatal development (Segment 3) studies (for overview see Seiler et al. 2004). However, these in vivo tests are time-consuming, laborious and expensive and, specifically, require the use of high numbers of laboratory animals.
Therefore, alternative in vitro screening methods have been developed to detect potential hazardous effects of chemicals or drugs on embryonic development, but until now, these test systems have not been accepted for regulatory purposes. These assays include the frog embryo teratogenesis assay (FETAX) on xenopus (Bantle et al. 1990), the chicken embryo toxicity screening test (CHEST) on chicken embryos (Jelinek et al. 1985; Boehn et al. 2009), the micromass (MM) assay using mouse embryonic mesenchymal cells (reviewed in Flint 1993; Brown et al. 1995; Spielmann 2005), and the mammalian whole embryo culture (WEC) assay using mouse (e.g. Sadler et al. 1982) or rat (Schmidt 1985; reviewed in Cockroft and Steele 1987). In addition, mammalian cells have been introduced as in vitro test systems for cytotoxicity, embryotoxicity or teratogenicity screening (Kimmel et al. 1982; Schwetz et al. 1991). Whereas the MM test and the WEC assay (based on in vitro culture of mammalian embryos) were validated and proposed as promising systems for screening of embryotoxic compounds, the CHEST assay was unable to distinguish between general and developmental toxicity, and the FETAX test has not been sufficiently validated (reviewed in Spielmann 2005). The most important drawback of all these assays is that they rely on somatic cells that not entirely reflect the reaction of embryonic cells to toxic compounds.
Early studies analysing cytotoxic effects of chemicals on murine EC, ES and EG cells
Tests for embryotoxicity include the analysis of toxic effects of drugs or environmental factors on embryonic cells or on cells of (early) embryonic developmental stages, whereas cytotoxicity assays also measure drug-induced alterations of metabolic pathways or structural integrity of the cells, which may or may not be directly related to cell death. Both cytotoxicity and embryotoxicity assays have been established with pluripotent stem cells.
Studies to determine cytotoxic effects of chemical compounds based on the colorimetric MTT test for cellular growth and survival (Mosmann 1983) were performed with murine EC (Atkinson et al. 1989), ES (Laschinski et al. 1991) and EG (Klemm et al. 2001b; Klemm et al. 2001a) cells. Undifferentiated mEC and mEG cells showed a higher sensitivity to selected embryotoxic agents when compared to mouse fibroblasts. Whereas in the MTT test, cell survival is determined indirectly by measuring alterations of metabolic activity following treatment with chemical compounds, the cloning efficiency test relies on the reproductive integrity of the cells and, therefore, should more directly reflect cytotoxic effects of test compounds. Consequently, cytotoxicity assays based on the reproductive integrity of cells (=clonogenicity) after application of mutagenic chemicals (ENU, MMC and MNNG) were established by using murine EC, EG and ES cells: a higher sensitivity of mEG cells in comparison with differentiated EPI-7 cells was detected, whereas both mEC cells and mESCs were less sensitive (Sehlmeyer et al. 1996b).
Embryotoxicity studies using mESCs
To overcome the limitations of cytotoxicity assays for the determination of embryotoxic effects of drugs or xenobiotics in vitro, the ESC-derived embryoid body (EB) model was introduced (Wobus et al. 1988; Wobus et al. 1991; Wobus et al. 1994a). ESCs differentiated in EBs for 5–7 days develop in vitro into progenitor cells of all three primary lineages of ectoderm, mesoderm and endoderm (reviewed in Wobus and Boheler 2005). The rationale of the EB model was that early developmental processes simulated by EB differentiation would be affected by treatment of undifferentiated PSCs with toxic substances and, as a consequence, specific differentiation processes would be inhibited, induced or accelerated.
At first, the well-known teratogen all-trans retinoic acid (RA) was applied at different concentrations and at specific time intervals of EB differentiation. It was found that RA affected the differentiation of mEC (Strickland and Mahdavi 1978; Edwards and McBurney 1983; Jones-Villeneuve et al. 1983) and mES (Strubing et al. 1995; Dani et al. 1997; Drab et al. 1997; Wobus et al. 1997; reviewed in Rohwedel et al. 1999; Rohwedel et al. 2001) cells in a concentration- and time-dependent manner. There is evidence that in vivo RA acts as morphogen forming a concentration-dependent gradient along embryonic axes, thus affecting the antero-posterior patterning of the body axis and the limbs (Kessel and Gruss 1991).
Such concentration- and stage-specific effects were also observed with mESCs in vitro. For example, high concentrations of RA applied to mESCs during early EB differentiation (day 0–2) revealed increased neural-specific transcript levels and significantly induced neuronal differentiation (Fraichard et al. 1995; Strubing et al. 1995), whereas RA application at lower concentrations applied between day 2 and 5 induced mesodermal, specifically skeletal muscle and partially cardiac differentiation (Wobus et al. 1994b). This is in line with developmental processes in the EB, where maximum expression levels of Brachyury (T) and expression of genes associated with dorsal mesoderm specification are observed at around day 3 of EB formation (Wobus et al. 1994b; Dani et al. 1997). Cardiac and vascular smooth muscle cells both originating (at least partially) from lateral plate mesoderm were induced by RA when applied at days 5–7 and 7–11, respectively (Wobus et al. 1994b; Drab et al. 1997; Wobus et al. 1997; Fig. 1). Obviously, the specific temporal response of the different cell types within the EB to RA correlated with RA receptor gene expression (reviewed in Rohwedel et al. 1999). These early studies provided the basis for the subsequent establishment of the so-called Embryonic Stem Cell Test (EST, see “Embryonic stem cell test (EST) with mESCs”).
Fig. 1.
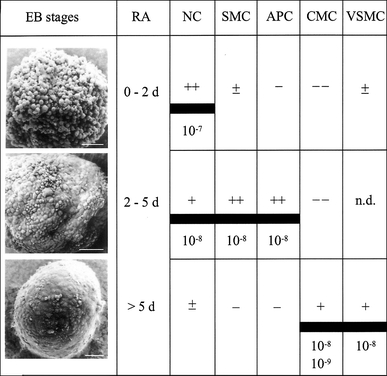
Stage-specific effects of all-trans retinoic acid (RA) on embryoid body (EB) differentiation of murine ES cells. Shown are scanning electron micrographs of 2d-, 5d- and 7d-EBs. ESC-derived EBs were treated with different RA concentrations (10−7, 10−8, 10−9 M) during defined cultivation periods (0–2, 2–5, >5 days). The different effects of RA on neuronal cell (NC), skeletal muscle cell (SMC), adipocyte (APC), cardiomyocyte (CMC) and vascular smooth muscle cell (VSMC) differentiation is demonstrated by inducing (+, ++, +++), inhibiting (−, −−) or no (±) effects. Bars = 50 mm (original data from Wobus et al. 1994b; Strubing et al. 1995; Dani et al. 1997; Drab et al. 1997; see also Rohwedel et al. 1999)
Similar distinct time- and development-specific effects of chemical compounds on ESC differentiation were also detected with lithium chloride (Schmidt et al. 2001) and suramin, a naphthylamine derivative of urea (Wiese et al. 2009). Lithium chloride applied to ESC-derived EBs at high concentration between days 5 and 15 caused reduced cardiac-α-MHC and skeletal muscle-specific MyoD, but slightly increased neural-specific synaptophysin and NFM transcript levels and led to the inhibition of cardiac and myogenic differentiation (Schmidt et al. 2001). High concentration of suramin between day 5 and 7 enhanced cardiac differentiation with specific induction of pacemaker-like cells, but inhibited neuronal, skeletal muscle and definitive endoderm differentiation (Wiese et al. 2009).
These studies demonstrated that the morphological evaluation of EB outgrowths after treatment with certain chemical compounds is only partially suitable to assess developmental toxicity since lineage-specific developmental effects are not perfectly mirrored. The data further showed that the developmental stage, at which substances were applied (undifferentiated ESCs vs. differentiating EBs), may affect the embryotoxic effects and that analysis of transcript levels of various lineage-specific genes may be a better indicator for developmental toxicity (Schmidt et al. 2001; reviewed in Rohwedel et al. 1999).
In addition to the well-established EST (see “Embryonic stem cell test (EST) with mESCs”), in vitro models based on differentiation of mESCs are generally accepted systems to test potential toxic effects of chemical compounds. For example, Lim and co-workers recently tested the cytotoxic effects of phthalate esters on mESC-derived neural progenitors and determined the neurotoxicity of mono-(2-ethylhexyl) phthalate, a degradation product of the commonly used commercial plasticizer for polyvinylchloride plastics (Lim et al. 2009). Meamar et al. (2010) found that 3,4-methylenedioxymetamphetamine (MDMA, ecstasy) inhibited neural and cardiac differentiation of murine ESCs at micromolar concentrations (Meamar et al. 2010).
In conclusion, the suitability of murine ESCs for toxicological analyses has been demonstrated in a broad range of studies on potential embryotoxic effects of specific compounds. However, the integration of such test systems into regulatory framework will require the standardization and validation of developmental toxicity tests using ESCs.
Embryonic stem cell test (EST) with mESCs
The mouse embryonic stem cell test (mEST) established by Spielmann and co-workers (Spielmann et al. 1997) is based on two assumptions. First, in vitro tests of basal cytotoxicity are sufficiently predictive for the rodent in vivo LD50 assay (Ekwall 1999; Spielmann et al. 1999) and, second, ESCs show alterations in their in vitro differentiation pattern when exposed to embryotoxic chemicals during EB differentiation (Wobus et al. 1988; Wobus et al. 1994b). The validated EST was proposed in 2002 as a scientifically validated in vitro method for detecting embryotoxicity (http://ecvam.jrc.it; see Spielmann 1998) and is based on the assessment of three toxicological endpoints and the comparison of data from three independent assays (see Fig. 2):
Morphological analysis of beating cardiomyocytes in EB outgrowths derived from mouse ESCs (line D3, Doetschman et al. 1985) and cultured in the presence of the test substance.
Cytotoxic effects on undifferentiated D3 ESCs.
Cytotoxic effects on NIH 3T3 fibroblasts following application of test substances.
Fig. 2.
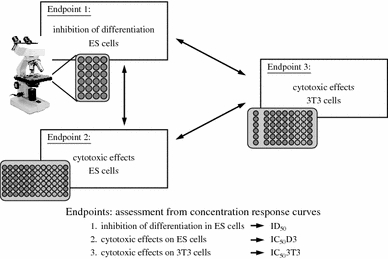
The embryonic stem cell test (EST) established for mESCs by Spielmann and co-workers. The EST comprises three parameters, the differentiation of mES cells (endpoint 1), cytotoxic effects on ES (D3) cells (endpoint 2) and on NIH 3T3 fibroblasts (endpoint 3). On the basis of these data, concentration response curves were established and the prediction for embryotoxic effects of compounds calculated (according to Seiler et al. 2006; see text for further information)
The test comprises the analysis of beating clusters in differentiating EBs (plated into single wells of microwell plates) and the determination of those concentrations of the test substance at which cardiac differentiation is inhibited by 50% (ID50). The cytotoxic effects on ES and 3T3 cells were estimated by determining those concentrations of the test substance at which proliferation was inhibited by 50% (IC50 D3 and IC50 3T3, respectively). On the basis of these data, concentration–response curves were generated and a biostatistical prediction model was established to assign the test compounds to three classes of embryotoxicity: non-embryotoxic, weakly embryotoxic and strongly embryotoxic (Genschow et al. 2002, 2004; Seiler et al. 2006). The EST was validated by using a set of 20 test compounds of known in vivo embryotoxicity. The embryotoxic potential of the test compounds was predicted with about 78% accuracy for non-embryotoxic and weakly embryotoxic compounds and 100% for strongly embryotoxic chemicals (Genschow et al. 2002, 2004). Together with the MM and rat WEC test, the mEST would be ready to be accepted by the regulatory commissions (Balls and Hellsten 2002); however, the Scientific Advisory Committee (ESAC) of the European Centre for the Validation of Alternative Methods (ECVAM) recognized that the three methods were still not qualified to replace the animal experiments stipulated in the present integrated test strategies (Spielmann 2009). For example, it was argued with respect to the mEST that data on only 20 chemicals were not sufficient to make final decisions on embryotoxicity. A new Integrated Project ReProTect was initiated within the European Union Framework Program 6, in which a new set of 13 non-classified chemicals was studied (Hareng et al. 2005). Since these test chemicals were not correctly classified by the EST, modifications and new endpoints were recommended for the EST (see Spielmann 2009).
Indeed, the primary (validated) mEST had several limitations. Since ESCs were differentiated as EBs in “hanging drops”, the test was (1) laborious and (2) time-consuming. (3) The substances were applied over the whole process of differentiation, (4) cardiac differentiation (formation of beating cardiomyocytes) was the only endpoint of the developmental analysis, and (5) no molecular parameters were considered (Schmidt et al. 2001; Piersma 2004; Seiler et al. 2006). It was recommended to include further endpoints, such as additional differentiation markers of ectoderm, mesoderm and endoderm lineages and the analysis of tissue-specific gene expression.
Based on previous studies that demonstrated lineage-dependent effects of RA on ESC differentiation (Wobus et al. 1994b) and the application of reporter gene constructs controlled by cardiac-specific promoters (Wobus et al. 1997; Kolossov et al. 1998), a battery of transgenic murine ESC lines expressing reporter genes controlled by different lineage-specific promoters was proposed as a test system for embryotoxicity (Rohwedel et al. 2001).
In addition, several studies that aimed on improving the EST by inclusion of new morphological and molecular endpoints were performed during the last years and methodical improvements were achieved. For example, serum replacement (SR) was substituted for foetal calf serum (FCS), and chemically defined media (CDM) were used in the EST (Seiler et al. 2006). Furthermore, reporter gene assays for developmental toxicity, specifically for cardiac toxicity (Bremer et al. 2001), have been included into the EST. The cardiac markers α-myosin heavy chain (MHC) and α-actinin were used instead of microscopical observation of beating EBs to determine cardiac differentiation after application of the test substances, and quantification of marker gene expression by FACS was performed after labelling of cells (Seiler et al. 2004; Seiler et al. 2006). The FACS-based EST (called “Molecular FACS-EST”) was successfully validated for assessing developmental toxicity (Buesen et al. 2009). While the “Molecular FACS-EST” showed the same sensitivity as the validated EST, the test duration was reduced and the test allowed for a more rapid and reproducible screening and for predicting developmental toxicity in vivo on the basis of in vitro data. Almost identical ID50 values were obtained for ten representative compounds of the three classes, indicating that values from FACS analyses can serve as new “toxicological endpoint” of the EST.
Furthermore, the inclusion of additional endpoints of differentiation, specifically of the neural lineage, was requested by researchers and authorities (Spielmann et al. 2006). The rationale of using such systems is to correctly classify substances that were not identified as embryotoxic in the conventional EST. The inclusion of parameters for neuronal differentiation into the mEST allowed, for example, the classification of methylmercury as a highly embryotoxic substance in accordance with in vivo data, whereas the conventional EST failed to correctly classify this compound (Stummann et al. 2007). To further increase the number of molecular markers as endpoints of embryotoxicity testing, real-time Taqman RT-PCR analyses have been adopted for the EST in a pilot study (zur Nieden et al. 2004).
Metabolic activation of specific chemical compounds was recognized as another potential factor to affect the experimental outcome of the EST and correct classification of test substances (Verwei et al. 2006). However, the S9 mix usually applied for metabolic activation of chemicals is toxic for ESCs and respective experiments have failed so far. Therefore, the consortium of the ReProTect project recommended other test systems, such as the Ames test, to analyse potential toxicity of compounds that require metabolic activation (Marx-Stoelting et al. 2009).
Peters and co-workers determined the “Relative Embryotoxic Potential” (REP) by using a modified EST screening system (Peters et al. 2008). In this study, 12 compounds were investigated in a modified EST performed in 96-well plates. Test substances were applied at day 3 of culture for 10 days, and the essay did not involve the preparation of EBs. This “Newly Revised High Throughput EST” allowed the analysis of a higher number of substances, while less manual work was required, and yielded data comparable to those obtained with the conventional EST.
At present, the EST is the only well-established in vitro test for developmental toxicity based on mammalian cells (Bremer and Hartung 2004). Present efforts to improve the EST are directed towards the application of additional lineage-specific markers to define additional toxicological endpoints. In addition to cardiac markers, markers of neuronal, bone and cartilage development are included. However, this will prolong the test duration from 7 or 10–30 or 32 days (Marx-Stoelting et al. 2009). In the future, novel molecular endpoints and reporter-based systems have to be included into the EST (see Rohwedel et al. 2001; Marx-Stoelting et al. 2009; Spielmann 2009). Such improvements would also be needed for the establishment of EST-like tests with human ESCs.
Screening of cytotoxic and embryotoxic effects using hESCs
The development of hESC-based in vitro systems for testing embryotoxicity of chemical compounds would be a significant progress. The use of hESCs would enhance the predictivity of in vitro assays and avoid problems associated with the interpretation of results from animal-based assays in a human context. For example, species-specific differences between mouse and human preimplantation development, such as in DNA methylation, DNA repair and expression of genes involved in drug metabolism may hamper the correct interpretation of animal studies for human beings (reviewed in Krtolica et al. 2009). The use of hESC-based test systems could avoid incorrect classification of chemicals due to inter-species variations and, consequently, would increase the safety of consumers and patients.
The question whether hESCs would be a suitable model for analysing developmental toxicity was investigated in several recent proof-of-concept studies (for overview see Table 3). In some of these studies, potential toxic effects of agents and noxes (such as certain chemical compounds, radiation, ethanol or cigarette smoke) on the viability and integrity of hESCs were investigated (Adler et al. 2008a; Zdravkovic et al. 2008; Krishnamoorthy et al. 2010; Lin et al. 2010; West et al. 2010; Wilson et al. 2010). This kind of studies may allow conclusions on the effects of certain hazards on the preimplantation embryo in vivo. In a panel of further studies, effects of well-known embryotoxic agents on the induction and progression of hESC differentiation were examined and endpoints suitable for the detection of embryotoxic effects on early stages of hESC differentiation were proposed (Adler et al. 2008b; Mehta et al. 2008; Flora and Mehta 2009; Stummann et al. 2009b). It is supposed that this kind of investigations might have implications for assessing potential risks even during later stages of embryonic and foetal development.
Table 3.
Published developmental toxicology studies involving hESCs- and/or hESC-derived cells
| Study | hESC lines | Noxes tested | Endpoints used/identified |
|---|---|---|---|
| Adler et al. (2008a) | SA002; SA002.5 | Known developmental toxicants (VPA, ATRA, 13-CRA); DMSO | hES cell viability (ATP content, resazurin reduction) |
| Adler et al. (2008b) | H1 | Known developmental toxicant (RA) | hES cell viability (resazurin reduction); mRNA levels of genes coding for stemness markers (Oct4, hTert); mRNA levels of genes coding for mesodermal and cardiac differentiation markers (Brachury; GATA-4, Nkx 2.5; MyH6)† |
| Flora et al. (2009) | ReliCell hES1 | Arsenic (potential reversal of arsenic effects by MiADSA were also investigated) | mRNA levels of stemness markers; mRNA levels of genes representative of the three germ layers |
| Krishnamoorthy et al. (2010) | H9; Bg02; abBG02 | Ethanol at low concentrations | CHRNA5 gene expression level |
| Lin et al. (2010) | H9 | Smoke from conventional and harm-reduced cigarettes | hESC morphology; hESC apoptosis |
| Metha et al. (2008) | ReliCell hES1 | Strong (busulfan; hydroxyurea), weak (caffeine, indomyacin) and non-embryotoxic (penicillin, saccharin) compounds | mRNA levels of genes coding for stemness markers; expression levels of genes representative of the three germ layers |
| Stummann et al. (2009) | H1 | Methylmercury (MeHg) | mRNA levels of genes coding for neural lineage markers in differentiating cells (NCAM; NeuroD; MAP2) |
| West et al. (2010) | H9 | Several drugs with known teratogenic effects in human | Abundance of certain small molecules (metabolites) as detected by mass spectrometry |
| Wilson et al. (2010) | H9 | γ-radiation (0.4–4 Gy) | hESC viability; global gene expression changes in hESCs |
| Zdravkovic et al. (2008) | H7; H9 | Nicotine exposure | Morphology and adhesion of hESCs; expression of stemness markers; hESC apoptosis |
More details are given in the text. † Effects of RA on cardiac differentiation of hESCs were not tested
For example, Adler and co-workers provided proof-of-concept that hESCs are a relevant in vitro model for developmental toxicity testing. In one study, the cytotoxic effects of well-known embryotoxicants [all-trans retinoic acid (ATRA), 13-cis RA (13CRA), valproic acid (VA) and dimethylsulfoxide (DMSO)] on hESCs, hESC-derived progenitors and human foreskin fibroblasts were investigated (Adler et al. 2008a). As observed in the respective murine cells, ATRA and 13CRA had a stronger cytotoxic effect on pluripotent stem cells than on fibroblasts, while mesenchymal progenitors derived from hESCs showed the highest sensitivity to both compounds. Interestingly, while both substances revealed comparable cytotoxic effects on human ESCs in this study, only ATRA but not 13CRA were shown to be cytotoxic in mouse ESCs in previous analyses, indicating the necessity of developing hESC-based assays for the assessment of human-specific developmental toxicity. In a second study, Adler and co-workers arranged the test system according to the EST, but used human instead of mouse ESCs (Adler et al. 2008b). Using two well-known developmental toxicants [5-fluorouracil (5-FU) and ATRA], cytotoxic effects similar to those previously observed in mouse systems were observed in hESCs and human fibroblasts. However, testing for potential cytotoxic effects on pluripotent stem cells is not sufficient to detect developmental toxicity of chemicals that affect developmental processes (reviewed in Rohwedel et al. 2001). Since the conventional EST based on mESCs depends on the microscopical observation and counting of beating areas after ESC differentiation and therefore cardiac differentiation is not precisely determined, the authors suggested several other potential endpoints for the analysis of toxic effects on differentiating hESCs. For example, markers of undifferentiated cells such as Oct4, hTert (human telomerase reverse transcriptase) and Dusp6 (dual specificity phosphatase 6) were shown to decrease reliably during early differentiation of hESCs, while Brachyury and GATA-4, markers of mesoderm and early cardiogenesis, were found to be suitable to monitor cardiac differentiation. The authors suggested that these markers may have the potential to serve as endpoints for developmental toxicity studies using a humanized EST (Adler et al. 2008b).
In a recent study, the effects of methylmercury (MeHg, a well-known developmental toxicant) on neural differentiation of hESCs were investigated (Stummann et al. 2009b). MeHg caused a marked decrease in mRNA levels of neural lineage-specific genes expressed in neural progenitors when added to hESCs at sub-cytotoxic levels from day 0 to day 12 of neural differentiation. In contrast, when added at later stages of neural differentiation (day 21–38), the effect of MeHg on mRNA levels of genes expressed in more mature neural cells was much less pronounced. These results are different to those obtained in studies with mouse ESCs (Stummann et al. 2007), in which MeHg exerted its repressive effect on neural differentiation probably rather by inhibition of maturation of neural precursor cells. Thus, there are possible species-specific differences in the mechanism of MeHg toxicity between man and rodents, which underline the need to establish human cell-based test systems for determining developmental toxicity in man.
While some studies using hESCs as model for early developmental toxicity are based on investigation of subtle differences in mRNA levels of lineage-specific genes, in one study changes in the abundance of small molecules in hESCs were investigated subsequent to exposure of cells to known developmental toxicants (West et al. 2010). Using a panel of well-defined human teratogens, biomarkers were identified that may be suited to predict developmental toxicity. For example, changes in the levels of asymmetric dimethylarginine (ADMA) and arginine were of some predictive value for the qualification of substances as teratogens or non-teratogens. Unfortunately, this study suffers from the lack of (non-hES) reference cells. Moreover, it remains to be determined whether changes in metabolite levels are also a reliable parameter to determine toxic effects on differentiating cells.
In another study, a cytotoxicity test system based on feeder-free hESCs and hESC-derived EBs was established, with human foreskin fibroblasts as reference cells (Mehta et al. 2008). Embryotoxic effects of several compounds (busulfan, hydroxyurea, indomethacin, caffeine, penicillin and saccharin) were tested using a cell proliferation assay. Developmental toxicity was assessed by quantitative RT-PCR analysis of alterations in transcript profiles of lineage-specific marker genes. The authors demonstrated proof-of-concept that hESCs can serve as a model to test embryotoxic effects of chemicals and that alterations in transcript levels of early lineage-specific markers are, to a certain degree, correlated with known embryotoxic effects caused by drugs or chemicals. More recently, the same group provided data on the reversal of arsenic-induced developmental toxicity by monoisoamyl dimercaptosuccinic acid (MiADMSA) in a human EB model (Flora and Mehta 2009). While arsenic at non-cytotoxic concentrations of 0.1 ng/ml significantly down-regulated expression of genes representative for all germ layers, this effect was abrogated by MiADMSA. Interestingly, these results were mirrored by the outcome of in vivo studies using pregnant Wister rats in which MiADMSA treatment reduced arsenic-induced visceral and skeletal defects in the offspring. Thus, the human EB model may be not only a suitable model to study effects of embryotoxic compounds, but may also be useful to identify drugs that can revert or prevent the effects of such compounds.
