

Abstract
Cystic fibrosis mostly follows a single Phe508 deletion in CFTR (cystic fibrosis transmembrane regulator) (CFTRΔF508), thereby causing premature fragmentation of the nascent protein with concomitant alterations of diverse cellular functions. We show that CK2, the most pleiotropic protein kinase, undergoes allosteric control of its different cellular forms in the presence of short CFTR peptides encompassing the Phe508 deletion: these CFTRΔF508 peptides drastically inhibit the isolated catalytic subunit (α) of the kinase and yet up-regulate the holoenzyme, composed of two catalytic and two non-catalytic (β) subunits. Remarkable agreement between in silico docking and our biochemical data point to different sites for the CFTRΔF508 peptide binding on isolated CK2α and on CK2β assembled into the holoenzyme, suggesting that CK2 targeting may be perturbed in cells expressing CFTRΔF508; this could shed light on some pleiotropic aspects of cystic fibrosis disease.
Keywords: allosteric regulation, cystic fibrosis, cystic fibrosis transmembrane regulator (CFTR), molecular modelling, protein kinase CK2
INTRODUCTION
Eukaryotic life is controlled by the reversible phosphorylation of proteins, such that ~50% of the human proteome (also referred to as the ‘phosphoproteome’) is phosphorylated at some time [1,2]. Such phosphotransferase reactions are catalysed by protein kinases, making up one of the largest families of enzymes, collectively referred to as the ‘kinome’ [3]. The specific contribution of the individual members (>500 in the human kinome) is non-linear, such that a few are dedicated enzymes impinging only on a paucity of protein targets, whereas the majority are more or less pleiotropic with many protein substrates subjected to their control. On the basis of available repertoires of substrates [4] and on a global phosphoproteome analysis [5], one single protein kinase is endowed with the most striking pleiotropy, possibly accounting for the generation of 10–20% of the entire phosphoproteome. This protein kinase is CK2, an acronym derived from the misnomer ‘casein kinase-2’, applied pragmatically merely to indicate one of the ubiquitous acidophilic serine/threonine protein kinases displaying high in vitro activity toward the artificial substrate casein, even though this is not among its many physiological targets [6]. CK2 appears to be endowed with an ample variety of cellular functions, with special reference to gene expression, protein synthesis and degradation, and signal transduction [7] and plays a key role as a pro-survival and an anti-apoptotic agent [8]. In particular, it has been suggested that the critical role of CK2 in the NF-κB (nuclear factor κB) and in the Wnt pathways [9] accounts for the death at mid-gestation of mice lacking the catalytic subunit α of CK2 [10]. Although loss of this CK2 catalytic subunit in development is lethal, its overexpression is capable of promoting tumorigenesis [11-14] and, more in general, alterations in CK2 level have been linked to a variety of pathological situations [15].
It is commonly held that the pathogenic potential of CK2 is related to its constitutive activity, which, in turn, may reflect its terrific pleiotropy [16]. However, this notion does not sit well with the fact that CK2 is equipped with a regulatory (β) subunit, whose obligate dimer status creates the backbone to form a heterotetrameric holoenzyme once combined with two catalytic subunits (α and/or α′). These latter catalytic moieties are active either alone or in combination with the β-subunits, with the caveat that the regulatory subunits appear to display a variable influence on the targeting of different substrates [17], despite themselves being unable to turn catalytic activity either on or off. This creates three different classes of CK2 substrates, depending on how β binding affects their phosphorylation by α. Consistent with the concept of CK2 being constitutively active lies the observation that, in all CK2α structures solved to date, regardless of species, crystal packing, ligands and quaternary structure, the regulatory key elements invariably adopt the conformation typical for active protein kinases [18]. Recently, a careful inspection of CK2α disclosed the existence of some dynamic regions therein [19], one of which has the potential to overlap a proposed binding pocket for small molecules located at the interface with the β-subunit [20]. It has been speculated that this represents an allosteric site, where some recently developed non-competitive inhibitors of the CK2 holoenzyme may bind [21]. On the other hand no physiological effector(s) capable of exploiting this putative, down-regulatory allosteric site are known. Recently, however, we reported that peptides encompassing the sequence of CFTR [CF (cystic fibrosis) transmembrane regulator] surrounding Phe508, a residue whose deletion is the commonest cause of cystic fibrosis, are powerful effectors of CK2 activity, displaying opposite effects on the holoenzyme as compared with the isolated catalytic subunits [22]. In particular, peptides bearing the Phe508 deletion (CFTRΔF508 peptides) were able to specifically inhibit CK2α in a non-competitive manner with respect to a specific phosphoacceptor substrate. Recently, Cohen et al. [23] described in vivo effects of these very same peptides in a developing lung model, such that Phe508-dependent changes in protein–protein interactions occur when such peptides are expressed using a peptide-encoding virus to infect the developing embryo. Importantly, in CF, the mechanism by which loss of Phe508 induces a multi-system disease that destroys many organs remains unknown. In the present study, we investigate the optimal features conferring inhibitory properties to these peptides. We show that they behave as allosteric effectors of CK2 by binding to two distinct sites, one of which may be located in a region of the upper lobe of the α-subunit, where it can both interfere with α/β inter-subunit interaction, altering the proper conformation of the ATP-binding domain. Our work may have immediate relevance to the amplification mechanism by which a single deleted amino acid in CFTR could cause so dramatic a change in the phenotype in ~1 in 3000 human infants, creating a multi-organ disease affecting networks of many hundreds of proteins and genes.
EXPERIMENTAL
Materials
Most of the reagents were purchased from Sigma. Purified wild-type and ΔF508 mutated recombinant murine NBD1 (nucleotide-binding domain 1 of CFTR; spanning sequence 389–673) were generously provided by the Philip J. Thomas laboratory (Southwestern Medical Center, University of Texas, Dallas, TX, U.S.A.). Recombinant α- and β-subunits of human protein kinase CK2 were expressed in Escherichia coli and purified as previously described [24].
Peptide synthesis
The CFTR-derived synthetic peptides were prepared by a solid-phase peptide synthesis method using an automatized peptide synthesizer (model 431-A; Applied Biosystems) and/or a multiple peptide robotic synthesizer (model Syro II; Multisyntech) as C-terminal acids on HMP resin (Applied Biosystems) or as C-terminal amides on Rink Amide PEGA {poly[acryloyl-bis(aminopropyl)poly(ethylene glycol)]} resin (Novabiochem) as previously described [22].
Phosphorylation assays
In vitro phosphorylation experiments were performed by incubating the CK2α catalytic subunit (8–28 nM) with peptide or protein substrates (final volume 25 μl) in a medium containing 50 mM Tris/HCl, pH 7.5, 12 mM MgCl2, 100 mM NaCl and 100 μM [γ-33P]ATP (specific radioactivity 2000–4000 c.p.m./pmol). The synthetic peptide RRRADDSDDDDD (100 μM) was used as a typical CK2α phosphoacceptor substrate, whereas a peptide derived from eIF2β (eukaryotic initiation factor 2β) [25] and calmodulin were also used in some experiments for other class effects. The autophosphorylation of the CK2 β-subunit was evaluated by incubating the CK2 holoenzyme in the presence of the phosphodonor nucleotide as described above and by subjecting samples to SDS/PAGE. The phosphate incorporated into peptide and protein substrates was determined as described previously [22,25]. Kinetic analysis and determination of inhibition constants were performed by testing the kinase in the absence or in the presence of increasing concentrations of inhibitor and by constructing the Lineweaver–Burk double-reciprocal plots of the data. Final data reported in Figures and Tables are calculated from the mean of at least three independent experiments with S.E.M. never exceeding 15%.
