

Abstract
The SNF2 family of ATPases acts in the context of chromatin to regulate transcription, replication, repair and recombination. Defects in SNF2 genes cause many human diseases. For example, mutations in SMARCAL1 (also named HARP) cause Schimke immuno-osseous dysplasia (SIOD); a multi-system disorder characterized by growth defects, immune deficiencies, renal failure and other complex phenotypes. Several groups including ours recently identified SMARCAL1 as a replication stress response protein. Importantly, SMARCAL1 localizes to stalled replication forks and this localization of SMARCAL1 activity prevents DNA damage accumulation during DNA replication. We determined that SIOD-related SMARCAL1 mutants could not prevent replication-associated DNA damage in cells in which endogenous SMARCAL1 was silenced, establishing the first link between SIOD and a defect in a specific biological activity. Here, we also report that cells from patients with SIOD exhibit elevated levels of DNA damage that can be rescued by re-introduction of wild-type SMARCAL1. Our data suggest that loss of SMARCAL1 function in patients may cause DNA replication-associated genome instability that contributes to the pleiotropic phenotypes of SIOD.
Key words: SMARCAL1, HARP, DNA replication, replication fork, RPA, DNA damage response, checkpoint, ATR, ATM, DNA-PK
Introduction
Genome maintenance defects cause cancer, premature aging and neurodegenerative diseases.1–4 DNA is a reactive chemical and is constantly subject to potentially deleterious lesions like DNA crosslinks, strand breaks and base modifications from both endogenous and exogenous sources. These lesions are particularly problematic during DNA replication, challenging the accurate duplication of the genome. The DNA damage response is a signal transduction pathway that recognizes these lesions and activates cellular programs that promote DNA repair, cell cycle arrest, and in cases where the damage burden is high, apoptosis. The replication stress response is a subset of the DNA damage response dedicated to preserving genome integrity during DNA replication. Several groups including ours recently defined SMARCAL1 as a new component of the replication stress response.5–9
SMARCAL1 (SWI/SNF-related, matrix associated, actin-dependent regulator of chromatin, subfamily A-like 1), also known as HARP (Hep-A related protein), is a member of the SNF2 family of proteins. Most SNF2 proteins act as chromatin remodelers within multi-protein complexes. Using the energy from ATP hydrolysis, they rearrange nucleosomes to facilitate gene regulation, replication, recombination and DNA repair. All SNF2 proteins contain seven motifs that are similar to those found in helicases which bind and hydrolyze ATP.10 SNF2 family members diverge in the regions flanking the SNF2 domain.11 These regions commonly dictate the functional specificity of the protein. For example, protein-protein interactions in the N-terminus of the SNF2 protein INO80 direct localization of INO80 to sites of double-stranded DNA breaks (DSB) where it functions to remodel nucleosomes and promote the recruitment of DNA processing factors.12
SMARCAL1 has an in vitro enzymatic activity distinct from other SNF2 family members. SMARCAL1 reanneals replication protein A (RPA)-stabilized, ssDNA bubbles in plasmid DNA.13 This annealing helicase activity depends on ATP hydrolysis. Where and when this activity is needed in cells had not been described until several recent reports placed SMARCAL1 at stalled replication forks. Combined with additional genetic data these results suggest that this activity is important at stalled replication forks to maintain genome integrity.
Identification of SMARCAL1 as a Replication Stress Response Protein
We identified SMARCAL1 in two functional genetic screens designed to identify novel genome maintenance activities in human cells.14 The rationale was as follows: deregulation of a genome maintenance activity by either RNAi depletion or cDNA overexpression will result in an increase in spontaneous DNA damage without the addition of genotoxic agents; the increase in DNA damage burden can be monitored by detecting the phosphorylation status of DNA damage response substrates. Both SMARCAL1 silencing and overexpression activate the DNA damage response specifically in replicating cells.5 In addition, silencing SMARCAL1 renders cells hypersensitive to replication stress agents like hydroxyurea (HU), camptothecin and aphidicolin.5,6 These data suggest that SMARCAL1 activity is critical for genome maintenance during DNA replication.
