

Abstract
Nuclear lamins are major components of the nuclear lamina, and play essential roles in supporting the nucleus and organizing nuclear structures. While a large number of clinically important mutations have been mapped to the LMNA gene in humans, very few mutations have been associated with the B-type lamins. We have shown that lamin B2-deficiency in mice results in severe brain abnormalities. While the early stages of forebrain development in lamin B2-deficient mice appear to be normal, cortical neurons fail to migrate and organize into proper layers within the cerebral cortex. The morphogenesis of the hippocampus and cerebellum is also severely impaired. These phenotypes are reminiscent of lissencephaly, a human brain developmental disorder characterized by an abnormal neuronal migration. Most mutations in lissencephaly patients affect cytoplasmic regulators of nuclear translocation, which is a crucial step in neuronal migration. The phenotypes of lamin B2-deficient mice suggest that lamin B2 may also play a key role in nuclear translocation. Potential mechanisms for lamin B2 involvement, which include mechanical and non-mechanical roles and participation in LINC complexes in the nuclear envelope, are discussed along with evidence that lamins B1 and B2 play distinct, cell-specific functions.
Key words: nuclear envelope, nuclear lamina, lamin B2, LINC complex, nesprin, SUN, lissencephaly, neuronal migration, cortical neurons, nuclear translocation
Nuclear Lamins in Health and Disease
The nuclear lamina provides structural support for the nucleus and interacts with many nuclear components, including nuclear pores and chromatin.1,2 The main components of the nuclear lamina are the lamins, a class of intermediate filament proteins found in all higher eukaryotes.3,4 Lamins are classified into two groups: A-type lamins, which are expressed in differentiated cells and B-type lamins, which are present in all cell types including stem cells.5,6 In mammals, the A-type lamins, predominantly lamin A and lamin C, are generated by alternative splicing of the LMNA transcript, whereas the somatic B-type lamins, lamin B1 and lamin B2, are encoded by two separate genes, LMNB1 and LMNB2.
A-type lamins have attracted tremendous interest with the discovery that mutations in LMNA cause a variety of severe human genetic diseases (e.g., muscular dystrophy, cardiomyopathy, peripheral neuropathy, partial lipodystrophy), generally grouped under the term laminopathies.7–9 Laminopathies are usually characterized by a late onset and often affect predominantly one tissue—skeletal and/or cardiac muscle, peripheral nerves or adipose tissue. One exception is Hutchinson-Gilford progeria syndrome (HGPS), a pediatric disorder presenting as premature aging.10,11 Patients with HGPS typically exhibit growth retardation, osteoporosis, alopecia and eventually die from complications of occlusive cardiovascular disease.7,8 Remarkably, however, the brain is not affected by this disease.
More than 300 human mutations have been identified in the LMNA gene. A large fraction of these mutations are missense mutations, but few insights have emerged between the location of a particular mutation and the disease phenotype.7,12 In particular, it is unclear why mutations in a gene that is so broadly expressed would give rise to diseases affecting only certain tissues.8 Part of the explanation could relate to abnormalities in heterochromatin organization and secondary effects on gene expression.13,14 Many but not all disease-causing LMNA mutations result in abnormal nuclear shape and defective nuclear lamina,15–17 and defects in lamins A and C cause motion-related fragility of the nuclear envelope.18,19
In contrast to the situation with LMNA, only two diseases have been linked to mutations in LMNB1 or LMNB2. Duplications of the LMNB1 gene result in an autosomal dominant leukodystrophy (ADLD), a neurodegenerative disease with widespread loss of myelin in the adult central nervous system.20–22 Also, one study reported an increased frequency of LMNB2 polymorphisms in patients with acquired partial lipodystrophy (Barraquer syndrome)23—but firm evidence for a bona fide disease association was limited.
The remarkable disparity in the frequency of disease association for the A- and B-type lamins led us to generate a mouse model for lamin B2-deficiency. Our goal was to determine if Lmnb2 was an essential gene for development or whether it could be redundant with Lmnb1. Complete redundancy of Lmnb1 and Lmnb2 seemed unlikely, as Lmnb1 deficiency is lethal at birth and homozygous mice displayed impaired growth and severe lung and bone defects.24 Our analysis of Lmnb2 knockout mice supported the notion that Lmnb1 and Lmnb2 have unique functions. The most striking result of our study was the discovery that lamin B2 is essential for neuronal migration in the developing brain.
