

Abstract
Amphiphlic block copolymers consisting of hydrophilic, poly(acrylic acid) randomly decorated with acrylate groups and hydrophobic, rubbery poly(n-butyl acrylate) self-assembled into well-defined micelles with an average diameter of ~21 nm. Radical polymerization of acrylamide in the presence of the crosslinkable micelles gave rise to hybrid, elastomeric hydrogels whose mechancial properties can be readily tuned by varying the BCM concentration.
Hydrogels are macroscopic, polymeric networks that imbibe large amount of water. Due to their biocompatibility, tissue-like viscoelasticity and structural similarity to the native extracelluar matrices (ECM), hydrogels are widely used in tissue engineering and drug delivery applications.1–3 Traditional hydrogels are derived from molecularly-dispersed, soluble precursors (monomers and multifunctional crosslinkers or macromers) that are randomly interconnected, lacking the structural complexity, mechanical integrity and functional diversity seen in the natural ECM.4 Novel hybrid hydrogels with hierarchical structures and robust mechanical properties have been synthesized using organic or inorganic particles of nano or micron dimensions as the constituent building blocks or multifunctional crosslinkers. For example, nanocomposite hydrogels with unprecedented mechanical strength have been constructed by initiating radical polymerization from the surface of clay nanoparticles. The unique mechanical properties were attributed to the reduced fluctuation in the crosslinking density and the cooperativity of the polymer chains connecting the same clays.5, 6 Soft hydrogel particles have also been exploited as the multifunctional, microscopic crosslinkers.7–10 Our group has created hyaluronic acid (HA)-based doubly crosslinked networks with densely crosslinked, nanoporous HA hydrogel particles covalently interconnected by a loose secondary network that is also HA-based. 11–15 Such hierarchically-structured hydrogels permit the controlled release of bone morphogenetic protein 2 (BMP-2) with reduced initial bursts over prolonged periods of time.12
Block copolymer micelles (BCMs) are yet another class of microscopic, spherical objects consisting of a segregated hydrophobic interior and a stealth, hydrophilic corona that stablizes the assembled structure and interacts with the aqueous environment.16 Unlike inorganic nanoparticles or covalently crosslinked hydrogel particles (microgels and nanogels), BCMs exhibit distinct core-shell structure, capable of sequestering hydrophobic molecules and modulating their release kinetics, thereby, extending their pharmacokinetics.17 Although water-dispersed BCMs have been extensively explored as drug delivery vehicles, they have not been investigated as multifunctional, microscopic crosslinkers for the formation of elastomeric hydrogels with desirable pharmacological activities. We hypothesize that strategic integration of well-defined hydrophobic microdomains within the hydrogel matrix will not only provide a local depot for therapeutically active molecules but also offer means for fine-tuning of hydrogel mechanics. The functional BCMs employed in our study are not for mechanical reinforcement purposes, as various groups have demonstrated with inorganic nanoparticles and polymeric microgels. Rather, through their self-assembled, core-shell structures, in combination with their covalent coupling with the hydrogel matrix, the BCMs offer ample opportunities for the enhancement of the biological activities of the hydrogels via the spatial and temperoal presentation of therapeutically active molecules.
In this proof-of-concept investigation, self-assembled BCMs consisting of rubbery, hydrophobic poly(n-butyl acrylate) (PnBA) and charged, hydrophilic poly(acrylic acid) (PAA) that were partially acrylated were utilized as covalent crosslinkers for the formation of poly(acrylamide) (PAAm)-based hydrogels (Scheme 1). Atom transfer radical polymerization (ATRP)18 was employed for the synthesis of the block copolymer precursors via the sequential polymerization of t-butyl acrylate (tBA) and n-butyl acrylate (nBA). The macroinitiator (PtBA-Br) with an average molecular weight of 12.5 kg/mol was synthesized by controlling the feed composition and the reaction time. The effectiveness of PtBA-Br to extend the second, PnBA block was confirmed collectively by the presence of the proton signals from the n-butyl groups in the 1H NMR spectrum for PtBA-b-PnBA (Figure S1–S2), as well as the shift of the GPC trace for the the diblock copolymer to a lower retention time in comparion to the macroinitiator (Figure S3). Subsequent hydrolysis of the t-butyl groups revealed the hydrophilic, PAA blocks (Figure S4). Partial esterification (20 mol%, Figure S5) of the PAA block with hydroxyethyl acrylate (HEA) resulted in a block copolymer that is radically crosslinkable. With a Mn of 11.2 kg/mol, the final product had a molecular composition of (PAA100-g-HEA20)-b-PnBA16.