When DNA lesions persist or occur during S phase, they can cause uncoupling of helicase and polymerase activities due to polymerase stalling.15 This functional uncoupling generates significant amounts of single-stranded DNA (ssDNA) as the helicase continues to unwind double-stranded DNA. The ssDNA binding protein, replication protein A (RPA), coats the DNA and recruits multiple replication stress response proteins to the stalled replication fork.16,17 Following replication stress, SMARCAL1 localizes to sites of stalled replication, co-localizing with RPA and other markers of stalled forks.5–7,9 The N-terminus of SMARCAL1 has sequence similarity to an RPA-binding region in the replication stress response protein TIPIN (Timeless interacting protein).18 Similar to TIPIN, this region of SMARCAL1 interacts with the 32 kDa subunit of RPA (RPA2).5,6 The RPA-interaction domain of SMARCAL1 is both necessary and sufficient to localize SMARCAL1 to stalled replication forks.5 Importantly, a SMARCAL1 mutant (ΔN) that does not bind RPA cannot rescue the replication-associated DNA damage observed in SMARCAL1-deficient cells.5,9 The ΔN-SMARCAL1 mutant still binds forked DNA in vitro and retains DNA stimulated ATPase activity and annealing helicase activity.5,13 Although the RPA interaction is not essential for SMARCAL1 enzymatic activities in vitro, the genome maintenance activities of SMARCAL1 in cells depend upon its interaction with RPA. It is likely that RPA functions to localize SMARCAL1 to its substrate at the replication fork.
If SMARCAL1 functions as an annealing helicase at stalled forks then we would expect to observe more ssDNA in SMARCAL1-silenced cells. Indeed, silencing SMARCAL1 leads to an increase in ssDNA especially in the presence of replication stress.5,9 Furthermore, SMARCAL1-silenced cells recover more slowly after resolution of the stalled fork since damage-dependent RPA phosphorylation persists in SMARCAL1-silenced cells after removing replication stress-inducing agents from the growth media,5 and fiber labeling indicates replication fork restart is slower.6 These results are consistent with a model in which SMARCAL1 anneals ssDNA at stalled forks, reduces the amount of ssDNA available for RPA binding, and promotes fork stability.
Why would cells require ssDNA annealing at stalled forks? The DNA at a replication fork is nucleosome-free and may have an increased propensity to form bubbles. Furthermore the 3′ and 5′ ends of nascent DNA strands could unwind from the template and form flaps especially during the process of polymerase switching at a DNA lesion. SMARCAL1 could function to reduce bubble and flap formation, an activity that would prevent inappropriate processing of the DNA. SMARCAL1 could also limit uncoupling of polymerase and the MCM helicase functioning as an anti-helicase directly at the fork to reduce ssDNA. Certainly other models are possible19 so further analysis of SMARCAL1 biochemical activities including the identification of its substrates will be essential to fully understand its genome maintenance activity.
Regulation of SMARCAL1
SMARCAL1 activity is likely highly regulated. Our data indicate that too much SMARCAL1 activity is equally challenging to cells as too little. Overexpression causes increased H2AX phosphorylation and damage signaling.5 One method of regulation is through phosphorylation by the phosphoinositide three-kinase-related kinase (PIKK) family of kinases including ataxia-telangiectasia mutated (ATM), ATM and Rad3-related (ATR), and DNA-dependent protein kinase (DNA-PK). In vitro, SMARCAL1 is a substrate of ATM, ATR and DNA-PK and all three contribute to SMARCAL1 phosphorylation in vivo following replication stress.5 The damage-induced phosphorylation of SMARCAL1 may be a mechanism by which the replication stress response regulates SMARCAL1 activity at replication forks.
Regulatory phosphorylation of other SNF2 family members has a variety of outcomes. Cyclin dependent kinase (CDK) phosphorylation of the SNF2 family protein, PICH (Plk1 interacting checkpoint helicase) is required to expose a Plk1 binding site in PICH and localize it to the centromere where it functions in the spindle checkpoint.20 Phosphorylation of SMARCAL1 could promote or limit its activity at the fork. Thus, identifying and functionally characterizing phosphorylation sites in SMARCAL1 will be important for understanding SMARCAL1 regulation.
SMARCAL1 and SIOD
Loss of SNF2-related activities can cause human diseases, including cancer. Mutations in the SNF2 family member ERCC6 cause Cockayne's syndrome type B, a genome instability syndrome characterized by dwarfism, mental retardation, and UV light sensitivity.21 Loss of function mutations in SMARCAL1 cause Schimke immuno-osseous dysplasia (SIOD).22 SIOD clinical phenotypes include growth retardation, renal failure, recurrent infections, cerebral ischemia, and altered skin pigmentation beginning in early childhood. Disease severity varies with some patients experiencing in utero onset and death within 5 years, while others have a later onset of symptoms and live until their early twenties.