Lamin B2 is Essential for Cortical Migration
Mice homozygous for the Lmnb2 knockout allele (Lmnb2-/-) appeared normal during development but died soon after birth.25 The most dramatic phenotype was profound disorganization of the layering of the cerebral cortex, reminiscent of lissencephaly in humans.26,27 Like lissencephaly, we found evidence of impaired neuronal migration in Lmnb2-deficient brains.25 The early stages of forebrain development were normal, but defects appeared as the cortical neurons initiated their radial migration. As neuronal progenitors exit the cell cycle and start their differentiation into neurons, they leave the ventricular zone and migrate along the glial fibers to form the cortical plate28 (Fig. 1). This process was abnormal in Lmnb2-/- embryos, with most neuronal cells accumulating in the intermediate zone (Fig. 1).25 Birthdating experiments and marker analysis for different layers of the cortex confirmed that most neurons failed to reach their target layers (Fig. 1). Morphogenesis of the cerebellum and hippocampus, which also involves neuronal migration and is sometimes perturbed in lissencephaly,26 was also profoundly abnormal in Lmnb2-/- embryos.25
Figure 1.
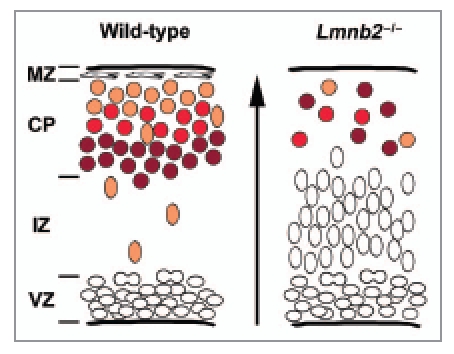
Neuronal migration defects in Lmnb2 knockout mice. Comparison of the migration of cortical neurons in wild-type (left) and lamin B2-deficient (Lmnb2-/-, right) brains at embryonic day 17.5. The arrow indicates the direction of migration. In wild-type brains (left), neuronal progenitors proliferate in the ventricular zone (VZ). Upon differentiation, the progenitors stop dividing and migrate across the intermediate zone (IZ) along the glial fibers. Neurons reaching the cortical plate (CP) intercalate between the marginal zone (MZ) and the neurons that arrived earlier. As a result, layers of neurons are formed, with the older neurons in the deeper layers of the cortical zone (dark red) and more recently differentiated cells (orange) located more superficially. In Lmnb2-deficient mice (right), most cells accumulate in the intermediate zone, and the reduced number of neurons observed in the cortical plate fail to organize into layers. Similar phenotypes are observed in other mouse models of lissencephaly.
The brain specificity of the Lmnb2 phenotype was surprising. LMNA mutations in humans affect primarily mesenchymal derivatives—muscle, heart, skin, bone and adipose tissue.8 Also, Lmna-deficiency results in muscular dystrophy in young mice,29 and Lmna knockout heterozygous mice manifest cardiomyopathy after one year of age.30 Lmnb1-/- mice die at birth and display severe lung and bone abnormalities and reduced growth.24 These differences in phenotypes led us to investigate whether differences in gene expression could underlie the brain-specific phenotype in Lmnb2 knockout mice.
In most previous studies, the two B-type lamins were not distinguished from each other and were assumed to be expressed in all cells.5,31 Röber et al.31 documented lamin B expression in all mouse tissues, whereas lamin A/C expression was confined to differentiating tissues starting at embryonic day 12, with a later onset in the brain (postnatal day 5). But interestingly, tissue specificity for lamin B1 and lamin B2 expression has been reported in a few studies.32,33 Broers et al.32 found that lamin B2 is expressed in most human adult tissues, while lamin B1 expression was more restricted to epithelia (of note, this survey did not include brain). Takamori et al.33 reported that lamin A/C, lamin B1 and lamin B2 were differentially expressed in the adult rat brain, and that the composition of the nuclear lamina varies during neuronal differentiation in the adult dentate gyrus and subventricular zone. We searched for differences in the expression patterns of lamins B1 and B2 in the developing mouse cortex. We found that both lamin B1 and lamin B2 were present in neuronal progenitors and differentiated neurons in wild-type embryos, at the same developmental stage affected in Lmnb2 mutants.25 However, we noted wide variation between cell types in the level of Lmnb2 expression (detected with a lacZ reporter gene inserted at the ATG of Lmnb2).25 During brain development, Lmnb2 was strongly expressed in neuronal progenitors in the ventricular zone; at later stages and in newborns, the β-galactosidase activity was detected in the upper layers of the cortex, hippocampus, cerebellum and olfactory bulbs.25 All of these structures develop rapidly, with neuronal migration playing an important role in their morphogenesis. This observation supports the concept that lamin B2 is required for neuronal migration, and that lamin B1 cannot compensate for the loss of lamin B2 in the developing brain.