Future challenges and perspectives for embryotoxicity studies using pluripotent stem cells
The recent developments in the improvement of the EST have underlined our previous requests to implement new strategies for embryotoxicity testing by using ESCs (1) by the analysis of tissue-specific genes via reporter gene expression, (2) automated high-throughput screening for changes in gene and protein expression patterns using microchip arrays for transcriptome and proteome analyses and (3) the application of human ESCs (see Rohwedel et al. 2001).
Until now, some progress has been made in the determination of endpoints suitable for determination of embryotoxicity in differentiating hESCs, and proof-of-concept was provided that hESC-based in vitro systems may be useful to predict human-specific developmental toxicity. However, there are numerous problems to be solved before hESC-based assays can be implemented into routine procedures for developmental toxicity testing of drugs and chemicals. These include (1) the establishment of reliable and reproducible differentiation procedures that can be performed in a high-throughput format. (2) The predictivity, sensitivity and specificity of the respective test systems have still to be shown for a wider panel of drugs and chemicals. So far, only few substances have been tested in the different approaches. (3) Furthermore, hESC-based systems may have the capacity to predict human-specific embryotoxic effects that cannot be measured with mouse cell systems due to species-specific differences. However, the superiority of hESC-based systems over existing in vitro tests for developmental toxicity has still to be shown, e.g. by performing thorough comparisons between established and hESC-based tests. In this context, the use of different hESC lines would be advantageous. (iv) Most importantly, drugs that were identified as embryotoxic only in late-stage animal toxicity studies or even after drug approval need to be tested in hESC-based in vitro models. Positive test results would be encouraging and would further stimulate the development in this field. In addition, it was discussed that the application of hiPSCs in embryotoxicity testing might be a promising tool as well (Heng et al. 2009). However, future work has to show whether hiPSCs will be applicable and offer advantages over hESCs in the field of developmental toxicology.
The potential of human pluripotent stem cell-derived cardiomyocytes in cardiac safety pharmacology and cardiotoxicity research
Requirements for safe and reliable in vitro models in cardiotoxicology
Cardiotoxicity is one of the leading reasons for drug attrition and is therefore critical for development and safety testing of new drugs. Within the process of drug development, all new compounds must be evaluated for sub-clinical cardiotoxicity (i.e. alterations of contractility, induction of arrhythmias, alteration on blood pressure and ischaemia) as potentially life-threatening events or effects that may result in cardiac morbidity (Stummann et al. 2009a). The major clinical manifestation of cardiotoxic effects includes drug-induced arrhythmias, contractility toxicity, ischaemia toxicity, secondary cardiotoxicity and valve toxicity. The requests for cardiotoxicity testing by the regulatory authorities are key requirements in pharmaceutical regulatory framework and are defined in the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) S4 guidelines and the “Guideline on Repeated Dose Toxicity” of the EMEA.
During recent years, a considerable number of drugs were withdrawn from the market due to unforeseen cardiotoxic side effects, and cardiotoxicity is one of the major reasons for late-stage attrition of drug candidates. The early detection of fatal side effects of new drugs can therefore prevent the continuation of a useless and cost-intensive developing process. Also, exclusion of compounds proving false-negative in sub-optimal test systems could be prevented by using suitable test systems for the detection of potential cardiotoxicity. It is important to note that many drugs that were withdrawn from the market due to cardiac side effects, including Cisapride, Droperidol or Vioxx, have had other than cardiovascular applications. A prominent example is the anti-inflammatory drug Vioxx (Merck), prescribed to more than 80 Million people (Topol 2004), that was withdrawn from the market in September 2004, because of increased risks of cardiac effects at high dosage and long-term use.
Among pro-arrhythmic cardiac side effects, drug-induced delayed repolarization of the ventricular action potential is very common. This delay becomes visible as a prolonged QT interval and has been associated with ventricular tachyarrhythmia such as life-threatening torsade de pointes (TdP). Most drugs that cause QT interval prolongation and TdP have been shown to block the α-subunit of the IKr channel encoded by the human ether-a-go-go-related gene (hERG). The IKr channel produces the rapid component of the delayed rectifier potassium current IKr which is involved in repolarization. Although the question whether QT prolongation is a surrogate biomarker for TdP is still under debate (Lee et al. 2010), safety assessment of drug candidates now has to include the demonstration that the drug does not prolong the human QT interval at therapeutic and supra-therapeutic doses (ICH E14, Clinical Evaluation of QT/QTc Interval Prolongation and Proarrhythmic Potential for Non-Antiarrhythmic Drugs). Therefore, cardiac safety pharmacology aims at the development of strategies to detect drug-induced unanticipated prolongation of the QT interval during early phases of drug development, but also other parameters were suggested to serve as potential hallmarks of TdP such as triangulation of the ECG shape, beat-to-beat variations or reverse use dependence.
To test for QT prolongation already in pre-clinical settings, several cell and tissue models are currently used (reviewed in Joshi et al. 2004). For example, some cellular in vitro systems are based on heterologous expression of the hERG channel in non-human (CHO) or human (HEK-239) cell lines. In these cell lines, potential interactions of drugs with the hERG channel can be detected by several methods. Although these cell-based systems are sensitive and can be used for high-throughput screening, they do not provide the human cardiac-specific environment and may result in false-positive results. Other commonly used models for cardiac safety pharmaco-toxicology include isolated cardiac tissues (such as papillary muscle or Purkinje fibres from guinea pigs or dogs) and primary cardiomyocytes, e.g. from rats or dogs. However, these systems are labour-intensive, sometimes highly variable and may raise ethical concerns due to the large animal numbers required. In vivo models applied in cardiac pharmaco-toxicology include hearts explanted from small animals, such as rats or guinea pigs (Langendorff perfused isolated heart, reviewed in Skrzypiec-Spring et al. 2007). Whereas these heart models resemble the complex in vivo situation better than other systems and offer the advantage of detecting cardiotoxic effects on the whole tissue levels, problems are related to the animal origin, inter-experimental variations and high costs. Altogether, none of these models alone is predictive enough to exclude cardiac side effects and, according to ICH guidance document ICH S7B, the conduction of ECG assays in conscious animals, usually dogs, are a minimal requirement for testing of new chemical entity (NCE) drug candidates.
A self-replicating cell source, such as pluripotent stem cells, with the potential to differentiate into cardiomyocytes would represent an alternative in vitro system for cardiotoxicity. Earlier studies on murine ESC-derived cardiomyocytes have already demonstrated the potential of ESCs for safety pharmacology and toxicology (reviewed in Boheler et al. 2002; Wobus and Boheler 2005). Murine ESCs differentiating into the cardiac lineage yield several specialized cell types of the heart, such as atrial-, ventricular-, sinus nodal- and Purkinje-like cells. Cardiac-specific genes as well as proteins, receptors and ion channels are expressed in a developmentally controlled manner, which closely recapitulates the developmental pattern of early cardiogenesis in vivo (for reviews see Boheler et al. 2002; Filipczyk et al. 2007). Genetic manipulation, addition of extrinsic factors and extracellular matrix proteins, engineering the microenvironment as well as improvements in cultivation protocols have been shown to trigger the cardiac differentiation process and to increase the efficiency of cardiac differentiation (for reviews see Filipczyk et al. 2007; Chen et al. 2008; Perino et al. 2008; Puceat 2008; Reinecke et al. 2008; Horton et al. 2009). However, only human cell-based in vitro systems would overcome the problem of inter-species differences that limit the use of mESCs for cardiotoxicity testing. So far, much progress has been made in the differentiation of hESCs towards cardiomyocytes, and this knowledge is now being applied to hiPSCs. In the following sections, recent strategies for the generation of cardiomyocytes from human pluripotent stem cells, their characterization and methods for their enrichment will be summarized, and the potential of these cardiomyocytes in safety pharmacology and toxicology will be discussed.
Production of hESC-derived cardiomyocytes for cardiotoxicity studies
Methods for cardiac differentiation of hESCs and characterization of cardiomyocytes
Since the first successful derivation of hESC-derived cardiomyocytes (Kehat et al. 2001), a large number of studies has been published on the generation of cardiac myocytes from hESCs. Three basic methods have been exploited and refined to differentiate hESCs to cardiac cells: EB formation, co-cultivation of hESC-derived cells with endoderm cells and differentiation in monolayer culture induced by specific signalling factors.
(i) The method most widely applied involves the formation of EBs by spontaneous aggregation of undifferentiated hESCs in suspension culture (Kehat et al. 2001), but also centrifugation-forced aggregation has been used (Burridge et al. 2007). After several days in suspension, the EBs are replated on specific matrices resulting in the formation of spontaneously beating clusters in the EB outgrowths, which were reported to be stable for up to 3 months (He et al. 2003). However, the number of beating EBs is highly variable and ranges from about 8 (Kehat et al. 2001) to 70% (Xu et al. 2002), but also lower efficiencies of cardiac cluster formation were reported (Denning et al. 2006). Similarly to mESCs, the number of contracting EBs from hESCs was shown to depend on several factors, including the cell line used (Mikkola et al. 2006), application of growth factors (Burridge et al. 2007; Pal and Khanna 2007) or size of EBs (Niebruegge et al. 2009; Mohr et al. 2010). The percentage of cardiac cells within the EBs (after dissociation) also exhibited substantial variation ranging from 2 to 70%. As previously observed in mESCs (Maltsev et al. 1993), contracting EBs also differed with respect to the cardiomyocyte composition: while different EBs showed various types of action potentials (APs), each EB was populated by a predominant type of cardiac myocytes giving rise to nodal-, atrial- or ventricular-like types of APs (He et al. 2003). It was suggested that the ability of hESC lines to produce chamber-specific derivatives to a different degree is due to subtle differences in the proteomes of undifferentiated hESCs (Moore et al. 2008a).
(ii) The second method involves the co-culture of hESCs with END-2 cells, visceral endoderm cells derived as permanent line from mEC cells, which provide inductive signals necessary for cardiac differentiation (Mummery et al. 2003). Omission of FCS and addition of ascorbic acid markedly improved the efficiency of cardiac differentiation and the number of cardiomyocytes (Passier et al. 2005). However, there are obvious differences in the amenability of hESCs to the differentiation induction. Even cell lines derived in the same laboratory under identical conditions showed marked differences in their potential to form beating areas using the END-2 co-culture system (0–9.4% of all aggregates; Pekkanen-Mattila et al. 2009). The END-2 effects were shown to be, at least partially, due to a rapid clearance of insulin from the culture medium and secretion of prostaglandin I2 (PGI2) by END-2 cells. Consequently, END-2-conditioned medium and a defined, insulin-free differentiation medium containing PGI2 also stimulated cardiac differentiation of hESCs. The efficiency of cardiac differentiation was further increased by addition of the p38 MAPK inhibitor SB203580 at low concentration. The number of beating EBs was as high as nearly 80% and the amount of cardiomyocytes was increased to up to 22% of the whole cell population (Graichen et al. 2008; Xu et al. 2008a).
(iii) More recently, methods for directed cardiac differentiation starting from FL-free monolayer hESC-derived colonies in defined media were developed. These methods are based on novel insights into molecular events during early human cardiogenesis partially derived from studies on gene expression patterns in differentiating hESCs (Beqqali et al. 2006). The differentiation protocols involve the addition of growth factors at specific time points of in vitro differentiation to mimic embryogenesis in vivo. In a first study, the differentiation was initiated by activin A, an inducer of mesendoderm, and BMP-4 specifying myocardial lineage commitment in hESCs. Resulting cell populations showed markers of early cardiac cells (Yao et al. 2006). Further development of this approach yielded consistently >30% cardiomyocytes that could be enriched to more than 80% by Percoll gradient centrifugation (Laflamme et al. 2007).
Based on studies of human and mouse heart development and mESCs, efforts were undertaken to derive cardiac progenitor cells from hESCs. Such cardiac-restricted, non-tumourigenic, expandable progenitor cell populations could serve as starting material for the production of defined cardiac cell types of high purity (reviewed in Perino et al. 2008). Keller and co-workers identified a population of KDRlow/C-Kitneg cells with the potential to develop into cardiac, smooth muscle and endothelial cells. Using a protocol based on the formation of a primitive-streak-like population followed by the induction of cardiac mesoderm, this approach resulted in a high fraction (up to 50%) of functional cardiomyocytes (Yang et al. 2008). Via lineage tracing in hESCs, Chien and co-workers identified a population of ISL1 + cells that could be expanded in vitro, purified to relative homogeneity and were able to develop at high efficiency into cells expressing genes of the major cardiac cell types (Bu et al. 2009).
Human ESC-derived cardiomyocytes have been analysed extensively with respect to molecular, cellular and functional characteristics including gene expression profiles, protein abundance, ultrastructure, electrophysiological features and pharmacological properties. During differentiation in vitro, hESC-derived cardiomyocytes progress towards an adult phenotype resulting in changes of structural and electrophysiological properties (Snir et al. 2003; Sartiani et al. 2007; Satin et al. 2008). Gene expression profiles of developing hESC-derived cardiomyocytes were found to be reminiscent of contractile cells in the developing heart, although many genes expressed in the adult heart are also expressed in differentiating cardiomyocytes (Beqqali et al. 2006; Synnergren et al. 2008). In a recent study a unique molecular signature characterized by a transcription factor network linked to the PPAR signalling pathway was identified (Xu et al. 2009), but so far, transcriptome profiling was only performed in cells differentiated up to 25 days.
Human ESCs differentiate into pacemaker-, atrial- and ventricular-like cells representing cardiomyocytes expressing multiple types of ventricular, atrial and nodal-like APs of the heart (He et al. 2003; Mummery et al. 2003). Cardiomyocytes derived from hESCs show spontaneous contractions indicating the presence of a functional excitation–contraction coupling system that is modulated via α- and β1/β2-adrenoceptors and muscarinic responses (Reppel et al. 2004; Norstrom et al. 2006; Brito-Martins et al. 2008). In addition, these cells exert functional Na+-, K+- and Ca2+-dependent ion channel activities.
However, there are some differences with respect to structural and electrophysiological properties of human ESC-derived cardiac cells in comparison with mature heart or mESC-derived cardiomyocytes: (1) Although developing hESC-derived cardiac cells show gross morphological changes coupled with a progressive increase in the amount and organization of contractile material (culminating in the presence of well-defined sarcomeres) and withdrawal from cell cycle, even after extended culture time they lack a developed T tubule system typical for cardiomyocytes of the adult heart. (2) In contrast to mESC-derived cardiomyocytes, limited binucleation was observed in cardiomyocytes differentiated from hESCs (Snir et al. 2003). (3) hESC-derived cardiomyocytes contained smooth muscle proteins typically for foetal, but not adult, human cardiac cells. (4) Electrophysiological properties showed differences; i.e. the inward rectifier current IK1 was absent from hESC-derived cardiomyocytes leading to a slightly depolarized membrane potential. As observed in foetal cardiomyocytes, hESC-derived cardiomyocytes exhibited the If channel and showed a low Vmax. Moreover, immaturity of intracellular Ca2+ handling of hESC-derived cardiomyocytes was reported, although to a different extent (Dolnikov et al. 2006; Liu et al. 2007; Satin et al. 2008; Zhu et al. 2009; Sedan et al. 2010).
Strategies for enrichment of hESC-derived cardiomyocytes
Efficient cardiac differentiation of hESCs is dependent on specific factors that critically affect cardiac differentiation, specifically, by concentration- and time-dependent activation of cardiac signalling pathways (reviewed in Filipczyk et al. 2007; Perino et al. 2008; Sarkar and Rao 2009). However, no single protocol has led to pure populations of functional human cardiomyocytes so far. Cell populations resulting from various differentiation strategies usually contain a mixture of different cell types. Therefore, several methods and procedures for the generation of pure cardiomyocyte populations by application of enrichment and/or selection procedures have been established.
At first, manual dissection of contracting cardiac cells (Kehat et al. 2001) and separation by Percoll density-gradient centrifugation (Xu et al. 2002; Xu et al. 2006) were used, which allowed a considerable enrichment of cardiomyocytes. However, these methods are either labour-intensive or disadvantageous with respect to insufficient purity and lack of scalability. More recent studies tried to identify surface markers endogenously expressed by cardiomyocytes, which could be used for cell sorting (Rust et al. 2009; Van Hoof et al. 2010). However, these methods did not yield cardiac cell populations of high purity, most likely due to low abundance of cardiac-specific proteins on the cell surface or to poor antigen–antibody avidity. Another recent method for enrichment of cardiomyocytes derived from hESCs or hiPSCs is based on the high abundance of mitochondria in developing cardiomyocytes (Hattori et al. 2010). Staining of differentiating EBs with a mitochondrial dye and subsequent FACS sorting resulted in cardiomyocyte populations of more than 99% purity. After washout of the dye, cells showed typical characteristics of ESC-derived cardiomyocytes and were able to integrate and survive in the murine heart.
Other methods for cardiomyocyte enrichment depend on genetic modification of hESCs and include positive selection for the expression of reporter and/or marker genes driven by cardiac-specific promoters in hESCs (lineage selection) or negative selection against proliferating cells in the heart cell population using a suicide gene approach. These strategies allowed an efficient enrichment of cardiomyocytes. For example, positive selection for cells expressing a puromycin-resistance gene driven by the cardiac-specific MYH6 promoter resulted in more than 90% α-actinin-positive cells, while negative selection against Herpes simplex virus thymidine kinase (HSV-TK) expression in proliferating (non-cardiac) cells yielded cell populations comprised of 33% cardiomyocytes (Anderson et al. 2007). In another study, negative selection against HSV-TK expression was combined with positive selection for cells expressing the neomycine-resistance gene driven by the mouse α-myosin heavy chain (MHC) promoter, which allowed for an enrichment of cardiomyocytes to nearly 100% purity. Notably, no tumour formation was detected after transplantation of enriched cells into SCID mice (Xu et al. 2008b). In another study, differentiation of hESCs expressing the eGFP reporter gene driven by the cardiac-specific human myosin light chain-2 V (MLC-2 V) promoter and subsequent FACS sorting of differentiated cells resulted in populations containing more than 90% cells that expressed cardiac-specific genes (Huber et al. 2007). Thus, lineage selection methods for cardiomyocyte enrichment seem to be efficient. Moreover, while the (viral or non-viral) genetic modification of cells might be a risk for in vivo applications because of possible tumour formation due to uncontrolled insertion of extrinsic genes, they should be of only minor importance for in vitro applications.
In summary, although in vitro derived, hESC-derived cardiomyocytes exhibit several characteristics of mature human cardiomyocytes, they resemble cardiomyocytes of a foetal stage. However, with respect to their electrophysiological properties, they show characteristics that make them a promising tool for safety pharmacology and drug development. Importantly, recent studies indicate that cardiomyocytes derived from hiPSCs have characteristics identical or very similar to those of hESC-derived cardiac cells (see “Human iPS cell-derived cardiomyocytes for in vitro pharmaco-toxicology”).
Application of hESC-derived cardiomyocytes in pharmaco-toxicology
In general, hPSC-derived cardiomyocytes could have significant advantages over in vitro and in vivo systems currently used in cardiac safety pharmacology. (1) Most importantly, hPSC-derived cardiomyocytes represent human cardiomyocytes that express the characteristic cardiac-specific genes, giving rise to human-specific proteins and signalling components. Cardiomyocytes display APs with cardiac-like morphologies and express genes for characteristic ion channels, such as the If pacemaker, Na+ and L-type Ca2+ and several potassium channels. (2) hPSC-derived cardiomyocytes are genetically identical and can be kept in culture for at least several weeks. It is expected that, in the near future, cardiomyocytes can be produced at large numbers and at reasonable high purity. (3) By use of hPSC-derived cardiomyocytes drug effects on electrophysiological properties can be analysed at the single cell level and in a network of inter-connected cells of cardiac clusters. (4) hPSCs are accessible to genetic modification, which are also present in the respective hPSC-derived cardiomyocytes. In addition, cardiomyocytes can be generated from hESCs derived from PGD embryos diagnosed for cardiac-specific mutations or from iPS cells derived from donors with an inherited cardiac disease or with a cardiac-specific phenotype representing a potential drug risk. (5) Since hPSC-derived cardiomyocytes are of human origin, the predictive value of toxic effects will not be restricted by species-specific differences inherent to animal-based test systems.
Most studies on cardiac differentiation of hPCSs were performed with hESCs, and the characterization of hESC-derived cardiomyocytes usually involved the pharmacological characterization as well as the investigation of cardio-active agents, such as ion channel blockers and substances with known pro-arrhythmogenic effects on cardiomyocytes (reviewed in Denning and Anderson 2008). However, information on the applicability of hESCs to safety pharmacology is still restricted. To assess the respective potential and the limitations of hESC-derived cardiomyocytes, sophisticated dose–response studies must be performed, including the investigation of drug responses on hESC-derived cardiomyocytes at concentrations measured in the plasma of patients under treatment. Moreover, side-to-side comparisons of drug responses of hESC-derived cardiomyocytes and currently used in vitro and in vivo models are required. Finally, a broad application of hPSC-based in vitro systems will require its use in HTP screening settings. In the recent past, several studies were published that aimed towards these goals. The main focus of this research was to present proof-of-concept that hESC-derived cardiomyocytes are able to mirror cardiac functions with respect to drug side effects and that these effects were at least comparable to those detected in currently used cell and tissue models.
The potential usefulness of hESC-derived cardiac cells for safety pharmacology was shown in several recent studies (Caspi et al. 2009; Braam et al. 2010; Jonsson et al. 2010; Liang et al. 2010; Otsuji et al. 2010; Pekkanen-Mattila et al. 2010; Peng et al. 2010). In general, hESC-derived cardiomyocytes were reactive to several drugs in a manner that resembles mature cardiomyocytes (Caspi et al. 2009). For example, E-4031 (a selective IKr blocker) and Sotalol (a class III anti-arrhythmic agent blocking IKs) prolonged action potential duration by mainly effecting terminal repolarization. Continuous blockade of IKr resulted in the development of after-depolarizations and treatment with class I and III anti-arrhythmic substances and cisapride led to a dose-dependent increase in the field potential duration. Administration of class I anti-arrhythmic agents (quinidine and propafenone) resulted in dose-dependent conduction slowing and reduced beating rate, while the gap junction uncoupler 1-heptanol caused slowing of conduction with higher concentrations leading to development of conduction blocks (Caspi et al. 2009). In another study (Braam et al. 2010), QT interval prolongation caused by several agents and drugs was measured by multi-electrode array (MEA) chips seeded with microdissected cardiac clusters consisting of mainly ventricle-like cells. MEA recordings showed dose-dependent QT interval prolongation after treatment with repolarization-prolonging anti-arrhythmic drugs. While anti-arrhythmic drugs caused QT interval prolongations at concentrations observed in the human plasma of patients under therapy with these drugs, several non-cardiac drugs associated with QT interval prolongation and TdP showed a significant effect on the AP duration only at concentrations above typical plasma levels, which underlines the necessity to define adequate safety margins. However, the hESC-based system showed an advantage over the classical hERG assays that would have led to a misclassification of some of the substances investigated. Moreover, the use of a six-well MEA system in this study could be a first step towards the establishment of test systems which allow for a higher throughput of test substances. In a further study, drug effects on parameters additional to AP prolongation were determined, the triangulation of the AP shape and short-term variability of polarization (STV; Jonsson et al. 2010). Both parameters are important for the assessment of a pro-arrhythmic potential, since changes are associated with the appearance of early after-depolarizations (EAD) as a trigger of TdP. The use of additional parameters in the risk assessment of TdP has been suggested since prolongation of AP alone is not considered to be an adequate predictor of pro-arrhythmic events (Hondeghem 2008). In the study by Jonsson and co-workers, E-4031 caused AP prolongation in ventricular-like hESC-derived cardiac clusters and importantly, occurrence of EADs was preceded by an increase in both triangulation and STV. Interestingly, there was a clear positive correlation between the ventricular phenotype of the cardiac clusters (beating frequency <50 bpm; action potential duration at 90% depolarization (APD90) >300 ms) and the occurrence of EADs, which are the cellular events underlying the onset of TdP in the heart.