Scheme 1.

Synthesis of crosslinkable block copolymer micelles (BCMs) and the formation of BCM-crosslinked, PAAm-based hydrogels. The hydrophobic, PnBA chains are shown in black, the HEA-modified PAA blocks are highlighted in orange and the radically polymerized PAAm chains are represented in blue.
The acrylated block copolymers readily assembled into spherical micelles via a solvent exchange process. Information regarding the critical micelle concentration (CMC), the onset of micellization of the block copolymers, was derived from steady-state fluorescence spectroscopy of pyrene.19 Typical emission spectra of aqueous solutions of pyrene at a concentration of 6.0×10−7 M in the presence of various amounts of BCMs are shown in Figure S6. The ratio between the intensities of the first (I1) and the third (I3) vibrational bands decreases as the polarity of the medium in which pyrene is dissolved increases.20 The I1/I3 values remain constant at low polymer concentrations, and decrease sharply as the polymer concentration exceeds a certain value. By locating the intersection between the two tangent lines, the CMC was found to be 0.005 mg/mL (Figure 1). The relatively low CMC value suggests that the micelles are thermodynamically stable.
Figure 1.

Determination of the CMC for P(AA100-g-HEA20)-b-PnBA16 employing the steady-state fluorescence spectroscopy of pyrene. The I1/I3 values remain constant at low polymer concentrations, and decrease sharply as the polymer concentration exceeds a certain value. By locating the intersection between the two tangent lines, the CMC was found to be 0.005 mg/mL.
Transmission electron microscope (TEM) revealed the presence of spherical nanoparticles with an estimated diameter of 21 nm (Figure 2A). The higher magnification image clearly shows the presence of distinct core-shell morphology manifested by the negative staining technique. Dynamic light scattering (DLS) analysis of BCMs dispersed in de-ionized water (Figure 2B) indicates a z-average diameter of 19 nm and a PDI of 0.26, in good agreement with the size estimation from TEM. Assuming that the radius of the PnBA core was determined by the contour length of the hydrophobic chains in all-trans configuration and the PAA chains in the shell were completely stretched, the micelle aggregation number was estimated to be roughly 10.16, 21
Figure 2.
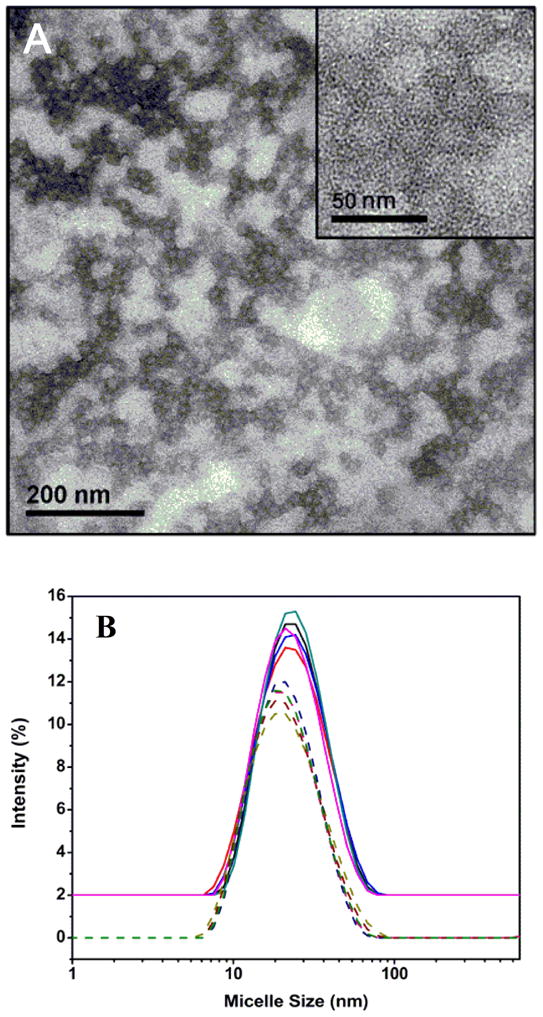
(A). Transmission electron micrograph of self-assembled BCMs negatively stained by uranyl acetate. Insert shows a higher magnification image. (B). DLS profiles of self-assembled BCMs in de-ionized H2O (dotted lines) and an aqueous AAm solution of 500 mg/mL (solid lines).