Nuclear Translocation, a Crucial Step in Neuronal Migration
Neuronal migration is essential for the patterning of the cerebral cortex. While the brain continues to develop after birth, a crucial step in patterning occurs at mid-gestation, with successive waves of neurons migrating from the ventricular zone to the cortical plate. Neurons that differentiate at the same time migrate together and stop at the same level in the cortical plate (Fig. 1). The final position of a neuron is a strong determinant for its maturation and the establishment of neuronal connections later within the cortex. As a result, the consequences of defective neuronal migration are quite severe, as illustrated by the pathology in lissencephaly.26,27
Timing is critical during neuronal migration and neurons move rapidly. To reach their destination, neurons move forward by multiple cycles of “nuclear translocation” in which the nucleus is pulled toward the centrosome by dynein, a minus-end directed motor moving along microtubules.34 Dynein is tightly regulated by cytoplasmic proteins, such as LIS1, NDE1 and NDEL1. Perturbations of those regulators impair neuronal migration and result in lissencephaly phenotypes.35,36 But until now, no nuclear protein, and in particular no component of the nuclear lamina, had been implicated in this process. The similarity in phenotypes observed in lissencephaly and lamin B2-deficient mice strongly suggested a role for lamin B2 in anchoring the nucleus to the microtubule network during neuronal migration. However, the localization of lamin B2 inside the nucleus strongly suggests that molecular partners would be required to link lamin B2 to cytoplasmic proteins. Obvious candidates are the LINC complexes that connect the nuclear compartment and cytoskeleton.
From the Fly Eye to the Mouse Brain: LINC Complexes and Nuclear Anchoring
LINC complexes LInk the Nucleus to the Cytoskeleton and are conserved from Drosophila and C. elegans to mammals.37–39 The central elements of a LINC complex are a KASH transmembrane protein in the outer nuclear membrane and a SUN protein in the inner nuclear membrane; the two proteins interact in the perinuclear space through their KASH and SUN domains (Fig. 2A and box). On the cytoplasmic side, KASH proteins interact with elements of the cytoskeleton (including actin, microtubules and intermediate filaments). On the nucleoplasmic side, SUN proteins are known to bind to lamins and other nuclear components. Thus far, the only documented interaction of SUN1/2 with lamin proteins in mammalian cells involves lamin A.39 Two major functions of the LINC complex are nuclear anchoring and nuclear migration. In mammals, research on KASH and SUN proteins has focused largely on their role in muscle differentiation, since SYNE1 and SYNE2—encoding the KASH domain proteins Syne-1/Nesprin 1 and Syne-2/Nesprin 2—are both associated with muscular dystrophy.40 Mouse studies have revealed that Syne-1/2 and Sun1/2 are required for nuclear anchoring in myocytes.41,42
Figure 2.