In addition, several recent studies aimed at the comparison of safety pharmacology assays currently used, namely those using canine or rabbit Purkinje fibres (PFs), with hESC-derived cardiomyocyte-based assays. For example, Jonsson and co-workers found a more pronounced ADP prolongation by E-4031 treatment of rabbit PFs than of hESC-derived cardiomyocytes, but triangulation and SVT values were comparable after treatment with this drug in both systems (Jonsson et al. 2010). For all parameters, baseline values were higher in the hESC-derived cardiac cells compared to rabbit PFs. This could be due to a lower and more heterogeneous coupling in hESC-derived cardiac clusters than in the PFs. In another study, the pharmacological profile of hESC-derived cardiomyocytes was compared to the profile of rabbit and canine PFs (Peng et al. 2010). According to this study, hESC-derived cardiomyocytes were highly sensitive to pharmacological blockage of a multitude of ion channels resulting in respective AP alterations. For most agents tested, the sensitivity of hESC-derived cardiac cells was higher than those of canine or rabbit PFs and species-specific differences were demonstrated (for example, IKs blockage by chromanol 293b that is absent in rabbit PFs; IKr blockage by terfenadine that is absent in canine PFs and less sensitive in rabbit PFs). Whether the higher sensitivity of hESC-derived cardiac cells observed in this study is due to species differences in ion channel expression, experimental settings, lack of diffusion barriers or uptake of substances by cytoplasmic structures (present in PFs, but absent in hESC-derived cells) remains to be determined. Clear advantages of the hESC-based assay compared to PFs were a higher stability and shorter duration, the latter probably due to more immediate drug effects in the hESC-derived cardiomyocytes.
In another recent study, the induction of early depolarizations by sotalol and quinidine was compared between mouse and human ESC-derived cardiomyocytes (Liang et al. 2010). While repolarization-prolonging anti-arrhythmica at relatively high concentrations provoked EADs in all mouse ESC-derived clusters tested, EADs were only occasionally observed in hESC-derived cardiomyocytes indicating either differences in the sensitivity of the specific ion channels or in different contribution of ion channels to the repolarization phase. Nevertheless, all drugs tested in this study showed similar effects on hESC-derived cardiac clusters as detected in earlier studies using animals or other in vitro test systems (Liang et al. 2010).
As discussed earlier, hPSC-derived cardiomyocytes pass through a process of maturation during in vitro cultivation. Accordingly, maturity increases with culture time and pharmacological analyses are usually performed on immature ESC-derived cardiac cells. However, despite their immature phenotype, ESC-derived cardiomyocytes showed close similarities to mature cardiomyocytes with respect to expression profile and functional characteristics, which qualifies hESC-derived cardiac cells for safety pharmaco-toxicology. It is still a matter of debate, whether appropriate drug testing will require more mature cardiomyocytes. Recently, Otsuji and colleagues demonstrated that hESC-derived cardiomyocytes selected for pacemaker function could be cultured for up to 1 year by continuous replating, which resulted in a more mature phenotype at the gene expression and functional levels (Otsuji et al. 2010). However, QT interval prolongation by E-4031 only occurred when hESC-derived cardiac cells were cultured as 3D aggregates, while cultivation under adhesive conditions led to cardiomyocytes with shortened QT intervals. The reason for this observation is not clear, but could be due to specific characteristics of the cell line used in this study (KhES-1). This cell line is characterized by an unusual low spontaneous formation of contractile EBs (less than 1%) and cardiac differentiation was only achieved by co-culture with END-2 cells.
Differences in the ability of hESC lines to develop into the cardiac lineage were already observed in previous studies. It should be noted that the number of hESC lines used for cardiac differentiation so far is very limited. The analysis of more than 70 original studies published on cardiac differentiation of hESCs between 2001 and 2009 revealed that only few cell lines (three of WiCell, two of ES Cell International and one of Harvard University) or their derivatives were used in more than three cardiac differentiation studies. Twenty-nine cell lines were used only in a single study, and their cardiac differentiation potential needs to be confirmed (Löser & Wobus, unpublished).
Until now, the evaluation of hPSC-derived cardiomyocytes in safety pharmacology focused mainly on the detection of unforeseen side effects on the electrophysiology of human heart cells. However, a major problem linked to the use of some cytostatic drugs is cardiac toxicity, which is an important complication of high-dose chemotherapy (reviewed in Albini et al. 2010). While some anti-cancer drugs exert their effects on the cardiovascular system rather in the periphery, for example, by increasing peripheral vascular resistance and blood pressure or by affecting the coagulation system, others directly damage cardiac myocytes. Mechanisms involved in anti-cancer therapy-induced heart damage include, for example, alterations of cellular signalling in cardiomyocytes (perturbing mitochondrial functions and activating apoptosis pathways), the stimulation of free radical production or massive histamine release resulting in conduction disturbances and arrhythmias. A prominent example of unexpected cardiac side effects of anti-cancer drugs is trastuzumab (Herceptin), a monoclonal antibody binding to the membrane protein HER-2 that is over-expressed in some cancers. Trastuzumab is approved for breast cancer therapy, but application resulted in heart failure in up to 4% and decreased cardiac function in up to 10% of patients, especially when co-administered with anthracyclines (Chien 2006). The cellular target of the cardiac side effects of trastuzumab is still unknown, and cardiotoxicity of this drug was not detected prior to its use in phase III clinical trials. In general, a reliable prediction of cardiotoxicity is hampered due to the still fragmentary understanding of the molecular basis underlying cardiotoxicity, the lack of recognized surrogate endpoints and the availability of adequate pre-clinical test systems.
In this respect, it is of interest to note that recent studies started to establish endpoints for cardiotoxicity that could be useful in toxicological test systems based on hESC-derived cardiac sells. For example, release of cardiac troponins into the mediums is a measure for drug-induced decrease in cardiac cell viability. Therefore, it was suggested to use human troponins I and II as well as cardiac natriuretic peptides as biomarkers to predict potential cardiac damage by chemotherapy (Dolci et al. 2008). Recently, the establishment of a hESC-derived cardiomyocyte-based assay was reported, in which the doxorubicin-induced release of troponin T and fatty acid binding protein 3 (FABP3) from cardiac cells was determined by surface plasmon resonance (SPR) biosensing (Andersson et al. 2010). The combination of additional markers with improved human cell-based in vitro systems will be helpful for cardiac toxicity screening of drugs such as anti-cancer agents. In addition, such improved in vitro systems could also facilitate the analysis of the mechanisms underlying cardiac side effects of anti-cancer drugs.
To obtain heart cells of a more mature phenotype, the generation of three-dimensional (3D) so-called engineered heart tissue (EHT) could be an option. Mechanical stretch as maturation-inducing factor has been included into this model. Several methods for the development of EHT using animal (mainly neonatal rat) cardiomyocytes were developed (Li et al. 2000; Shimizu et al. 2002; Zimmermann et al. 2002). The generation of 3D EHTs from hESC-derived cardiac tissue was also performed by seeding hESC-derived cardiomyocytes either alone or in combination with endothelial progenitor cells and embryonic fibroblasts on degradable scaffolds (Caspi et al. 2007; Lesman et al. 2010). However, these studies were performed with the major goal of analysing the engraftment, viability and functionality of human EHTs after transplantation into immune-suppressed animals, and not in respect of the establishment of models for drug screening or safety pharmacology. Interestingly, in a recent study, fibrin-based mini-EHTs containing neonatal rat heart cells were applied to detect changes in relaxation time and contractile forces following drug treatment (Hansen et al. 2010). Of note, contractile activity in these experiments was measured in a 24-well scale using an automated detection system. Further studies have to show whether similar approaches can be developed for hESC-derived cardiac cells.
Human iPS cell-derived cardiomyocytes for in vitro pharmaco-toxicology
With the successful cardiac differentiation of hiPSCs, the use of hiPSC-derived cardiomyocytes for pharmaco-toxicological studies comes into view. Cardiomyocytes were differentiated from hiPSCs by both the classical EB method and by co-culture with END2 cells (Yokoo et al. 2009; Zhang et al. 2009; Zwi et al. 2009; Freund et al. 2010). In addition, ISL1+ progenitor cells isolated from hiPSCs were induced to differentiate into the major cardiac cell types (Moretti et al. 2010a).
Cardiomyocytes differentiated from hiPSCs were found to be very similar to those derived from hESCs with respect to morphology, gene expression patterns and electrophysiological features. The cardiomyocytes showed APs of nodal-, ventricular- and atrial-like phenotypes, responsiveness to pharmacological stimulation and sensitivity to ion channel blockers. In one study, hiPSC-derived cardiac cells even exhibited a higher maturation grade with respect to calcium signalling than cardiomyocytes derived from hESCs (Gai et al. 2009). Moreover, cardioactive drugs had the same effect on contractility and beating frequency in hiPSC- and hESC-derived cardiomyocytes, and several drugs caused the expected changes of QT-interval duration (Yokoo et al. 2009; Zwi et al. 2009).
Although the number of hiPSC lines investigated with respect to cardiac differentiation is still rather low and the impact of factors such as residual expression of reprogramming genes on cardiac differentiation remains to be determined, it is obvious that hiPSC-derived cardiomyocytes will be a valuable source for cardiac cells in drug discovery and safety pharmacology. This has also attracted the interest of the pharmaceutical industry. For example, the Madison-based company Cellular Dynamics International (CDI) has launched hiPSC-derived iCell™ Cardiomyocytes designed to aid drug discovery and improve the predictability of compound efficacy and toxicity (Cellular Dynamics, Press release of Dec 12th, 2009). Moreover, there is some hope that in the future patient-specific hiPSCs could be commonly used in safety pharmacology for selecting optimal medication for specific patients or patient groups. For example, there may be different causes for a higher susceptibility of certain patients to drug-induced QT interval prolongation and TdP, e.g. specific mutations in genes coding for ion channels, and genetic testing for respective subtypes would be difficult and erroneous (Tester et al. 2006). In these cases, the use of patient-specific hiPSC-derived cardiomyocytes could be an attractive way to detect patient-specific risks, such as prolonged QT intervals following treatment with certain drugs.
Apart from their potential role in safety pharmacology screening, cardiac cells derived from patients may also be useful to investigate the pathophysiology of cardiac disorders. For example, hiPSCs generated from patients with LEOPARD syndrome were successfully differentiated into cardiomyocytes (Carvajal-Vergara et al. 2010). Although there were some drawbacks with respect to the variability of the cell populations, cardiac cells showed some characteristics consistent with cardiac hypertrophy commonly found in patients with this syndrome. In addition, phosphorylation of certain proteins possibly involved in the diseased phenotype was altered. Recently, cardiac cells were also derived from two patients with an inherited form of the LQT syndrome (Moretti et al. 2010b). These cells showed disease-specific anomalies in the duration of action potentials, the action potential rate adaptation and IKs currents. It was shown that in the specific mutant channel trafficking to the plasma membrane was suppressed in a dominant-negative manner. These first studies clearly underline the potential value of patient-specific hiPSC-derived cardiomyocytes for a deeper understanding of disease mechanisms and for the identification of novel drug targets.
Future challenges of cardiotoxicity testing using human pluripotent stem cells
It was shown over the past years that functional cardiomyocytes can be obtained from both hESCs and hiPSCs. These cells are of human origin, express the genes for important ion channels, are responsive to external stimuli and show the anticipated response to a broad range of pharmaceutically active agents. Therefore, cardiomyocytes derived from hPSCs may offer the basis for the development of novel, human-specific platforms for assessing the safety of novel compounds. However, to make hPSC-derived cardiomyocytes widely applicable to screening procedures and to gain the acceptance of regulatory authorities, several problems have to be solved:
(i) Efficient differentiation of hPCSs and enrichment of cardiomyocytes
Although there is much progress in cardiac differentiation of hPSCs, the understanding of the differentiation process and the characteristics of defined human cardiac progenitor cells is still limited and further research in this field is required. A deeper understanding of factors and signalling pathways involved in cardiac differentiation is the basis for the development of improved differentiation protocols. In the longer term, reliable, efficient and standardized differentiation protocols have to be established that can be reproduced with different pluripotent cell lines in various laboratories. Since hPSC differentiation usually results in a mixture of more and less mature cardiomyocytes of different cardiac phenotypes (Pekkanen-Mattila et al. 2010), strategies for enrichment of cells of a specific phenotype and a certain degree of maturity will be needed.
(ii) Maturation of stem cell-derived cardiomyocytes
So far, hPSC-derived cardiomyocytes resemble in some aspects a foetal, relatively immature cardiac phenotype. Although currently available cells show a panel of electrophysiological and pharmacological similarities to the human heart, the generation of cells with a phenotype more similar to human adult cardiomyocytes will be necessary to recapitulate the situation in the human heart. One possible approach to obtain more mature cardiomyocytes could be culturing the cells for extended periods of time which is, however, labour-intensive (Otsuji et al. 2010). In addition, mechanical loading was shown to stimulate maturation of cardiomyocytes derived from mouse ESCs cultured on elastic polymer scaffolds (Gwak et al. 2008). Furthermore, it was shown recently that non-cardiomyocytes within the differentiating cell population drive ion channel development and electrophysiological maturation of hESC-derived cardiomyocytes (Kim et al. 2010a). It will be interesting to figure out the nature of these cells and the instructive signals that promote cardiomyocyte maturation.
(iii) Scaling up hESC differentiation
Current methods for differentiation of hESCs to cardiomyocytes suffer from a poor scalability. For example, EB generation and protocols for extended culture are labour-intensive and difficult to scale up. In addition, differentiation is still costly due to the requirement of expensive growth factors. Thus, protocols for production of hPSC-derived cardiomyocytes are required in which manual labour is minimized and costs are significantly reduced, e.g. by using more cost-effective defined chemical compounds. Efforts to scale up cardiac differentiation in either stirred bioreactors (Niebruegge et al. 2009) or suspended microcarrier (Lecina et al. 2010) cultures have been initiated but need further development and standardization.
(iv) Comparison with currently used standards
In recent studies, proof-of-principle was provided that hPSC-derived cardiomyocytes are sensitive to a panel of agents with well-known effects on the human heart, and, in some studies, cardiac electrophysiology was compared to that observed in existing animal-derived test systems. However, thorough side-by-side comparisons of hESC-derived cardiomyocytes and established in vitro and in vivo models for drug testing have still to be done. This will be the prerequisite to assess the potential and limitations of hESC-based systems for detection of cardiotoxicity. Ideally, the proof of the usefulness of hESC-derived cardiomyocytes would be the identification of agents as cardiotoxic that had failed in late stages of drug development or in clinical trials.
In summary, the use of hPSC-derived cardiomyocytes for safety testing is a promising option, but requires more efficient and reproducible differentiation protocols that allow for the production of mature cardiomyocytes at large numbers. The hPSC-derived cardiomyocytes have to show essential characteristics of human heart cells, should be readily accessible to cryopreservation and must be suitable for use in HTP screening platforms.
The potential of hepatocytes generated from human pluripotent stem cells for safety pharmacology and toxicology
The need to test drug-induced hepatotoxicity
Drug-induced liver injury (DILI) is a serious complication of clinical routine therapy and among the most frequent reasons for withdrawal from the market of approved drugs. DILI is the major cause of acute liver failure and of liver transplantations in Western countries. In a recent search of several DILI registries, 298 approved drugs were identified that caused liver injury as a side effect. A total of 265 of these drugs were associated with acute liver failure and six have been suspended or withdrawn due to hepatotoxicity either in the US or in Europe (Suzuki et al. 2010). In addition, also herbal products have been widely associated with hepatotoxicity (reviewed in Seeff 2007).
The mechanisms by which drugs and their metabolites exert liver toxicity are complex and only partially understood. Mechanisms for DILI include cell stress imposed by reactive metabolites (e.g. depletion of glutathione or binding to enzymes and lipids), direct targeting of mitochondrial functions (e.g. ATP depletion or inhibition of β-oxidation causing steatosis) and immune reactions triggered by drugs or their metabolites. These processes ultimately result in apoptotic or necrotic cell death (reviewed in Russmann et al. 2009). While some drugs cause a reproducible and dose-dependent liver damage in several animal species and usually can be detected in pre-clinical development, a large proportion of DILI in man occurs infrequently, partially only after long-time treatment, without obvious dose dependency and in an unpredictable fashion (“idiosyncratic” hepatotoxicity). Idiosyncratic hepatotoxicity may depend on many individual “risk” factors such as genetic predisposition, individual differences in liver metabolism or environmental factors (reviewed in Chalasani and Bjornsson 2010).
Currently, there is no in vitro system that mirrors the complex functions of the human liver. Considerable differences among species in the activities of drug metabolizing enzymes and the production of hepatotoxic metabolites in a species-specific manner hamper the use of classical pre-clinical animal models and animal cell-based in vitro systems for the prediction of hepatic toxicity in humans. Hepatotoxic effects may become only apparent in human beings, and unexpected human liver toxicity is one of the main causes for failure of promising drug candidates in development. To assess potential DILI in drug development, human cell systems are needed that reproduce fundamental processes involved in hepatotoxicity and can predict human liver toxicity with high reliability.
To date, isolated primary human hepatocytes (pHHs) are the most suitable in vitro cell models for drug metabolism and hepatocyte toxicity studies (reviewed in Hewitt et al. 2007). Hepatocytes are the major cell type of the liver and, when cultured under appropriate conditions, retain many physiological functions of liver cells, but only for limited time. pHHs can be used to investigate many aspects of DILI, such as cell viability, formation of toxic drug metabolites, impairment of mitochondrial function or inhibition of the biliary transport system. The major problems that limit the use of pHHs in standard hepatocyte toxicity testing are the limited availability and the restricted in vitro proliferation capacity. In general, the use of pHHs is highly expensive, and supply with freshly isolated hepatocytes is unpredictable. In addition, cultured primary hepatocytes lose their differentiated phenotype (=dedifferentiation) even under optimized culture conditions, and metabolic functions critical for investigation of drug metabolism and toxicity decline over time. Moreover, there are considerable variations between hepatocyte preparations due to a variety of factors such as health, diet and medication of the donor, genetic polymorphisms between donors and differences in the epigenetic status of relevant genes resulting in diversity of metabolic phenotypes. Notably, expression of genes for cytochrome P450 proteins (CYP450s) critical for phase I metabolism strongly varies between hepatocyte preparations from different donors which may confound data interpretation.
As an alternative to primary hepatocytes, human hepatocellular carcinoma cell lines that exhibit partial liver cell characteristics have been frequently used for the investigation of certain aspects of liver metabolism and toxicity (reviewed in Donato et al. 2008), but their use is limited due to low expression of crucial hepatocyte-specific genes and no or only low enzyme activities of the CYP450 family members that are critical for phase I metabolism.
In addition, attempts were undertaken to derive functional hepatic cells from somatic stem cells from outside the liver. First data on the contribution of un-fractionated bone marrow-derived stem cells to liver regeneration were published in 1999 (Petersen et al. 1999), but the concept of trans-differentiation was challenged by others (Terada et al. 2002; Ying et al. 2002). Still, there are many reports describing hepatic differentiation of other human cells, for example, blood monocytes (Ehnert et al. 2008) and mesenchymal stem cells (MSCs; reviewed in Ochiya et al. 2010). According to these reports, hepatocyte-like cells expressed a panel of liver-specific genes and proteins. However, although liver functions after transplantation of such cells into rodents could be improved in certain experimental settings, their functional characteristics were limited in most cases. Moreover, a broader use of MSC-derived hepatocytes in screening studies for hepatotoxicity has not been reported so far, obviously because of restricted in vitro proliferation and differentiation potential and lack of standardization. It has to be shown whether and to what extent these hepatocyte-like cells may be able to be used as human in vitro cell system.
Consequently, there is a strong need for human-specific in vitro test systems that mirror critical characteristics of liver cells, and present efforts are directed towards the establishment of PSC-derived hepatocyte-like cells. Here, the generation and characterization of hepatocyte-like cells from hESCs and hiPSCs will be presented.
Generation of hepatocyte-like cells from hESCs
In early studies, mouse ESCs were differentiated spontaneously or by addition of growth factors (Abe et al. 1996), which resulted in the formation of hepatocytic cells expressing hepatocyte-specific genes (reviewed in Lavon and Benvenisty 2005). More advanced protocols for the in vitro generation of hepatocyte-like cells from ESCs were predominantly based on data of mouse liver development (reviewed in Si-Tayeb et al. 2010a). In brief, during gastrulation definitive endoderm (DE) develops from epiblast cells that transit the most anterior region of the primitive streak. Formation of DE in the early embryo strongly depends on transforming growth factor (TGF)-β and Wnt-signalling. Further specification to cells of the hepatic epithelial lineages involves signalling from the developing cardiac mesoderm and the septum transversum, which provide FGF and BMP signalling, respectively. Cells then start to proliferate and to invade the stroma of the septum transversum mesenchyme thereby forming the liver bud composed of bi-potent cells that express early hepatocytic and biliary markers. Colonization of the liver bud by hematopoietic cells and autocrine signalling further stimulate liver development. Induction of Notch signalling promotes differentiation along the cholangiocytic lineage, while hepatocyte growth factor (HGF) excreted by the septum transversum and non-parenchymal liver cells supports differentiation into foetal hepatocytes. Regional specification of hepatocytes is regulated by a complex network of transcription factors including β-catenin and hepatocyte nuclear factor (HNF)-4α, and further maturation is stimulated, for example, by the action of oncostatin M (OSM) mainly produced by hematopoietic cells and of glucocorticoids.
Many efforts have been undertaken to obtain mature and metabolically active hepatocytes from hESCs, and up to now, there are reports from more than 20 research groups describing the establishment and optimization of differentiation protocols (reviewed in Baxter et al. 2010). Most strategies rely on the in vitro recapitulation of developmental processes and involve the application of soluble growth factors, co-culture with hepatic and non-hepatic cell types and reconstruction of an extracellular matrix (ECM). In addition, a positive effect of histone deacetylase inhibitors such as sodium butyrate on hepatic differentiation was observed. In some cases, these approaches were supplemented by introduction of reporter genes driven by liver-specific promoters into hESCs or hepatic progenitors to trace differentiated cells. In general, the majority of recent protocols for hepatocyte differentiation of hESCs include three major steps: (1) the differentiation of hESCs into DE cells, followed by (2) the specification into hepatic progenitor cells and (3) maturation into hepatocyte-like cells (see Baxter et al. 2010).
Methods for hepatic differentiation of hESCs were established to mimic processes of lineage commitment during embryogenesis by use of multi-stage protocols starting from either hESC-derived EBs or monolayer cultures. EB formation in the presence of serum and addition of growth factors (Rambhatla et al. 2003; Shirahashi et al. 2004; Schwartz et al. 2005; Baharvand et al. 2006; Duan et al. 2007; Chiao et al. 2008; Basma et al. 2009) mimic processes taking place during gastrulation, thereby providing signals necessary for the induction of hepatocyte development. Subsequent plating of cells and treatment with exogenous factors resulted in the generation of cells exhibiting features of hepatic cells. However, the process of EB formation is spontaneous, stochastic and results in the development of diverse cell types. Alternatively, staged protocols starting from monolayer cultures were exploited more recently. These protocols usually involve as a first step the generation of DE cells from hESCs, which are characterized by DE-specific Sox-17 and FoxA2 (HNF-3α) co-expression (D’Amour et al. 2005). DE formation has been obtained by cultivation of hESCs for usually 3–4 days at low serum in the presence of activin A alone (Agarwal et al. 2008) or in combination with insulin–transferrin–selenium (ITS; Cai et al. 2007; Song et al. 2009), by the addition of sodium butyrate (Hay et al. 2008b; Duan et al. 2010), Wnt3a (Hay et al. 2008a) or FGF2 (Brolen et al. 2010). In some protocols, inhibition of activin effects by serum was antagonized by phosphatidylinositol 3-kinase (PI3 K) inhibitors (McLean et al. 2007; Shiraki et al. 2008). Efficient DE formation was also accomplished by combining activin A, BMP2, FGF2 and a PI3 K inhibitor (Touboul et al. 2010). Subsequently, in most protocols hepatic specification from DE was shown to be stimulated by supplementation with FGF and BMP proteins for several days. Finally, the development of hepatic progenitors to more mature hepatocytes was achieved by treatment with additional growth factors such as hepatocyte growth factor (HGF), dexamethasone (Dex) and oncostatin M (OSM).
Cell-to-cell interactions are important both in the developing and the mature adult liver. Therefore, co-culture of differentiating liver cells with different cell types was tested for its capacity to improve hepatic differentiation of hESCs. For example, co-culture with mouse embryonic stromal (STO) cells allowed for the expansion of hepatic progenitor cells (Zhao et al. 2009a), and co-culture with a mesoderm-derived mouse cell line (M15) favoured expression of liver cell markers in differentiated cells (Shiraki et al. 2008). Similarly, co-cultivation with a murine mesenchymal cell line derived form foetal liver (MLSgt20) promoted hepatic maturation of alpha-foetoprotein (AFP)-positive cells (Ishii et al. 2010). In one study, human foetal liver stromal cells genetically manipulated to secret human FGF-2 were used as feeder cells for hepatic specification of DE cells (Pei et al. 2009). In addition to co-culture with human and non-human cells, growth of differentiating cells on extracellular matrices has been shown to be beneficial for hepatic differentiation. For example, laminin, Matrigel and collagen matrices were tested under 2D conditions (Ishii et al. 2008) with the best effects observed with Matrigel. To mimic 3D conditions of hepatic differentiation, collagen type 1 scaffolds as well as poly-amino-urethane (PAU)-coated polytetrafluoroethylene (PTFE) fabrics have been used (Baharvand et al. 2006; Soto-Gutierrez et al. 2006).
