

Abstract
Tumor cells survive under conditions of nutrient deprivation by mechanisms that are not fully understood. The MUC1 oncoprotein is aberrantly overexpressed by most human carcinomas and blocks oxidative stress-induced death. The present studies show that MUC1 inhibits the induction of necrosis in response to the deprivation of glucose. MUC1 suppressed glucose deprivation-induced increases in reactive oxygen species (ROS) and thereby depletion of ATP and cell death. Cells respond to oxidative stress and energy depletion with the induction of autophagy. Our results demonstrate that MUC1 blocks depletion of ATP and sustains growth of glucose-deprived cells by a mechanism sensitive to the autophagy inhibitor, 3-methyladenine. Silencing expression of ATG7, a protein essential for the formation of autophagic vacuoles, also attenuated the MUC1-sustained increases in ATP and growth in response to glucose deprivation. Moreover, we found that MUC1 stimulates AMPK activation and thereby promotes lysosomal turnover of LC3-II, a marker of starvation-induced autophagic activity. These results indicate that MUC1 suppresses glucose deprivation-induced increases in ROS and thereby promotes ATP production and survival. The findings also indicate that the overexpression of MUC1 as found in human cancers could provide a survival advantage in micro-environments with low glucose levels.
Keywords: MUC1, glucose deprivation, oxidative stress, autophagy, ATG7, LC3-II, AMP-activated protein kinase
Introduction
Solid tumors are dependent on angiogenesis to sustain a supply of oxygen and nutrients. In the absence of sufficient new blood vessel formation, tumor cells need to adapt to environments that are hypoxic and deprived of nutrients. Activation of the hypoxia-inducible transcription factor 1α signaling pathway is a well characterized response of tumor cells to hypoxic conditions (1,2). As an integral part of the response to hypoxia, tumor cells use glycolysis as a primary mechanism for ATP production (3,4). Paradoxically, however, hypoxic environments are also often associated with depletion of glucose (5–7). Thus, like that for hypoxia, adaptation to glucose deprivation is an essential response for tumor cell survival. Glucose deprivation has been associated with increases in reactive oxygen species (ROS) (8,9) and depletion of ATP (10–12). In turn, intracellular ATP depletion promotes the induction of necrosis (13,14). These findings indicate that tumor cells can adapt to glucose deprivation by suppressing ROS-dependent ATP depletion and death.
Autophagy is a process in which intracellular proteins and organelles are targeted to lysosomes for recycling and energy production (15). Nutrient deprivation is associated with the activation of autophagy and, in turn, survival or the induction of cell death. Autophagy is also activated in the cellular response to oxidative stress (16–19). Studies of cancer cells deprived of nutrients have demonstrated that activation of autophagy is dependent on cell context (20). In this regard, activation of the Akt→mTOR pathway, as found in diverse cancers, is associated with suppression of autophagy (20). Conversely, inhibition of Akt promotes activation of the AMP-activated protein kinase (AMPK) (21) and thereby autophagy (22,23). Other studies have shown that Bcl-2, the BH3-only BINIP3 and the Bcl-2-interacting HSPIN1 protein regulate the induction of autophagy in transformed cells (24–26). However, little is known about what dictates whether autophagy results in survival or death of cancer cells. Cells deficient in Beclin 1, a protein involved in autophagic vesicle formation, are not resistant to cell death, consistent with autophagy being a survival pathway (27,28). In addition, recent work has shown that survival of solid tumors is promoted by autophagy and that inhibition of both autophagy and apoptosis results in necrosis (29). These findings indicate that autophagy is activated by certain tumor cells as a survival strategy in response to nutrient deprivation and depletion of ATP.