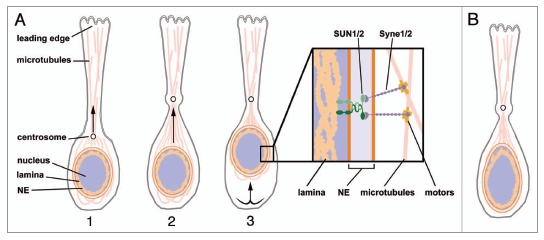
Nuclear movement during neuronal migration. (A) Nuclear translocation during neuronal migration in a wild-type neuron. Neuronal progression is achieved by the succession of three steps. (1) After extension of the leading process, the centrosome positioned in front of the nucleus is pulled in the direction of the leading edge. (2) The nucleus moves toward the centrosome by contraction of the microtubules. (3) The trailing process retracts, and as a result the whole neuron is moved forward. This sequence is repeated many times during cortical migration. Box: Model for a LINC complex spanning the neuron nuclear envelope, based on the similarities between phenotypes in Lmnb2, Syne-1/2 and Sun1/2 mutant mice. A complex involving lamin B2, SUN1/2 and Syne1/2 proteins might provide anchoring of the nucleus to the network of microtubules. Syne1/2 interaction with the microtubules is mediated by binding to motors that move along the microtubules.38 (B) A possible mechanism for defective neuronal migration in the setting of lamin B2 deficiency. Lamin B2 deficiency might impair the mechanical properties of nuclei in neurons, and an increase in nuclear deformability could prevent nuclear translocation and adversely affect neuronal migration. NE, nuclear envelope.
The importance of mammalian LINC complexes is not restricted to muscle cells. Mutations in SYNE1 have been shown to cause cerebellar ataxia in humans.43 Recently, Zhang et al.44 have obtained direct evidence for LINC function in the brain by reassessing the perinatal lethality of Sun1/Sun2 double mutant mice and of mice expressing truncated forms of Syne-1 and Syne-2. They showed that the loss of both SUN1 and SUN2 or the deletion of the KASH domain in both Syne-1/Nesprin 1 and Syne-2/Nesprin 2, resulted in severe defects in neurogenesis and neuronal migration.44 In addition, Zhang et al.44 showed that SUN1 and SUN2 proteins form a complex with Syne-2/Nesprin 2 and play redundant functions in anchoring the nucleus to the centrosome in glial cells.
The striking similarities in brain phenotypes between the Sun1/2-deficient mice, the Syne-1/2 mutants and our Lmnb2-/- mice suggest the existence of a neuronal LINC complex linking lamin B2 to the microtubule network (Fig. 2A). As noted earlier, only lamin A has been reported to interact with SUN1/2 in mammalian cells,39 but Drosophila B-type lamin has been described as an element of a LINC complex required for eye development.45,46 Nuclear migration is essential for photoreceptor differentiation in the fly, and a failure of the nucleus to reach an apical position results in abnormal eye development. Patterson et al.45 showed that this process depends on a connection between centrosome and nucleus, and that this connection requires Lamin Dm0 (the Drosophila B-type lamin) and Klarsicht, a founder protein of the KASH family.45 Further studies have shown that the interaction of Klarsicht with Lamin Dm0 is mediated by the SUN protein Klaroid.46
These studies in Drosophila support the hypothesis that mammalian lamin B2 might be part of a LINC complex. However, until a direct interaction between lamin B2 and one of the SUN proteins is demonstrated, other mechanisms and molecular partners cannot be excluded.
Perturbations of Nuclear Lamina and Altered Nuclear Mechanics
Another model to explain the Lmnb2 neuronal phenotype could involve impaired nuclear mechanics (Fig. 2B). Several LMNA mutations affect the stiffness of the nuclear envelope.17,18 Lamin B1 deficiency does not perturb the nuclear mechanics in fibroblasts,18 but nevertheless causes severe nuclear shape abnormalities.24 Also, the nuclei of lamin B1-deficient fibroblasts spin inside the cell, a striking phenotype that almost certainly indicates a defect in nucleus anchoring.47 (It is not known whether this finding is specific for cultured fibroblasts, nor whether the nuclei of lamin B2-deficient fibroblasts spin).
Taken together, these observations might help to explain the sensitivity of neurons to an absence of lamin B2. Several studies have reported that migrating neurons display irregular nuclear shape,48,49 most likely a consequence of the substantial deformation forces that accompany neuronal migration. It seems possible that lamin B2 might play a key role in supporting nuclear shape, and the absence of lamin B2 might result in increased deformability of the nucleus (Fig. 2B). Increased deformability of the lamina might significantly delay the migration of neurons, resulting in disorganized cortical layers. This model remains to be tested.