Molecular modelling
For the computer-aided protein–protein docking stages, the human CK2 α-subunit, the human CK2α2β2 tetramer and CFTRΔF508 peptide (amino acids 500–518) were retrieved from the PDB (PDB code: 1JWH and 1XMJ respectively). Hydrogen atoms were added to the protein structure using standard geometries with the MOE program (Molecular Operating Environment, http://www.chemcomp.com/software.htm). To minimize contacts between hydrogens, the structures were subjected to Amber99 force field minimization until the rms (root mean square) of conjugate gradient was <0.1 kcal · mol−1 ·Å−1 (1 kcal≈4.184 kJ; 1 Å=0.1 nm), keeping the heavy atoms fixed at their crystallographic positions. The wild-type CFTR, CFTRΔF508 and CFTRΔI507 (CFTR with Ile507 deleted) peptides were built starting from the crystallographic structure 1XMJ and were submitted to MOE molecular dynamic approach for 1 ns at a constant temperature of 300 K within a previously minimized water system. After this initial in silico approach, a set of protein–protein docking experiments was performed. Protein–protein docking is a computational tool useful in predicting the three-dimensional structure of protein complexes from the co-ordinates of its subunits. We have adopted a two-stage approach, which recently demonstrated good efficacy; in the first stage, the two monomers were treated as rigid bodies and all the rotational and translational degrees of freedom were fully explored using a cluster of protein–protein docking algorithms (Zdock; Gramm and Echer); in the second stage, a small number of structures deriving from the initial stage was refined in the docking zone using side-chain rotamers and an energy-minimization strategy [25,27]. The final complexes were submitted to a new molecular dynamic step at the same condition described before to validate their stability over time.
RESULTS
Structural features of CK2α inhibitory peptides derived from CFTR
Figure 1 illustrates the dose-dependent effect of wild-type or various mutated peptides encompassing the CFTR(500–518) sequence (see Figure 2, peptide 1), on the activity of the CK2 catalytic subunit (α) assayed with the α-specific peptide substrate RRRADDSDDDDD. At low CFTR peptide concentrations, inhibition by the wild-type peptide (open circles) is not apparent (being instead replaced by slight stimulation), but, nonetheless, inhibition initiates at concentrations beyond 50 μM. Deletion of Phe508 is itself sufficient to convert the peptide into a powerful inhibitor with an IC50 of approx. 15 μM (compare open circles with filled squares). By contrast, the deletion of Ile507 (open squares), also responsible for CF on one allele by promoting unfolding and premature degradation [28], but nonetheless extremely rare (<0.1%) as compared with Phe508 (70–90%) [29], causes a loss in inhibitory efficacy. Also of note in Figure 1 is the effect of mutating Val510 to aspartate either in the context of a wild-type peptide or its ΔF508 equivalent, the latter combination having been shown to partially restore functionality of CFTRΔF508 [30]; in our hands this V510D mutation significantly abrogates the inhibitory efficacy of the CFTRΔF508 peptide, yet has a diametrically opposed effect when present in the wild-type peptide, whose moderate inhibitory efficiency is increased almost equalling that of the CFTRΔF508 peptide. These data demonstrate that the sequence around the common mutation in CFTR has residue-dependent regulatory relevance to CK2α function. Recently, Treharne et al. [31] have demonstrated that CK2 can control CFTR itself in an Phe508-related manner, thus adding further evidence to our proposed link between CFTR and CK2 through this Phe508 region.
Figure 1. Sequence-determined variability of inhibition of CK2α by short CFTR peptides straddling the Phe508 region of wild-type or mutant CFTR.

The percentage change in CK2α activity (ordinate, relative to no-peptide control as 100%) was determined against the specific synthetic peptide RRRADDSDDDDD as phosphorylatable substrate. A bi-directional effect on CK2α occurs between Phe508 and negative charge compared with hydrophobicity at amino acid 510. For this and subsequent Figures, CK2α activity was determined as described in the Experimental section and all data points are calculated from the mean of at least three independent experiments with the S.E.M. never exceeding 15% (not shown for clarity).
Figure 2. IC50 values for the inhibition of CK2α by CFTR-derived peptides.
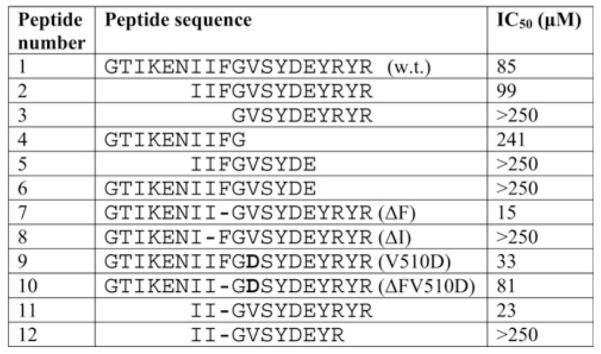
The parent peptide (1, w.t.) reproduces the 500–518 sequence of human CFTR. It belongs to the NBD1 domain of CFTR shown in Figure 3. The parentheses contain the simplified acronyms used to indicate the background of the peptides that are reported. Bold residues indicate mutations and hyphens indicate missing residues.
The peptides used for the experiment in Figure 1 encompass a region of CFTR NBD1 centred on an exposed loop bridging two helices [32], also including two short ‘wings’ belonging to these helices (Figure 3). To gain information about which elements within the peptides might be important for their efficacy towards CK2α activity, shortened derivatives were synthesized bearing deletions in their N- and/or C-termini. These are listed together with the ‘parent’ peptides in Figure 2 where their IC50 values, reflecting the ability of each to inhibit CK2α, are also reported. These data lead us to conclude that, although the first six N-terminal residues (GTIKEN) are dispensable, as their removal does not compromise inhibition by either the wild-type or the ΔF508 peptides (compare peptides 1 and 2 with peptides 7 and 11 respectively), any further removal of the II doublet is dramatically detrimental as it fully abrogates their inhibitory potential (compare peptide 2 with peptide 3). This is consistent with the crucial role of Ile507, whose deletion is sufficient to suppress inhibition (see Figure 1). In sharp contrast, the whole C-terminal portion of the peptides (generating in full-size NBD1 the initiation point of a new helix) appears to be strictly required for efficient inhibition, since the removal of the C-terminal doublet YR from a ΔF508 N-terminally shortened peptide abrogates inhibition, at least up to 250 μM (compare peptide 12 with peptide 11 in Figure 2).
Figure 3. Parent peptide and NBD1 domain structure.

The peptides used for the experiment in Figure 1 encompass a region of CFTR NBD1 centred on an exposed loop bridging two helices [32], also including two short ‘wings’ belonging to these helices (left). The w.t. peptide of Figure 2 is shown at the bottom. The parent peptide (peptide number 1 in Figure 2) belongs to the NBD1 domain of CFTR schematically represented in the right cartoon (in practice, the two NBDs are thought to create a dimeric sandwich trapping two nucleotides).
Kinetics are consistent with an allosteric mechanism of inhibition of CK2α by CFTRΔF508 peptides
The CFTR peptides used in the present study include a potential CK2 phosphoacceptor site (Ser511) which is not phosphorylated by CK2 to any appreciable extent [22]. It also includes three tyrosine residues, one of which (Tyr512) could represent a target for the tyrosine kinase Syk, owing to nearby acidic determinants [33]. We have found that the CFTR(500–518) peptide is unaffected by the Src family protein kinases Lyn and c-Fgr, whereas it is slowly phosphorylated by Syk ([34] and E. Tibaldi, A.M. Brunati, M.A. Pagano, O. Marin and L.A. Pinna, unpublished work) and that a derivative of this peptide in which Tyr512 was replaced by phosphotyrosine underwent a weak phosphorylation by CK2 at Ser511 [22]. We wondered whether the inhibition of CK2α by these peptides was due to a pseudosubstrate effect, resulting in competition with respect to the phosphoacceptor substrate. To check this point and, more in general, to gain information about the mechanism underlying inhibition of CK2α by CFTR peptides, competition kinetics were performed with respect to either the phosphoacceptor substrate or the co-substrate ATP. It was previously found that inhibition of phosphorylation by the CFTRΔF508 peptide was not competitive with respect to the specific CK2 peptide substrate RRRADDSDDDDD [22]. Similar results have been now obtained using the more physiological target calmodulin as the phosphoacceptor substrate (Figure 4A); again inhibition by the CFTRΔF508 peptide is not competitive with respect to this phosphoacceptor substrate. On the other hand, competition experiments with respect to the co-substrate ATP (Figure 4B) gave rise to bi-phasic kinetics: at concentrations around its Km value (15 μM), which are those generally adopted for this kind of kinetics, ATP is typically non-competitive against inhibition by the CFTR peptide, with Vmax values drastically diminished by the CFTR peptide, whereas Km values are similar. If, however, the ATP concentration is increased above 100 μM, this tends to attenuate inhibition, disclosing a mixed mechanism of inhibition: the double reciprocal plot of this part of the curve reveals both increased Km and decreased Vmax values, suggesting that the binding sites for the peptide and for ATP, albeit physically separated, are functionally linked. These data are consistent with an allosteric mechanism of inhibition whereby, upon binding of the CFTR peptide, the affinity for ATP is drastically decreased. We also observe that the inhibitory efficacy of the peptide can be overcome by ATP only if the peptide concentration is much lower than its IC50 value (2.5 μM compared with 15 μM): with 10 μM peptide in fact the Vmax remains 2.5-fold lower than in its absence, showing that under these conditions the catalytic mechanism is impaired even if the ATP concentration is raised to saturating level.