MUC1 is a heterodimeric mucin-type glycoprotein that is aberrantly overexpressed by diverse human carcinomas (30). The MUC1 N-terminal subunit (MUC1-N) is the mucin component of the dimer that consists of variable numbers of O-glycosylated tandem repeats (31,32). MUC1-N is tethered to the cell membrane by the MUC1 C-terminal transmembrane subunit (MUC1-C) (33). MUC1-C is also targeted to the nucleus and mitochondria of transformed cells (34–38). MUC1-C includes a cytoplasmic domain that functions as a substrate for c-Src, glycogen synthase kinase 3β, protein kinase Cδ and c-Abl (33,39–42). In addition, the MUC1-C cytoplasmic domain binds directly to and stabilizes the Wnt pathway effector β-catenin (39,43,44). MUC1-C also interacts directly with the IκB kinase β (IKKβ) and thereby activates the NF-κB p65 signaling pathway (45). Overexpression of MUC1-C protects cells against increased intracellular oxidant levels and H2O2-induced cell death by a mechanism involving in part activation of the FOXO3a transcription factor and the upregulation of catalase (46,47). Moreover, the MUC1-C cytoplasmic domain is sufficient for inducing transformation (34,44,48).
The present study demonstrates that MUC1 blocks the death response of human carcinoma cells to glucose deprivation. We show that MUC1 attenuates glucose deprivation-induced increases in ROS and depletion of ATP. The results also demonstrate that MUC1 promotes the induction of autophagy as a mechanism for maintaining ATP production and growth.
Materials and methods
Cell culture
Human HCT116/vector and HCT116/MUC1 colon cancer cells (35,49) were cultured in Dulbecco’s modified Eagle’s medium containing 25 or 1 mM glucose, 10% heat-inactivated, dialyzed fetal bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin and 2 mM L-glutamine. The cells were treated with 500 U/ml catalase (Sigma), 10 mM N-acetyl cysteine (NAC; Sigma), 10 mM 3-methyladenine (3-MA; Sigma), 50 μM chloroquine (CQ; Sigma) or 10 μM compound C (Calbiochem). Cell number was determined by trypan blue staining.
Immunoblot analysis
Cells were lysed as described (46) and analyzed by immunoblotting with anti-MUC1-C (Ab5; NeoMarkers Inc., Fremont, CA), anti-β-actin (Sigma), anti-MAP1LC3 (NanoTools; Freiburg, Germany), anti-phospho-Akt (anti-p-Akt) anti-Akt, anti-phospho-AMPKα (Thr-172) (p-AMPK) and anti-AMPKα (Cell Signaling Inc.). Antigen-antibody complexes were visualized by enhanced chemiluminescence (ECL; Amersham Biosciences).
Measurement of H2O2 levels
Cells were incubated with 5 μM DCFH-DA (Molecular Probes) for 20 min at 37°C. Fluorescence of oxidized DCF was measured at an excitation wavelength of 480 nm and an emission wavelength of 525 nm using a flow cytometer (BD Biosciences).
Measurement of ATP levels
ATP levels were measured using an ATP determination kit (Molecular Probes). Protein concentration was determined by the Bio-Rad protein assay.
Assessment of necrosis
Cells were incubated in 1 μg/ml propidium iodide/phosphate-buffered saline for 5 min at room temperature and then monitored by flow cytometry.
Reverse transcription polymerase chain reaction (RT-PCR)
Total cellular RNA was extracted in TRIzol dissolved in RNase-free water and incubated for 10 min at 55°C. ATG7 specific primers (5′-TTCACTAAAACAGATTGAAG-3′) and (5′-GTTTGAGATTTAGATCCACT-3′) were designed to amplify a 500-bp fragment. Primers for β-actin were used as a control (50). The RNA was reversed transcribed and amplified using SuperScript One-Step RT-PCR with Platinum Taq (Invitrogen Inc.). Amplified fragments were analyzed by electrophoresis in 2% agarose gels.
Silencing of ATG7
Cells were seeded at 2×105/60 mm dish. After 24 h, the cells were transfected with control siRNA or ATG7 siRNA pools (siGenome Smart pool reagents; Dharmacon RNA Technologies) in the presence of oligofectamine (Invitrogen).