Because hESC-derived hepatocyte differentiation is still not efficient, enrichment strategies may be necessary to obtain hepatocyte cultures of adequate purity, but lineage selection has been applied only rarely. This might be due to the fact that the hepatocyte identity can be hardly linked to the exclusive expression of a single gene, but rather to a specific transcriptional network. For example, the selection of cells expressing an AFP promoter-driven reporter gene resulted in hepatic cell populations that showed only low levels of liver-specific transcripts, such as albumin, tyrosine aminotransferase and tryptophan 2, 3-deoxygenase (Ishii et al. 2008). This is probably due to the fact that AFP gene expression is not restricted to DE cells from which hepatocytes arise, but also to extra-embryonic endoderm. Other selection strategies for hepatic cells were based on “genetic” approaches, such as the use of albumin or alpha-1-antitrypsin (AAT) promoter-driven reporter systems (Lavon et al. 2004; Duan et al. 2007). On the other hand, a selection strategy based on FACS sorting of differentiated cells for the presence of the surface asialoglycoprotein receptor 1 (ASGPR1) allowed the enrichment of hepatocyte-like cells. Secretion of albumin, urea and AAT as well as expression and activity of the CYP450 proteins by sorted cells reached levels near to those observed in pHHs (Basma et al. 2009).
Characteristics of hESC-derived hepatocyte-like cells
In the past, derivation of hepatocytes from diverse (stem) cell types has been reported. Hengstler et al. addressed the problem of over-interpreting stem cell-derived cells as “hepatocytes” and suggested a minimal set of functions that are necessary to define “true” hepatocytes: (1) metabolism of xenobiotics and endogenous substances (hormones and ammonia), (2) synthesis and secretion of albumin, clotting factors, complement, transporter proteins, bile, lipids and lipoproteins and (3) storage of glucose (glycogen), fat soluble vitamins A, D, E and K, folate, vitamin B12, copper and iron (Hengstler et al. 2005). Furthermore, the comparison of stem cell-derived hepatocyte-like cells with primary hepatocytes was suggested to be mandatory.
More recently, a list of criteria for characterization of “liver cells” derived from various stem cell sources has been recommended (Sancho-Bru et al. 2009). Although these criteria aim at the production of pure and standardized hepatocyte populations for future clinical use, they may be also applicable to the characterization of hepatocytes for in vitro toxicology applications. The list includes parameters for characterization of cells during stages of hepatic differentiation, criteria for ultrastructural evaluation as well as a panel of functional tests. Since several “hepatocyte” markers and functions are also exhibited by other than liver cells (e.g. expression of certain CYP450 genes, glycogen storage, etc.), the authors recommend to demonstrate co-expression of hepatocyte markers in the same cell and to perform several functional tests to unambiguously prove hepatocyte identity of the differentiated cell population (Sancho-Bru et al. 2009).
Cells derived from hESCs have been analysed in vitro for hepatic properties, including morphological and ultrastructural features as well as gene expression and functional characteristics. In some studies, the ability of hESC-derived hepatocyte-like cells to repopulate mouse livers was also investigated (Agarwal et al. 2008; Basma et al. 2009; Touboul et al. 2010). Although, in general, hepatocyte-like cells exhibited some typical hepatic characteristics, quantitative comparative analyses of hESC-derived hepatic cells with pHHs for metabolic functions critical for drug metabolism were only performed in a few studies.
Morphological analyses revealed the polygonal shape and distinct round nuclei typical for hepatocytes in hESC-derived cells. In addition, hepatocyte-specific ultrastructural characteristics, such as prominent nucleoli, well-developed Golgi apparatus, abundance of mitochondria and lysosomes, glycogen granules and rough, smooth endoplasmic reticulum as well as cell-junctional complexes and biliary canaliculi were observed (Baharvand et al. 2006; Baharvand et al. 2008; Shiraki et al. 2008; Basma et al. 2009; Pei et al. 2009; Sasaki et al. 2009).
The process of hepatic differentiation of hESCs has been characterized by analysing transcripts and proteins typical for specific stages of hepatocyte development (reviewed in Snykers et al. 2009). The differentiation of DE cells was determined by transcript levels of FoxA2, Sox17 and partially CXCR4. Presence of hepatocyte progenitors was verified by the presence of AFP at transcript and protein levels, usually in combination with albumin, a marker of more mature hepatic cells. In one study, bi-potential hepatic progenitor cells derived from sorted N-cadherin-positive hepatic endoderm and expressing AFP (but not albumin) were generated. These cells showed some proliferative capacity as determined by Ki67 gene expression and expressed the putative hepatoblast markers EpCAM and CD133 as well as the bile duct lineage markers KRT7 and KRT19 (Zhao et al. 2009a). Finally, in most studies maturation of hepatocytes was verified by the presence of combinations of liver-specific transcripts and proteins, such as albumin, α1-antitrypsin, certain cytokeratins, HNF-4, tyrosine aminotransferase (TAT), CYP450s, transthyretin (TTR) and others (for overview see Snykers et al. 2009).
However, the definition of the hepatic identity of hESC-derived cells has been complicated by the fact that the protocols used for hepatic differentiation generally resulted in a mixture of cell types. Some markers considered as “liver-specific”, such as AFP, are also present in other cell types. AFP gene expression declines during hepatocyte differentiation and is absent from mature adult hepatocytes. However, in several studies, AFP gene expression was still detected at the end of the in vitro differentiation process, indicating that the hESC-derived hepatocyte-like cells represent an immature phenotype (Baharvand et al. 2006; Hay et al. 2007; Hay et al. 2008b; Ishii et al. 2008). Other genes, such as those for CK18 or TTR, are also expressed at several stages of hepatic development in vitro, and without the analysis of additional markers the correct assignment of the cell population to a specific developmental stage is not possible. Therefore, detection of simultaneous expression of hepatocyte-specific genes, such as transcription factors and/or hepatic enzymes, is necessary to ensure a genuine hepatic phenotype of the differentiated cells.
To determine the functionality of hESC-derived hepatocyte-like cells, tests for liver-specific metabolic functions were performed, frequently including storage of glycogen, albumin secretion and uptake and release of indocyanine green (ICG; reviewed in Baxter et al. 2010). In several studies, hepatocyte-like cells were tested for other liver cell functions, for example urea synthesis and secretion, secretion of additional serum proteins such as α1-antitrypsin (AAT) or coagulation factors, low-density lipoprotein (LDL) uptake, gluconeogenesis or entry of a pseudotyped hepatitis C virus into cells (Schwartz et al. 2005; Soto-Gutierrez et al. 2006; Cai et al. 2007; Baharvand et al. 2008; Fletcher et al. 2008; Hay et al. 2008b; Basma et al. 2009; Zhao et al. 2009a) .
However, a validated standardized protocol for differentiation of hESCs toward hepatocytes is currently not available, exact parameters that define the functionality of hepatic cells are lacking and the data differ among studies. Another problem is the high variability of cellular reference systems used by different authors and the poor description of these systems in some studies. Reference systems frequently used are hepatoma cell lines (such as HepG2, HuH7 and Alexander cells), foetal and adult liver cell extracts, and rat or human pHHs. Although pHHs were regularly used, data on the origin of pHHs (e.g. freshly isolated vs. cryoconserved, individual donor vs. hepatocyte pool, in-house prepared cells vs. commercially acquired cells, etc.), methods for hepatocyte cultivation and data on age, purity and characteristics of the specific hepatocyte preparation are often lacking. These problems complicate the comparison of hESC-derived hepatocyte-like cells with respect to their liver cell characteristics and hamper the assessment, whether specific protocols may be useful for efficient differentiation of functional hepatocytes from hESCs.
In summary, there has been some progress in the establishment of protocols for the generation of hepatocyte-like cells from hESCs over the past decade. It is still a matter of debate to what degree stem cell-derived hepatocytes will have to resemble primary human hepatocytes before they can be used to reliably predict metabolite generation and (de)toxification (Hengstler et al. 2005). In any case, the establishment of a validated and standardized battery of tests to assess the identity and functionality of hepatocyte-like cells would be a major advance in this field (see also below).
Derivation of hepatocyte-like cells from hiPSCs
The development of reprogramming strategies offers the opportunity to generate hepatic cells from hiPSCs, which could have a major impact on human disease research, regenerative medicine and drug development. hiPSC-derived hepatic cells from different ethnic groups and polymorphic variants as well as from patients suffering from specific metabolic diseases would allow the investigation of individual drug risks that are not accessible with other techniques. Moreover, patient-specific hiPSC-derived hepatocytes would be available for cell therapeutic applications.
The generation of hepatocyte-like cells from hiPSCs was shown in several studies (Song et al. 2009; Liu et al. 2010; Si-Tayeb et al. 2010b; Sullivan et al. 2010; Touboul et al. 2010). Independent of the origin of donor cells and reprogramming factors, hiPSCs were generally able to differentiate into hepatocyte-like cells that expressed genes in a liver-specific manner and exhibited certain hepatocyte functions. In these proof-of-concept studies, the efficiency of differentiation towards hepatocyte-like cells was comparable to hESCs and only subtle differences in gene expression and metabolic capacities of differentiated progeny were observed between hESCs und hiPSCs. Minor differences were also observed in hepatocyte-like cells derived from different hiPSC lines probably due to the heterogeneity of donor cells used for hiPSC generation. However, expression levels of genes for liver-specific enzymes and metabolic activities were only marginally tested, and a more comprehensive and comparative analysis of hiPSC-derived hepatic cells with respect to important liver cell characteristics remains to be done.
In general, hiPSCs have been mainly produced from fibroblasts. In one study, however, pHHs were exploited as starting material for hiPSC generation and subsequent hepatic differentiation (Liu et al. 2010). When compared to MSC-derived hiPSCs, reprogramming of pHHs resulted in the accelerated formation of hiPSC colonies, but the hepatic differentiation capacity of hepatocyte-derived hiPSCs did not differ from that of hESC- and fibroblast-derived hiPSCs. However, an extensive analysis of hepatic characteristics as well as quantitative investigations of differentiation capacities has not been performed. In view of recent results of the epigenetic memory of iPS cells (Kim et al. 2010b), it will be interesting to explore whether hepatocyte-derived hiPSCs might allow for a more effective generation of hepatocyte-like cells, which exhibit a higher maturation level as hESC-derived hepatocyte-like cells. In addition, use of pHH-derived hiPSCs for generating patient-specific hepatocytes may also be of interest for developing disease models, since, for example, somatic mutations important for development and progression of liver cell tumours are acquired in the liver only and cannot be mirrored by cells generated from fibroblast-derived hiPSCs (Bluteau et al. 2002).
Recently, hiPSCs were generated from patients with inherited metabolic disorders of the liver (Rashid et al. 2010). The cells were differentiated into the hepatic lineage resulting in hepatocyte-like cells that showed some features of mature hepatocytes and, most importantly, exhibited molecular characteristics of the diseased phenotypes (α1-antitrypsin deficiency, familial hypercholesterolaemia and glycogen storage disease type 1α, respectively). This proof-of-concept study provided evidence that multiple liver disease-specific cell lines can be generated and that hepatocytes differentiated from these hiPSCs may be useful for modelling liver diseases in vitro and in a patient-specific manner.
Requirements for the use of pluripotent stem cell-derived hepatocyte-like cells in safety pharmacology and toxicology
As mentioned earlier, drug-induced liver injury is a multi-factorial disorder commonly due to CYP450-dependent formation of reactive metabolites that directly exert hepatotoxicity or form adducts with liver proteins triggering an immune response. In addition, hepatotoxicity can be caused by interference with mitochondrial function, inhibition of xenobiotic metabolism or disturbance of bile acid transport. In many cases, liver toxicity only occurs after prolonged exposure to drugs. It is still a matter of debate whether hepatocytes in vitro might be able to model chronic hepatotoxicity. In any case, hepatocyte populations of reproducible quality, which can be cultured for prolonged periods of time and which show liver cell-specific functions comparable to those of pHHs would be a suitable in vitro system to investigate potential hepatotoxic drug effects.
One of the most critical parameters for the potential use of hPSC-derived hepatocyte-like cells in safety pharmacology and toxicology is their ability to metabolize drugs by phase I enzymes of the CYP450 family. In addition, the presence and functionality of phase II enzymes involved in drug conjugation and of transporter proteins responsible for elimination and clearance of drug metabolites as well as for uptake of drugs will be important for the functionality of stem cell-derived hepatocyte-like cells in vitro. So far, data on the activity of these enzymes in hPSC-derived hepatocyte-like cells are still fragmentary. While in some earlier studies the activity of certain CYP450 subtypes in hESC-derived hepatocyte-like cells was determined (Rambhatla et al. 2003; Schwartz et al. 2005), data on CYP450 gene expression patterns are lacking. In some studies, CYP450 expression at the transcript and protein level was analysed and metabolic activity of the CYP450 enzymes was determined in several studies.
Obviously, there are broad differences in CYP450 gene expression between hESC-derived hepatocyte-like cells generated in different studies. An example is the expression of the Cyp3A4 gene whose product plays a central role in metabolism of aromatic amines, oestrogen compounds and certain drugs. In some studies, CYP3A4 transcripts were reported to be present at up to 1,000-fold lower abundance in hESC-derived hepatocyte-like cells than in human liver or pHHs even after CYP induction (Ek et al. 2007; Shiraki et al. 2008; Brolen et al. 2010), whereas Basma and colleagues found an only threefold lower Cyp3A4 transcript level in the hESC-derived progeny compared to pHHs (Basma et al. 2009). This discrepancy is probably not solely due to the hepatocyte enrichment step performed in the latter study, because sorting for ASGPR1-positive cells only yielded about twofold enrichment of hepatocyte-like cells according to albumin secretion data. Other reasons may include the use of different hESC lines (KhES-1, SA02 or SA167 and H1, respectively), different protocols used for in vitro differentiation or, most likely, the quality of pHHs used as reference cells.
In several studies, metabolic activities of CYP450 enzymes present in hESC-derived hepatocyte-like cells were analysed (Hay et al. 2008a; Basma et al. 2009; Moore and Moghe 2009; Zhao et al. 2009a; Brolen et al. 2010; Touboul et al. 2010). CYP450 activity was generally detected, but with considerable variations among studies. This can be illustrated by data on Cyp1A2 activity that was determined in at least six studies by either EROD assay or measurement of phenacetin metabolization. In an early study on hepatic differentiation of hESCs, CYP1A2 activity in hESC-derived hepatocyte-like cells was shown to be comparable to that in pHHs (Rambhatla et al. 2003). Interestingly, the results of the EROD assay presented in this study were comparable to those reported very recently by another group who applied a more specific differentiation protocol involving several hepatic growth factors (Moore and Moghe 2009). In two studies using the EROD assay under similar conditions, Cyp1A2 activity was shown to be 4 and 40% of that measured in pHHs, respectively (Hay et al. 2008a; Basma et al. 2009). In contrast, in another study involving a multi-stage differentiation protocol (Brolen et al. 2010) phenacetin metabolization by Cyp1A was only slightly higher in hESC-derived hepatocyte-like cells than in HepG2 cells. HepG2 cells are known to exert about 100-fold lover Cyp1A2 activity than pHHs (Westerink and Schoonen 2007). In addition to differences in cell lines and the differentiation protocol used in these studies, specificities in the experimental settings and the quality of the reference cells may account for the differences in the reported CYP450 activities in relation to pHHs.
It should be noted that standardized data on the characteristics and the quality of the pHHs used as a reference system are lacking from most studies. Therefore, the reasons for the differences observed in gene expression and enzyme activities of liver cell proteins remain elusive. In many cases, only comparison within their own studies was of interest to the authors (e.g. production of hepatocyte enzymes at different stages of the differentiation process). Consequently, data were often given in a format (e.g. μg/well), which makes it impossible to compare different studies and to draw conclusions on the suitability of a specific protocol for deriving functional hepatocytes from hPSCs. Therefore, presenting data in a format suitable for inter-study comparisons would be desirable. Moreover, data for assessment of the quality of the reference cells (usually pHHs) should be included. For example, urea secretion has been suggested as a reproducible and easy to measure factor with significance for quality of hepatocytes used in bioreactors (Pless et al. 2006).
While some efforts were undertaken to prove the presence and functionality of phase I enzymes in hESC-derived hepatocyte-like cells, the presence and activity of phase II enzymes and transporter protein activity in these cells was investigated in only a few studies. For example, Söderdahl et al. investigated the presence of glutathione S-transferases (GSTs) in hepatocyte-like cells (Söderdahl et al. 2007). While presence of the GSTA1-1 subunit was clearly shown in hESC-derived hepatic cells, GSTM-1 was only weakly expressed in hepatocyte-like cells derived from four of five hESC lines and GSTP-1 gene expression was not detectable. However, general GST activity as measured by catalytic activity towards 1-chloro-2,4-dinitrobenzene (CDNB) was comparable to that in pHHs and clearly exceeded GST activity of the hepatoma cell line HepG2.
In a recent study, a staged protocol was used to obtain hepatocyte-like cells of relatively high purity (about 90% expressing ALB or AAT, and 60% are ASGPR1-positive at days 20–22; Duan et al. 2010). Differentiated cells expressed the genes coding for phase I and phase II liver enzymes, transporter proteins and nuclear receptors critical in regulating the expression of metabolizing enzymes. The major pathways of bufuralol metabolism were shown to be active in these cells and several previously unknown metabolic pathways for bufuralol metabolization were identified. Although metabolites of bufuralol were produced in hESC-derived hepatocyte-like cells to a lower extent than in pHHs, hydroxylation of phenacetin (Cyp1A2) and bufuralol (CYP2D6) was performed at a comparable rate at 48 h post-drug addition. These data are encouraging since, for the first time, it was shown that hESC-derived hepatic cells indeed have the capacity to metabolize drugs in a manner comparable to pHHs.
Until now, there are only limited data available on long-term cultivation of hPSC-derived hepatocyte-like cells. In only a few studies, cells were differentiated for more than 20–30 days and data on stability of gene expression or metabolic function after prolonged culture were only occasionally reported. However, this would be important, because detection of drug effects might require the application over an extended period of time or reiterated exposure. In a few studies, extended differentiation times resulted in increased hepatocyte-specific transcript and protein levels suggesting further maturation in vitro. For example, AAT secretion by hepatocyte-like cells increased nearly sixfold from day 31–43 after starting differentiation although secretion of both albumin and AAT were low in comparison with pHHs (Shirahashi et al. 2004). In another report, albumin transcript levels doubled, whereas AFP gene expression markedly dropped between day 18 and 50 of hepatocyte differentiation (Shiraki et al. 2008). Moreover, Cyp3A4 transcript levels increased about 10-fold in this period. Although both albumin and Cyp3A4 levels were quite low when compared to foetal and adult human hepatocytes in this study, differentiation and culture for extended periods of time may allow for further hepatocyte maturation.
On the other hand, it will be critical to investigate whether hPSC-derived hepatocyte-like cells will undergo de-differentiation and lose their mature phenotype when cultured for several days or weeks. Recently, Kheatni and Bhatia developed a miniaturized, multi-well culture system with an optimized microscale architecture that facilitated extended culture of pHHs for several weeks as determined by analysis of several liver cell-specific functions including phase I/phase II enzyme gene expression (Khetani and Bhatia 2008). Application of such a system to hepatic differentiation of hPSCs could not only allow for longer cultivation, but also help to scale down cell culture to formats required for HTP-based methods in the pharmaceutical industry. However, cryopreservation of hPSC-derived hepatocytes has not been reported yet.
Future challenges of the application of pluripotent stem cell-derived hepatocytes in toxicology
Initial progress has been achieved in the establishment of test systems for hepatotoxicity using hepatic cells derived from hPSCs. However, the following problems have to be solved before these cells can be applied in routine pharmaco-toxicology.
(i) Immature phenotype of hPSC-derived hepatocytes
The immature phenotype and inadequate expression of genes coding for enzymes critical for liver cell metabolism is one of the main problems of hPSC-derived hepatocyte-like cells. To establish novel models for predicting liver cell toxicity with higher confidence than other in vitro systems, further improvements in differentiation protocols are necessary. Since murine ESCs fully mature after transplantation into syngeneic animals, it is likely that also hESCs have the full hepatic developmental potential. Therefore, reproduction of a liver-specific environment may be necessary for hepatocyte maturation in vitro. This may be obtained by ECM-supported 3D-co-culture systems. For example, improved survival and differentiation of foetal human hepatocytes into hepatocyte-like cells have been shown in 3D culture systems under constant perfusion (Ring et al. 2010; Schmelzer et al. 2010). Co-culture with liver epithelial cells and/or vascular endothelial cells may also have a positive impact on ESC-derived liver cell differentiation and maturation (e.g. Moore et al. 2008b). In addition, it will be necessary to establish culture conditions that facilitate the development and maintenance of mature phenotypes for a period long enough to perform pharmacological and toxicological studies.
(ii) Lack of defined and validated endpoints of hepatic differentiation and maturation
The application of hPSC-derived hepatocytes will require defined endpoints for hepatic differentiation and maturation in vitro. Parameters and functional tests appropriate to standardize hepatocyte differentiation and to compare differentiation procedures have been proposed, but are not validated until now. Validation studies should include liver-specific gene expression and several functional tests including analyses of metabolizing enzyme activities.
(iii) Requirement of standardized reference systems for differentiated hepatic cells
At present, pHHs are well-established reference cells, but their metabolic capacity can strongly differ among preparations (Hewitt et al. 2007). Consequently, it is currently difficult to assess to what extent hPSC-derived hepatocyte-like cells exhibit characteristics of liver cells when compared to pHHs. Therefore, standardized and validated parameters defining the quality of reference hepatocytes are needed. In this context, it may be of interest to establish a database of global pHH expression profiles that could be useful to classify specific populations of hepatocyte-like cells. A corresponding system has been successfully developed for pluripotent hES and hiPS cells from diverse sources and laboratories that can be used to assess the pluripotent phenotype (“stemness”) of human stem cells (Müller et al. 2008).
(iv) Generation of hepatocytes at high numbers
To meet the requirements of the pharmaceutical industry, differentiation and cultivation of hepatocyte-like cells have to be scaled up. Human ESCs can be cultured in stirred-suspension bioreactors (reviewed in Kehoe et al. 2010) and an experimental approach for the differentiation of hESCs into definitive endoderm in a microcarrier bioreactor has been reported (Lock and Tzanakakis 2009). It will be necessary to explore similar approaches for the differentiation of hESCs and hiPSCs into the hepatic lineage. For HTP applications, test systems also need to be miniaturized.
(v) High variability of hES and hiPS cell lines and lack of standardization
Studies on hepatic differentiation of hESCs were performed using only about 20 of the more then 1,000 existing hESC lines, and only eight lines were used in more than one single study so far. Similarly, only a limited number of hiPSC lines were tested for their hepatic differentiation potential. Several hESC lines showed a different potential for hepatic differentiation (Ek et al. 2007). It is still an open question, whether protocols established and optimized for a specific cell line can be successfully applied to other cell lines. Since provision of cell lines representing a multitude of metabolic phenotypes would be one of the utmost advantages of using hPSC-derived hepatocytes, investigation of this question is of major interest.
In conclusion, the recent progress in the establishment of hepatic differentiation of human ES and iPS cells is encouraging with the perspective to implement novel human cell-based in vitro assays for toxicology. The test systems may have the potential to predict hepatotoxicity already in early phases of drug development. They would reduce drug attrition, may lead to cost reduction in development of novel drugs and to improved safety profiles of pharmaceuticals. However, before such systems can be implemented into routine toxicity testing, robust and reproducible hepatic differentiation protocols have to be developed and the problems discussed above have to be solved.
Future Perspectives
Since the establishment of the first human pluripotent stem cell lines more than a decade ago the therapeutic potential of stem cells has been extensively discussed. Only in the more recent past, the unique potential of pluripotent stem cells in drug research and toxicology was realized. Pluripotent human stem cells could offer many benefits to this field, especially the perspective of an unlimited supply with human cells of well-defined origin, homogeneous characteristics and relevance to the human situation. While toxicological assays using murine PSCs are already performed routinely, human PSCs are still under investigation with respect to their suitability for these applications. However, proof-of-principle has been provided that these cells could make a valuable contribution to the technology platforms in safety pharmacology and toxicology. Especially in the field of developmental toxicology, hESCs represent a unique human-specific test system, although methodical questions remain to be solved, relevant endpoints must be identified and HTP screening methods still have to be developed.