Figure 4. Kinetic analysis of the mode of inhibition of CK2α activity towards substrates induced by the CFTRΔF508 peptide.

(A) The double reciprocal plot of the phosphorylation of increasing concentrations of the CK2α substrate calmodulin either in the absence (■) or in the presence (▲) of 160 μM peptide reveals no change in Km (abscissa intercept) but an approx. 3-fold decline in Vmax (ordinate intercept) induced by the peptide consistent with a non-competitive action. (B) As ATP concentrations rise, the double reciprocal plots reveal two phases. Note the abrupt increase in the slope of the double reciprocal plot induced by sub-maximal concentrations of peptide as reflected in alterations in both Vmax and Km.
Inhibition of CK2α by CFTRΔF508 peptide is abrogated by the CK2 β-subunit and by full-length NBD1
Since the inhibitory effect of the CFTR peptides was not observed when the specific peptide substrate was phosphorylated by CK2 holoenzyme [22], we reasoned that the peptide-binding site could be located on the surface of CK2α facing the β-subunit where it may no longer have been accessible once the heterotetrameric holoenzyme had formed. This point was investigated further by evaluating the inhibitory efficacy of increasing amounts of the CFTRΔF508 peptide either directly on CK2α or on CK2α previously pre-incubated with a 10-fold molar excess of β-subunit. As shown in Figure 5(A), pre-treatment with the β-subunit fully protects the α-subunit from the inhibitory effect of the CFTRΔF508 peptide. This is consistent with the idea that the CK2 α-subunit site, where the CFTRΔF508 peptide exerts its down-regulation on the catalytic subunit in isolation, is now either occupied by the β-subunit or is, in some unknown manner, no longer accessible once the β-subunit is bound to the α-subunit (note that the peptide also binds the β-subunit, see below). The interactions occurring between CK2 subunits are known from the crystal structure of CK2 holoenzyme [35], in particular, an exposed region of the upper lobe of the α-subunit is implicated in making multiple contacts with diverse elements within the β-subunits. Especially important in this respect appears to be the C-terminal segment of the β-subunit encompassing residues 182–203: a peptide reproducing this region was previously shown to interact with the N-terminal region of the α-subunit [36] and to partially mimic some of the effects of the full-size β-subunit [37]. We therefore took advantage of the same peptide to check whether it might compete against inhibition by the CFTRΔF508 peptide. As shown in Figure 5(B), the β-peptide (onset at 50 μM) by itself exerts a modest stimulation of CK2α catalytic activity as previously observed [37]. Albeit starting from a lower baseline, such a stimulatory effect is much more pronounced if the experiment is performed with CK2α pre-inhibited by the concomitant presence of the CFTRΔF508 peptide. As a result (Figure 5C), inhibition by the CFTRΔF508 peptide is significantly reduced by increasing the concentrations of the β-peptide, consistent with competition between the two peptides for the same or overlapping site(s). This permitted us to map the allosteric site to the upper lobe of CK2α, precisely to the β-strands 1 and 2. Note that the relatively modest effect of the β-peptide as compared with the full-length β-subunit closely reflects the different affinity for the α-subunit, showing a KD value of 8.45 μM and 40 nM respectively ([22] and see Supplementary Table S1 at http://www.BiochemJ.org/bj/426/bj4260019add.htm). A very high affinity of the β-subunit for the α-subunit may also account for the fact that the CFTRΔF508 peptide fails to dissociate the holoenzyme, as revealed by size-exclusion chromatography experiments revealing no detectable generation of isolated CK2 subunits from the holoenzyme treated with 100-fold molar excess of either the usual CFTRΔF508(500–518) peptide (see Supplementary Figure S1B at http://www.BiochemJ.org/bj/426/bj4260019add.htm) or a longer version of it (500–523) characterized by higher water solubility (results not shown). The observation, however, that treatment with either peptide results in broadening of the peak corresponding to the holoenzyme (compare Supplementary Figures S1A and S1B) suggests that the peptide affects the overall conformation of the tetramer, possibly weakening the inter-subunit interactions {as previously observed by SPR (surface plasmon resonance) experiments using a similar CFTRΔF508 peptide [22]}.
Figure 5. CK2α inhibition by the CFTRΔF508 peptide is counteracted by the β-subunit and by NBD1ΔF508.

CK2α activity was determined, as described in the Experimental section, against the specific synthetic peptide RRRADDSDDDDD as the phosphorylatable substrate. (A) The dose-dependent inhibitory power of the CFTRΔF508 peptide was evaluated either against CK2α alone or on CK2α pre-incubated with a 10-fold molar excess of CK2β to create the holoenzyme. Note the loss of peptide-induced inhibition when the holoenzyme is present. (B) The ability of the β-peptide(181–203) to stimulate the activity of CK2α was evaluated either in the absence (circles) or in the presence (triangles) of 40 μM CFTRΔF508 peptide. Note that the degree of inhibition of CK2α induced by the CFTRΔF508 peptide (compare zero values on abscissa) is relieved as the β-subunit concentration increases. The percentage change of the resulting decrease in inhibition is plotted in (C). (D) The inhibitory effect of increasing concentrations of the CFTRΔF508 peptide on CK2α activity was determined either in the absence or in the presence of recombinant NBD1ΔF508 full-length protein (50 μM). Controls with NBD1ΔF508 alone (in the absence of the peptide substrate) were run, whose phosphorylation was <5% of that of the peptide, and subtracted from this. Controls of CK2α activity tested either in the absence or in the presence of NBD1ΔF508 revealed no significant effect, i.e. the 100% absolute values were similar (results not shown). Thus the inhibitory effect of the short peptide towards CK2α was dramatically reduced by the full-length protein domain that by itself had no effect on the kinase activity.
Next, we determined whether full-length NBD1 competes for a peptide derived from itself. Since we have previously reported that recombinant purified full-length NBD1ΔF508 strongly interacts with CK2α [22], a pertinent question is whether binding occurs through the structural elements reproduced by the ΔF508 peptide (residues 500–518; Figure 2). To answer this question, competition experiments were designed aimed at assessing whether recombinant purified NBD1ΔF508 can overcome the inhibitory effect of a peptide reproducing its own Phe508-deleted 500–518 stretch. A technical problem in running this experiment arises from the fact that the concentrated stock of NBD1 is dissolved in a high ATP concentration (2 mM) in order to maintain its proper conformation for folding studies, a circumstance which could lead to isotopic dilution of radiolabelled ATP in the kinase assay. To circumvent this problem, the whole experiment was performed at 350 μM ATP either in the absence or presence of NBD1ΔF508. Under these conditions, phosphorylation of NBD1ΔF508 was negligible as compared with that of the peptide substrate (see legend of Figure 5); therefore substrate radiolabelling is fully accounted for by the peptide.