Results
MUC1 protects against increases in ROS and loss of viability in response to glucose deprivation
Glucose deprivation is associated with the induction of oxidative stress (8,9). Other studies have demonstrated that MUC1 suppresses reactive oxygen species (ROS) levels constitutively and in the response to H2O2 treatment (46,47). HCT116/vector and HCT116/MUC1 cells were thus studied to assess the effects of MUC1 on ROS levels as a consequence of glucose deprivation. The results demonstrate that glucose deprivation is associated with ROS accumulation in HCT116/vector cells and that this response is suppressed in HCT116/MUC1 cells (Fig. 1A). Quantification of the flow cytometry data in the separately isolated clones confirmed that MUC1 suppresses glucose deprivation-induced increases in ROS (Fig. 1B). To assess the effects of MUC1 on cell viability in the response to glucose deprivation, HCT116/vector and HCT116/MUC1 cells were cultured in the presence of 25 or 1 mM glucose for 24 h. Analysis by PI staining demonstrated that glucose deprivation of HCT116/vector cells is associated with loss of cell membrane integrity as found in the induction of necrosis (Fig. 1C). Notably, however, this response to glucose deprivation was substantially decreased by MUC1 expression (Fig. 1C). Similar results were obtained in separately isolated HCT116/vector and HCT116/MUC1 clones (Fig. 1C) and in multiple experiments (Fig. 1D), indicating that the observed effects of MUC1 are not due to clonal selection. These findings indicate that glucose deprivation-induced oxidative stress and necrosis is suppressed by a MUC1-dependent mechanism.
Figure 1.

MUC1 blocks increases in ROS and necrosis of HCT116 cells in response to glucose deprivation. (A) HCT116/vector and HCT116/MUC1 cells were cultured in 25 or 1 mM glucose for 24 h. The cells were incubated with DCFH-DA for 20 min. Fluorescence of oxidized DCF was measured by flow cytometry. (B) The results are expressed as the relative ROS level (mean ± SD of three separate experiments) for cells in 1 mM glucose (open bars) compared to that in 25 mM glucose (solid bars). (C) HCT116/vector (clones A and B) and HCT116/MUC1 (clones A and B) cells were cultured in 25 or 1 mM glucose for 24 h. The cells were stained with PI and analyzed by flow cytometry. (D) The results are expressed as the percentage (mean ± SD of three separate experiments) necrotic cells for 25 mM (solid bars) or 1 mM (open bars) glucose.
MUC1 suppresses ROS-induced ATP depletion in the response to glucose deprivation
Glucose deprivation of HCT116/vector cells was associated with pronounced decreases in ATP at 48 and 72 h (Fig. 2A). Significantly, however, ATP levels were decreased at 24 h and then returned toward baseline by 72 h in the response of HCT116/MUC1 cells to glucose deprivation (Fig. 2A). Previous studies showed that MUC1 suppresses ROS levels in part by upregulating catalase expression (46). To determine if increases in ROS are responsible for the glucose deprivation-induced decreases in ATP, we treated the HCT116/vector cells with catalase. Analysis of H2O2 levels showed that catalase suppresses glucose deprivation-induced increases in ROS (Fig. 2B) and these findings were confirmed in repetitive experiments (Fig. 2C). Importantly, catalase also attenuated the glucose deprivation-induced decreases in ATP (Fig. 2D).
Figure 2.

MUC1 blocks ROS-induced depletion of ATP in the response to glucose deprivation. (A) HCT116/vector (open bars) and HCT116/MUC1 (solid bars) cells were cultured in 1 mM glucose for 0, 24, 48 and 72 h. Intracellular ATP levels are expressed as mean ± SD of three separate determinations relative to that at 0 h. (B–D) HCT116/vector cells were cultured in 25 mM glucose (solid bars), 1 mM glucose (open bars) or 1 mM glucose and 500 U/ml catalase (shaded bars) for 24 h. The cells were incubated with DCFH-DA and analyzed by flow cytometry (B). The results are expressed as the relative ROS level (mean ± SD of three separate experiments) compared to that obtained in 25 mM glucose (C). Intracellular ATP levels are expressed as mean ± SD of three experiments relative to that at 0 h (D).