Defining Specific Functions for Lamin B1 and Lamin B2
B-type lamins are commonly considered as a single entity, but the analysis of Lmnb1 and Lmnb2 mutant mice has proven otherwise. Although both Lmnb1 and Lmnb2 mutants die soon after birth, Lmnb2-deficient embryos were normal in size, and the lungs and bones developed normally25-in contrast to the Lmnb1 knockout phenotype.24 It is unknown if the brain is affected by Lmnb1 deficiency, but the reduced size of the head of Lmnb1 mutants provides a hint that brain pathology might exist. Studies with cultured cells also support distinct functions for lamin B1 and lamin B2. Vergnes et al.24 found that Lmnb1-deficient embryonic fibroblasts displayed severe abnormalities of the nuclear envelope with numerous blebs,24 while Lmnb2-deficient fibroblasts have a normal nuclear morphology.24,25 In addition, lamin B1-deficient fibroblasts develop severe polyploidy, exhibit slow growth and enter senescence prematurely.24 These defects could relate to the proposed role of lamin B1 in mitotic spindle assembly.50 In contrast, lamin B2-deficient fibroblasts have a normal chromosome count and proliferate normally.25 These phenotypic differences support the idea that Lmnb1 and Lmnb2 play unique functions in mammals. However, additional studies will be required to understand specific functions of these two lamins and to identify binding partners in different tissues.
Concluding Remarks and Perspectives
Studies of human mutations and a variety of mutant mouse models have greatly expanded our understanding of the A-type lamins, although more work is needed to understand why different mutations in LMNA cause different disease phenotypes. By comparison, B-type lamins have been neglected. The finding of neuronal migration defects in lamin B2-deficient mice promises to change this situation, and should energize efforts to better understand the functions of both lamin B1 and lamin B2. We hope that future efforts will involve human geneticists, as we strongly suspect that “LMNB2 diseases” exist in humans. If the mouse is a useful guide, LMNB2 mutations will likely be uncovered in human patients with neurodevelopmental disorders, and not with the spectrum of diseases typically associated with LMNA mutations.
Acknowledgements
This work was supported by National Institutes of Health Grants AG035626, HL76839, HL86683, GM66152; a March of Dimes Grant (6-FY2007-1012), the Ellison Medical Foundation and an American Heart Association Scientist Development Grant (0835489N).
Footnotes
Previously published online: www.landesbioscience.com/journals/autophagy/article/12830
References
- 1.Stewart CL, Roux KJ, Burke B. Blurring the boundary: The nuclear envelope extends its reach. Science. 2007;318:1408–1412. doi: 10.1126/science.1142034. [DOI] [PubMed] [Google Scholar]
- 2.Prokocimer M, Davidovich M, Nissim-Rafinia M, Wiesel-Motiuk N, Bar D, Barkan R, et al. Nuclear lamins: Key regulators of nuclear structure and activities. J Cell Mol Med. 2009;13:1059–1085. doi: 10.1111/j.1582-4934.2008.00676.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gerace L, Blum A, Blobel G. Immunocytochemical localization of the major polypeptides of the nuclear pore complex-lamina fraction. Interphase and mitotic distribution. J Cell Biol. 1978;79:546–566. doi: 10.1083/jcb.79.2.546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aebi U, Cohn J, Buhle L, Gerace L. The nuclear lamina is a meshwork of intermediate-type filaments. Nature. 1986;323:560–564. doi: 10.1038/323560a0. [DOI] [PubMed] [Google Scholar]