Hybrid hydrogels were prepared by radical polymerization of AAm in the presence of crosslinkable BCMs. Throughout our study, the amount of the initiator (APS), accelerator (TEMED) and monomer (AAm) were kept constant, while the concentration of the crosslinkers was systematically varied (Table S1). PAAm gels crosslinked by N,N′-methylene bisacrylamide (MBA) were included as the controls. Samples (or controls) were designated as S (or C)-x-BCM (or MBA), where S and C stand for sample and control, respectively, and x represents the concentration of the crosslinkers (in mg/mL) employed. The addition of AAm to the BCM dispersion did not alter the average size and the overall size distributions of the particles, as evidenced by DLS (Figure 2B). Therefore, the micelle structures are preserved in the final hybrid hydrogels. Radical polymerization of AAm with 15 mg/mL BCMs (Figure 3A) gave rise to a viscoelastic solid that did not flow. In the absence of any crosslinkers, overnight incubation of AAm with APS/TEMED led to a viscous solution that readily flowed (Figure 3B). GPC analysis of the resulting PAAm revealed an Mn of 400 kg/mol and a PDI of 1.46, reflecting the characteristics of a traditional radical polymerization reaction.22 1H NMR was employed to gain qualitative understanding of the efficiency of the crosslinking reaction. To this end, S-10-BCM gels were immersed in a concentrated HCl solution at an elevated temperature for 24 h. Such treatment essentially hydrolyzes the ester linkages at the crosslinking points and the amide bonds in PAAm, giving rise to a soluble mixture containing PAA and various low molecular weight byproducts that include acrylic acid from the unreacted acrlyate moities, if any, on BCMs (Supporting Information).23 While the viny protons (~6.0 ppm) from HEA after the acid treatment are clearly present (date not shown), those from S-10-BCM gels can be hardly distinguished from the background noise (Figure S7). Quantitative analysis regarding the vinyl group conversion during crosslinking is impossible due to the uncertainties associated with the peak assignment and the low intensity of the vinyl protons. Our results, although qualitative in nature, confirm the high conversion of the acrylate groups and the efficient coupling of BCMs to the PAAm chains during the radical crosslinking.
Figure 3.

Vial inversion experiment demonstrating the crosslinkability the BCMs. Radical polymerization of AAm with 15 mg/mL BCMs (A) gave rise to a viscoelastic solid that did not flow. In the absence of any crosslinkers (B), overnight incubation of AAm with APS/TEMED led to a viscous solution that flew. GPC analysis of the resulting PAAm revealed an Mn of 400 kg/mol and a PDI of 1.46.