As shown in Figure 5(D), the addition of 50 μM NBD1ΔF508 abrogates the inhibitory efficacy of the ΔF508 peptide, consistent with the view that the protein and the peptide bind to the same CK2 site, most likely through the same structural elements considering that the Phe508 deletion is essential for both high-affinity binding of NBD1 [22] and efficient inhibition by the peptide (see Figures 1 and 2). Note that the outcome of this experiment implies that the binding of full-length CFTRΔF508 is not promoting by itself any appreciable inhibition of CK2α. This point was also independently assessed by control experiments where no significant inhibition of CK2α could be observed by raising the NBD1ΔF508 concentration up to 20 μM (results not shown), a concentration sufficient to display a protective effect against inhibition by the ΔF508 peptide. A possible explanation for the differential efficacy of the full-size protein compared with the peptide derived from it could reside in the greater rigidity of the folded NBD1ΔF508 structure [32] as opposed to the ΔF508 peptide, whose disordered structure in water solution is more prone to adopt the most favourable conformation to adapt to elements of the allosteric site which are not essential for binding but are responsible for its functional consequences. This view point was corroborated by modelling presented below.
A second binding site for the CFTRΔF508 peptide is present in the β-subunit of CK2
Although a CK2α-binding site for CFTR peptides located at the interface between the α- and the β-subunits would account for the abrogation of the inhibitory efficacy of these peptides upon holoenzyme formation (see above), it is not sufficient to explain why the very same peptides display a stimulatory effect on the CK2 holoenzyme when this is assayed with the majority of its established protein substrates [22]. A number of lines of experimental evidence support the view that such a stimulatory effect is mediated by a different CK2 region that is also able to bind the CFTR peptides, discretely located in the N-terminal domain of the β-subunit. First, it should be recalled that the β-subunit is also able to interact with NBD1 in a manner which is enhanced by the Phe508 deletion [22]. Secondly, the ΔF508 peptide is much more effective than the wild-type peptide in preventing CK2 holoenzyme autophosphorylation which takes place at the N-terminal end of the β-subunit (S2 and S3) and which is dependent on the integrity of an acidic cluster located somewhat down-stream in the same region [38]. Such an effect of the CFTR peptides (wild-type and mutant) is illustrated in Figure 6(A) using the phosphorylation of the β-subunit as a read out. Thirdly, we have shown previously [22] that stimulation of CK2 holoenzyme by the CFTRΔF508 peptide is especially evident with substrates belonging to class II, whose phosphorylation is usually prevented instead of being stimulated by the β subunit [17]. Interestingly, such a down-regulation is mediated by the same acidic cluster which is required for autophosphorylation of the β-subunit [39]. This point is exemplified in Figure 6(B) by calmodulin, a typical class II substrate, whose phosphorylation by the CK2 holoenzyme is almost negligible under basal conditions but undergoes a 30-fold enhancement upon addition of 80 μM CFTRΔF508 peptide. Note, however, that if calmodulin is replaced by the eIF2β(1–22) peptide, a class III substrate whose phosphorylation entirely relies on the β-subunit, providing thereby its N-terminal segment structural elements are important for substrate recognition in this case [25], the effect of the CFTRΔF508 peptide now becomes inhibitory (Figure 6C). Thus three different signals could potentially be generated merely by the loss of one phenylalanine residue. As in the case of down-regulation of CK2α, the efficacy on CK2 holoenzyme is also restricted to the CFTR peptides, whereas full-length NBD1ΔF508 has no appreciable effect (results not shown). The most likely explanation is the same discussed above, i.e. the greater rigidity of the NBD1 structure present, as compared with the derived peptide, hampering its adaptation to the responsive elements present on the kinase. We cannot of course exclude the possibility that the in vivo state of the NBD1 might be different and our purified preparation might not reflect that state (phosphorylation, ubiquitination, etc. as described in [40]).
Figure 6. Variable effects of the interaction of CK2 holoenzyme with the wild-type and ΔF508 CFTR-derived peptides.

Autophosphorylation of the CK2 β-subunit and assays on holoenzyme activity towards substrates (peptide and protein) were performed as described in the Experimental section. (A) Inhibition of the CK2 β-subunit autophosphorylation by increasing concentrations of either wild-type or ΔF508 CFTR peptides. (B) Stimulation of the phosphorylation of calmodulin induced by increasing concentrations of either wild-type or ΔF508 CFTR peptides (contrast with the Vmax decline in Figure 3A). Note the enhanced relative potency of the Phe508-deleted peptide towards calmodulin phosphorylation (compare open bars) when holoenzyme is present against its contrasting effects on CK2α alone. However, in (C), the very same peptide has inhibitory actions towards the phosphorylation of the eIF2β-derived peptide as the concentrations of this CFTRΔF508 peptide rise. Note no effect when eIF2β-derived peptide (class III substrate) is replaced by another peptide (class I substrate) showing selectivity of action towards different substrates.
In summary, all available data obtained with the CK2 holoenzyme can be explained assuming an interaction between the CFTRΔF508 peptide and the N-terminal tail of the β-subunit which is also responsible for autophosphorylation, down-regulation of class II substrate phosphorylation and recruitment of class III substrates. The more direct evidence that this region of the β-subunit indeed mediates the stimulatory efficacy of the CFTRΔF508 peptide was provided by experiments where polylysine was used to evoke calmodulin phosphorylation (Figure 7). The binding site for polylysine as well as for other polybasic polypeptides, which, unlike polyamines, are able to activate CK2 holoenzyme toward calmodulin and other class II substrates, has been previously mapped to the region encompassing the 55–64 acidic cluster of the β-subunit [41], suspected here to also be implicated in CFTR peptide binding. As shown in Figure 7, polylysine exerts a dramatic stimulatory effect on calmodulin phosphorylation, 20-fold more pronounced than that exerted by the CFTRΔF508 peptide. Interestingly, if both polylysine and the CFTRΔF508 peptide are present together, instead of observing an additive affect on stimulation, the phosphorylation falls 5-fold with respect to that triggered by polylysine alone, reaching a value intermediate between those observed upon separate addition of each of the two effectors. This is precisely what one would expect assuming a competition of the two variably effective stimulators (polylysine and the CFTRΔF508 peptide) for the same site, or at least their binding to two overlapping sites. Modelling further corroborates such a conclusion, as illustrated in the following section.
Figure 7. The stimulatory efficacy of polylysine on CK2 holoenzyme is reduced by the CFTRΔF peptide.
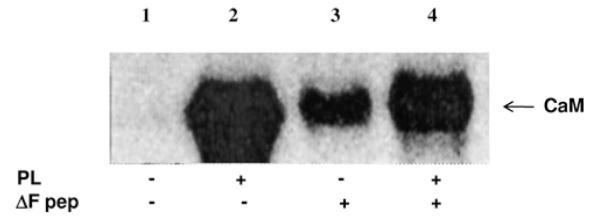
Calmodulin (CaM) underwent phosphorylation by CK2 holoenzyme in the absence (lane 1) or presence of either 330 nM polylysine (PL), 80 μM CFTRΔF peptide (lanes 2 and 3 respectively) or both (lane 4). The stimulatory effect of polylysine observed in lane 2 was attenuated by the CFTRΔF peptide (lane 4), which by itself is capable of enhancing phosphorylation of CaM (lane 3), albeit much less efficiently than polylysine. These results argue for a competition between polylysine and the CFTRΔF peptide for the same anchoring site.