- 5.Dechat T, Pfleghaar K, Sengupta K, Shimi T, Shumaker DK, Solimando L, et al. Nuclear lamins: Major factors in the structural organization and function of the nucleus and chromatin. Genes Dev. 2008;22:832–853. doi: 10.1101/gad.1652708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hutchison CJ. Lamins: Building blocks or regulators of gene expression? Nat Rev Mol Cell Biol. 2002;3:848–858. doi: 10.1038/nrm950. [DOI] [PubMed] [Google Scholar]
- 7.Worman HJ, Fong LG, Muchir A, Young SG. Laminopathies and the long strange trip from basic cell biology to therapy. J Clin Invest. 2009;119:1825–1836. doi: 10.1172/JCI37679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dauer WT, Worman HJ. The nuclear envelope as a signaling node in development and disease. Dev Cell. 2009;17:626–638. doi: 10.1016/j.devcel.2009.10.016. [DOI] [PubMed] [Google Scholar]
- 9.Burke B, Stewart CL. Life at the edge: The nuclear envelope and human disease. Nat Rev Mol Cell Biol. 2002;3:575–585. doi: 10.1038/nrm879. [DOI] [PubMed] [Google Scholar]
- 10.Eriksson M, Brown WT, Gordon LB, Glynn MW, Singer J, Scott L, et al. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature. 2003;423:293–298. doi: 10.1038/nature01629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de Sandre-Giovannoli A, Bernard R, Cau P, Navarro C, Amiel J, Boccaccio I, et al. Lamin A truncation in Hutchinson-Gilford progeria. Science. 2003;300:2055. doi: 10.1126/science.1084125. [DOI] [PubMed] [Google Scholar]
- 12.Wilson KL, Zastrow MS, Lee KK. Lamins and disease: Insights into nuclear infrastructure. Cell. 2001;104:647–650. [PubMed] [Google Scholar]
- 13.Hutchison CJ, Alvarez-Reyes M, Vaughan OA. Lamins in disease: Why do ubiquitously expressed nuclear envelope proteins give rise to tissue-specific disease phenotypes? J Cell Sci. 2001;114:9–19. doi: 10.1242/jcs.114.1.9. [DOI] [PubMed] [Google Scholar]
- 14.Misteli T, Scaffidi P. Genome instability in progeria: When repair gets old. Nat Med. 2005;11:718–719. doi: 10.1038/nm0705-718. [DOI] [PubMed] [Google Scholar]
- 15.Bonne G, Di Barletta MR, Varnous S, Bécane HM, Hammouda EH, Merlini L, et al. Mutations in the gene encoding lamin A/C cause autosomal dominant Emery-Dreifuss muscular dystrophy. Nat Genet. 1999;21:285–288. doi: 10.1038/6799. [DOI] [PubMed] [Google Scholar]
- 16.Muchir A, Medioni J, Laluc M, Massart C, Arimura T, van der Kooi AJ, et al. Nuclear envelope alterations in fibroblasts from patients with muscular dystrophy, cardiomyopathy and partial lipodystrophy carrying lamin A/C gene mutations. Muscle Nerve. 2004;30:444–450. doi: 10.1002/mus.20122. [DOI] [PubMed] [Google Scholar]
- 17.Dahl KN, Scaffidi P, Islam MF, Yodh AG, Wilson KL, Misteli T. Distinct structural and mechanical properties of the nuclear lamina in Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci USA. 2006;103:10271–10276. doi: 10.1073/pnas.0601058103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lammerding J, Schulze PC, Takahashi T, Kozlov S, Sullivan T, Kamm RD, et al. Lamin A/C deficiency causes defective nuclear mechanics and mechanotransduction. J Clin Invest. 2004;113:370–378. doi: 10.1172/JCI19670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Broers JL, Peeters EA, Kuijpers HJ, Endert J, Bouten CV, Oomens CW, et al. Decreased mechanical stiffness in LMNA-/- cells is caused by defective nucleo-cytoskeletal integrity: Implications for the development of laminopathies. Hum Mol Genet. 2004;13:2567–2580. doi: 10.1093/hmg/ddh295. [DOI] [PubMed] [Google Scholar]
- 20.Padiath QS, Saigoh K, Schiffmann R, Asahara H, Yamada T, Koeppen A, et al. Lamin B1 duplications cause autosomal dominant leukodystrophy. Nat Genet. 2006;38:1114–1123. doi: 10.1038/ng1872. [DOI] [PubMed] [Google Scholar]