With respect to cardio- and hepatotoxicology the establishment of reproducible and inexpensive differentiation protocols that are amenable to HTP platforms is still a challenge. However, specifically for cardio- and hepatotoxicology, but also for neurotoxicology studies patient- and disease-specific hiPSCs offer enormous opportunities for the development of in vitro systems that can significantly improve drug safety and contribute to acceleration of drug development.
One of the most important problems related to the use of iPS cells in pharmaco-toxicology, the potentially tumourigenic status of pluripotent stem cells, will eventually be overcome by recent innovations. Instead of reprogramming to pluripotency, several studies demonstrated lineage reprogramming or transcription factor-based trans-differentiation of cells from one lineage to the other. This direct lineage reprogramming could avoid the potentially tumourigenic state of ES or iPS cells and the contamination of reprogrammed cells by residual pluripotent stem cells (reviewed in Wobus 2010). Such direct conversion of fibroblasts into skeletal muscle cells by the master regulator gene MyoD was already shown in the late 1980s (Davis et al. 1987). Recently, murine fibroblasts could be reprogrammed by specific transcription factors into neural (Vierbuchen et al. 2010) or cardiac (Ieda et al. 2010) cells. Previous studies have shown the conversion of B lymphocytes into macrophages by CEP/B expression (Xie et al. 2004). In addition, a switch from the exocrine to the endocrine lineage has been demonstrated in vivo by adenovirus transfection of acinar cells by transcription factors involved in pancreatic differentiation (Pdx1, Ngn3, MafA) resulting in the induction of insulin-producing beta cells (Zhou et al. 2008). This new strategy may also be an attractive alternative for the generation of human “induced cardiomyocytes” (as discussed in Passier and Mummery 2010) or “induced hepatocytes”. The prospect of producing specialized human cells without the need to transiently reprogramme somatic cells into a pluripotent, potentially tumourigenic state is very promising.
Another strategy would be partial reprogramming of specialized cells to a state of higher plasticity (dedifferentiation), resembling progenitor cells with multi-potent capacities. Such cell systems would allow developmental toxicological analyses also on tissue-specific progenitor cell populations.
Future studies have to analyse the preconditions and consequences of these reprogramming strategies for human cells, such as the role of the cell type of origin, epigenetic status of donor cells as well as the stability and functionality of the trans-differentiated or dedifferentiated cells.
Acknowledgments
We thank Anke Guhr, Jacqueline Schirm and Steven Posselt for valuable help in performing searches of literature databases.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Contributor Information
Anna M. Wobus, Phone: +49-39482-5256, FAX: +49-39482-5481, Email: wobusam@ipk-gatersleben.de
Peter Löser, Email: loeserp@rki.de.
References
- Andersson H et al (2010) Assaying cardiac biomarkers for toxicity testing using biosensing and cardiomyocytes derived from human embryonic stem cells. J Biotechnol 150:175–181 [DOI] [PubMed]
- Abe K, et al. Endoderm-specific gene expression in embryonic stem cells differentiated to embryoid bodies. Exp Cell Res. 1996;229:27–34. doi: 10.1006/excr.1996.0340. [DOI] [PubMed] [Google Scholar]
- Abuin A, Zhang H, Bradley A. Genetic analysis of mouse embryonic stem cells bearing Msh3 and Msh2 single and compound mutations. Mol Cell Biol. 2000;20:149–157. doi: 10.1128/mcb.20.1.149-157.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adler S, Lindqvist J, Uddenberg K, Hyllner J, Strehl R. Testing potential developmental toxicants with a cytotoxicity assay based on human embryonic stem cells. Altern Lab Anim. 2008;36:129–140. doi: 10.1177/026119290803600204. [DOI] [PubMed] [Google Scholar]
- Adler S, Pellizzer C, Hareng L, Hartung T, Bremer S. First steps in establishing a developmental toxicity test method based on human embryonic stem cells. Toxicol In Vitro. 2008;22:200–211. doi: 10.1016/j.tiv.2007.07.013. [DOI] [PubMed] [Google Scholar]
- Agarwal S, Holton KL, Lanza R. Efficient differentiation of functional hepatocytes from human embryonic stem cells. Stem Cells. 2008;26:1117–1127. doi: 10.1634/stemcells.2007-1102. [DOI] [PubMed] [Google Scholar]
- Albini A, Pennesi G, Donatelli F, Cammarota R, De Flora S, Noonan DM. Cardiotoxicity of anticancer drugs: the need for cardio-oncology and cardio-oncological prevention. J Natl Cancer Inst. 2010;102:14–25. doi: 10.1093/jnci/djp440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amabile G, Meissner A. Induced pluripotent stem cells: current progress and potential for regenerative medicine. Trends Mol Med. 2009;15:59–68. doi: 10.1016/j.molmed.2008.12.003. [DOI] [PubMed] [Google Scholar]
- Anderson D, Self T, Mellor IR, Goh G, Hill SJ, Denning C. Transgenic enrichment of cardiomyocytes from human embryonic stem cells. Mol Ther. 2007;15:2027–2036. doi: 10.1038/sj.mt.6300303. [DOI] [PubMed] [Google Scholar]
- Andrews PW. From teratocarcinomas to embryonic stem cells. Philos Trans R Soc Lond B Biol Sci. 2002;357:405–417. doi: 10.1098/rstb.2002.1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anon Teratology Society position paper: recommendations for vitamin A use during pregnancy. Teratology. 1987;35:269–275. doi: 10.1002/tera.1420350215. [DOI] [PubMed] [Google Scholar]
- Atkinson KA, Hulme LM, Clothier RH, Balls M. In vivo cytotoxicity of 24 chemicals to mouse teratocarcinoma cells. ATLA Alt Lab Anim. 1989;17:34–37. [Google Scholar]
- Baharvand H, Hashemi SM, Kazemi Ashtiani S, Farrokhi A. Differentiation of human embryonic stem cells into hepatocytes in 2D and 3D culture systems in vitro. Int J Dev Biol. 2006;50:645–652. doi: 10.1387/ijdb.052072hb. [DOI] [PubMed] [Google Scholar]
- Baharvand H, Hashemi SM, Shahsavani M. Differentiation of human embryonic stem cells into functional hepatocyte-like cells in a serum-free adherent culture condition. Differentiation. 2008;76:465–477. doi: 10.1111/j.1432-0436.2007.00252.x. [DOI] [PubMed] [Google Scholar]
- Baker M. Testing time for stem cells. Nature. 2010;463:719. doi: 10.1038/463719a. [DOI] [PubMed] [Google Scholar]
- Balls M, Hellsten E. Statement of the scientific validity of the embryonic stem cell test (EST)–an in Vitro test for embryotoxicity. Altern Lab Anim. 2002;30:265–268. [PubMed] [Google Scholar]
- Bantle JA, Fort DJ, Rayburn JR, DeYoung DJ, Bush SJ. Further validation of FETAX: evaluation of the developmental toxicity of five known mammalian teratogens and non-teratogens. Drug Chem Toxicol. 1990;13:267–282. doi: 10.3109/01480549009032286. [DOI] [PubMed] [Google Scholar]
- Basma H, et al. Differentiation and transplantation of human embryonic stem cell-derived hepatocytes. Gastroenterology. 2009;136:990–999. doi: 10.1053/j.gastro.2008.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxter MA, et al. Generating hepatic cell lineages from pluripotent stem cells for drug toxicity screening. Stem Cell Res. 2010;5:4–22. doi: 10.1016/j.scr.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Nun IF, Benvenisty N. Human embryonic stem cells as a cellular model for human disorders. Mol Cell Endocrinol. 2006;252:154–159. doi: 10.1016/j.mce.2006.03.034. [DOI] [PubMed] [Google Scholar]
- Beqqali A, Kloots J, Ward-van Oostwaard D, Mummery C, Passier R. Genome-wide transcriptional profiling of human embryonic stem cells differentiating to cardiomyocytes. Stem Cells. 2006;24:1956–1967. doi: 10.1634/stemcells.2006-0054. [DOI] [PubMed] [Google Scholar]
- Bluteau O, et al. Bi-allelic inactivation of TCF1 in hepatic adenomas. Nat Genet. 2002;32:312–315. doi: 10.1038/ng1001. [DOI] [PubMed] [Google Scholar]
- Blyszczuk P, et al. Expression of Pax4 in embryonic stem cells promotes differentiation of nestin-positive progenitor and insulin-producing cells. Proc Natl Acad Sci USA. 2003;100:998–1003. doi: 10.1073/pnas.0237371100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehn S, et al. Modification and evaluation of the chicken embryotoxicity screening test (CHEST) as in vitro test system for embryotoxicity. Toxicol Lett. 2009;189:S82. [Google Scholar]
- Boheler KR, Czyz J, Tweedie D, Yang HT, Anisimov SV, Wobus AM. Differentiation of pluripotent embryonic stem cells into cardiomyocytes. Circ Res. 2002;91:189–201. doi: 10.1161/01.res.0000027865.61704.32. [DOI] [PubMed] [Google Scholar]
- Boland MJ, et al. Adult mice generated from induced pluripotent stem cells. Nature. 2009;461:91–94. doi: 10.1038/nature08310. [DOI] [PubMed] [Google Scholar]
- Braam SR, Tertoolen L, van de Stolpe A, Meyer T, Passier R, Mummery CL. Prediction of drug-induced cardiotoxicity using human embryonic stem cell-derived cardiomyocytes. Stem Cell Res. 2010;4:107–116. doi: 10.1016/j.scr.2009.11.004. [DOI] [PubMed] [Google Scholar]
- Bradley A, Evans M, Kaufman MH, Robertson E. Formation of germ-line chimaeras from embryo-derived teratocarcinoma cell lines. Nature. 1984;309:255–256. doi: 10.1038/309255a0. [DOI] [PubMed] [Google Scholar]
- Bremer S, Hartung T. The use of embryonic stem cells for regulatory developmental toxicity testing in vitro–the current status of test development. Curr Pharm Des. 2004;10:2733–2747. doi: 10.2174/1381612043383700. [DOI] [PubMed] [Google Scholar]
- Bremer S, Vogel R. Pluripotent stem cells of the mouse as a potential in vitro model for mammalian germ cells. Sister chromatid exchanges induced by MMC and ENU in undifferentiated cell lines compared to differentiated cell lines. Mutat Res. 1999;444:97–102. doi: 10.1016/s1383-5718(99)00090-x. [DOI] [PubMed] [Google Scholar]
- Bremer S, et al. Establishment of an in vitro reporter gene assay for developmental cardiac toxicity. Toxicol In Vitro. 2001;15:215–223. doi: 10.1016/s0887-2333(01)00007-8. [DOI] [PubMed] [Google Scholar]
- Brinster RL. The effect of cells transferred into the mouse blastocyst on subsequent development. J Exp Med. 1974;140:1049–1056. doi: 10.1084/jem.140.4.1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brito-Martins M, Harding SE, Ali NN. beta(1)- and beta(2)-adrenoceptor responses in cardiomyocytes derived from human embryonic stem cells: comparison with failing and non-failing adult human heart. Br J Pharmacol. 2008;153:751–759. doi: 10.1038/sj.bjp.0707619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brolen G, et al. Hepatocyte-like cells derived from human embryonic stem cells specifically via definitive endoderm and a progenitor stage. J Biotechnol. 2010;145:284–294. doi: 10.1016/j.jbiotec.2009.11.007. [DOI] [PubMed] [Google Scholar]
- Brons IG, et al. Derivation of pluripotent epiblast stem cells from mammalian embryos. Nature. 2007;448:191–195. doi: 10.1038/nature05950. [DOI] [PubMed] [Google Scholar]
- Brown NA, et al. Screening chemicals for reproductive toxicity: the current alternatives. The report and recommendations of an ECVAM/ETS Workshop (ECVAM Workshop 12) ATLA Alt Lab Anim. 1995;23:868–882. [Google Scholar]
- Bu L, et al. Human ISL1 heart progenitors generate diverse multipotent cardiovascular cell lineages. Nature. 2009;460:113–117. doi: 10.1038/nature08191. [DOI] [PubMed] [Google Scholar]
- Buecker C, et al. A murine ESC-like state facilitates transgenesis and homologous recombination in human pluripotent stem cells. Cell Stem Cell. 2010;6:535–546. doi: 10.1016/j.stem.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buesen R, et al. Embryonic stem cell test remastered: comparison between the validated EST and the new molecular FACS-EST for assessing developmental toxicity in vitro. Toxicol Sci. 2009;108:389–400. doi: 10.1093/toxsci/kfp012. [DOI] [PubMed] [Google Scholar]
- Burridge PW, et al. Improved human embryonic stem cell embryoid body homogeneity and cardiomyocyte differentiation from a novel V-96 plate aggregation system highlights interline variability. Stem Cells. 2007;25:929–938. doi: 10.1634/stemcells.2006-0598. [DOI] [PubMed] [Google Scholar]
- Cai J, et al. Directed differentiation of human embryonic stem cells into functional hepatic cells. Hepatology. 2007;45:1229–1239. doi: 10.1002/hep.21582. [DOI] [PubMed] [Google Scholar]
- Cao T, Lu K, Fu X, Heng BC. Differentiated fibroblastic progenies of human embryonic stem cells for toxicology screening. Cloning Stem Cells. 2008;10:1–10. doi: 10.1089/clo.2007.0049. [DOI] [PubMed] [Google Scholar]
- Carvajal-Vergara X, et al. Patient-specific induced pluripotent stem-cell-derived models of LEOPARD syndrome. Nature. 2010;465:808–812. doi: 10.1038/nature09005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspi O, et al. Tissue engineering of vascularized cardiac muscle from human embryonic stem cells. Circ Res. 2007;100:263–272. doi: 10.1161/01.RES.0000257776.05673.ff. [DOI] [PubMed] [Google Scholar]
- Caspi O, et al. In vitro electrophysiological drug testing using human embryonic stem cell derived cardiomyocytes. Stem Cells Dev. 2009;18:161–172. doi: 10.1089/scd.2007.0280. [DOI] [PubMed] [Google Scholar]
- Cervantes RB, Stringer JR, Shao C, Tischfield JA, Stambrook PJ. Embryonic stem cells and somatic cells differ in mutation frequency and type. Proc Natl Acad Sci USA. 2002;99:3586–3590. doi: 10.1073/pnas.062527199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalasani N, Bjornsson E. Risk factors for idiosyncratic drug-induced liver injury. Gastroenterology. 2010;138:2246–2259. doi: 10.1053/j.gastro.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Wu L, Wang ZZ. Extrinsic regulation of cardiomyocyte differentiation of embryonic stem cells. J Cell Biochem. 2008;104:119–128. doi: 10.1002/jcb.21604. [DOI] [PubMed] [Google Scholar]
- Chiao E, et al. Isolation and transcriptional profiling of purified hepatic cells derived from human embryonic stem cells. Stem Cells. 2008;26:2032–2041. doi: 10.1634/stemcells.2007-0964. [DOI] [PubMed] [Google Scholar]
- Chien KR. Herceptin and the heart–a molecular modifier of cardiac failure. N Engl J Med. 2006;354:789–790. doi: 10.1056/NEJMp058315. [DOI] [PubMed] [Google Scholar]
- Chin MH, et al. Induced pluripotent stem cells and embryonic stem cells are distinguished by gene expression signatures. Cell Stem Cell. 2009;5:111–123. doi: 10.1016/j.stem.2009.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung Y, et al. Human embryonic stem cell lines generated without embryo destruction. Cell Stem Cell. 2008;2:113–117. doi: 10.1016/j.stem.2007.12.013. [DOI] [PubMed] [Google Scholar]
- Cockroft DL, Steele CE. Postimplantation Embryo Culture and its Application to Problems in Teratology. In: Atterwill CK, Steele CE, editors. In vitro methods in toxicology. Cambridge: Cambridge University Press; 1987. [Google Scholar]
- Colman A, Dreesen O. Pluripotent stem cells and disease modeling. Cell Stem Cell. 2009;5:244–247. doi: 10.1016/j.stem.2009.08.010. [DOI] [PubMed] [Google Scholar]
- Conrad S, et al. Generation of pluripotent stem cells from adult human testis. Nature. 2008;456:344–349. doi: 10.1038/nature07404. [DOI] [PubMed] [Google Scholar]
- Czyz J, et al. High frequency electromagnetic fields (GSM signals) affect gene expression levels in tumor suppressor p53-deficient embryonic stem cells. Bioelectromagnetics. 2004;25:296–307. doi: 10.1002/bem.10199. [DOI] [PubMed] [Google Scholar]
- Czyz J, Nikolova T, Schuderer J, Kuster N, Wobus AM. Non-thermal effects of power-line magnetic fields (50 Hz) on gene expression levels of pluripotent embryonic stem cells-the role of tumour suppressor p53. Mutat Res. 2004;557:63–74. doi: 10.1016/j.mrgentox.2003.09.011. [DOI] [PubMed] [Google Scholar]
- D’Amour KA, Agulnick AD, Eliazer S, Kelly OG, Kroon E, Baetge EE. Efficient differentiation of human embryonic stem cells to definitive endoderm. Nat Biotechnol. 2005;23:1534–1541. doi: 10.1038/nbt1163. [DOI] [PubMed] [Google Scholar]
- Dani C, et al. Differentiation of embryonic stem cells into adipocytes in vitro. J Cell Sci. 1997;110(Pt 11):1279–1285. doi: 10.1242/jcs.110.11.1279. [DOI] [PubMed] [Google Scholar]
- Davis RL, Weintraub H, Lassar AB. Expression of a single transfected cDNA converts fibroblasts to myoblasts. Cell. 1987;51:987–1000. doi: 10.1016/0092-8674(87)90585-x. [DOI] [PubMed] [Google Scholar]
- Davis RP, et al. Targeting a GFP reporter gene to the MIXL1 locus of human embryonic stem cells identifies human primitive streak-like cells and enables isolation of primitive hematopoietic precursors. Blood. 2008;111:1876–1884. doi: 10.1182/blood-2007-06-093609. [DOI] [PubMed] [Google Scholar]
- Denning C, Anderson D. Cardiomyocytes from human embryonic stem cells as predictors of cardiotoxicity. Drug Discov Today Therap Strat. 2008;5:223–232. [Google Scholar]
- Denning C, et al. Common culture conditions for maintenance and cardiomyocyte differentiation of the human embryonic stem cell lines, BG01 and HUES-7. Int J Dev Biol. 2006;50:27–37. doi: 10.1387/ijdb.052107cd. [DOI] [PubMed] [Google Scholar]
- Di Domenico AI, Christodoulou I, Pells SC, McWhir J, Thomson AJ. Sequential genetic modification of the hprt locus in human ESCs combining gene targeting and recombinase-mediated cassette exchange. Cloning Stem Cells. 2008;10:217–230. doi: 10.1089/clo.2008.0016. [DOI] [PubMed] [Google Scholar]
- Dimos JT, et al. Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons. Science. 2008;321:1218–1221. doi: 10.1126/science.1158799. [DOI] [PubMed] [Google Scholar]
- Doetschman TC, Eistetter H, Katz M, Schmidt W, Kemler R. The in vitro development of blastocyst-derived embryonic stem cell lines: formation of visceral yolk sac, blood islands and myocardium. J Embryol Exp Morphol. 1985;87:27–45. [PubMed] [Google Scholar]
- Dolci A, Dominici R, Cardinale D, Sandri MT, Panteghini M. Biochemical markers for prediction of chemotherapy-induced cardiotoxicity: systematic review of the literature and recommendations for use. Am J Clin Pathol. 2008;130:688–695. doi: 10.1309/AJCPB66LRIIVMQDR. [DOI] [PubMed] [Google Scholar]
- Dolgin E. Gene flaw found in induced stem cells. Nature. 2010;464:663. doi: 10.1038/464663a. [DOI] [PubMed] [Google Scholar]
- Dolnikov K, et al. Functional properties of human embryonic stem cell-derived cardiomyocytes: intracellular Ca2 + handling and the role of sarcoplasmic reticulum in the contraction. Stem Cells. 2006;24:236–245. doi: 10.1634/stemcells.2005-0036. [DOI] [PubMed] [Google Scholar]
- Donato MT, Lahoz A, Castell JV, Gomez-Lechon MJ. Cell lines: a tool for in vitro drug metabolism studies. Curr Drug Metab. 2008;9:1–11. doi: 10.2174/138920008783331086. [DOI] [PubMed] [Google Scholar]
- Donehower LA, et al. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356:215–221. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- Drab M, et al. From totipotent embryonic stem cells to spontaneously contracting smooth muscle cells: a retinoic acid and db-cAMP in vitro differentiation model. FASEB J. 1997;11:905–915. doi: 10.1096/fasebj.11.11.9285489. [DOI] [PubMed] [Google Scholar]
- Duan Y, et al. Differentiation and enrichment of hepatocyte-like cells from human embryonic stem cells in vitro and in vivo. Stem Cells. 2007;25:3058–3068. doi: 10.1634/stemcells.2007-0291. [DOI] [PubMed] [Google Scholar]
- Duan Y, et al. Differentiation and characterization of metabolically functioning hepatocytes from human embryonic stem cells. Stem Cells. 2010;28:674–686. doi: 10.1002/stem.315. [DOI] [PubMed] [Google Scholar]
- Durcova-Hills G, Tang F, Doody G, Tooze R, Surani MA (2008) Reprogramming primordial germ cells into pluripotent stem cells. PLoS One 3(10):e3531 [DOI] [PMC free article] [PubMed]
- Eastmond DA, et al. Mutagenicity testing for chemical risk assessment: update of the WHO/IPCS Harmonized Scheme. Mutagenesis. 2009;24:341–349. doi: 10.1093/mutage/gep014. [DOI] [PubMed] [Google Scholar]
- Ebert AD, Svendsen CN. Human stem cells and drug screening: opportunities and challenges. Nat Rev Drug Discov. 2010;9:367–372. doi: 10.1038/nrd3000. [DOI] [PubMed] [Google Scholar]
- Ebert AD, et al. Induced pluripotent stem cells from a spinal muscular atrophy patient. Nature. 2009;457:277–280. doi: 10.1038/nature07677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards MK, McBurney MW. The concentration of retinoic acid determines the differentiated cell types formed by a teratocarcinoma cell line. Dev Biol. 1983;98:187–191. doi: 10.1016/0012-1606(83)90348-2. [DOI] [PubMed] [Google Scholar]
- Eggan K, et al. Hybrid vigor, fetal overgrowth, and viability of mice derived by nuclear cloning and tetraploid embryo complementation. Proc Natl Acad Sci USA. 2001;98:6209–6214. doi: 10.1073/pnas.101118898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehling UH. Differential spermatogenic response of mice to the induction of mutations by antineoplastic drugs. Mutat Res. 1974;26:285–295. doi: 10.1016/s0027-5107(74)80026-6. [DOI] [PubMed] [Google Scholar]
- Ehling UH, Machemer L, Buselmaier W. Standard protocol for the dominant lethal test on male mice set up by the work group ‘Dominant Lethal Mutations of the ad hoc Committee Chemogenetics’. Arch Toxicol. 1978;39:173–185. doi: 10.1007/BF00368226. [DOI] [PubMed] [Google Scholar]
- Ehnert S, Nussler AK, Lehmann A, Dooley S. Blood monocyte-derived neohepatocytes as in vitro test system for drug metabolism. Drug Metab Dispos. 2008;36:1922–1929. doi: 10.1124/dmd.108.020453. [DOI] [PubMed] [Google Scholar]
- Ek M, et al. Expression of drug metabolizing enzymes in hepatocyte-like cells derived from human embryonic stem cells. Biochem Pharmacol. 2007;74:496–503. doi: 10.1016/j.bcp.2007.05.009. [DOI] [PubMed] [Google Scholar]
- Ekwall B. Overview of the final MEIC results: II. The in vitro–in vivo evaluation, including the selection of a practical battery of cell tests for prediction of acute lethal blood concentrations in humans. Toxicol In Vitro. 1999;13:665–673. doi: 10.1016/s0887-2333(99)00061-2. [DOI] [PubMed] [Google Scholar]
- European Group on Ethics in Science and New Technologies (2007) Recommendations on the ethical review of hESC FP7 research projects—Opinion No 22, http://ec.europa.eu/european_group_ethics/publications/docs/opinion_22_final_follow_up_en.pdf
- Evans MJ. The isolation and properties of a clonal tissue culture strain of pluripotent mouse teratoma cells. J Embryol Exp Morphol. 1972;28:163–176. [PubMed] [Google Scholar]