MUC1 suppresses ROS-induced necrosis in the response to glucose deprivation
Depletion of ATP is associated with the induction of necrosis (13,14). In this regard, catalase treatment also suppressed the necrotic response to glucose deprivation (Fig. 3A and B). In further support for a ROS-mediated mechanism, treatment with the radical scavenger NAC blocked glucose deprivation-induced necrosis of HCT116/vector cells (Fig. 3C and D). These findings indicate that MUC1 suppresses ROS-induced ATP depletion and necrosis in the response to glucose deprivation.
Figure 3.


MUC1 blocks ROS-induced necrosis in the response to glucose deprivation. (A and B) HCT116/vector and HCT116/MUC1 cells were cultured in 25, 1 or 1 mM glucose and 500 U/ml catalase for 24 h. The cells were stained with PI and analyzed by flow cytometry (A). The results obtained with the HCT116/vector cells cultured in 25 mM glucose (solid bars), 1 mM glucose (open bars) or 1 mM glucose and catalase (shaded bars) are expressed as the percentage (mean ± SD of three experiments) necrotic cells (B). (C and D) HCT116/vector and HCT116/MUC1 cells were cultured in 25, 1 or 1 mM glucose and 10 mM NAC for 24 h. The cells were stained with PI and analyzed by flow cytometry (C). The results obtained with the indicated cells cultured in 25 mM glucose (solid bars), 1 mM glucose (open bars) or 1 mM glucose and NAC (shaded bars) are expressed as the percentage (mean ± SD of three experiments) necrotic cells (D).
MUC1 promotes autophagy
Cells respond to oxidative stress and energy depletion with the induction of autophagy (16–18). To determine whether the MUC1-dependent regulation of ATP levels is due to autophagy, cells were deprived of glucose in the presence of 3-MA, an inhibitor of autophagy (51). Treatment of glucose-deprived HCT116/MUC1 cells with 3-MA was associated with a progressive decrease in ATP levels (Fig. 4A) and cell number (Fig. 4B). ATG7 is an essential protein for the formation of autophagic vacuoles (52). To determine whether ATG7 is necessary for MUC1-induced increases in ATP levels, we transfected the HCT116/MUC1 cells with non-specific (NS) siRNA or ATG7 siRNA pools. Analysis of the transfectants confirmed that ATG7 expression is reduced by the ATG7 siRNA (Fig. 4C). Moreover, consistent with involvement of autophagy, silencing ATG7 was associated with attenuation of ATP levels (Fig. 4D, left) and proliferation (Fig. 4D, right) in the response of HCT116/MUC1 cells to glucose deprivation. Microtubule-associated protein-1 light chain-3 (LC3), associates with autophagosomes, is activated in part by ATG7 and is cleaved to generate the soluble LC3-I form (53,54). In turn, LC3-I is modified to the membrane-bound LC3-phospholipid conjugate (LC3-II). Lysosomal turnover of LC3-II is thus used as a measure of starvation-induced autophagic activity (55,56). To determine whether MUC1 affects the turnover of LC3-II, the HCT116 cells were treated with chloroquine (CQ), a lysosomal acidification inhibitor, to block lysosomal proteolysis. LC3-II levels were substantially increased in CQ-treated HCT116/MUC1 cells, as compared to that in HCT116/vector cells (Fig. 4E). These results indicate that MUC1 promotes the induction of autophagy and thereby ATP production and proliferation.
Figure 4.