Molecular modelling
To gain information about the possible modes of interaction between the CFTR peptides and CK2, the docking procedure described in the Experimental section was exploited. We first focused on the interactions with the catalytic subunit which are responsible for allosteric down-regulation of its activity (see Figure 4) and which are counteracted by the β-subunit and by a β-peptide encompassing residues 182–203 (Figures 5A and 5B). As shown in Figure 8(A), the docking procedure maps the interacting region of the α-subunit to its upper lobe where hydrophobic interactions take place between the Ile–Ile (506–507) doublet of the CFTR wild-type peptide and a small superficial cleft of α formed by Ile69, Leu41 and Phe54. Note that the same residues are also involved in the interactions with the full size β-subunit and with the β-peptide (PDB code 1DS5). In addition, a π-π stacking occurs between peptide Tyr515 and CK2α Phe54 just at the end of the glycine loop; this could lead to a weak alteration of the loop conformation resulting in slight inhibition of catalytic activity. The same hydrophobic interactions are maintained if the wild-type peptide is replaced by the CFTRΔF508 equivalent (Figure 8B): in this case, however, a couple of additional electrostatic interactions are also observed, which could account for both tighter binding and more drastic inhibitory efficacy. In fact, owing to the Phe508 deletion, the C-terminal carboxyl of the peptide now moves back approx. 4 Å, making an important electrostatic interaction with the side chain of Lys44, whereas Arg518 becomes electrostatically bonded to Glu52. Both Lys44 and Glu52 participate in the glycine loop, and their interaction with the CFTRΔF508 peptide is expected to hamper ATP binding to the loop, thus accounting for the observed increase in Km for ATP (see Figure 4B). Interestingly, the deletion of Ile507 fails to confer to the CFTR peptide the increased binding efficacy observed upon deletion of Phe508: as shown in Figure 8(C), the remaining isoleucine residue occupies a position intermediate between those occupied by the two isoleucine residues (506 and 507) in both the wild-type and the CFTRΔF508 peptides. Consequently, it is exposed towards the solvent, making contacts with two other residues of the peptide (Val510 and Tyr515) leaving unoccupied the hydrophobic α cleft which otherwise provides an important anchorage to the peptide (compare Figure 8C with Figures 8A and 8B). The behaviour of the three CFTR peptides as highlighted by molecular docking also reflects in comparable outcome of protein–protein DS (docking sampling); this parameter is a percentage value of how much the preferred peptide pose is sampled with respect to the others. Whereas with CFTRΔI507 DS is only 1.5% (only six poses out of 400 total), with CFTR wild-type and ΔF508, DS rises to 8.7% (35/400) and 23.7% (95/400) respectively.
Figure 8. Molecular docking of wild-type and mutated CFTR peptides by CK2α and by CK2 holoenzyme.

In (A), (B) and (C), the main molecular contacts between CK2α (green), CFTR peptides (red) and CK2β (blue; D only) are shown as complexes between CK2 α-subunit and CFTR peptides (wild-type, ΔF508 or ΔI507). Using Glu52/Ile69 (green) as a CK2α fixed reference points, note the different contacts for Phe54 and Leu41 (both green) against differently folded CFTR peptides in (A), (B) and (C). In (D), the interactions between the β-subunit of the CK2 holoenzyme and the CFTRΔF508 peptide are highlighted. Dotted red areas denote hydrophobic interactions, blue dotted lines indicate salt bridges. The DS parameter for each complex, calculated as described in the Experimental section and discussed in the text, is also shown.
We next moved to the CK2 holoenzyme with an aim to map the site responsible for up-regulation of its activity towards the majority of its substrates (with special reference to Class II subtype) and for inhibition of autophosphorylation. In agreement with the expectation based on biochemical data (Figures 6A–6C), the protein–protein docking strategy has localized this site on the N-terminal region of the β-subunit (see Figure 8D). In this region, a series of water-exposed residues encompassing amino acids 33–61 define a particular surface characterized by a very small hydrophobic motif generated by Phe34, Leu36 and Leu39 and an extended highly charged platform including Lys33, Arg47, Asp50, Glu57, Asp59, Glu60 and Glu61. All of these residues display a striking complementarity with the CFTR peptide sequences. In particular, the CFTRΔF508 peptide uses its Ile–Ile 506–507 doublet to make a hydrophobic contact with the β-subunit residues Phe34 and Leu36: this is followed by a strong electrostatic network involving Asp513 and Glu514 (bonded to Lys33 and Arg47 respectively) and Arg516/Arg518 interacting with Glu57 and Glu61. These interactions explain the complexity of the biochemical data shown in Figures 6(A)–6(C) since, remarkably, β-subunit residues engaged with the CFTRΔF508 peptide are those previously demonstrated to be responsible for autophosphorylation, down-regulation of Class II substrate phosphorylation and recruitment of Class III substrates. Interestingly, and, once more in agreement with experimental data, the interactions with the β-subunit are partially lost if the CFTRΔF508 peptide is replaced by the wild-type one: insertion of Phe508 in fact modifies the electrostatic network downstream, causing the failure of Glu514 and Arg518 to interact with Lys33 and with Glu61 respectively. Also consistent with the modelling is the loss of stimulatory activity caused by the deletion of the C-terminal doublet (Tyr517–Arg518).
DISCUSSION
The most disconcerting feature of CK2 probably comprises its lack of stringent regulation. Although the CK2 holoenzyme is endowed with two non-catalytic β-subunits, these do not seem to act as sensu stricto regulatory elements since they are not required to either turn on [as cyclins do with CDKs (cyclin-dependent kinases)] or turn off [as R subunits do with PKA (protein kinase A)] the catalytic subunits. Furthermore, as pointed out by Raaf et al. [18], in all of the numerous crystal structures of CK2 α-subunit solved thus far, either alone or in combination with the β-subunits and with a variety of ligands, are invariably in the active conformation.
The β-subunits, on the other hand, besides providing a docking platform for interacting proteins and modulators [42], also affect the stability of the kinase and, more importantly, control its targeting of protein substrates, which, accordingly, have been assigned to three classes [18]: class I includes the majority of substrates, whose phosphorylation is efficiently performed irrespective of the presence of β-subunits; class II is composed of substrates, exemplified by calmodulin, whose phosphorylation is drastically inhibited upon formation of the holoenzyme; whereas substrates belonging to class III are those, such as eIF2β, whose phosphorylation is conversely crucially dependent on the β-subunit, being readily performed by the holoenzyme, but never by the isolated catalytic subunits. It has been proposed that these very different behaviours are at least partially accounted for on one hand by the potential of the β-subunit to act as a pseudo-substrate inhibitor by virtue of its highly acidic N-terminal segment also harbouring an autophosphorylation site at Ser2 and Ser3 [38,43], yet on the other by the requirement of the β-subunit to generate the binding site for a limited number of substrates which are not effectively recognized by the catalytic subunit alone [25,44].
Whether in vivo the formation of CK2 holoenzyme is a reversible reaction or not is still an open question [7]. On one hand, the very high-affinity binding between catalytic and non-catalytic subunits (KD = 40 nM) [22] argues against dissociation of the holoenzyme under physiological conditions; on the other, it has been recently shown that an exchange between assembled and ‘free’ subunits can take place, albeit very slowly [45]. In any case, the existence in cells of CK2 catalytic subunits not combined with the β-subunits has been reported from time to time (e.g. [46-48]). Especially pertinent to the outcome of our work is the striking abundance of CK2α not accompanied by a concomitant rise in CK2β observed in CEM drug-resistant cells (R-CEM) as compared with CEM drug-sensitive (S-CEM) cells [49], given that in this case the elevated CK2α was not paralleled by any increase in catalytic activity measured with the specific peptide substrate. This suggests that, in R-CEM cells, CK2α is in an inactive form.
Interestingly it has been recently shown that a flexible region of CK2α exists at the interface with the β-subunit [20], which could be induced to adopt an inactive conformation, should there be a ligand able to bind to it instead of to the β-subunit [18]. This region overlaps fairly well the allosteric site where, based on the biochemical data and molecular docking analysis reported in the present study, we propose that the binding of the CFTRΔF508 peptide takes place, inducing or possibly stabilizing an inactive conformation of the catalytic subunit (see Figure 8). According to our modelling, inactivation is mediated by a displacement of the glycine loop due to interactions with the C-terminal residues of the CFTRΔF508 peptide (see Figure 8 and comments in the text). These interactions are much weaker with the wild-type CFTR peptide and with the ΔI507 equivalent, thus accounting for failure of these peptides to inhibit CK2α as drastically as the CFTRΔF508 peptide (see Figure 3).
Collectively, our data are consistent with the scenario depicted in Figure 9, where in the presence of CFTR fragments bearing the Phe508 deletion, newly synthesized CK2α is sequestered in an inactive complex less prone to readily interact with the β-subunits. There is abundant evidence that CFTR degrades in the proteasome when Phe508 is deleted, but the same fate is reported to a lesser degree for wild-type CFTR as well [40].