To determine whether loss of the genome maintenance function of SMARCAL1 at stalled replication forks contributes to the SIOD disease, we took advantage of a point mutation in SMARCAL1 (R764Q) identified in a patient with severe SIOD symptoms. This mutation inactivates SMARCAL1 ATPase and annealing helicase activities.13 Our data indicated that the mutation did not perturb SMARCAL1 localization to stalled replication forks. However, expression of the mutant protein in cells in which the endogenous allele is silenced using RNAi fails to complement the loss of SMARCAL1 genome maintenance activity.5,9 Also, SIOD-derived patient cells progress into mitosis more slowly than cells complemented with the wild-type gene after release from a thymidine block.6 These data established the first link between the SIOD disease and a defect in the replication stress response.
If SIOD were due to a loss of the genome maintenance activities of SMARCAL1, we would expect to observe an increase in DNA damage during S-phase in SIOD patient cells. Indeed, fibroblasts derived from an SIOD patient exhibit elevated levels of γH2AX, which can be rescued by introduction of wild-type SMARCAL1 (Fig. 1A). Neither the RPA binding-mutant (ΔN) nor the R764Q SMARCAL1 mutant can decrease the amount of γH2AX. Finally, re-introduction of SMARCAL1 to SIOD fibroblasts reduces RPA phosphorylation following HU treatment compared to cells expressing only the vector (Fig. 1B). These results are consistent with the idea that SMARCAL1 acts as an annealing helicase at stalled forks to limit the amount of ssDNA.
Figure 1.
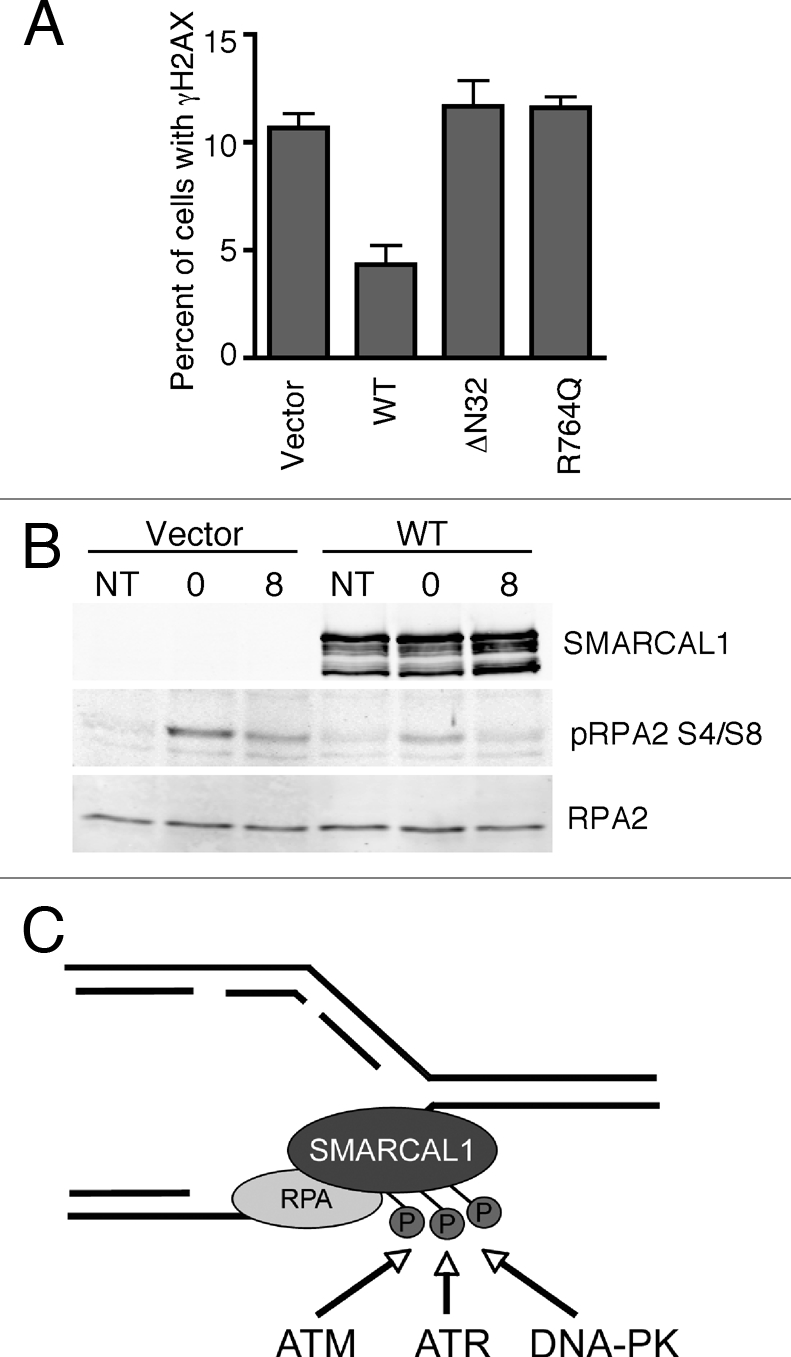
SIOD patient fibroblasts exhibit increased DNA damage signaling. (A) SIOD patient-derived human fibroblasts stably expressing vector only, WT, ΔN, or R764Q SMARCAL1 were cultured on coverslips and stained with antibodies to γH2AX and appropriate secondary antibodies. The percentage of cells with γH2AX staining was scored; error bars are standard deviation 300 cells were counted with each cell type analyzed in triplicate. (B) Cells with an integrated empty vector (Vector) or vector expressing wild-type SMARCAL1 (WT) were left untreated (NT) or treated with 2 mM hydroxyurea (HU) for 5 h then released into normal growth media for 0 or 8 h as indicated. Total cell lysates were prepared and immunoblotted with antibodies to SMARCAL1, phosphorylated RPA2, or total RPA2. (C) Model for SMARCAL1 regulation at stalled replication forks. DNA polymerase and helicase activities are uncoupled at replication forks in response to DNA lesions or agents like HU that interfere with DNA synthesis. Uncoupling causes the formation of ssDNA that is rapidly coated with RPA. An interaction between the N-terminus of SMARCAL1 and RPA2 localizes SMARCAL1 to the stalled fork where it is phosphorylated by DNA-damage activated kinases.
Conclusions