- Evans MJ, Kaufman MH. Establishment in culture of pluripotential cells from mouse embryos. Nature. 1981;292:154–156. doi: 10.1038/292154a0. [DOI] [PubMed] [Google Scholar]
- Fassler R, et al. Differentiation and integrity of cardiac muscle cells are impaired in the absence of beta 1 integrin. J Cell Sci. 1996;109(Pt 13):2989–2999. doi: 10.1242/jcs.109.13.2989. [DOI] [PubMed] [Google Scholar]
- Favor J, Sund M, Neuhauser-Klaus A, Ehling UH. A dose-response analysis of ethylnitrosourea-induced recessive specific-locus mutations in treated spermatogonia of the mouse. Mutat Res. 1990;231:47–54. doi: 10.1016/0027-5107(90)90175-4. [DOI] [PubMed] [Google Scholar]
- Feng Q, et al. Hemangioblastic derivatives from human induced pluripotent stem cells exhibit limited expansion and early senescence. Stem Cells. 2010;28:704–712. doi: 10.1002/stem.321. [DOI] [PubMed] [Google Scholar]
- Filipczyk AA, Passier R, Rochat A, Mummery CL. Regulation of cardiomyocyte differentiation of embryonic stem cells by extracellular signalling. Cell Mol Life Sci. 2007;64:704–718. doi: 10.1007/s00018-007-6523-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher J, et al. The inhibitory role of stromal cell mesenchyme on human embryonic stem cell hepatocyte differentiation is overcome by Wnt3a treatment. Cloning Stem Cells. 2008;10:331–339. doi: 10.1089/clo.2007.0094. [DOI] [PubMed] [Google Scholar]
- Flint OP (1993) In vitro tests for teratogens: desirable endpoints, test batteries and current status of the micromass teratogen test. Reprod Toxicol(7 Suppl 1):103–111 [DOI] [PubMed]
- Flora SJ, Mehta A. Monoisoamyl dimercaptosuccinic acid abrogates arsenic-induced developmental toxicity in human embryonic stem cell-derived embryoid bodies: comparison with in vivo studies. Biochem Pharmacol. 2009;78:1340–1349. doi: 10.1016/j.bcp.2009.07.003. [DOI] [PubMed] [Google Scholar]
- Fraichard A, Chassande O, Bilbaut G, Dehay C, Savatier P, Samarut J. In vitro differentiation of embryonic stem cells into glial cells and functional neurons. J Cell Sci. 1995;108(Pt 10):3181–3188. doi: 10.1242/jcs.108.10.3181. [DOI] [PubMed] [Google Scholar]
- Freund C, Davis RP, Gkatzis K, Ward-van Oostwaard D, Mummery CL. The first reported generation of human induced pluripotent stem cells (iPS cells) and iPS cell-derived cardiomyocytes in the Netherlands. Neth Heart J. 2010;18:51–54. [PMC free article] [PubMed] [Google Scholar]
- Gai H, et al. Generation and characterization of functional cardiomyocytes using induced pluripotent stem cells derived from human fibroblasts. Cell Biol Int. 2009;33:1184–1193. doi: 10.1016/j.cellbi.2009.08.008. [DOI] [PubMed] [Google Scholar]
- Genschow E et al (2002) The ECVAM international validation study on in vitro embryotoxicity tests: results of the definitive phase and evaluation of prediction models. European centre for the validation of alternative methods. Altern Lab Anim 30:151–176 [DOI] [PubMed]
- Genschow E, et al. Validation of the embryonic stem cell test in the international ECVAM validation study on three in vitro embryotoxicity tests. Altern Lab Anim. 2004;32:209–244. doi: 10.1177/026119290403200305. [DOI] [PubMed] [Google Scholar]
- German Stem Cell Act (Act ensuring protection of embryos in connection with the importation and utilization of human embryonic stem cells) (2002) BGBlI 2277 http://www.bmj.bund.de/files/-/1146/Stammzellgesetz%20englisch.pdf
- Gilbert N. Crucial data on REACH not disclosed. Nature. 2010;464:1116–1117. doi: 10.1038/4641116a. [DOI] [PubMed] [Google Scholar]
- Graichen R, et al. Enhanced cardiomyogenesis of human embryonic stem cells by a small molecular inhibitor of p38 MAPK. Differentiation. 2008;76:357–370. doi: 10.1111/j.1432-0436.2007.00236.x. [DOI] [PubMed] [Google Scholar]
- Green H, Easley K, Iuchi S. Marker succession during the development of keratinocytes from cultured human embryonic stem cells. Proc Natl Acad Sci USA. 2003;100:15625–15630. doi: 10.1073/pnas.0307226100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan K, et al. Pluripotency of spermatogonial stem cells from adult mouse testis. Nature. 2006;440:1199–1203. doi: 10.1038/nature04697. [DOI] [PubMed] [Google Scholar]
- Guenou H, et al. Human embryonic stem-cell derivatives for full reconstruction of the pluristratified epidermis: a preclinical study. Lancet. 2009;374:1745–1753. doi: 10.1016/S0140-6736(09)61496-3. [DOI] [PubMed] [Google Scholar]
- Guenther MG, et al. Chromatin structure and gene expression programs of human embryonic and induced pluripotent stem cells. Cell Stem Cell. 2010;7:249–257. doi: 10.1016/j.stem.2010.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gwak SJ, et al. The effect of cyclic strain on embryonic stem cell-derived cardiomyocytes. Biomaterials. 2008;29:844–856. doi: 10.1016/j.biomaterials.2007.10.050. [DOI] [PubMed] [Google Scholar]
- Hanna J, et al. Human embryonic stem cells with biological and epigenetic characteristics similar to those of mouse ESCs. Proc Natl Acad Sci USA. 2010;107:9222–9227. doi: 10.1073/pnas.1004584107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen A, et al. Development of a drug screening platform based on engineered heart tissue. Circ Res. 2010;107:35–44. doi: 10.1161/CIRCRESAHA.109.211458. [DOI] [PubMed] [Google Scholar]
- Hareng L, Pellizzer C, Bremer S, Schwarz M, Hartung T. The integrated project ReProTect: a novel approach in reproductive toxicity hazard assessment. Reprod Toxicol. 2005;20:441–452. doi: 10.1016/j.reprotox.2005.04.003. [DOI] [PubMed] [Google Scholar]
- Hattori F, et al. Nongenetic method for purifying stem cell-derived cardiomyocytes. Nat Methods. 2010;7:61–66. doi: 10.1038/nmeth.1403. [DOI] [PubMed] [Google Scholar]
- Hay DC, Zhao D, Ross A, Mandalam R, Lebkowski J, Cui W. Direct differentiation of human embryonic stem cells to hepatocyte-like cells exhibiting functional activities. Cloning Stem Cells. 2007;9:51–62. doi: 10.1089/clo.2006.0045. [DOI] [PubMed] [Google Scholar]
- Hay DC, et al. Highly efficient differentiation of hESCs to functional hepatic endoderm requires ActivinA and Wnt3a signaling. Proc Natl Acad Sci USA. 2008;105:12301–12306. doi: 10.1073/pnas.0806522105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay DC, et al. Efficient differentiation of hepatocytes from human embryonic stem cells exhibiting markers recapitulating liver development in vivo. Stem Cells. 2008;26:894–902. doi: 10.1634/stemcells.2007-0718. [DOI] [PubMed] [Google Scholar]
- He JQ, Ma Y, Lee Y, Thomson JA, Kamp TJ. Human embryonic stem cells develop into multiple types of cardiac myocytes: action potential characterization. Circ Res. 2003;93:32–39. doi: 10.1161/01.RES.0000080317.92718.99. [DOI] [PubMed] [Google Scholar]
- Hendrickx AG. Teratogenicity of Retinoids in Rodents, Primates and Humans. J Toxicol Sci. 1998;23:272. [Google Scholar]
- Heng BC, Richards M, Shu Y, Gribbon P. Induced pluripotent stem cells: a new tool for toxicology screening? Arch Toxicol. 2009;83:641–644. doi: 10.1007/s00204-009-0414-2. [DOI] [PubMed] [Google Scholar]
- Hengstler JG, et al. Generation of human hepatocytes by stem cell technology: definition of the hepatocyte. Expert Opin Drug Metab Toxicol. 2005;1:61–74. doi: 10.1517/17425255.1.1.61. [DOI] [PubMed] [Google Scholar]
- Hewitt NJ, et al. Primary hepatocytes: current understanding of the regulation of metabolic enzymes and transporter proteins, and pharmaceutical practice for the use of hepatocytes in metabolism, enzyme induction, transporter, clearance, and hepatotoxicity studies. Drug Metab Rev. 2007;39:159–234. doi: 10.1080/03602530601093489. [DOI] [PubMed] [Google Scholar]
- Hockemeyer D, et al. Efficient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-finger nucleases. Nat Biotechnol. 2009;27:851–857. doi: 10.1038/nbt.1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hondeghem LM. QT prolongation is an unreliable predictor of ventricular arrhythmia. Heart Rhythm. 2008;5:1210–1212. doi: 10.1016/j.hrthm.2008.05.006. [DOI] [PubMed] [Google Scholar]
- Horton RE, Millman JR, Colton CK, Auguste DT. Engineering microenvironments for embryonic stem cell differentiation to cardiomyocytes. Regen Med. 2009;4:721–732. doi: 10.2217/rme.09.48. [DOI] [PubMed] [Google Scholar]
- Hu BY, et al. Neural differentiation of human induced pluripotent stem cells follows developmental principles but with variable potency. Proc Natl Acad Sci USA. 2010;107:4335–4340. doi: 10.1073/pnas.0910012107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huangfu D, et al. Induction of pluripotent stem cells by defined factors is greatly improved by small-molecule compounds. Nat Biotechnol. 2008;26:795–797. doi: 10.1038/nbt1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber I, et al. Identification and selection of cardiomyocytes during human embryonic stem cell differentiation. Faseb J. 2007;21:2551–2563. doi: 10.1096/fj.05-5711com. [DOI] [PubMed] [Google Scholar]
- Hübner K, et al. Derivation of oocytes from mouse embryonic stem cells. Science. 2003;300:1251–1256. doi: 10.1126/science.1083452. [DOI] [PubMed] [Google Scholar]
- Ieda M, et al. Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell. 2010;142:375–386. doi: 10.1016/j.cell.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Society for Stem Cell Research (ISSCR) (2006) Guidelines for the Conduct of Human Embryonic Stem Cell Research, http://www.isscr.org/guidelines/ISSCRhESCguidelines2006.pdf [PubMed]
- Irion S, Luche H, Gadue P, Fehling HJ, Kennedy M, Keller G. Identification and targeting of the ROSA26 locus in human embryonic stem cells. Nat Biotechnol. 2007;25:1477–1482. doi: 10.1038/nbt1362. [DOI] [PubMed] [Google Scholar]
- Ishii T, et al. Effects of extracellular matrixes and growth factors on the hepatic differentiation of human embryonic stem cells. Am J Physiol Gastrointest Liver Physiol. 2008;295:G313–G321. doi: 10.1152/ajpgi.00072.2008. [DOI] [PubMed] [Google Scholar]
- Ishii T, et al. In vitro hepatic maturation of human embryonic stem cells by using a mesenchymal cell line derived from murine fetal livers. Cell Tissue Res. 2010;339:505–512. doi: 10.1007/s00441-009-0906-7. [DOI] [PubMed] [Google Scholar]
- Jakob H, Boon T, Gaillard J, Nicolas J, Jacob F. Teratocarcinoma of the mouse: isolation, culture and properties of pluripotential cells. Ann Microbiol (Paris) 1973;124:269–282. [PubMed] [Google Scholar]
- Jalving M, Schepers H. Induced pluripotent stem cells: will they be safe? Curr Opin Mol Ther. 2009;11:383–393. [PubMed] [Google Scholar]
- Jelinek R, Peterka M, Rychter Z. Chick embryotoxicity screening test—130 substances tested. Indian J Exp Biol. 1985;23:588–595. [PubMed] [Google Scholar]
- Jensen J, Hyllner J, Bjorquist P. Human embryonic stem cell technologies and drug discovery. J Cell Physiol. 2009;219:513–519. doi: 10.1002/jcp.21732. [DOI] [PubMed] [Google Scholar]
- Jha A (2007) Stem cell bank for drug testing may cut animal experiments. In: The Guardian, p 11
- Jones-Villeneuve EM, Rudnicki MA, Harris JF, McBurney MW. Retinoic acid-induced neural differentiation of embryonal carcinoma cells. Mol Cell Biol. 1983;3:2271–2279. doi: 10.1128/mcb.3.12.2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson MK, et al. Quantified proarrhythmic potential of selected human embryonic stem cell-derived cardiomyocytes. Stem Cell Res. 2010;4:189–200. doi: 10.1016/j.scr.2010.02.001. [DOI] [PubMed] [Google Scholar]
- Joshi A, Dimino T, Vohra Y, Cui C, Yan GX (2004) Preclinical strategies to assess QT liability and torsadogenic potential of new drugs: the role of experimental models. J Electrocardiol 37(Suppl):7–14 [DOI] [PubMed]
- Kahan BW, Ephrussi B. Developmental potentialities of clonal in vitro cultures of mouse testicular teratoma. J Natl Cancer Inst. 1970;44:1015–1036. [PubMed] [Google Scholar]
- Kaji K, Norrby K, Paca A, Mileikovsky M, Mohseni P, Woltjen K. Virus-free induction of pluripotency and subsequent excision of reprogramming factors. Nature. 2009;458:771–775. doi: 10.1038/nature07864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang L, Wang J, Zhang Y, Kou Z, Gao S. iPS cells can support full-term development of tetraploid blastocyst-complemented embryos. Cell Stem Cell. 2009;5:135–138. doi: 10.1016/j.stem.2009.07.001. [DOI] [PubMed] [Google Scholar]
- Kehat I, et al. Human embryonic stem cells can differentiate into myocytes with structural and functional properties of cardiomyocytes. J Clin Invest. 2001;108:407–414. doi: 10.1172/JCI12131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kehoe DE, Jing D, Lock LT, Tzanakakis ES. Scalable stirred-suspension bioreactor culture of human pluripotent stem cells. Tissue Eng Part A. 2010;16:405–421. doi: 10.1089/ten.tea.2009.0454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr DA, et al. Human embryonic germ cell derivatives facilitate motor recovery of rats with diffuse motor neuron injury. J Neurosci. 2003;23:5131–5140. doi: 10.1523/JNEUROSCI.23-12-05131.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessel M, Gruss P. Homeotic transformations of murine vertebrae and concomitant alteration of Hox codes induced by retinoic acid. Cell. 1991;67:89–104. doi: 10.1016/0092-8674(91)90574-i. [DOI] [PubMed] [Google Scholar]
- Khan IF, et al. Engineering of human pluripotent stem cells by AAV-mediated gene targeting. Mol Ther. 2010;18:1192–1199. doi: 10.1038/mt.2010.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khetani SR, Bhatia SN. Microscale culture of human liver cells for drug development. Nat Biotechnol. 2008;26:120–126. doi: 10.1038/nbt1361. [DOI] [PubMed] [Google Scholar]
- Kim D, et al. Generation of human induced pluripotent stem cells by direct delivery of reprogramming proteins. Cell Stem Cell. 2009;4:472–476. doi: 10.1016/j.stem.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim C, et al. Non-cardiomyocytes influence the electrophysiological maturation of human embryonic stem cell-derived cardiomyocytes during differentiation. Stem Cells Dev. 2010;19:783–795. doi: 10.1089/scd.2009.0349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K, et al. Epigenetic memory in induced pluripotent stem cells. Nature. 2010;467:285–290. doi: 10.1038/nature09342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimmel GL, Smith K, Kochhar DM, Pratt RM. Overview of in vitro teratogenicity testing: aspects of validation and application to screening. Teratog Carcinog Mutagen. 1982;2:221–229. doi: 10.1002/1520-6866(1990)2:3/4<221::aid-tcm1770020304>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- Klemm M, Genschow E, Pohl I, Barrabas C, Liebsch M, Spielmann H. Permanent embryonic germ cell lines of BALB/cJ mice–an in vitro alternative for in vivo germ cell mutagenicity tests. Toxicol In Vitro. 2001;15:447–453. doi: 10.1016/s0887-2333(01)00050-9. [DOI] [PubMed] [Google Scholar]
- Klemm M, Genschow E, Pohl I, Barrabas C, Liebsch M, Spielmann H. Permanent embryonic mouse germ cell-lines, an in vitro alternative to in vivo germ cell mutagenicity tests. ALTEX. 2001;18:127–130. [PubMed] [Google Scholar]
- Knoepfler PS. Deconstructing stem cell tumorigenicity: a roadmap to safe regenerative medicine. Stem Cells. 2009;27:1050–1056. doi: 10.1002/stem.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko K, et al. Induction of pluripotency in adult unipotent germline stem cells. Cell Stem Cell. 2009;5:87–96. doi: 10.1016/j.stem.2009.05.025. [DOI] [PubMed] [Google Scholar]
- Ko K et al (2010) Human adult germline stem cells in question. Nature 465:E1 (discussion E3) [DOI] [PubMed]
- Kola I, Landis J. Can the pharmaceutical industry reduce attrition rates? Nat Rev Drug Discov. 2004;3:711–715. doi: 10.1038/nrd1470. [DOI] [PubMed] [Google Scholar]
- Kolossov E, et al. Functional characteristics of ESC-derived cardiac precursor cells identified by tissue-specific expression of the green fluorescent protein. J Cell Biol. 1998;143:2045–2056. doi: 10.1083/jcb.143.7.2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnamoorthy M, et al. Low ethanol concentration alters CHRNA5 RNA levels during early human development. Reprod Toxicol. 2010;30:489–492. doi: 10.1016/j.reprotox.2010.04.011. [DOI] [PubMed] [Google Scholar]
- Krizhanovsky V, Lowe SW. Stem cells: the promises and perils of p53. Nature. 2009;460:1085–1086. doi: 10.1038/4601085a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krtolica A, Ilic D, Genbacev O, Miller RK. Human embryonic stem cells as a model for embryotoxicity screening. Regen Med. 2009;4:449–459. doi: 10.2217/rme.09.13. [DOI] [PubMed] [Google Scholar]
- Labosky PA, Barlow DP, Hogan BL (1994) Embryonic germ cell lines and their derivation from mouse primordial germ cells. Ciba Found Symp 182:157–168 (discussion 168–178) [DOI] [PubMed]
- Laflamme MA, et al. Cardiomyocytes derived from human embryonic stem cells in pro-survival factors enhance function of infarcted rat hearts. Nat Biotechnol. 2007;25:1015–1024. doi: 10.1038/nbt1327. [DOI] [PubMed] [Google Scholar]
- Laschinski G, Vogel R, Spielmann H. Cytotoxicity test using blastocyst-derived euploid embryonal stem cells: a new approach to in vitro teratogenesis screening. Reprod Toxicol. 1991;5:57–64. doi: 10.1016/0890-6238(91)90111-r. [DOI] [PubMed] [Google Scholar]
- Laustriat D, Gide J, Peschanski M. Human pluripotent stem cells in drug discovery and predictive toxicology. Biochem Soc Trans. 2010;38:1051–1057. doi: 10.1042/BST0381051. [DOI] [PubMed] [Google Scholar]
- Lavon N, Benvenisty N. Study of hepatocyte differentiation using embryonic stem cells. J Cell Biochem. 2005;96:1193–1202. doi: 10.1002/jcb.20590. [DOI] [PubMed] [Google Scholar]
- Lavon N, Yanuka O, Benvenisty N. Differentiation and isolation of hepatic-like cells from human embryonic stem cells. Differentiation. 2004;72:230–238. doi: 10.1111/j.1432-0436.2004.07205002.x. [DOI] [PubMed] [Google Scholar]
- Lecina M, Ting S, Choo A, Reuveny S, Oh S (2010) Scalable platform for human embryonic stem cell differentiation to cardiomyocytes in suspended microcarrier cultures. Tissue Eng Part C Methods 16:1609–1619 [DOI] [PubMed]
- Lee G, Studer L. Induced pluripotent stem cell technology for the study of human disease. Nat Methods. 2010;7:25–27. doi: 10.1038/nmeth.f.283. [DOI] [PubMed] [Google Scholar]
- Lee N, Authier S, Pugsley MK, Curtis MJ. The continuing evolution of torsades de pointes liability testing methods: is there an end in sight? Toxicol Appl Pharmacol. 2010;243:146–153. doi: 10.1016/j.taap.2009.12.002. [DOI] [PubMed] [Google Scholar]
- Leist M, et al. The biological and ethical basis of the use of human embryonic stem cells for in vitro test systems or cell therapy. ALTEX. 2008;25:163–190. [PubMed] [Google Scholar]
- Lesman A, et al. Transplantation of a tissue-engineered human vascularized cardiac muscle. Tissue Eng Part A. 2010;16:115–125. doi: 10.1089/ten.TEA.2009.0130. [DOI] [PubMed] [Google Scholar]
- Li RK, et al. Construction of a bioengineered cardiac graft. J Thorac Cardiovasc Surg. 2000;119:368–375. doi: 10.1016/S0022-5223(00)70193-0. [DOI] [PubMed] [Google Scholar]
- Liang H, et al. Human and murine embryonic stem cell-derived cardiomyocytes serve together as a valuable model for drug safety screening. Cell Physiol Biochem. 2010;25:459–466. doi: 10.1159/000303051. [DOI] [PubMed] [Google Scholar]
- Lim CK, et al. Differential cytotoxic effects of mono-(2-ethylhexyl) phthalate on blastomere-derived embryonic stem cells and differentiating neurons. Toxicology. 2009;264:145–154. doi: 10.1016/j.tox.2009.08.015. [DOI] [PubMed] [Google Scholar]
- Lin S, Fonteno S, Weng JH, Talbot P. Comparison of the toxicity of smoke from conventional and harm reduction cigarettes using human embryonic stem cells. Toxicol Sci. 2010;118:202–212. doi: 10.1093/toxsci/kfq241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Fu JD, Siu CW, Li RA. Functional sarcoplasmic reticulum for calcium handling of human embryonic stem cell-derived cardiomyocytes: insights for driven maturation. Stem Cells. 2007;25:3038–3044. doi: 10.1634/stemcells.2007-0549. [DOI] [PubMed] [Google Scholar]
- Liu H, Ye Z, Kim Y, Sharkis S, Jang YY. Generation of endoderm-derived human induced pluripotent stem cells from primary hepatocytes. Hepatology. 2010;51:1810–1819. doi: 10.1002/hep.23626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lock LT, Tzanakakis ES. Expansion and differentiation of human embryonic stem cells to endoderm progeny in a microcarrier stirred-suspension culture. Tissue Eng Part A. 2009;15:2051–2063. doi: 10.1089/ten.tea.2008.0455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombardo A, et al. Gene editing in human stem cells using zinc finger nucleases and integrase-defective lentiviral vector delivery. Nat Biotechnol. 2007;25:1298–1306. doi: 10.1038/nbt1353. [DOI] [PubMed] [Google Scholar]
- Lorico A, Rappa G, Flavell RA, Sartorelli AC. Double knockout of the MRP gene leads to increased drug sensitivity in vitro. Cancer Res. 1996;56:5351–5355. [PubMed] [Google Scholar]
- Löser P, Schirm J, Guhr A, Wobus AM, Kurtz A. Human embryonic stem cell lines and their use in international research. Stem Cells. 2010;28:240–246. doi: 10.1002/stem.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maherali N, et al. Directly reprogrammed fibroblasts show global epigenetic remodeling and widespread tissue contribution. Cell Stem Cell. 2007;1:55–70. doi: 10.1016/j.stem.2007.05.014. [DOI] [PubMed] [Google Scholar]
- Maltsev VA, Rohwedel J, Hescheler J, Wobus AM. Embryonic stem cells differentiate in vitro into cardiomyocytes representing sinusnodal, atrial and ventricular cell types. Mech Dev. 1993;44:41–50. doi: 10.1016/0925-4773(93)90015-p. [DOI] [PubMed] [Google Scholar]
- Marion RM, et al. Telomeres acquire embryonic stem cell characteristics in induced pluripotent stem cells. Cell Stem Cell. 2009;4:141–154. doi: 10.1016/j.stem.2008.12.010. [DOI] [PubMed] [Google Scholar]
- Martin GR. Isolation of a pluripotent cell line from early mouse embryos cultured in medium conditioned by teratocarcinoma stem cells. Proc Natl Acad Sci USA. 1981;78:7634–7638. doi: 10.1073/pnas.78.12.7634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marx-Stoelting P, et al. A review of the implementation of the embryonic stem cell test (EST). The report and recommendations of an ECVAM/ReProTect Workshop. Altern Lab Anim. 2009;37:313–328. doi: 10.1177/026119290903700314. [DOI] [PubMed] [Google Scholar]