Figure 9. Schematic illustration of the proposed dual effect of CFTR fragments on CK2α and on CK2 holoenzyme.
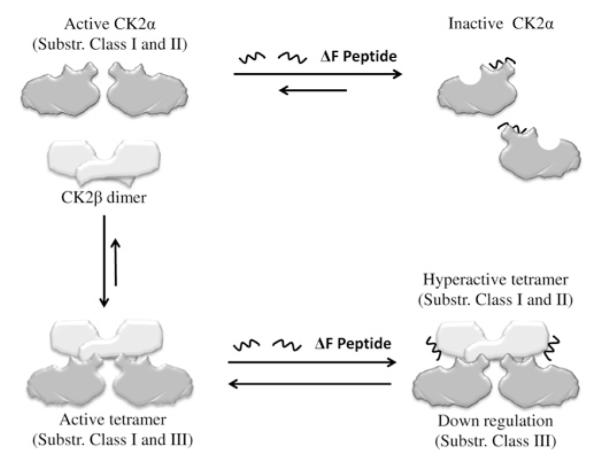
The binding of CFTRΔF508 peptides to the CK2 α-subunit allosteric site disclosed by Figure 3 locks CK2α in an inactive conformation and hampers the formation of the active holoenzyme (new steady state based on competition between CFTR peptides and CK2β outlined in Figure 4). The effect of binding of CFTRΔF508 peptides to the β-subunit within the CK2 holoenzyme is variable depending on the class of substrate affected (see Figure 5), but with most substrates is expected to induce activation [22]. In this model, as concentrations of the CFTR peptide with Phe508 deleted rise, calmodulin, and more in general class II substrates, would be hyper-phosphorylated whereas phosphorylation of eIF2β and other class III targets would be inhibited.
To note that, in this manner, the CFTRΔF508 fragments not only promote allosteric inhibition of ‘free’ CK2α, immediately resulting in down-regulation of class II substrates, but will also induce, by subtracting α from the 2α+β2↔α2β2 equilibrium, a gradual decrease in CK2 holenzyme, expected to reduce the phosphorylation of the remaining targets of CK2 as well. This latter effect, however, could be overcome, or at least attenuated, by the binding of the CFTRΔF508 peptide to its second allosteric site highlighted in the present study, located at the N-terminus of CK2β, whose occupancy results in the up-regulation of CK2 holoenzyme toward all its substrates [22], with the exception of those belonging to class III (e.g. the eIF2β peptide, see Figure 6C). Thus CK2 activity could be fine tuned.
Assuming that a mechanism similar to that schematically depicted in Figure 9 is operating in R-CEM cells (regardless of the nature of the allosteric effector responsible therein, see [50] for possible candidates), this could account for the puzzling observation that in those cells, despite the fact that the increase in CK2α was not accompanied by any increase in activity toward the specific peptide substrate, nevertheless the phosphorylation of some endogenous targets of CK2 was significantly enhanced [49].
Our data from the present study may also provide a breakthrough toward the understanding of some enigmatic aspects of CF pathology, which are difficult to reconcile with the loss of function of CFTR, notably alterations in the inflammatory responses and a 6-fold excess of bowel cancer in young patients, some of whom are in their teenage years [51]. Considering the pleiotropy of CK2 and its implication in pathological events such as inflammation, infections and tumorigenesis [15], the hypothesis that some of the derangements observed in CF pathology are caused by CK2 through the generation of biologically active CFTR peptides will deserve attention in future experiments given that CFTR is rapidly degraded by the proteasome, which in turn predicts 8–12 mer peptides of varying composition based on its known catalytic properties. Pertinent to this may be the results obtained with peptides, including the ΔI507 deletion. This deletion, otherwise very similar to the ΔF508 one as far as its consequences on the stability of CFTR is concerned [28], nevertheless sharply differentiates in its population prevalence which is negligible compared with ΔF508 [29]. This observation suggests that the ΔI507 mutation is unable to provide the selective advantage of the ΔF508 mutation (note this only pertains to heterogygotes), although both promote the same loss of function of CFTR in a CF disease context where loss of ion channel function plays a critical role. Our data are consistent with the hypothesis that the selective advantage of the CFTRΔF508 mutation in heterozygotes is due to a gain, rather than a loss, of function. Furthermore, that deregulation of CK2 mediates this effect is corroborated by our data showing that the CFTRΔI507 peptide is unable to exert the same efficacy of the CFTRΔF508 peptide on CK2 activity.
We are aware that for the time being a serious limit to our working hypothesis arises from the paucity of information about the endogenous generation and actual concentration in CF cells of CFTRΔF508 fragments of the right size and composition, that might equate to our in vitro work with their synthetic derivatives. Yet it is now almost axiomatic that approximately two-thirds of newly synthesized wild-type CFTR is proteasomally degraded, a figure which rises to over 90% for the phenylalanine-deleted mutant [40]. Interestingly, this apparently wanton destruction does not occur with other ABC proteins such as P-glycoprotein, which is efficiently processed without degradation, suggesting that CFTR is somehow a special case. For example, CFTR is even targeted for destruction through ubiquitination prior to its complete synthesis on the ribosome [40].
Evidence is now available that the same CFTR peptides used in this in vitro study, when expressed by viral infection in a fetal animal model, do display biological effects, notably enhancement of Wnt reporter gene expression, which may well be mediated by CK2 [23]. Other caveats arise from the extremely variable dynamics of CFTR expression (and degradation), with peaks of 50-fold more mRNA found in fetal life compared with adult lung [52], leaving open the possibility that under special circumstances the proposed degraded peptide concentration might be higher than expected. We must also consider the crucial importance that the conformation of such peptides appears to have in order to display biological activity toward CK2. This latter point is highlighted by the failure of the very same 500–518 sequence, which in the peptide is quite effective, to display any similar efficacy on both the CK2 α-subunit and CK2 holoenzyme once it is incorporated into the whole NBD1 domain and by a number of unpublished observations (by O. Marin, M.A. Pagano, F. Meggio and L.A. Pinna) undertaken in our laboratories showing that, by just changing the method of solubilization of the peptides (e.g. DMSO compared with water), their efficacy on CK2 sometimes is dramatically impaired.
In his wide-ranging review, one of the founding fathers of the CFTR field, John Riordan [40], recently commented that it remains controversial as to whether CF really is a pleiotropic disease. It is clear that strong views are held on either side of the debate (CFTR as a channelopathy, CFTR in the innate immune system, etc.). Our aim is to provide a testable route to measure the degree of CF pleiotropy and our future efforts will be directed to assess the presence of these peptides and to monitor corresponding alterations in CK2 activity and targeting in cells where these peptides may well be generated. Regardless of the outcome of these future studies, it should be underlined that our present data substantially contribute to the definition of allosteric sites where cellular ligands other than CFTR peptides may also bind to modulate CK2 activity. Pertinent to this may be the intriguing observation that the sequence surrounding the Phe508 deletion in CFTR is fairly well conserved in a number of proteins (G. Cozza, M.A. Pageno, F. Meggio and L.A. Pinna, unpublished work), with special reference to some histones which stimulate CK2 holoenzyme by binding to the N-terminal region of the β-subunit [41]. It should be highlighted in particular that histone H4, which has been shown to be a powerful stimulator of the CK2 holoenzyme [41], shares remarkable similarity with the CFTRΔF508 peptide (see Supplementary Figure S2 at http://www.BiochemJ.org/bj/426/bj4260019add.htm), whereas this is not the case with respect to histone H1 whose efficacy on CK2 holoenzyme activity was found to be negligible [41]. This discloses the possibility that a number of protein and peptide ligands sharing the motif found around the deleted Phe508 of CFTR could function as modulators of CK2 activity.
Supplementary Material
ACKNOWLEDGEMENTS
We thank the Molecular Modelling Section (MMS) co-ordinated by Professor S. Moro.