It is worth emphasizing that the hydrogels investigated here are distinctly different from the well-known, thermal responsive hydrogels, in which gelation occurs through the physical association of BCMs, via either a jammed micelle24 or micellar bridges mechanism.25 In our case, the BCMs are multifunctional, nanoscale objects that covalently immobilize PAAm chains in the macroscopic hydrogel matrix. The sol fraction (SF) and the swelling ratio (SW) values (Table S1) for BCM-crossslinked hydrogels decreased with an increase of BCM concentration, indicating that the crosslinking density is directly related to BCM concentration. A similar trend was observed from the control gels crosslinked by MBA. Although C-2-MBA and S-15-BCM contained the same amount of crosslinkable units in feed, the former swelled to a much lesser extent than the latter. The BCMs are nanoscale objects with multiple crosslinkable units whereas MBA is a low molecular weight, difunctional reagent. It is likely that the acrylate groups in BCMs and the acrylamide groups in MBA exhibit different reactivity. It is also plausible that some acrylate groups were not readily accessible or have been consumed by intramicellular crosslinking. Prolonged incubation (up to 2 months) of the hydrogels in water at 37 °C did not cause significant changes in hydrogel wet weight, suggesting the hydrogels are stably crosslinked and are devoid of diffusible polymers and monomers.
The fully hydrated, BCM-crosslinked hydrogels were subjected to uniaxial tensile tests and the representative stress-strain curves were included in Figure 4. All BCM-crosslinked hydrogels displayed a sigmoid curve characteristic of elastomeric materials with low modulus, high resilience and large deformation before fracture. The BCM-crosslinked gels exhibited an initial compliant response, followed by a rollover to a stiffer behavior at approximately 270%, 292% and 366% strain for S-15-BCM, S-10-BCM and S-7.5-BCM, respectively (Figure 4A). The initial compliant responses correlate wtih the rubber elasticity theory where the energy is consumed in extending the Gaussion polymer chains in the network. The finite extensibility of PAAm chains is reached at high extension, leading to a rapid stress built-up. 26, 27 Figure 4B shows typical cyclic stress-strain curves for S-10-BCM gels at 100 mm/min at various strains and cycles. The BCM-crosslinked hydrogels are highly elastomeric, capable of responding to cyclic strains as high as 350% rapidly and reversibly. Complete recovery to their original length in multiple extension-retraction experiments (up to 10) was observed. Remarkally, the hybrid hydrogels exhibited close-to-zero hysteresis for all cyclic tests at various maximum stains, and the loading/unloading curves overlapped perfectly irrespective to the previous loading history. The absence of hysteresis means that the energy introduced to the hydrogels during the extension process was stored elastically and was completely recovered when the force was relaxed. In other words, no energy was permanently dissipated during the deformation. Therefore, partial bond breakage or alteration of microstructures are not present in the BCM-crosslinked gels. The overlapping loading/unloading curves suggest that the recovery is rapid and highly efficient.
Figure 4.

(A). Representative stress-strain curves for BCM-crosslinked hydrogels (filled symbols) along with the MBA-crosslinked controls (open symbols). Fully swollen samples were stretched at a rate of 100 mm/min until failure. (B). Representative extension-retraction cycles for S-10-BCM at a loading rate of 100 mm/min. Loading and unloading cycles were performed in immediate succession on each sample. Red squares: cycle 1 stress-stress profile at a εmax of 120%; black triangles: cycle 2 stress-stress profile at a εmax of 250%; blue circles: cycle 3 stress-stress profile a εmax of 350%; purple inverted triangles: cycle 10 stress-strain profile at a εmax of 350%.
The Young’s modulus was calculated from the slope of the initial linear region of the stress-strain (<30% strain) curves. Figure 5A shows that as the crosslinker concentration increases, the Young’s modulus increases steadily, commensurate with the swelling ratio results. There is a direct correlation of the BCM concentration and the mechanical properties of the hybrid hydrogels. S-15-BCM exhibited the highest tensile strength with an average stress at break of 152.5 ± 7.8 kPa (Figure 5B), whereas S-7.5-BCM can be extended up to 479 ± 30% (Figure 5C). Overall, higher BCM concentration resulted in stiffer gels with higher ultimate tensile strength, but lower elongation to break (Figure 5B and 5C). Analysis of the control gels revealed similar MBA-dependent Young’s modulus. While C-2-MBA is significantly stiffer, and undergoes a brittle failure at a maximum strain of 56%, other control gels prepared with a lower crosslinking density (C-0.5-MBA and C-0.2-MBA) were more pliable.