MUC1 promotes autophagy in the response to glucose deprivation. (A and B) HCT116/MUC1 cells were cultured in 1 mM glucose and the absence (solid bars and squares) or presence (open bars and squares) of 10 mM 3-MA for 0–72 h. Intracellular ATP levels are expressed as mean ± SD of three separate experiments relative to that at 0 h (A). Cell number as assessed by trypan blue staining is expressed as the mean ± SD of three separate experiments (B). (C and D) HCT116/MUC1 cells were transfected with a non-specific (NS) siRNA or ATG7 siRNA for 5 days. Total cellular RNA was amplified with ATG7- and β-actin-specific primers (C). Intracellular ATP levels for ATG7 siRNA transfected cells (open bars) relative to that obtained with the NS siRNA transfected cells (solid bars) are expressed as mean ± SD of three separate experiments (D, left). Numbers of NS siRNA (solid bars) and ATG7 siRNA (open bars) transfected cells as assessed by trypan-blue staining is expressed as the mean ± SD of three separate experiments (D, right). (E) HCT116/vector and HCT116/MUC1 cells were cultured in 1 mM glucose for 24 h in the absence and presence of 50 μM CQ for the last 10 h. Lysates were immunoblotted with the indicated antibodies.
MUC1 promotes autophagy by a AMPK-dependent mechanism in the response to glucose deprivation
Previous work showed that MUC1 constitutively suppresses Akt activity in HCT116 (47). Glucose deprivation of HCT116/vector cells was associated with increases in phospho-Akt levels at 24 h before the onset of necrosis (Fig. 5A). By contrast, phospho-Akt levels were suppressed in HCT116/MUC1 cells that were deprived of glucose for 24 h (Fig. 5A). In addition, compared to that found in HCT116/vector cells at 24 h, phospho-Akt levels remained lower in HCT116/MUC1 cells at 48 and 72 h of glucose deprivation (Fig. 5A). Akt inhibits activation of the AMPK (21) that has been shown to stimulate autophagy in yeast and mammalian cells (22,23). Consequently, we asked if MUC1 affects activation of AMPK by glucose deprivation. Consistent with the suppression of phospho-Akt, the results show that AMPK is activated in HCT116 cells by a MUC1-dependent mechanism (Fig. 5B). To determine whether the activation of AMPK confers the induction of autophagy, HCT116/MUC1 cells were deprived of glucose in the presence of CQ alone or CQ and compound C, a specific AMPK inhibition (57). Activation of AMPK by glucose deprivation was comparable in the absence and presence of CQ (Fig. 5C). As expected, the addition of compound C was associated with inhibition of AMPK activity (Fig. 5C). The results also demonstrate that inhibition of AMPK with compound C blocks glucose deprivation-induced increases in LC3-II levels in HCT116/MUC1 cells (Fig. 5D). These findings indicate that MUC1 promotes the induction of autophagy in a survival response to glucose deprivation by an AMPK-dependent mechanism.
Figure 5.
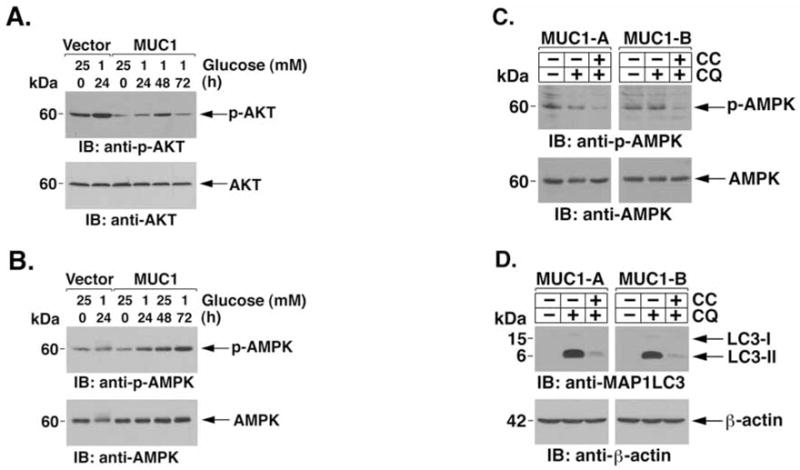
MUC1 promotes autophagy in response to glucose deprivation by an AMPK-dependent mechanism. (A and B) HCT116/vector-A and HCT116/MUC1-A cells were cultured in the presence of 25 or 1 mM glucose for the indicated times. Lysates were immunoblotted with the indicated antibodies. (C and D) HCT116/MUC1 cells were cultured in 1 mM glucose in the absence or presence of 10 μM compound C for 24 h. CQ was added as indicated during the last 10 h. Lysates were immunoblotted with the indicated antibodies.