FUNDING This work was supported by the Italian Cystic Fibrosis Research Foundation [grant number FFC#4/2007] with the contribution of Banca Popolare di Verona e Novara and Fondazione Giorgio Zanotto, by the Associazione Italiana per la Ricerca sul Cancro (AIRC) and by the European Commission [grant number PRO-KINASERESEARCH 503467 (to L.A.P.)] and by the Wellcome Trust [grant number 069150/z/02/z (to A.M.)]. We are grateful to the Wellcome Trust for ongoing support to the Pinna and Mehta laboratories.
Abbreviations used
- CF
cystic fibrosis
- CFTR
cystic fibrosis transmembrane regulator
- CFTRΔF508
CFTR with Phe508 deleted
- CFTRΔI507
CFTR with Ile507 deleted
- DS
docking sampling
- eIF2β
eukaryotic initiation factor 2β
- NBD1
nucleotide-binding domain 1 of CFTR
- R-CEM
CEM drug-resistant cells.
REFERENCES
- 1.Olsen JV, Blagoev B, Gnad F, Macek B, Kumar C, Mortensen P, Mann M. Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell. 2006;127:635–648. doi: 10.1016/j.cell.2006.09.026. [DOI] [PubMed] [Google Scholar]
- 2.Villén J, Beausoleil SA, Gerber SA, Gygi SP. Large-scale phosphorylation analysis of mouse liver. Proc. Natl. Acad. Sci. U.S.A. 2007;104:1488–1493. doi: 10.1073/pnas.0609836104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 4.Meggio F, Pinna LA. One-thousand-and-one substrates of protein kinase CK2? FASEB J. 2003;17:349–368. doi: 10.1096/fj.02-0473rev. [DOI] [PubMed] [Google Scholar]
- 5.Salvi M, Sarno S, Cesaro L, Nakamura H, Pinna LA. Extraordinary pleiotropy of protein kinase CK2 revealed by weblogo phosphoproteome analysis. Biochim. Biophys. Acta Mol. Cell Res. 2009;1793:847–859. doi: 10.1016/j.bbamcr.2009.01.013. [DOI] [PubMed] [Google Scholar]
- 6.Pinna LA. A historical view of protein kinase CK2. Cell. Mol. Biol. Res. 1994;40:383–390. [PubMed] [Google Scholar]
- 7.Litchfield DW. Protein kinase CK2: structure, regulation and role in cellular decisions of life and death. Biochem. J. 2003;369:1–15. doi: 10.1042/BJ20021469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ahmed K, Gerber DA, Cochet C. Joining the cell survival squad: an emerging role for protein kinase CK2. Trends Cell Biol. 2002;12:226–230. doi: 10.1016/s0962-8924(02)02279-1. [DOI] [PubMed] [Google Scholar]
- 9.Dominguez I, Sonenshein GE, Seldin DC. Protein kinase CK2 in health and disease: CK2 and its role in Wnt and NF-κB signalling: linking development and cancer. Cell. Mol. Life Sci. 2009;66:1850–1857. doi: 10.1007/s00018-009-9153-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lou DY, Dominguez I, Toselli P, Landesman-Bollag E, O’Brien C, Seldin DC. The α catalytic subunit of protein kinase CK2 is required for mouse embryonic development. Mol. Cell. Biol. 2008;28:131–139. doi: 10.1128/MCB.01119-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Seldin DC, Leder P. Casein kinase IIα transgene-induced murine lymphoma: relation to theileriosis in cattle. Science. 1995;267:894–897. doi: 10.1126/science.7846532. [DOI] [PubMed] [Google Scholar]
- 12.Kelliher MA, Seldin DC, Leder P. Tal-1 induces T cell acute lymphoblastic leukemia accelerated by casein kinase IIα. EMBO J. 1996;15:5160–5166. [PMC free article] [PubMed] [Google Scholar]
- 13.Landesman-Bollag E, Channavajhala PL, Cardiff RD, Seldin DC. P53 deficiency and misexpression of protein kinase CK2α collaborate in the development of thymic lymphomas in mice. Oncogene. 1998;16:2965–2974. doi: 10.1038/sj.onc.1201854. [DOI] [PubMed] [Google Scholar]
- 14.Landesman-Bollag E, Romieu-Mourez R, Song DH, Sonenshein GE, Cardiff RD, Seldin DC. Protein kinase CK2 in mammary gland tumorigenesis. Oncogene. 2001;20:3247–3257. doi: 10.1038/sj.onc.1204411. [DOI] [PubMed] [Google Scholar]
- 15.Guerra B, Issinger O-G. Protein kinase CK2 in human diseases. Curr. Med. Chem. 2008;15:1870–1886. doi: 10.2174/092986708785132933. [DOI] [PubMed] [Google Scholar]
- 16.Pinna LA. The raison d’etre of constitutively active protein kinases: the lesson of CK2. Acc. Chem. Res. 2003;36:378–384. doi: 10.1021/ar020164f. [DOI] [PubMed] [Google Scholar]
- 17.Pinna LA. Protein kinase CK2: a challenge to canons. J. Cell Sci. 2002;115:3873–3878. doi: 10.1242/jcs.00074. [DOI] [PubMed] [Google Scholar]
- 18.Raaf J, Issinger OG, Niefind K. First inactive conformation of CK2α, the catalytic subunit of protein kinase CK2. J. Mol. Biol. 2009;386:1212–1221. doi: 10.1016/j.jmb.2009.01.033. [DOI] [PubMed] [Google Scholar]
- 19.Niefind K, Raaf J, Issinger OG. Protein kinase CK2 in health and disease. Protein kinase CK2: from structures to insights. Cell. Mol. Life Sci. 2009;66:1800–1816. doi: 10.1007/s00018-009-9149-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Raaf J, Brunstein E, Issinger O-G, Niefind K. The CK2α/CK2β interface of human protein kinase CK2 harbors a binding pocket for small molecules. Chem. Biol. 2008;15:111–117. doi: 10.1016/j.chembiol.2007.12.012. [DOI] [PubMed] [Google Scholar]
- 21.Prudent R, Cochet C. New protein kinase CK2 inhibitors: jumping out of the catalytic box. Chem. Biol. 2009;16:112–120. doi: 10.1016/j.chembiol.2009.01.004. [DOI] [PubMed] [Google Scholar]
- 22.Pagano MA, Arrigoni G, Marin O, Sarno S, Meggio F, Treharne KJ, Mehta A, Pinna LA. Modulation of protein kinase CK2 activity by fragments of CFTR encompassing F508 may reflect functional links with cystic fibrosis pathogenesis. Biochemistry. 2008;47:7925–7936. doi: 10.1021/bi800316z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cohen JC, Killeen E, Chander A, Takemaru K, Larson JE, Treharne KJ, Mehta A. Small interfering peptide (siP) for in vivo examination of the developing lung interactonome. Dev. Dyn. 2009;238:386–393. doi: 10.1002/dvdy.21834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sarno S, Vaglio P, Meggio F, Issinger O-G, Pinna LA. Protein kinase CK2 mutants defective in substrate recognition. Purification and kinetic analysis. J. Biol. Chem. 1996;271:10595–10601. doi: 10.1074/jbc.271.18.10595. [DOI] [PubMed] [Google Scholar]
- 25.Poletto G, Vilardell J, Marin O, Pagano MA, Cozza G, Sarno S, Falques A, Itarte E, Pinna LA, Meggio F. The regulatory β subunit of protein kinase CK2 contributes to the recognition of the substrate consensus sequence. A study with an eIF2β-derived peptide. Biochemistry. 2008;47:8317–8325. doi: 10.1021/bi800216d. [DOI] [PubMed] [Google Scholar]
- 26.Reference deleted
- 27.Cozza G, Moro S, Gotte G. Elucidation of the ribonuclease A aggregation process mediated by 3D domain swapping: a computational approach reveals possible new multimeric structures. Biopolymers. 2008;89:26–39. doi: 10.1002/bip.20833. [DOI] [PubMed] [Google Scholar]