Figure 5.


Young’s modulus (A), ultimate stress (B) and ultimate strain (C) as a function of hydrogel composition. Results were reported as an average of 5 repeats ± standard deviation.
With a glass transition temperature of −4928 to −55 °C,20 the hydrophobically associated PnBA chains in the micelle core are conformationally flexible at ambient temperature. Moreover, the self-assembled micellar structures, consisting of collapsed, hydrophobically associated PnBA chains, are effectively locked-in by their covalent coupling with the PAAm chain in the hydrogel matrix. The open question is whether the hydrophobically segregated PnBA chains can be linearized or unraveled after the PAAm chains have lost most of their conformational freedom. The unraveling of the hydrophobically associated PnBA chains may occur, however, its contribution is diminished since the PAAm network is densely crosslinked and the overall BCM content is low. Our results show that the mechanical properties of the BCM-crosslinked gels can be readily tuned simply by varying the crosslinker concentration.
The robust mechanical properties observed on the BCM-crosslinked gels confirm that their high stability. These gels are unique in that the crosslinking points are consisted of self-assembled block copolymers that are reinforced by the PAAm chains. Our motivation for using BCMs as the microscopic crosslinkers arises from their well-defined core-shell structure that allows for the sequestration of hydrophobic drug molecules. The covalent coupling between BCMs and the PAAm network essentially locks in the assembled structures and permits the precise control over the diffusion of the sequestered drug molecules. The connectivity also ensures that the external forces be readily transmitted to the immobilized BCMs, allowing the drug release kinetics to be further modulated by mechanical stimulations.
In conclusion, amphiphilic block copolymers consisting of rubbery, hydrophobic cores and charged, hydrophilic segments partially functionalized with acrylate groups were synthesized and characterized. These polymers self-assembled into discrete block copolymer micelles with an average diameter of ~21 nm and a CMC of 0.005 g/L. The multifunctional BCMs are capable of crosslinking acrylamide monomers radically, forming hybrid hydrogels containing hydrophobically associated microdomains at the junction points. The BCM-crosslinked hydrogels are highly elastomeric and their mechanical properties can be readily tuned by varying the BCM concentration. These novel hydrogels present unique opportunities for the controlled release of hydrophobic drug molecules by their sequestration into the micelle core prior to the hydrogel formation.
Supplementary Material
Acknowledgments
We are grateful for the financial support from the National Science Foundation (DMR: 0643226 to XJ), National Institutes of Health (NIDCD: 3R01DC008965-03S2 to XJ) and the University of Delaware New Faculty Startup Funds. The authors wish to acknowledge Dr. Michael E. Mackay and Mr. Jonathan E. Seppala for their generous help with the mechanical testing.