Discussion
MUC1 blocks glucose deprivation-induced necrosis
MUC1 is normally expressed on the apical borders of secretory epithelial cells (30). Carcinoma cells that have lost polarity aberrantly express MUC1 at high levels over the entire cell surface (30). Overexpression of MUC1 is associated with targeting of MUC1-C, the C-terminal subunit, to the nucleus and mitochondria, and protection of cells against stress-induced apoptosis (35,36,38,46,47). Cancer cells succumb by mechanisms of apoptosis, necrosis and autophagy (58). The present studies demonstrate that MUC1 blocks the induction of necrosis in the response of HCT116 cells to glucose deprivation. Necrosis is the end result of ATP depletion that occurs with loss of nutrients (13,14,59). In this context, like the induction of necrosis, glucose deprivation was associated with a marked depletion of ATP in the absence, but not in the presence, of MUC1. Moreover, glucose deprivation of cells expressing MUC1 was associated with increases in ATP, indicating that MUC1 has a functional role in regulating energy balance. These findings also indicated that MUC1 blocks glucose deprivation-induced necrosis by attenuating ATP depletion.
MUC1 attenuates ROS-induced depletion of ATP and death in response to glucose deprivation
Glucose deprivation is associated with increased mitochondrial generation of ROS and oxidative stress-induced cell death (8,9,60,61). In turn, increases in ROS contribute to the depletion of ATP by disrupting mitochondrial function. Notably, MUC1 suppresses basal and H2O2-induced intracellular ROS levels in part by the upregulation of catalase (46,47). Treatment of the MUC1-null HCT116/vector cells with exogenous catalase attenuated glucose deprivation-induced increases in ROS. Catalase also attenuated glucose deprivation-induced ATP depletion and necrosis, indicating that increases in ROS are in large part responsible for these responses. These results thus support a model in which MUC1 upregulates catalase and potentially other enzymes, such as GPx1 and SOD (46), involved in the defense against oxidative stress and thereby attenuates increases in ROS that deplete ATP and induce necrosis. MUC1 may also suppress ROS production by other mechanisms. For example, MUC1-C is targeted to the mitochondrial outer membrane and blocks stress-induced decreases in the mitochondrial transmembrane potential (35,37,47). Thus, MUC1-C may directly suppress mitochondrial dysfunction and the increased generation of ROS.
MUC1 promotes autophagy in the survival response to glucose deprivation
Autophagy is activated in the cellular response to nutrient deprivation (15). A fundamental question for cancer cells is whether autophagy functions as a survival or death response. Increasing evidence has linked oxidative stress to the induction of autophagy (16–19,62). In addition, autophagic cell death is induced by degradation of catalase and ROS accumulation (63). Our findings that MUC1 protects against glucose deprivation-induced ROS accumulation and depletion of ATP suggested that this response could be mediated in part by autophagy. In this regard, we found that ATP levels are sustained by a MUC1-dependent mechanism in glucose-deprived cells, supporting the involvement of a process, such as autophagy, that generates ATP. Indeed, treatment with 3-MA or silencing ATG7 was associated with abrogation of the effects of MUC1 on sustaining ATP levels and growth in the response to glucose deprivation. We also found that MUC1 expression is associated with increased lysosomal turnover of LC3-II, a marker of autophagic activity (55,56). Our results indicate that the mechanism by which MUC1 promotes autophagy in the response to glucose deprivation involves stimulation of AMPK activity. In this regard, MUC1 suppresses activation of the PI3K→phospho-Akt pathway in HCT116 cells (47). Suppression of PI3K→Akt signaling is associated with activation of AMPK and thereby autophagy (20–23). Consistent with these findings, the results show that MUC1 stimulates AMPK activation in the response to glucose deprivation and that AMPK is necessary for the induction of autophagy. Our findings also indicate that MUC1 promotes autophagy in a survival, as opposed to a death, response to glucose deprivation. Autophagy induces cell death by ROS accumulation (63). The present results support a model in which MUC1 suppresses glucose deprivation-induced increases in ROS and thereby promotes ATP production and survival.