- 28.Kerem BS, Zielenski J, Markiewicz D, Bozon D, Gazit E, Yahav J, Kennedy D, Riordan JR, Collins FS, Rommens JM, Tsui LC. Identification of mutations in regions corresponding to the two putative nucleotide (ATP)-binding folds of the cystic fibrosis gene. Proc. Natl. Acad. Sci. U.S.A. 1990;87:8447–8451. doi: 10.1073/pnas.87.21.8447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bobadilla JL, Macek M, Jr, Fine JP, Farrell PM. Cystic fibrosis: a worldwide analysis of CFTR mutations - correlation with incidence data and application to screening. Hum. Mut. 2002;19:575–606. doi: 10.1002/humu.10041. [DOI] [PubMed] [Google Scholar]
- 30.Wang Y, Loo TW, Bartlett MC, Clarke DM. Correctors promote maturation of cystic fibrosis transmembrane conductance regulator (CFTR)-processing mutants by binding to the protein. J. Biol. Chem. 2007;282:33247–33251. doi: 10.1074/jbc.C700175200. [DOI] [PubMed] [Google Scholar]
- 31.Treharne KJ, Xu Z, Chen JH, Best OG, Cassidy DM, Gruenert DC, Hegyi P, Gray MA, Sheppard DN, Kunzelmann K, Mehta A. Inhibition of protein kinase CK2 closes the CFTR Cl- channel, but has no effect on the cystic fibrosis mutant ΔF508-CFTR. Cell. Physiol. Biochem. 2009;24:347–360. doi: 10.1159/000257427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lewis HA, Buchanan SG, Burley SK, Conners K, Dickey M, Dorwart M, Fowler R, Gao X, Guggino WB, Hendrickson WA, et al. Structure of nucleotide-binding domain 1 of the cystic fibrosis transmembrane conductance regulator. EMBO J. 2004;23:282–293. doi: 10.1038/sj.emboj.7600040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pinna LA, Ruzzene M. How do protein kinases recognize their substrates? Biochim. Biophys. Acta. 1996;1314:191–225. doi: 10.1016/s0167-4889(96)00083-3. [DOI] [PubMed] [Google Scholar]
- 34.Pinna LA. Protein kinase CK2: an overview, European Cystic Fibrosis Society and The Physiological Society Joint Conference; Carvoeiro, Portugal. 19–23 April 2006; 2006. Abstract S6.1. [Google Scholar]
- 35.Niefind K, Guerra B, Ermakowa I, Issinger O-G. Crystal structure of human protein kinase CK2: insights into basic properties of the CK2 holoenzyme. EMBO J. 2001;20:5320–5331. doi: 10.1093/emboj/20.19.5320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Battistutta R, Sarno S, De Moliner E, Marin O, Issinger O-G, Zanotti G, Pinna LA. The crystal structure of the complex of Zea mays α subunit with a fragment of human β subunit provides the clue to the architecture of protein kinase CK2 holoenzyme. Eur. J. Biochem. 2000;267:5184–5190. doi: 10.1046/j.1432-1327.2000.01587.x. [DOI] [PubMed] [Google Scholar]
- 37.Sarno S, Marin O, Boschetti M, Pagano MA, Meggio F, Pinna LA. Cooperative modulation of protein kinase CK2 by separate domains of its regulatory β-subunit. Biochemistry. 2000;39:12324–12329. doi: 10.1021/bi0011431. [DOI] [PubMed] [Google Scholar]
- 38.Boldyreff B, Meggio F, Pinna LA, Issinger O-G. Efficient autophosphorylation and phosphorylation of the β-subunit by casein kinase-2 require the integrity of an acidic cluster 50 residues downstream from the phosphoacceptor site. J. Biol. Chem. 1994;269:4827–4831. [PubMed] [Google Scholar]
- 39.Sarno S, Vaglio P, Marin O, Meggio F, Issinger O-G, Pinna LA. Basic residues in the 74–83 and 191–198 segments of protein kinase CK2 catalytic subunit are implicated in negative but not in positive regulation by the β-subunit. Eur. J. Biochem. 1997;248:290–295. doi: 10.1111/j.1432-1033.1997.00290.x. [DOI] [PubMed] [Google Scholar]
- 40.Riordan JR. CFTR function and prospects for therapy. Annu. Rev. Biochem. 2008;77:701–726. doi: 10.1146/annurev.biochem.75.103004.142532. [DOI] [PubMed] [Google Scholar]
- 41.Meggio F, Boldyreff B, Issinger OG, Pinna LA. Casein kinase 2 down-regulation and activation by polybasic peptides are mediated by acidic residues in the 55–64 region of the β-subunit. A study with calmodulin as phosphorylatable substrate. Biochemistry. 1994;33:4336–4442. doi: 10.1021/bi00180a030. [DOI] [PubMed] [Google Scholar]
- 42.Bibby AC, Litchfield DW. The multiple personalities of the regulatory subunit of protein kinase CK2: CK2 dependent and CK2 independent roles reveal a secret identity for CK2β. Int. J. Biol. 2005;1:67–79. doi: 10.7150/ijbs.1.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Boldyreff B, Meggio F, Pinna LA, Issinger OG. Reconstitution of normal and hyperactivated forms of casein kinase-2 by variably mutated β-subunits. Biochemistry. 1993;32:12672–12677. doi: 10.1021/bi00210a016. [DOI] [PubMed] [Google Scholar]
- 44.Marin O, Sarno S, Boschetti M, Pagano MA, Meggio F, Ciminale V, D’Agostino DM, Pinna LA. Unique features of HIV-1 Rev protein phosphorylation by protein kinase CK2 (‘casein kinase-2’) FEBS Lett. 2000;481:63–67. doi: 10.1016/s0014-5793(00)01971-2. [DOI] [PubMed] [Google Scholar]
- 45.Laudet B, Moucadel V, Prudent R, Filhol O, Wong YS, Royer D, Cochet C. Identification of chemical inhibitors of protein-kinase CK2 subunit interaction. Mol. Cell. Biochem. 2008;316:63–69. doi: 10.1007/s11010-008-9821-6. [DOI] [PubMed] [Google Scholar]
- 46.Stalter G, Siemer S, Becht E, Ziegler M, Remberger K, Issinger OG. Asymmetric expression of protein kinase CK2 subunits in human kidney tumors. Biochim. Biophys. Res. Commun. 1994;202:141–147. doi: 10.1006/bbrc.1994.1904. [DOI] [PubMed] [Google Scholar]
- 47.Lüscher B, Litchfield DW. Biosynthesis of casein kinase II in lymphoid cell lines. Eur. J. Biochem. 1994;220:521–526. doi: 10.1111/j.1432-1033.1994.tb18651.x. [DOI] [PubMed] [Google Scholar]
- 48.Guerra B, Siemer S, Boldyreff B, Issinger OG. Protein kinase CK2: evidence for a protein kinase CK2β subunit fraction, devoid of the catalytic CK2α subunit, in mouse brain and testicles. FEBS Lett. 1999;462:353–357. doi: 10.1016/s0014-5793(99)01553-7. [DOI] [PubMed] [Google Scholar]
- 49.Di Maira G, Brustolon F, Bertacchini J, Tosoni K, Marmiroli S, Pinna LA, Ruzzene M. Pharmacological inhibition of protein kinase CK2 reverts the multidrug resistance phenotype of a CEM cell line characterized by high CK2 level. Oncogene. 2007;26:6915–6926. doi: 10.1038/sj.onc.1210495. [DOI] [PubMed] [Google Scholar]
- 50.Treharne KJ, Cassidy D, Goddard C, Colledge WH, Cassidy A, Mehta A. Epithelial IgG and its relationship to the loss of F508 in the common mutant form of the cystic fibrosis transmembrane conductance regulator. FEBS Lett. 2009;583:2493–2499. doi: 10.1016/j.febslet.2009.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mehta A. Cystic fibrosis as a bowel cancer syndrome and the potential role of CK2. Mol. Cell. Biochem. 2008;316:169–175. doi: 10.1007/s11010-008-9815-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Broackes-Carter FC, Mouchel N, Gill D, Hyde S, Bassett J, Harris A. Temporal regulation of CFTR expression during ovine lung development: implications for CF gene therapy. Hum. Mol. Genet. 2002;11:125–131. doi: 10.1093/hmg/11.2.125. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.