We conclude that SMARCAL1 is a replication stress protein regulated by both its RPA-interaction leading to fork localization and by phosphorylation catalyzed by the ATM, ATR and DNA-PK kinases (Fig. 1C). SMARCAL1 acts as an annealing helicase at forks where the polymerase and helicase activities have been uncoupled due to DNA lesions or other perturbations. This activity is important to prevent degeneration of the fork into a double-strand break. The absence of this activity contributes to the human disease Schimke immuno-osseous dysplasia.
Acknowledgements
This work is supported by National Cancer Institute grant R01CA136933 to D.C.
Abbreviations
- SMARCAL1
SWI/SNF-related, matrix associated, actin-dependent regulator of chromatin, subfamily A-like 1
- RPA
replication protein A
- ssDNA
single-stranded DNA
- SIOD
Schimke immuno-osseous dysplasia
- HU
hydroxyurea
Footnotes
Previously published online: www.landesbioscience.com/journals/nucleus/article/11739
References
- 1.Hartwell LH, Kastan MB. Cell cycle control and cancer. Science. 1994;266:1821–1828. doi: 10.1126/science.7997877. [DOI] [PubMed] [Google Scholar]
- 2.Kastan MB, Bartek J. Cell cycle checkpoints and cancer. Nature. 2004;432:316–323. doi: 10.1038/nature03097. [DOI] [PubMed] [Google Scholar]
- 3.McKinnon PJ, Caldecott KW. DNA strand break repair and human genetic disease. Annu Rev Genomics Hum Genet. 2007;8:37–55. doi: 10.1146/annurev.genom.7.080505.115648. [DOI] [PubMed] [Google Scholar]
- 4.Barzilai A, Biton S, Shiloh Y. The role of the DNA damage response in neuronal development, organization and maintenance. DNA Repair. 2008;7:1010–1027. doi: 10.1016/j.dnarep.2008.03.005. [DOI] [PubMed] [Google Scholar]
- 5.Bansbach CE, Betous R, Lovejoy CA, Glick GG, Cortez D. The annealing helicase SMARCAL1 maintains genome integrity at stalled replication forks. Genes Dev. 2009;23:2405–2414. doi: 10.1101/gad.1839909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ciccia A, Bredemeyer AL, Sowa ME, Terret ME, Jallepalli PV, Harper JW, Elledge SJ. The SIOD disorder protein SMARCAL1 is an RPA-interacting protein involved in replication fork restart. Genes Dev. 2009;23:2415–2425. doi: 10.1101/gad.1832309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Postow L, Woo EM, Chait BT, Funabiki H. Identification of SMARCAL1 as a component of the DNA damage response. J Biol Chem. 2009;284:35951–35961. doi: 10.1074/jbc.M109.048330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yusufzai T, Kong X, Yokomori K, Kadonaga JT. The annealing helicase HARP is recruited to DNA repair sites via an interaction with RPA. Genes Dev. 2009;23:2400–2404. doi: 10.1101/gad.1831509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yuan J, Ghosal G, Chen J. The annealing helicase HARP protects stalled replication forks. Genes Dev. 2009;23:2394–2399. doi: 10.1101/gad.1836409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Flaus A, Martin DM, Barton GJ, Owen-Hughes T. Identification of multiple distinct Snf2 subfamilies with conserved structural motifs. Nucleic Acids Res. 