Does the MUC1-mediated response to glucose deprivation represent a physiologic defense mechanism that is exploited by human tumors?
MUC1 is part of the mucous barrier that physically protects epithelial cells from stress-induced damage. With shedding of MUC1-N, the mucin component of the heterodimer, into the mucous barrier, MUC1-C remains at the cell surface to signal stress to the interior of the cell. The epithelial stress response is associated with loss of polarity and activation of a repair and survival program (64). Thus, under physiologic conditions, targeting of MUC1-C to the interior of the cell could contribute to protection of epithelial cells against depletion of ATP by promoting autophagy in the presence of a decrease in availability of nutrients. In carcinoma cells, MUC1-C is constitutively expressed at high levels in the cytosol, nucleus and mitochondria. Human tumors that overexpress MUC1 may therefore have a survival advantage to glucose deprivation by exploiting mechanisms, such as autophagy, that evolved for the survival and repair of epithelia.
Acknowledgments
This work was supported by Grants CA42802 and CA97098 awarded by the National Cancer Institute.
Abbreviations
- MUC1
mucin 1
- MUC1-N
MUC1 N-terminal subunit
- MUC1-C
MUC1 C-terminal subunit
- ROS
reactive oxygen species
- AMPK
AMP-activated protein kinase
- NAC
N-acetyl cysteine
- 3-MA
3-methyladenine
- LC3
microtubule-associated protein-1 light chain-3
- CQ
chloroquine
References
- 1.Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3:721–732. doi: 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- 2.Harris AL. Hypoxia - a key regulatory factor in tumour growth. Nat Rev Cancer. 2002;2:38–47. doi: 10.1038/nrc704. [DOI] [PubMed] [Google Scholar]
- 3.Dang CV, Semenza GL. Oncogenic alterations of metabolism. Trends Biochem Sci. 1999;24:68–72. doi: 10.1016/s0968-0004(98)01344-9. [DOI] [PubMed] [Google Scholar]
- 4.Seagroves TN, Ryan HE, Lu H, et al. Transcription factor HIF-1 is a necessary mediator of the pasteur effect in mammalian cells. Mol Cell Biol. 2001;21:3436–3444. doi: 10.1128/MCB.21.10.3436-3444.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Helmlinger G, Yuan F, Dellian M, Jain RK. Interstitial pH and pO2 gradients in solid tumors in vivo: high-resolution measurements reveal a lack of correlation. Nat Med. 1997;3:177–182. doi: 10.1038/nm0297-177. [DOI] [PubMed] [Google Scholar]
- 6.Griffiths JR, McIntyre DJ, Howe FA, Stubbs M. Why are cancers acidic? A carrier-mediated diffusion model for H+ transport in the interstitial fluid. Novartis Found Symp. 2001;240:46–62. doi: 10.1002/0470868716.ch4. discussion 62–67, 152–153. [DOI] [PubMed] [Google Scholar]
- 7.Secomb TW, Hsu R, Braun RD, Ross JR, Gross JF, Dewhirst MW. Theoretical simulation of oxygen transport to tumors by three-dimensional networks of microvessels. Adv Exp Med Biol. 1998;454:629–634. doi: 10.1007/978-1-4615-4863-8_74. [DOI] [PubMed] [Google Scholar]
- 8.Jelluma N, Yang X, Stokoe D, Evan GI, Dansen TB, Haas-Kogan DA. Glucose withdrawal induces oxidative stress followed by apoptosis in glioblastoma cells but not in normal human astrocytes. Mol Cancer Res. 2006;4:319–330. doi: 10.1158/1541-7786.MCR-05-0061. [DOI] [PubMed] [Google Scholar]
- 9.Ahmad IM, Aykin-Burns N, Sim JE, et al. Mitochondrial O2*− and H2O2 mediate glucose deprivation-induced stress in human cancer cells. J Biol Chem. 2005;280:4254–4263. doi: 10.1074/jbc.M411662200. [DOI] [PubMed] [Google Scholar]