- 21.Meijer I, Simoes-Lopes A, Laurent S, Katz T, St-Onge J, Verlaan D, et al. A novel duplication confirms the involvement of 5q23.2 in autosomal dominant leukodystrophy. Arch Neurol. 2008;65:1496–1501. doi: 10.1001/archneur.65.11.1496. [DOI] [PubMed] [Google Scholar]
- 22.Brussino A, Vaula G, Cagnoli C, Mauro A, Pradotto L, Daniele D, et al. A novel family with Lamin B1 duplication associated with adult-onset leucoencephalopathy. J Neurol Neurosurg Psychiatry. 2009;80:237–240. doi: 10.1136/jnnp.2008.147330. [DOI] [PubMed] [Google Scholar]
- 23.Hegele RA, Cao H, Liu DM, Costain GA, Charlton-Menys V, Rodger NW, et al. Sequencing of the reannotated LMNB2 gene reveals novel mutations in patients with acquired partial lipodystrophy. Am J Hum Genet. 2006;79:383–389. doi: 10.1086/505885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vergnes L, Peterfy M, Bergo MO, Young SG, Reue K. Lamin B1 is required for mouse development and nuclear integrity. Proc Natl Acad Sci USA. 2004;101:10428–10433. doi: 10.1073/pnas.0401424101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Coffinier C, Chang SY, Nobumori C, Tu Y, Farber EA, Toth JI, et al. Abnormal development of the cerebral cortex and cerebellum in the setting of lamin B2 deficiency. Proc Natl Acad Sci USA. 2010;107:5076–5081. doi: 10.1073/pnas.0908790107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gupta A, Tsai LH, Wynshaw-Boris A. Life is a journey: A genetic look at neocortical development. Nat Rev Genet. 2002;3:342–355. doi: 10.1038/nrg799. [DOI] [PubMed] [Google Scholar]
- 27.Wynshaw-Boris A. Lissencephaly and LIS1: Insights into the molecular mechanisms of neuronal migration and development. Clin Genet. 2007;72:296–304. doi: 10.1111/j.1399-0004.2007.00888.x. [DOI] [PubMed] [Google Scholar]
- 28.Bielas S, Higginbotham H, Koizumi H, Tanaka T, Gleeson JG. Cortical neuronal migration mutants suggest separate but intersecting pathways. Annu Rev Cell Dev Biol. 2004;20:593–618. doi: 10.1146/annurev.cellbio.20.082503.103047. [DOI] [PubMed] [Google Scholar]
- 29.Sullivan T, Escalante-Alcalde D, Bhatt H, Anver M, Bhat N, Nagashima K, et al. Loss of A-type lamin expression compromises nuclear envelope integrity leading to muscular dystrophy. J Cell Biol. 1999;147:913–919. doi: 10.1083/jcb.147.5.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wolf CM, Wang L, Alcalai R, Pizard A, Burgon PG, Ahmad F, et al. Lamin A/C haploinsufficiency causes dilated cardiomyopathy and apoptosis-triggered cardiac conduction system disease. J Mol Cell Cardiol. 2008;44:293–303. doi: 10.1016/j.yjmcc.2007.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Röber RA, Weber K, Osborn M. Differential timing of nuclear lamin A/C expression in the various organs of the mouse embryo and the young animal: A developmental study. Development. 1989;105:365–378. doi: 10.1242/dev.105.2.365. [DOI] [PubMed] [Google Scholar]
- 32.Broers JL, Machiels BM, Kuijpers HJ, Smedts F, van den Kieboom R, Raymond Y, et al. A- and B-type lamins are differentially expressed in normal human tissues. Histochem Cell Biol. 1997;107:505–517. doi: 10.1007/s004180050138. [DOI] [PubMed] [Google Scholar]
- 33.Takamori Y, Tamura Y, Kataoka Y, Cui Y, Seo S, Kanazawa T, et al. Differential expression of nuclear lamin, the major component of nuclear lamina, during neurogenesis in two germinal regions of adult rat brain. Eur J Neurosci. 2007;25:1653–1662. doi: 10.1111/j.1460-9568.2007.05450.x. [DOI] [PubMed] [Google Scholar]
- 34.Solecki DJ, Govek EE, Tomoda T, Hatten ME. Neuronal polarity in CNS development. Genes Dev. 2006;20:2639–2647. doi: 10.1101/gad.1462506. [DOI] [PubMed] [Google Scholar]
- 35.Wynshaw-Boris A, Gambello MJ. LIS1 and dynein motor function in neuronal migration and development. Genes Dev. 2001;15:639–651. doi: 10.1101/gad.886801. [DOI] [PubMed] [Google Scholar]