Notes and References
- 1.Langer R, Tirrell DA. Nature. 2004;428:487–492. doi: 10.1038/nature02388. [DOI] [PubMed] [Google Scholar]
- 2.Lutolf MP, Gilbert PM, Blau HM. Nature. 2009;462:433–441. doi: 10.1038/nature08602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Slaughter BV, Khurshid SS, Fisher OZ, Khademhosseini A, Peppas NA. Adv Mater. 2009;21:3307–3329. doi: 10.1002/adma.200802106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bohidar HB, Dubin P, Osade Y. Polymer Gels: Fundamentals and Applications (ACS Symposium Series) American Chemical Society; Washington, DC: 2003. [Google Scholar]
- 5.Haraguchi K, Farnworth R, Ohbayashi A, Takehisa T. Macromolecules. 2003;36:5732–5741. [Google Scholar]
- 6.Tanaka Y, Gong JP, Osada Y. Prog Polym Sci. 2005;30:1–9. [Google Scholar]
- 7.Gan TT, Zhang YJ, Guan Y. Biomacromolecules. 2009;10:1410–1415. doi: 10.1021/bm900022m. [DOI] [PubMed] [Google Scholar]
- 8.Bencherif SA, Siegwart DJ, Srinivasan A, Horkay F, Hollinger JO, Washburn NR, Matyjaszewski K. Biomaterials. 2009;30:5270–5278. doi: 10.1016/j.biomaterials.2009.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang T, Xu HG, Jiao KX, Zhu LP, Brown HR, Wang HL. Adv Mater. 2007;19:1622–1626. [Google Scholar]
- 10.Qin XP, Zhao F, Liu YK, Wang HY, Feng SY. Colloid Polym Sci. 2009;287:621–625. [Google Scholar]
- 11.Jha AK, Hule RA, Jiao T, Teller SS, Clifton RJ, Duncan RL, Pochan DJ, Jia XQ. Macromolecules. 2009;42:537–546. doi: 10.1021/ma8019442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jha AK, Malik MS, Farach-Carson MC, Duncan RL, Jia X. Soft Matter. 2010 doi: 10.1039/C1030SM00101E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jia XQ, Kiick KL. Macromol Biosci. 2009;9:140–156. doi: 10.1002/mabi.200800284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jia XQ, Yeo Y, Clifton RJ, Jiao T, Kohane DS, Kobler JB, Zeitels SM, Langer R. Biomacromolecules. 2006;7:3336–3344. doi: 10.1021/bm0604956. [DOI] [PubMed] [Google Scholar]
- 15.Sahiner N, Jha AK, Nguyen D, Jia XQ. J Biomater Sci-Polym Ed. 2008;19:223–243. doi: 10.1163/156856208783432462. [DOI] [PubMed] [Google Scholar]
- 16.Colombani O, Ruppel M, Burkhardt M, Drechsler M, Schumacher M, Gradzielski M, Schweins R, Muller AHE. Macromolecules. 2007;40:4351–4362. [Google Scholar]
- 17.Davis ME, Chen Z, Shin DM. Nat Rev Drug Discov. 2008;7:771–782. doi: 10.1038/nrd2614. [DOI] [PubMed] [Google Scholar]
- 18.Coessens V, Pintauer T, Matyjaszewski K. Prog Polym Sci. 2001;26:337–377. [Google Scholar]
- 19.Astafieva I, Zhong XF, Eisenberg A. Macromolecules. 1993;26:7339–7352. [Google Scholar]
- 20.Colombani O, Ruppel M, Schubert F, Zettl H, Pergushov DV, Muller AHE. Macromolecules. 2007;40:4338–4350. [Google Scholar]
- 21.Eghbali E, Colombani O, Drechsler M, Muller AHE, Hoffmann H. Langmuir. 2006;22:4766–4776. doi: 10.1021/la053272u. [DOI] [PubMed] [Google Scholar]
- 22.Odian G. Principles of Polymerization. John Wiley & Sons, Inc; Hoboken, NJ: 2004. [Google Scholar]
- 23.March J. Advanced Organic Chemistry. John Wiley & Sons Inc; New York: 1992. [Google Scholar]
- 24.Wanka G, Hoffmann H, Ulbricht W. Macromolecules. 1994;27:4145–4159. [Google Scholar]
- 25.Mortensen K, Brown W, Jorgensen E. Macromolecules. 1994;27:5654–5666. [Google Scholar]
- 26.Anseth KS, Bowman CN, Brannon-Peppas L. Biomaterials. 1996;17:1647–1657. doi: 10.1016/0142-9612(96)87644-7. [DOI] [PubMed] [Google Scholar]
- 27.Meissner B. Polymer. 2000;41:7827–7841. [Google Scholar]
- 28.Sperling LH. Physical Polymer Science. Wiley-Interscience; New York: 2001. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.