- Matsui Y, Zsebo K, Hogan BL. Derivation of pluripotential embryonic stem cells from murine primordial germ cells in culture. Cell. 1992;70:841–847. doi: 10.1016/0092-8674(92)90317-6. [DOI] [PubMed] [Google Scholar]
- Mayshar Y, et al. Identification and classification of chromosomal aberrations in human induced pluripotent stem cells. Cell Stem Cell. 2010;7:521–531. doi: 10.1016/j.stem.2010.07.017. [DOI] [PubMed] [Google Scholar]
- McBurney MW, Jones-Villeneuve EM, Edwards MK, Anderson PJ. Control of muscle and neuronal differentiation in a cultured embryonal carcinoma cell line. Nature. 1982;299:165–167. doi: 10.1038/299165a0. [DOI] [PubMed] [Google Scholar]
- McLean AB, et al. Activin a efficiently specifies definitive endoderm from human embryonic stem cells only when phosphatidylinositol 3-kinase signaling is suppressed. Stem Cells. 2007;25:29–38. doi: 10.1634/stemcells.2006-0219. [DOI] [PubMed] [Google Scholar]
- Meamar R, Karamali F, Sadeghi HM, Etebari M, Nasr-Esfahani MH, Baharvand H. Toxicity of ecstasy (MDMA) towards embryonic stem cell-derived cardiac and neural cells. Toxicol In Vitro. 2010;24:1133–1138. doi: 10.1016/j.tiv.2010.03.005. [DOI] [PubMed] [Google Scholar]
- Mehta A, Konala VB, Khanna A, Majumdar AS. Assessment of drug induced developmental toxicity using human embryonic stem cells. Cell Biol Int. 2008;32:1412–1424. doi: 10.1016/j.cellbi.2008.08.012. [DOI] [PubMed] [Google Scholar]
- Mikkola M, et al. Distinct differentiation characteristics of individual human embryonic stem cell lines. BMC Dev Biol. 2006;6:40. doi: 10.1186/1471-213X-6-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsui K, et al. Gene targeting in human pluripotent stem cells with adeno-associated virus vectors. Biochem Biophys Res Commun. 2009;388:711–717. doi: 10.1016/j.bbrc.2009.08.075. [DOI] [PubMed] [Google Scholar]
- Mohr JC, et al. The microwell control of embryoid body size in order to regulate cardiac differentiation of human embryonic stem cells. Biomaterials. 2010;31:1885–1893. doi: 10.1016/j.biomaterials.2009.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore RN, Moghe PV. Expedited growth factor-mediated specification of human embryonic stem cells toward the hepatic lineage. Stem Cell Res. 2009;3:51–62. doi: 10.1016/j.scr.2009.04.004. [DOI] [PubMed] [Google Scholar]
- Moore JC, et al. Distinct cardiogenic preferences of two human embryonic stem cell (hESC) lines are imprinted in their proteomes in the pluripotent state. Biochem Biophys Res Commun. 2008;372:553–558. doi: 10.1016/j.bbrc.2008.05.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore RN, et al. Enhanced differentiation of embryonic stem cells using co-cultivation with hepatocytes. Biotechnol Bioeng. 2008;101:1332–1343. doi: 10.1002/bit.21987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moretti A, et al. Mouse and human induced pluripotent stem cells as a source for multipotent Isl1 + cardiovascular progenitors. Faseb J. 2010;24:700–711. doi: 10.1096/fj.09-139477. [DOI] [PubMed] [Google Scholar]
- Moretti A et al (2010b) Patient-specific induced pluripotent stem-cell models for long-QT syndrome. N Engl J Med 363:1397–1409 [DOI] [PubMed]
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- Müller FJ, et al. Regulatory networks define phenotypic classes of human stem cell lines. Nature. 2008;455:401–405. doi: 10.1038/nature07213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mummery C, et al. Differentiation of human embryonic stem cells to cardiomyocytes: role of coculture with visceral endoderm-like cells. Circulation. 2003;107:2733–2740. doi: 10.1161/01.CIR.0000068356.38592.68. [DOI] [PubMed] [Google Scholar]
- Murry CE, Keller G. Differentiation of embryonic stem cells to clinically relevant populations: lessons from embryonic development. Cell. 2008;132:661–680. doi: 10.1016/j.cell.2008.02.008. [DOI] [PubMed] [Google Scholar]
- Nagy A, Nagy K. The mysteries of induced pluripotency: where will they lead? Nat Methods. 2010;7:22–24. doi: 10.1038/nmeth.f.292. [DOI] [PubMed] [Google Scholar]
- Nagy A, Rossant J, Nagy R, Abramow-Newerly W, Roder JC. Derivation of completely cell culture-derived mice from early-passage embryonic stem cells. Proc Natl Acad Sci USA. 1993;90:8424–8428. doi: 10.1073/pnas.90.18.8424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama M. Homologous recombination in human iPS and ESCs for use in gene correction therapy. Drug Discov Today. 2010;15:198–202. doi: 10.1016/j.drudis.2010.01.006. [DOI] [PubMed] [Google Scholar]
- Narwani K, et al. Human embryonic stem cells from aneuploid blastocysts identified by pre-implantation genetic screening. In Vitro Cell Dev Biol Anim. 2010;46:309–316. doi: 10.1007/s11626-010-9303-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Institutes of Health (NIH) (2009) National Institutes of Health Guidelines on Human Stem Cell Research, http://stemcells.nih.gov/policy/2009guidelines.htm
- Nau H. Correlation of transplacental and maternal pharmacokinetics of retinoids during organogenesis with teratogenicity. Methods Enzymol. 1990;190:437–448. doi: 10.1016/0076-6879(90)90050-b. [DOI] [PubMed] [Google Scholar]
- Nau H (1993) Embryotoxicity and teratogenicity of topical retinoic acid. Skin Pharmacol 6(Suppl 1):35–44 [DOI] [PubMed]
- Newman AM, Cooper JB. Lab-specific gene expression signatures in pluripotent stem cells. Cell Stem Cell. 2010;7:258–262. doi: 10.1016/j.stem.2010.06.016. [DOI] [PubMed] [Google Scholar]
- Niebruegge S, et al. Generation of human embryonic stem cell-derived mesoderm and cardiac cells using size-specified aggregates in an oxygen-controlled bioreactor. Biotechnol Bioeng. 2009;102:493–507. doi: 10.1002/bit.22065. [DOI] [PubMed] [Google Scholar]
- Nishikawa S, Goldstein RA, Nierras CR. The promise of human induced pluripotent stem cells for research and therapy. Nat Rev Mol Cell Biol. 2008;9:725–729. doi: 10.1038/nrm2466. [DOI] [PubMed] [Google Scholar]
- Norstrom A, Akesson K, Hardarson T, Hamberger L, Bjorquist P, Sartipy P. Molecular and pharmacological properties of human embryonic stem cell-derived cardiomyocytes. Exp Biol Med (Maywood) 2006;231:1753–1762. doi: 10.1177/153537020623101113. [DOI] [PubMed] [Google Scholar]
- Ochiya T, Yamamoto Y, Banas A. Commitment of stem cells into functional hepatocytes. Differentiation. 2010;79:65–73. doi: 10.1016/j.diff.2009.10.002. [DOI] [PubMed] [Google Scholar]
- OECD (2008) Guidance document on mammalian reproductive toxicity testing and assessment. Series on testing and assessment no. 43. ENV/JM/MONO 16. In: OECD OfEC-oaD (ed), Paris
- Okita K, Ichisaka T, Yamanaka S. Generation of germline-competent induced pluripotent stem cells. Nature. 2007;448:313–317. doi: 10.1038/nature05934. [DOI] [PubMed] [Google Scholar]
- Otsuji TG, Minami I, Kurose Y, Yamauchi K, Tada M, Nakatsuji N. Progressive maturation in contracting cardiomyocytes derived from human embryonic stem cells: Qualitative effects on electrophysiological responses to drugs. Stem Cell Res. 2010;4:201–213. doi: 10.1016/j.scr.2010.01.002. [DOI] [PubMed] [Google Scholar]
- Pal R, Khanna A. Similar pattern in cardiac differentiation of human embryonic stem cell lines, BG01 V and ReliCellhES1, under low serum concentration supplemented with bone morphogenetic protein-2. Differentiation. 2007;75:112–122. doi: 10.1111/j.1432-0436.2006.00123.x. [DOI] [PubMed] [Google Scholar]
- Papaioannou VE, Gardner RL, McBurney MW, Babinet C, Evans MJ. Participation of cultured teratocarcinoma cells in mouse embryogenesis. J Embryol Exp Morphol. 1978;44:93–104. [PubMed] [Google Scholar]
- Park IH, et al. Disease-specific induced pluripotent stem cells. Cell. 2008;134:877–886. doi: 10.1016/j.cell.2008.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passier R, Mummery C. Getting to the heart of the matter: direct reprogramming to cardiomyocytes. Cell Stem Cell. 2010;7:139–141. doi: 10.1016/j.stem.2010.07.004. [DOI] [PubMed] [Google Scholar]
- Passier R, et al. Increased cardiomyocyte differentiation from human embryonic stem cells in serum-free cultures. Stem Cells. 2005;23:772–780. doi: 10.1634/stemcells.2004-0184. [DOI] [PubMed] [Google Scholar]
- Pei H, et al. Lineage restriction and differentiation of human embryonic stem cells into hepatic progenitors and zone 1 hepatocytes. Tissue Eng Part C Methods. 2009;15:95–104. doi: 10.1089/ten.tec.2008.0234. [DOI] [PubMed] [Google Scholar]
- Pekkanen-Mattila M, et al. Substantial variation in the cardiac differentiation of human embryonic stem cell lines derived and propagated under the same conditions—a comparison of multiple cell lines. Ann Med. 2009;41:360–370. doi: 10.1080/07853890802609542. [DOI] [PubMed] [Google Scholar]
- Pekkanen-Mattila M, et al. Human embryonic stem cell-derived cardiomyocytes: demonstration of a portion of cardiac cells with fairly mature electrical phenotype. Exp Biol Med (Maywood) 2010;235:522–530. doi: 10.1258/ebm.2010.009345. [DOI] [PubMed] [Google Scholar]
- Peng S, Lacerda AE, Kirsch GE, Brown AM, Bruening-Wright A. The action potential and comparative pharmacology of stem cell-derived human cardiomyocytes. J Pharmacol Toxicol Methods. 2010;61:277–286. doi: 10.1016/j.vascn.2010.01.014. [DOI] [PubMed] [Google Scholar]
- Pera MF, Tam PP (2010) Extrinsic regulation of pluripotent stem cells. Nature 465:713–720 [DOI] [PubMed]
- Perino MG, Yamanaka S, Li J, Wobus AM, Boheler KR. Cardiomyogenic stem and progenitor cell plasticity and the dissection of cardiopoiesis. J Mol Cell Cardiol. 2008;45:475–494. doi: 10.1016/j.yjmcc.2008.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters AK, Steemans M, Hansen E, Mesens N, Verheyen GR, Vanparys P. Evaluation of the embryotoxic potency of compounds in a newly revised high throughput embryonic stem cell test. Toxicol Sci. 2008;105:342–350. doi: 10.1093/toxsci/kfn126. [DOI] [PubMed] [Google Scholar]
- Petersen BE, et al. Bone marrow as a potential source of hepatic oval cells. Science. 1999;284:1168–1170. doi: 10.1126/science.284.5417.1168. [DOI] [PubMed] [Google Scholar]
- Peura TT, Bosman A, Stojanov T. Derivation of human embryonic stem cell lines. Theriogenology. 2007;67:32–42. doi: 10.1016/j.theriogenology.2006.09.031. [DOI] [PubMed] [Google Scholar]
- Peura T, Bosman A, Chami O, Jansen RP, Texlova K, Stojanov T. Karyotypically normal and abnormal human embryonic stem cell lines derived from PGD-analyzed embryos. Cloning Stem Cells. 2008;10:203–216. doi: 10.1089/clo.2007.0062. [DOI] [PubMed] [Google Scholar]
- Phillips BW, Crook JM. Pluripotent human stem cells: a novel tool in drug discovery. BioDrugs. 2010;24:99–108. doi: 10.2165/11532270-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Piersma AH. Validation of alternative methods for developmental toxicity testing. Toxicol Lett. 2004;149:147–153. doi: 10.1016/j.toxlet.2003.12.029. [DOI] [PubMed] [Google Scholar]
- Pless G, et al. Evaluation of primary human liver cells in bioreactor cultures for extracorporeal liver support on the basis of urea production. Artif Organs. 2006;30:686–694. doi: 10.1111/j.1525-1594.2006.00285.x. [DOI] [PubMed] [Google Scholar]
- Polo JM, et al. Cell type of origin influences the molecular and functional properties of mouse induced pluripotent stem cells. Nat Biotechnol. 2010;28:848–855. doi: 10.1038/nbt.1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prelle K, Zink N, Wolf E. Pluripotent stem cells—model of embryonic development, tool for gene targeting, and basis of cell therapy. Anat Histol Embryol. 2002;31:169–186. doi: 10.1046/j.1439-0264.2002.00388.x. [DOI] [PubMed] [Google Scholar]
- Puceat M. Protocols for cardiac differentiation of embryonic stem cells. Methods. 2008;45:168–171. doi: 10.1016/j.ymeth.2008.03.003. [DOI] [PubMed] [Google Scholar]
- Rambhatla L, Chiu CP, Kundu P, Peng Y, Carpenter MK. Generation of hepatocyte-like cells from human embryonic stem cells. Cell Transplant. 2003;12:1–11. doi: 10.3727/000000003783985179. [DOI] [PubMed] [Google Scholar]
- Rashid ST, et al. Modeling inherited metabolic disorders of the liver using human induced pluripotent stem cells. J Clin Invest. 2010;120:3127–3136. doi: 10.1172/JCI43122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinecke H, Minami E, Zhu WZ, Laflamme MA. Cardiogenic differentiation and transdifferentiation of progenitor cells. Circ Res. 2008;103:1058–1071. doi: 10.1161/CIRCRESAHA.108.180588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reppel M, Boettinger C, Hescheler J. Beta-adrenergic and muscarinic modulation of human embryonic stem cell-derived cardiomyocytes. Cell Physiol Biochem. 2004;14:187–196. doi: 10.1159/000080326. [DOI] [PubMed] [Google Scholar]
- Resnick JL, Bixler LS, Cheng L, Donovan PJ. Long-term proliferation of mouse primordial germ cells in culture. Nature. 1992;359:550–551. doi: 10.1038/359550a0. [DOI] [PubMed] [Google Scholar]
- Reubinoff BE, Pera MF, Fong CY, Trounson A, Bongso A. Embryonic stem cell lines from human blastocysts: somatic differentiation in vitro. Nat Biotechnol. 2000;18:399–404. doi: 10.1038/74447. [DOI] [PubMed] [Google Scholar]
- Ring A et al (2010) Hepatic maturation of human fetal hepatocytes in four-compartment three-dimensional perfusion culture. Tissue Eng Part C Methods 16:835–845 [DOI] [PubMed]
- Rohwedel J, Horak V, Hebrok M, Fuchtbauer EM, Wobus AM. M-twist expression inhibits mouse embryonic stem cell-derived myogenic differentiation in vitro. Exp Cell Res. 1995;220:92–100. doi: 10.1006/excr.1995.1295. [DOI] [PubMed] [Google Scholar]
- Rohwedel J, et al. Loss of beta1 integrin function results in a retardation of myogenic, but an acceleration of neuronal, differentiation of embryonic stem cells in vitro. Dev Biol. 1998;201:167–184. doi: 10.1006/dbio.1998.9002. [DOI] [PubMed] [Google Scholar]
- Rohwedel J, Guan K, Wobus AM. Induction of cellular differentiation by retinoic acid in vitro. Cells Tissues Organs. 1999;165:190–202. doi: 10.1159/000016699. [DOI] [PubMed] [Google Scholar]
- Rohwedel J, Guan K, Hegert C, Wobus AM. Embryonic stem cells as an in vitro model for mutagenicity, cytotoxicity and embryotoxicity studies: present state and future prospects. Toxicol In Vitro. 2001;15:741–753. doi: 10.1016/s0887-2333(01)00074-1. [DOI] [PubMed] [Google Scholar]
- Rowntree RK, McNeish JD. Induced pluripotent stem cells: opportunities as research and development tools in 21st century drug discovery. Regen Med. 2010;5:557–568. doi: 10.2217/rme.10.36. [DOI] [PubMed] [Google Scholar]
- Ruby KM, Zheng B. Gene targeting in a HUES line of human embryonic stem cells via electroporation. Stem Cells. 2009;27:1496–1506. doi: 10.1002/stem.73. [DOI] [PubMed] [Google Scholar]
- Russell LB, Russell WL. Pathways of radiation effects in the mother and the embryo. Cold Spring Harb Symp Quant Biol. 1954;19:50–59. doi: 10.1101/sqb.1954.019.01.009. [DOI] [PubMed] [Google Scholar]
- Russell LB, Selby PB, von Halle E, Sheridan W, Valcovic L. Use of the mouse spot test in chemical mutagenesis: interpretation of past data and recommendations for future work. Mutat Res. 1981;86:355–379. doi: 10.1016/0165-1110(81)90011-7. [DOI] [PubMed] [Google Scholar]
- Russmann S, Kullak-Ublick GA, Grattagliano I. Current concepts of mechanisms in drug-induced hepatotoxicity. Curr Med Chem. 2009;16:3041–3053. doi: 10.2174/092986709788803097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rust W, Balakrishnan T, Zweigerdt R. Cardiomyocyte enrichment from human embryonic stem cell cultures by selection of ALCAM surface expression. Regen Med. 2009;4:225–237. doi: 10.2217/17460751.4.2.225. [DOI] [PubMed] [Google Scholar]
- Sadler TW, Horton WE, Warner CW. Whole embryo culture: a screening technique for teratogens? Teratog Carcinog Mutagen. 1982;2:243–253. doi: 10.1002/1520-6866(1990)2:3/4<243::aid-tcm1770020306>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- Saha K, Jaenisch R. Technical challenges in using human induced pluripotent stem cells to model disease. Cell Stem Cell. 2009;5:584–595. doi: 10.1016/j.stem.2009.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai K, et al. Efficient integration of transgenes into a defined locus in human embryonic stem cells. Nucleic Acids Res. 2010;38:e96. doi: 10.1093/nar/gkp1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancho-Bru P, et al. Stem and progenitor cells for liver repopulation: can we standardise the process from bench to bedside? Gut. 2009;58:594–603. doi: 10.1136/gut.2008.171116. [DOI] [PubMed] [Google Scholar]
- Sarkar P, Rao BM. Molecular aspects of cardiac differentiation in embryonic stem cells. Crit Rev Biomed Eng. 2009;37:283–320. doi: 10.1615/critrevbiomedeng.v37.i4-5.10. [DOI] [PubMed] [Google Scholar]
- Sartiani L, Bettiol E, Stillitano F, Mugelli A, Cerbai E, Jaconi ME. Developmental changes in cardiomyocytes differentiated from human embryonic stem cells: a molecular and electrophysiological approach. Stem Cells. 2007;25:1136–1144. doi: 10.1634/stemcells.2006-0466. [DOI] [PubMed] [Google Scholar]
- Sartipy P, Bjorquist P, Strehl R, Hyllner J. Pluripotent human stem cells as novel tools in drug discovery and toxicity testing. IDrugs. 2006;9:702–705. [PubMed] [Google Scholar]
- Sasaki K, et al. Hepatocyte differentiation from human ESCs using the simple embryoid body formation method and the staged-additional cocktail. Sci World J. 2009;9:884–890. doi: 10.1100/tsw.2009.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satin J, et al. Calcium handling in human embryonic stem cell-derived cardiomyocytes. Stem Cells. 2008;26:1961–1972. doi: 10.1634/stemcells.2007-0591. [DOI] [PubMed] [Google Scholar]
- Schmelzer E, et al. Three-dimensional perfusion bioreactor culture supports differentiation of human fetal liver cells. Tissue Eng Part A. 2010;16:2007–2016. doi: 10.1089/ten.TEA.2009.0569. [DOI] [PubMed] [Google Scholar]
- Schmidt BP. Teratogenicity testing of new drugs with the postimplantation embryo culture system. In: Homburger F, editor. Concepts in toxicology. Basel: Karger Publ; 1985. pp. 46–57. [Google Scholar]
- Schmidt MM, Guan K, Wobus AM. Lithium influences differentiation and tissue-specific gene expression of mouse embryonic stem (ES) cells in vitro. Int J Dev Biol. 2001;45:421–429. [PubMed] [Google Scholar]
- Schnerch A, Cerdan C, Bhatia M. Distinguishing between mouse and human pluripotent stem cell regulation: the best laid plans of mice and men. Stem Cells. 2010;28:419–430. doi: 10.1002/stem.298. [DOI] [PubMed] [Google Scholar]
- Schwartz RE, Linehan JL, Painschab MS, Hu WS, Verfaillie CM, Kaufman DS. Defined conditions for development of functional hepatic cells from human embryonic stem cells. Stem Cells Dev. 2005;14:643–655. doi: 10.1089/scd.2005.14.643. [DOI] [PubMed] [Google Scholar]
- Schwetz BA, Morrissey RE, Welsch F, Kavlock RA. In vitro teratology. Environ Health Perspect. 1991;94:265–268. doi: 10.1289/ehp.94-1567960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedan O, et al. Human embryonic stem cell-derived cardiomyocytes can mobilize 1, 4, 5-inositol trisphosphate-operated [Ca2 +]i stores: the functionality of angiotensin-II/endothelin-1 signaling pathways. Ann NY Acad Sci. 2010;1188:68–77. doi: 10.1111/j.1749-6632.2009.05085.x. [DOI] [PubMed] [Google Scholar]
- Seeff LB (2007) Herbal hepatotoxicity. Clin Liver Dis 11:577–596 (vii) [DOI] [PubMed]
- Sehlmeyer U, Wobus AM. Lower mutation frequencies are induced by ENU in undifferentiated embryonic cells than in differentiated cells of the mouse in vitro. Mutat Res. 1994;324:69–76. doi: 10.1016/0165-7992(94)90070-1. [DOI] [PubMed] [Google Scholar]
- Sehlmeyer U, Wobus AM. MNNG is less cytotoxic and mutagenic in undifferentiated embryonic cells than in differentiated cells of the mouse in vitro. In Vitro Toxicol J Mol Cell Toxicol. 1995;8:121–127. [Google Scholar]
- Sehlmeyer U, Meister A, Beisker W, Wobus AM. Low mutagenic effects of mitomycin C in undifferentiated embryonic P19 cells are correlated with efficient cell cycle control. Mutat Res. 1996;354:103–112. doi: 10.1016/0027-5107(96)00047-4. [DOI] [PubMed] [Google Scholar]
- Sehlmeyer U, Rohwedel J, Wobus AM. Primordial germ cell-derived embryonic germ cells of the mouse-in vitro model for cytotoxicity studies with chemical mutagens. Toxicol In Vitro. 1996;10:755–763. doi: 10.1016/s0887-2333(96)00054-9. [DOI] [PubMed] [Google Scholar]
- Seifinejad A, Tabebordbar M, Baharvand H, Boyer LA, Salekdeh GH. Progress and promise towards safe induced pluripotent stem cells for therapy. Stem Cell Rev. 2010;6:297–306. doi: 10.1007/s12015-010-9121-x. [DOI] [PubMed] [Google Scholar]
- Seiler A, Visan A, Buesen R, Genschow E, Spielmann H. Improvement of an in vitro stem cell assay for developmental toxicity: the use of molecular endpoints in the embryonic stem cell test. Reprod Toxicol. 2004;18:231–240. doi: 10.1016/j.reprotox.2003.10.015. [DOI] [PubMed] [Google Scholar]
- Seiler A, et al. Current status of the embryonic stem cell test: the use of recent advances in the field of stem cell technology and gene expression analysis. ALTEX: Alternativen zu Tierexperimenten. 2006;23:393–399. [Google Scholar]
- Shamblott MJ, et al. Derivation of pluripotent stem cells from cultured human primordial germ cells. Proc Natl Acad Sci USA. 1998;95:13726–13731. doi: 10.1073/pnas.95.23.13726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Desponts C, Do JT, Hahm HS, Scholer HR, Ding S. Induction of pluripotent stem cells from mouse embryonic fibroblasts by Oct4 and Klf4 with small-molecule compounds. Cell Stem Cell. 2008;3:568–574. doi: 10.1016/j.stem.2008.10.004. [DOI] [PubMed] [Google Scholar]
- Shimizu T, et al. Fabrication of pulsatile cardiac tissue grafts using a novel 3-dimensional cell sheet manipulation technique and temperature-responsive cell culture surfaces. Circ Res. 2002;90:e40. doi: 10.1161/hh0302.105722. [DOI] [PubMed] [Google Scholar]
- Shirahashi H, et al. Differentiation of human and mouse embryonic stem cells along a hepatocyte lineage. Cell Transplant. 2004;13:197–211. doi: 10.3727/000000004783984016. [DOI] [PubMed] [Google Scholar]
- Shiraki N, Umeda K, Sakashita N, Takeya M, Kume K, Kume S. Differentiation of mouse and human embryonic stem cells into hepatic lineages. GenESCs. 2008;13:731–746. doi: 10.1111/j.1365-2443.2008.01201.x. [DOI] [PubMed] [Google Scholar]