- 36.Vallee RB, Tsai JW. The cellular roles of the lissencephaly gene LIS1 and what they tell us about brain development. Genes Dev. 2006;20:1384–1393. doi: 10.1101/gad.1417206. [DOI] [PubMed] [Google Scholar]
- 37.Méjat A, Misteli T. LINC complexes in health and disease. Nucleus. 2010;1:40–52. doi: 10.4161/nucl.1.1.10530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fridkin A, Penkner A, Jantsch V, Gruenbaum Y. SUN-domain and KASH-domain proteins during development, meiosis and disease. Cell Mol Life Sci. 2009;66:1518–1533. doi: 10.1007/s00018-008-8713-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Burke B, Roux KJ. Nuclei take a position: Managing nuclear location. Dev Cell. 2009;17:587–597. doi: 10.1016/j.devcel.2009.10.018. [DOI] [PubMed] [Google Scholar]
- 40.Zhang Q, Bethmann C, Worth NF, Davies JD, Wasner C, Feuer A, et al. Nesprin-1 and -2 are involved in the pathogenesis of Emery Dreifuss muscular dystrophy and are critical for nuclear envelope integrity. Hum Mol Genet. 2007;16:2816–2833. doi: 10.1093/hmg/ddm238. [DOI] [PubMed] [Google Scholar]
- 41.Zhang X, Xu R, Zhu B, Yang X, Ding X, Duan S, et al. Syne-1 and Syne-2 play crucial roles in myonuclear anchorage and motor neuron innervation. Development. 2007;134:901–908. doi: 10.1242/dev.02783. [DOI] [PubMed] [Google Scholar]
- 42.Lei K, Zhang X, Ding X, Guo X, Chen M, Zhu B, et al. SUN1 and SUN2 play critical but partially redundant roles in anchoring nuclei in skeletal muscle cells in mice. Proc Natl Acad Sci USA. 2009;106:10207–10212. doi: 10.1073/pnas.0812037106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gros-Louis F, Dupre N, Dion P, Fox MA, Laurent S, Verreault S, et al. Mutations in SYNE1 lead to a newly discovered form of autosomal recessive cerebellar ataxia. Nat Genet. 2007;39:80–85. doi: 10.1038/ng1927. [DOI] [PubMed] [Google Scholar]
- 44.Zhang X, Lei K, Yuan X, Wu X, Zhuang Y, Xu T, et al. SUN1/2 and Syne/Nesprin-1/2 complexes connect centrosome to the nucleus during neurogenesis and neuronal migration in mice. Neuron. 2009;64:173–187. doi: 10.1016/j.neuron.2009.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Patterson K, Molofsky AB, Robinson C, Acosta S, Cater C, Fischer JA. The functions of Klarsicht and nuclear lamin in developmentally regulated nuclear migrations of photoreceptor cells in the Drosophila eye. Mol Biol Cell. 2004;15:600–610. doi: 10.1091/mbc.E03-06-0374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kracklauer MP, Banks SM, Xie X, Wu Y, Fischer JA. Drosophila klaroid encodes a SUN domain protein required for Klarsicht localization to the nuclear envelope and nuclear migration in the eye. Fly. 2007;1:75–85. doi: 10.4161/fly.4254. [DOI] [PubMed] [Google Scholar]
- 47.Ji JY, Lee RT, Vergnes L, Fong LG, Stewart CL, Reue K, et al. Cell nuclei spin in the absence of lamin B1. J Biol Chem. 2007;282:20015–20026. doi: 10.1074/jbc.M611094200. [DOI] [PubMed] [Google Scholar]
- 48.Tsai JW, Bremner KH, Vallee RB. Dual subcellular roles for LIS1 and dynein in radial neuronal migration in live brain tissue. Nat Neurosci. 2007;10:970–979. doi: 10.1038/nn1934. [DOI] [PubMed] [Google Scholar]
- 49.Goodchild RE, Kim CE, Dauer WT. Loss of the dystonia-associated protein torsinA selectively disrupts the neuronal nuclear envelope. Neuron. 2005;48:923–932. doi: 10.1016/j.neuron.2005.11.010. [DOI] [PubMed] [Google Scholar]
- 50.Tsai MY, Wang S, Heidinger JM, Shumaker DK, Adam SA, Goldman RD, et al. A mitotic lamin B matrix induced by RanGTP required for spindle assembly. Science. 2006;311:1887–1893. doi: 10.1126/science.1122771. [DOI] [PubMed] [Google Scholar]