2006;34:2887–2905. doi: 10.1093/nar/gkl295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eisen JA, Sweder KS, Hanawalt PC. Evolution of the SNF2 family of proteins: subfamilies with distinct sequences and functions. Nucleic Acids Res. 1995;23:2715–2723. doi: 10.1093/nar/23.14.2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Conaway RC, Conaway JW. The INO80 chromatin remodeling complex in transcription, replication and repair. Trends Biochem Sci. 2009;34:71–77. doi: 10.1016/j.tibs.2008.10.010. [DOI] [PubMed] [Google Scholar]
- 13.Yusufzai T, Kadonaga JT. HARP is an ATP-driven annealing helicase. Science. 2008;322:748–750. doi: 10.1126/science.1161233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lovejoy CA, Xu X, Bansbach CE, Glick GG, Zhao R, Ye F, et al. Functional genomic screens identify CINP as a genome maintenance protein. Proc Natl Acad Sci USA. 2009;106:19304–19309. doi: 10.1073/pnas.0909345106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Byun TS, Pacek M, Yee MC, Walter JC, Cimprich KA. Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes Dev. 2005;19:1040–1052. doi: 10.1101/gad.1301205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fanning E, Klimovich V, Nager AR. A dynamic model for replication protein A (RPA) function in DNA processing pathways. Nucleic Acids Res. 2006;4:4126–4137. doi: 10.1093/nar/gkl550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu X, Vaithiyalingam S, Glick GG, Mordes DA, Chazin WJ, Cortez D. The basic cleft of RPA70N binds multiple checkpoint proteins including RAD9 to regulate ATR signaling. Mol Cell Biol. 2008;28:7345–7353. doi: 10.1128/MCB.01079-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Unsal-Kacmaz K, Chastain PD, Qu PP, Minoo P, Cordeiro-Stone M, Sancar A, Kaufmann WK. The human Tim/Tipin complex coordinates an Intra-S checkpoint response to UV that slows replication fork displacement. Mol Cell Biol. 2007;27:3131–3142. doi: 10.1128/MCB.02190-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Driscoll R, Cimprich KA. HARPing on about the DNA damage response during replication. Genes Dev. 2009;23:2359–2365. doi: 10.1101/gad.1860609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baumann C, Korner R, Hofmann K, Nigg EA. PICH, a centromere-associated SNF2 family ATPase, is regulated by Plk1 and required for the spindle checkpoint. Cell. 2007;128:101–114. doi: 10.1016/j.cell.2006.11.041. [DOI] [PubMed] [Google Scholar]
- 21.Mallery DL, Tanganelli B, Colella S, Steingrimsdottir H, van Gool AJ, Troelstra C, et al. Molecular analysis of mutations in the CSB (ERCC6) gene in patients with Cockayne syndrome. Am J Hum Genet. 1998;62:77–85. doi: 10.1086/301686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boerkoel CF, Takashima H, John J, Yan J, Stankiewicz P, Rosenbarker L, et al. Mutant chromatin remodeling protein SMARCAL1 causes Schimke immuno-osseous dysplasia. Nat Genet. 2002;30:215–220. doi: 10.1038/ng821. [DOI] [PubMed] [Google Scholar]