- Si-Tayeb K, Lemaigre FP, Duncan SA. Organogenesis and development of the liver. Dev Cell. 2010;18:175–189. doi: 10.1016/j.devcel.2010.01.011. [DOI] [PubMed] [Google Scholar]
- Si-Tayeb K, et al. Highly efficient generation of human hepatocyte-like cells from induced pluripotent stem cells. Hepatology. 2010;51:297–305. doi: 10.1002/hep.23354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skrzypiec-Spring M, Grotthus B, Szelag A, Schulz R. Isolated heart perfusion according to Langendorff—still viable in the new millennium. J Pharmacol Toxicol Methods. 2007;55:113–126. doi: 10.1016/j.vascn.2006.05.006. [DOI] [PubMed] [Google Scholar]
- Smith AG, et al. Inhibition of pluripotential embryonic stem cell differentiation by purified polypeptides. Nature. 1988;336:688–690. doi: 10.1038/336688a0. [DOI] [PubMed] [Google Scholar]
- Snir M, et al. Assessment of the ultrastructural and proliferative properties of human embryonic stem cell-derived cardiomyocytes. Am J Physiol Heart Circ Physiol. 2003;285:H2355–H2363. doi: 10.1152/ajpheart.00020.2003. [DOI] [PubMed] [Google Scholar]
- Snykers S, De Kock J, Rogiers V, Vanhaecke T. In vitro differentiation of embryonic and adult stem cells into hepatocytes: state of the art. Stem Cells. 2009;27:577–605. doi: 10.1634/stemcells.2008-0963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Söderdahl T, et al. Glutathione transferases in hepatocyte-like cells derived from human embryonic stem cells. Toxicol In Vitro. 2007;21:929–937. doi: 10.1016/j.tiv.2007.01.021. [DOI] [PubMed] [Google Scholar]
- Soldner F, et al. Parkinson’s disease patient-derived induced pluripotent stem cells free of viral reprogramming factors. Cell. 2009;136:964–977. doi: 10.1016/j.cell.2009.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Z, et al. Efficient generation of hepatocyte-like cells from human induced pluripotent stem cells. Cell Res. 2009;19:1233–1242. doi: 10.1038/cr.2009.107. [DOI] [PubMed] [Google Scholar]
- Song H, Chung SK, Xu Y. Modeling disease in human ESCs using an efficient BAC-based homologous recombination system. Cell Stem Cell. 2010;6:80–89. doi: 10.1016/j.stem.2009.11.016. [DOI] [PubMed] [Google Scholar]
- Sonoda E, Sasaki MS, Morrison C, Yamaguchi-Iwai Y, Takata M, Takeda S. Sister chromatid exchanges are mediated by homologous recombination in vertebrate cells. Mol Cell Biol. 1999;19:5166–5169. doi: 10.1128/mcb.19.7.5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soto-Gutierrez A, et al. Differentiation of human embryonic stem cells to hepatocytes using deleted variant of HGF and poly-amino-urethane-coated nonwoven polytetrafluoroethylene fabric. Cell Transplant. 2006;15:335–341. doi: 10.3727/000000006783981945. [DOI] [PubMed] [Google Scholar]
- Spielmann H (1998) Reproduction and development. Environ Health Perspect 106(Suppl 2):571–576 [DOI] [PMC free article] [PubMed]
- Spielmann H. Predicting the risk of developmental toxicity from in vitro assays. Toxicol Appl Pharmacol. 2005;207:375–380. doi: 10.1016/j.taap.2005.01.049. [DOI] [PubMed] [Google Scholar]
- Spielmann H. The way forward in reproductive/developmental toxicity testing. Altern Lab Anim. 2009;37:641–656. doi: 10.1177/026119290903700609. [DOI] [PubMed] [Google Scholar]
- Spielmann H, Pohl I, Döring B, Liebsch M, Moldenhauer F (1997) The embryonic stem cell test, an in vitro embryotoxicity test using two permanent mouse cell lines: 3 T 3 fibroblasts and embryonic stem cells. In Vitro Toxicology
- Spielmann H, Genschow E, Liebsch M, Halle W. Determination of the starting dose for acute oral toxicity (LD50) testing in the up and down procedure (UDP) from cytotoxicity data. ATLA Alt Lab Anim. 1999;27:957–966. doi: 10.1177/026119299902700609. [DOI] [PubMed] [Google Scholar]
- Spielmann H, et al. The practical application of three validated in vitro embryotoxicity tests. The report and recommendations of an ECVAM/ZEBET workshop (ECVAM workshop 57) Altern Lab Anim. 2006;34:527–538. doi: 10.1177/026119290603400504. [DOI] [PubMed] [Google Scholar]
- Stadtfeld M, Nagaya M, Utikal J, Weir G, Hochedlinger K. Induced pluripotent stem cells generated without viral integration. Science. 2008;322:945–949. doi: 10.1126/science.1162494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadtfeld M, et al. Aberrant silencing of imprinted genes on chromosome 12qF1 in mouse induced pluripotent stem cells. Nature. 2010;465:175–181. doi: 10.1038/nature09017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens LC. Experimental production of testicular teratomas in mice. Proc Natl Acad Sci USA. 1964;52:654–661. doi: 10.1073/pnas.52.3.654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens LC. The development of transplantable teratocarcinomas from intratesticular grafts of pre- and postimplantation mouse embryos. Dev Biol. 1970;21:364–382. doi: 10.1016/0012-1606(70)90130-2. [DOI] [PubMed] [Google Scholar]
- Stevens LC, Little CC. Spontaneous testicular teratomas in an inbred strain of mice. Proc Natl Acad Sci USA. 1954;40:1080–1087. doi: 10.1073/pnas.40.11.1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart CL, Gadi I, Bhatt H. Stem cells from primordial germ cells can reenter the germ line. Dev Biol. 1994;161:626–628. doi: 10.1006/dbio.1994.1058. [DOI] [PubMed] [Google Scholar]
- Stojkovic M, Lako M, Strachan T, Murdoch A. Derivation, growth and applications of human embryonic stem cells. Reproduction. 2004;128:259–267. doi: 10.1530/rep.1.00243. [DOI] [PubMed] [Google Scholar]
- Strickland S, Mahdavi V. The induction of differentiation in teratocarcinoma stem cells by retinoic acid. Cell. 1978;15:393–403. doi: 10.1016/0092-8674(78)90008-9. [DOI] [PubMed] [Google Scholar]
- Strubing C, Ahnert-Hilger G, Shan J, Wiedenmann B, Hescheler J, Wobus AM. Differentiation of pluripotent embryonic stem cells into the neuronal lineage in vitro gives rise to mature inhibitory and excitatory neurons. Mech Dev. 1995;53:275–287. doi: 10.1016/0925-4773(95)00446-8. [DOI] [PubMed] [Google Scholar]
- Stummann TC, Hareng L, Bremer S. Embryotoxicity hazard assessment of methylmercury and chromium using embryonic stem cells. Toxicology. 2007;242:130–143. doi: 10.1016/j.tox.2007.09.022. [DOI] [PubMed] [Google Scholar]
- Stummann TC, et al. Report and recommendations of the workshop of the European centre for the validation of alternative methods for drug-induced cardiotoxicity. Cardiovasc Toxicol. 2009;9:107–125. doi: 10.1007/s12012-009-9045-3. [DOI] [PubMed] [Google Scholar]
- Stummann TC, Hareng L, Bremer S. Hazard assessment of methylmercury toxicity to neuronal induction in embryogenesis using human embryonic stem cells. Toxicology. 2009;257:117–126. doi: 10.1016/j.tox.2008.12.018. [DOI] [PubMed] [Google Scholar]
- Sullivan GJ, et al. Generation of functional human hepatic endoderm from human induced pluripotent stem cells. Hepatology. 2010;51:329–335. doi: 10.1002/hep.23335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K, et al. Highly efficient transient gene expression and gene targeting in primate embryonic stem cells with helper-dependent adenoviral vectors. Proc Natl Acad Sci USA. 2008;105:13781–13786. doi: 10.1073/pnas.0806976105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki A, et al. Drugs associated with hepatotoxicity and their reporting frequency of liver adverse events in VigiBase: unified list based on international collaborative work. Drug Saf. 2010;33:503–522. doi: 10.2165/11535340-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Synnergren J, et al. Cardiomyogenic gene expression profiling of differentiating human embryonic stem cells. J Biotechnol. 2008;134:162–170. doi: 10.1016/j.jbiotec.2007.11.011. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Takahashi K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Tenzen T, Zembowicz F, Cowan CA. Genome modification in human embryonic stem cells. J Cell Physiol. 2010;222:278–281. doi: 10.1002/jcp.21948. [DOI] [PubMed] [Google Scholar]
- Terada N, et al. Bone marrow cells adopt the phenotype of other cells by spontaneous cell fusion. Nature. 2002;416:542–545. doi: 10.1038/nature730. [DOI] [PubMed] [Google Scholar]
- Tesar PJ, et al. New cell lines from mouse epiblast share defining features with human embryonic stem cells. Nature. 2007;448:196–199. doi: 10.1038/nature05972. [DOI] [PubMed] [Google Scholar]
- Tester DJ, et al. Allelic dropout in long QT syndrome genetic testing: a possible mechanism underlying false-negative results. Heart Rhythm. 2006;3:815–821. doi: 10.1016/j.hrthm.2006.03.016. [DOI] [PubMed] [Google Scholar]
- Thomas KR, Capecchi MR. Site-directed mutagenesis by gene targeting in mouse embryo-derived stem cells. Cell. 1987;51:503–512. doi: 10.1016/0092-8674(87)90646-5. [DOI] [PubMed] [Google Scholar]
- Thomson JA, et al. Embryonic stem cell lines derived from human blastocysts. Science. 1998;282:1145–1147. doi: 10.1126/science.282.5391.1145. [DOI] [PubMed] [Google Scholar]
- Thyagarajan B, et al. Creation of engineered human embryonic stem cell lines using phiC31 integrase. Stem Cells. 2008;26:119–126. doi: 10.1634/stemcells.2007-0283. [DOI] [PubMed] [Google Scholar]
- Topol EJ. Failing the public health–rofecoxib, Merck, and the FDA. N Engl J Med. 2004;351:1707–1709. doi: 10.1056/NEJMp048286. [DOI] [PubMed] [Google Scholar]
- Touboul T, et al. Generation of functional hepatocytes from human embryonic stem cells under chemically defined conditions that recapitulate liver development. Hepatology. 2010;51:1754–1765. doi: 10.1002/hep.23506. [DOI] [PubMed] [Google Scholar]
- Toyooka Y, Tsunekawa N, Akasu R, Noce T. Embryonic stem cells can form germ cells in vitro. Proc Natl Acad Sci USA. 2003;100:11457–11462. doi: 10.1073/pnas.1932826100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trosko JE, Chang CC. Factors to consider in the use of stem cells for pharmaceutic drug development and for chemical safety assessment. Toxicology. 2010;270:18–34. doi: 10.1016/j.tox.2009.11.019. [DOI] [PubMed] [Google Scholar]
- Tzimas G, Sass JO, Wittfoht W, Elmazar MM, Ehlers K, Nau H. Identification of 9, 13-dicis-retinoic acid as a major plasma metabolite of 9-cis-retinoic acid and limited transfer of 9-cis-retinoic acid and 9, 13-dicis-retinoic acid to the mouse and rat embryos. Drug Metab Dispos. 1994;22:928–936. [PubMed] [Google Scholar]
- Urbach A, Schuldiner M, Benvenisty N. Modeling for Lesch-Nyhan disease by gene targeting in human embryonic stem cells. Stem Cells. 2004;22:635–641. doi: 10.1634/stemcells.22-4-635. [DOI] [PubMed] [Google Scholar]
- Van Hoof D, et al. Identification of cell surface proteins for antibody-based selection of human embryonic stem cell-derived cardiomyocytes. J Proteome Res. 2010;9:1610–1618. doi: 10.1021/pr901138a. [DOI] [PubMed] [Google Scholar]
- Van Sloun PP, et al. The role of nucleotide excision repair in protecting embryonic stem cells from genotoxic effects of UV-induced DNA damage. Nucleic Acids Res. 1999;27:3276–3282. doi: 10.1093/nar/27.16.3276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaziri H, et al. Spontaneous reversal of the developmental aging of normal human cells following transcriptional reprogramming. Regen Med. 2010;5:345–363. doi: 10.2217/rme.10.21. [DOI] [PubMed] [Google Scholar]
- Verwei M, van Burgsteden JA, Krul CA, van de Sandt JJ, Freidig AP. Prediction of in vivo embryotoxic effect levels with a combination of in vitro studies and PBPK modelling. Toxicol Lett. 2006;165:79–87. doi: 10.1016/j.toxlet.2006.01.017. [DOI] [PubMed] [Google Scholar]
- Vierbuchen T, Ostermeier A, Pang ZP, Kokubu Y, Sudhof TC, Wernig M. Direct conversion of fibroblasts to functional neurons by defined factors. Nature. 2010;463:1035–1041. doi: 10.1038/nature08797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel R (1993) In vitro approach to fertility research: genotoxicity tests on primordial germ cells and embryonic stem cells. Reprod Toxicol 7(Suppl 1):69–73 [DOI] [PubMed]
- Vojnits K, Bremer S. Challenges of using pluripotent stem cells for safety assessments of substances. Toxicology. 2010;270:10–17. doi: 10.1016/j.tox.2009.12.003. [DOI] [PubMed] [Google Scholar]
- Wernig M, et al. In vitro reprogramming of fibroblasts into a pluripotent ES-cell-like state. Nature. 2007;448:318–324. doi: 10.1038/nature05944. [DOI] [PubMed] [Google Scholar]
- West PR, Weir AM, Smith AM, Donley EL, Cezar GG. Predicting human developmental toxicity of pharmaceuticals using human embryonic stem cells and metabolomics. Toxicol Appl Pharmacol. 2010;247:18–27. doi: 10.1016/j.taap.2010.05.007. [DOI] [PubMed] [Google Scholar]
- Westerink WM, Schoonen WG. Cytochrome P450 enzyme levels in HepG2 cells and cryopreserved primary human hepatocytes and their induction in HepG2 cells. Toxicol In Vitro. 2007;21:1581–1591. doi: 10.1016/j.tiv.2007.05.014. [DOI] [PubMed] [Google Scholar]
- Wiese C et al (2009) Differentiation induction of mouse embryonic stem cells into sinus node-like cells by suramin. Int J Cardiol [DOI] [PMC free article] [PubMed]
- Williams RL, et al. Myeloid leukaemia inhibitory factor maintains the developmental potential of embryonic stem cells. Nature. 1988;336:684–687. doi: 10.1038/336684a0. [DOI] [PubMed] [Google Scholar]
- Wilson KD, et al. Effects of ionizing radiation on self-renewal and pluripotency of human embryonic stem cells. Cancer Res. 2010;70:5539–5548. doi: 10.1158/0008-5472.CAN-09-4238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wobus AM (2010) The Janus face of pluripotent stem cells—connection between pluripotency and tumourigenicity. BioEssays (in press) [DOI] [PubMed]
- Wobus AM, Boheler KR. Embryonic stem cells: prospects for developmental biology and cell therapy. Physiol Rev. 2005;85:635–678. doi: 10.1152/physrev.00054.2003. [DOI] [PubMed] [Google Scholar]
- Wobus AM, Grosse R, Schoneich J. Specific effects of nerve growth factor on the differentiation pattern of mouse embryonic stem cells in vitro. Biomed Biochim Acta. 1988;47:965–973. [PubMed] [Google Scholar]
- Wobus AM, Wallukat G, Hescheler J. Pluripotent mouse embryonic stem cells are able to differentiate into cardiomyocytes expressing chronotropic responses to adrenergic and cholinergic agents and Ca2 + channel blockers. Differentiation. 1991;48:173–182. doi: 10.1111/j.1432-0436.1991.tb00255.x. [DOI] [PubMed] [Google Scholar]
- Wobus AM, Kleppisch T, Maltsev V, Hescheler J. Cardiomyocyte-like cells differentiated in vitro from embryonic carcinoma cells P19 are characterized by functional expression of adrenoceptors and Ca2 + channels. In Vitro Cell Dev Biol Anim. 1994;30A:425–434. doi: 10.1007/BF02631310. [DOI] [PubMed] [Google Scholar]
- Wobus AM, Rohwedel J, Maltsev V, Hescheler J. In vitro differentiation of embryonic stem cells into cardiomyocytes or skeletal muscle cells is specifically modulated by retinoic acid. Roux’s Arch Dev Biol. 1994;204:36–45. doi: 10.1007/BF00189066. [DOI] [PubMed] [Google Scholar]
- Wobus AM, et al. Retinoic acid accelerates embryonic stem cell-derived cardiac differentiation and enhances development of ventricular cardiomyocytes. J Mol Cell Cardiol. 1997;29:1525–1539. doi: 10.1006/jmcc.1997.0433. [DOI] [PubMed] [Google Scholar]
- Woltjen K, et al. piggyBac transposition reprograms fibroblasts to induced pluripotent stem cells. Nature. 2009;458:766–770. doi: 10.1038/nature07863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu W, Welsh MJ (1996) Expression of the 25-kDa heat-shock protein (HSP27) correlates with resistance to the toxicity of cadmium chloride, mercuric chloride, cis-platinum(II)-diammine dichloride, or sodium arsenite in mouse embryonic stem cells transfected with sense or antisense HSP27 cDNA. Toxicol Appl Pharmacol 141:330–339 [DOI] [PubMed]
- Xie H, Ye M, Feng R, Graf T. Stepwise reprogramming of B cells into macrophages. Cell. 2004;117:663–676. doi: 10.1016/s0092-8674(04)00419-2. [DOI] [PubMed] [Google Scholar]
- Xu C, Police S, Rao N, Carpenter MK. Characterization and enrichment of cardiomyocytes derived from human embryonic stem cells. Circ Res. 2002;91:501–508. doi: 10.1161/01.res.0000035254.80718.91. [DOI] [PubMed] [Google Scholar]
- Xu C, Police S, Hassanipour M, Gold JD. Cardiac bodies: a novel culture method for enrichment of cardiomyocytes derived from human embryonic stem cells. Stem Cells Dev. 2006;15:631–639. doi: 10.1089/scd.2006.15.631. [DOI] [PubMed] [Google Scholar]
- Xu XQ, et al. Chemically defined medium supporting cardiomyocyte differentiation of human embryonic stem cells. Differentiation. 2008;76:958–970. doi: 10.1111/j.1432-0436.2008.00284.x. [DOI] [PubMed] [Google Scholar]
- Xu XQ, et al. Highly enriched cardiomyocytes from human embryonic stem cells. Cytotherapy. 2008;10:376–389. doi: 10.1080/14653240802105307. [DOI] [PubMed] [Google Scholar]
- Xu XQ, Soo SY, Sun W, Zweigerdt R. Global expression profile of highly enriched cardiomyocytes derived from human embryonic stem cells. Stem Cells. 2009;27:2163–2174. doi: 10.1002/stem.166. [DOI] [PubMed] [Google Scholar]
- Xue H, et al. A Targeted Neuroglial Reporter Line Generated by Homologous Recombination in Human Embryonic Stem Cells. Stem Cells. 2009;27:1836–1846. doi: 10.1002/stem.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, et al. Human cardiovascular progenitor cells develop from a KDR + embryonic-stem-cell-derived population. Nature. 2008;453:524–528. doi: 10.1038/nature06894. [DOI] [PubMed] [Google Scholar]
- Yao S, et al. Long-term self-renewal and directed differentiation of human embryonic stem cells in chemically defined conditions. Proc Natl Acad Sci USA. 2006;103:6907–6912. doi: 10.1073/pnas.0602280103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying QL, Nichols J, Evans EP, Smith AG. Changing potency by spontaneous fusion. Nature. 2002;416:545–548. doi: 10.1038/nature729. [DOI] [PubMed] [Google Scholar]
- Ylä-Outinen L, Heikkila J, Skottman H, Suuronen R, Aanismaa R, Narkilahti S (2010) Human cell-based micro electrode array platform for studying neurotoxicity. Front Neuroengineering 3 [DOI] [PMC free article] [PubMed]
- Yokoo N, et al. The effects of cardioactive drugs on cardiomyocytes derived from human induced pluripotent stem cells. Biochem Biophys Res Commun. 2009;387:482–488. doi: 10.1016/j.bbrc.2009.07.052. [DOI] [PubMed] [Google Scholar]
- Yu J, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–1920. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- Yu J, et al. Human induced pluripotent stem cells free of vector and transgene sequences. Science. 2009;324:797–801. doi: 10.1126/science.1172482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaehres H, Lensch MW, Daheron L, Stewart SA, Itskovitz-Eldor J, Daley GQ. High-efficiency RNA interference in human embryonic stem cells. Stem Cells. 2005;23:299–305. doi: 10.1634/stemcells.2004-0252. [DOI] [PubMed] [Google Scholar]
- Zalzman M, et al. Zscan4 regulates telomere elongation and genomic stability in ESCs. Nature. 2010;464:858–863. doi: 10.1038/nature08882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zdravkovic T, Genbacev O, LaRocque N, McMaster M, Fisher S. Human embryonic stem cells as a model system for studying the effects of smoke exposure on the embryo. Reprod Toxicol. 2008;26:86–93. doi: 10.1016/j.reprotox.2008.07.004. [DOI] [PubMed] [Google Scholar]
- Zeng X, Rao MS. Controlled genetic modification of stem cells for developing drug discovery tools and novel therapeutic applications. Curr Opin Mol Ther. 2008;10:207–213. [PubMed] [Google Scholar]
- Zeng X, et al. An in vitro model of human dopaminergic neurons derived from embryonic stem cells: MPP + toxicity and GDNF neuroprotection. Neuropsychopharmacology. 2006;31:2708–2715. doi: 10.1038/sj.npp.1301125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, et al. Derivation of human embryonic stem cells from developing and arrested embryos. Stem Cells. 2006;24:2669–2676. doi: 10.1634/stemcells.2006-0377. [DOI] [PubMed] [Google Scholar]
- Zhang J, et al. Functional cardiomyocytes derived from human induced pluripotent stem cells. Circ Res. 2009;104:e30–e41. doi: 10.1161/CIRCRESAHA.108.192237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao D, et al. Derivation and characterization of hepatic progenitor cells from human embryonic stem cells. PLoS One. 2009;4:e6468. doi: 10.1371/journal.pone.0006468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao XY, et al. iPS cells produce viable mice through tetraploid complementation. Nature. 2009;461:86–90. doi: 10.1038/nature08267. [DOI] [PubMed] [Google Scholar]
- Zhou W, Freed CR. Adenoviral gene delivery can reprogram human fibroblasts to induced pluripotent stem cells. Stem Cells. 2009;27:2667–2674. doi: 10.1002/stem.201. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Brown J, Kanarek A, Rajagopal J, Melton DA. In vivo reprogramming of adult pancreatic exocrine cells to beta-cells. Nature. 2008;455:627–632. doi: 10.1038/nature07314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, et al. Generation of induced pluripotent stem cells using recombinant proteins. Cell Stem Cell. 2009;4:381–384. doi: 10.1016/j.stem.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu WZ, Santana LF, Laflamme MA. Local control of excitation-contraction coupling in human embryonic stem cell-derived cardiomyocytes. PLoS One. 2009;4:e5407. doi: 10.1371/journal.pone.0005407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann WH, et al. Tissue engineering of a differentiated cardiac muscle construct. Circ Res. 2002;90:223–230. doi: 10.1161/hh0202.103644. [DOI] [PubMed] [Google Scholar]
- Zou J, et al. Gene targeting of a disease-related gene in human induced pluripotent stem and embryonic stem cells. Cell Stem Cell. 2009;5:97–110. doi: 10.1016/j.stem.2009.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- zur Nieden NI, Kempka G, Ahr HJ. Molecular multiple endpoint embryonic stem cell test–a possible approach to test for the teratogenic potential of compounds. Toxicol Appl Pharmacol. 2004;194:257–269. doi: 10.1016/j.taap.2003.09.019. [DOI] [PubMed] [Google Scholar]
- Zwaka TP, Thomson JA. Homologous recombination in human embryonic stem cells. Nat Biotechnol. 2003;21:319–321. doi: 10.1038/nbt788. [DOI] [PubMed] [Google Scholar]
- Zwi L, et al. Cardiomyocyte differentiation of human induced pluripotent stem cells. Circulation. 2009;120:1513–1523. doi: 10.1161/CIRCULATIONAHA.109.868885. [DOI] [PubMed] [Google Scholar]