- 10.Xu RH, Pelicano H, Zhou Y, et al. Inhibition of glycolysis in cancer cells: a novel strategy to overcome drug resistance associated with mitochondrial respiratory defect and hypoxia. Cancer Res. 2005;65:613–621. [PubMed] [Google Scholar]
- 11.Buzzai M, Bauer DE, Jones RG, et al. The glucose dependence of Akt-transformed cells can be reversed by pharmacologic activation of fatty acid beta-oxidation. Oncogene. 2005;24:4165–4173. doi: 10.1038/sj.onc.1208622. [DOI] [PubMed] [Google Scholar]
- 12.Laderoute KR, Amin K, Calaoagan JM, et al. 5′-AMP-activated protein kinase (AMPK) is induced by low-oxygen and glucose deprivation conditions found in solid-tumor microenvironments. Mol Cell Biol. 2006;26:5336–5347. doi: 10.1128/MCB.00166-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eguchi Y, Shimizu S, Tsujimoto Y. Intracellular ATP levels determine cell death fate by apoptosis or necrosis. Cancer Res. 1997;57:1835–1840. [PubMed] [Google Scholar]
- 14.Leist M, Single B, Castoldi AF, Kuhnle S, Nicotera P. Intracellular adenosine triphosphate (ATP) concentration: a switch in the decision between apoptosis and necrosis. J Exp Med. 1997;185:1481–1486. doi: 10.1084/jem.185.8.1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290:1717–1721. doi: 10.1126/science.290.5497.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim E, Sohn S, Kwon H, et al. Sodium selenite induces superoxide-mediated mitochondrial damage and subsequent autophagic cell death in malignant glioma cells. Cancer Res. 2007;67:6314–6324. doi: 10.1158/0008-5472.CAN-06-4217. [DOI] [PubMed] [Google Scholar]
- 17.Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007;3:371–373. doi: 10.1038/sj.emboj.7601623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gao M, Yeh P, Lu Y, et al. OSU-03012, a novel celecoxib derivative, induces reactive oxygen species-related autophagy in hepatocellular carcinoma. Cancer Res. 2008;68:9348–9357. doi: 10.1158/0008-5472.CAN-08-1642. [DOI] [PubMed] [Google Scholar]
- 19.Kiffin R, Bandyopadhyay U, Cuervo AM. Oxidative stress and autophagy. Antioxid Redox Signal. 2006;8:152–162. doi: 10.1089/ars.2006.8.152. [DOI] [PubMed] [Google Scholar]
- 20.Kondo Y, Kanzawa T, Sawaya R, Kondo S. The role of autophagy in cancer development and response to therapy. Nat Rev Cancer. 2005;5:726–734. doi: 10.1038/nrc1692. [DOI] [PubMed] [Google Scholar]
- 21.Hahn-Windgassen A, Nogueira V, Chen CC, Skeen JE, Sonenberg N, Hay N. Akt activates the mammalian target of rapamycin by regulating cellular ATP level and AMPK activity. J Biol Chem. 2005;280:32081–32089. doi: 10.1074/jbc.M502876200. [DOI] [PubMed] [Google Scholar]
- 22.Meley D, Bauvy C, Houben-Weerts JH, et al. AMP-activated protein kinase and the regulation of autophagic proteolysis. J Biol Chem. 2006;281:34870–34879. doi: 10.1074/jbc.M605488200. [DOI] [PubMed] [Google Scholar]
- 23.Wang Z, Wilson WA, Fujino MA, Roach PJ. Antagonistic controls of autophagy and glycogen accumulation by Snf1p, the yeast homolog of AMP-activated protein kinase, and the cyclin-dependent kinase Pho85p. Mol Cell Biol. 2001;21:5742–5752. doi: 10.1128/MCB.21.17.5742-5752.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vande Velde C, Cizeau J, Dubik D, et al. BNIP3 and genetic control of necrosis-like cell death through the mitochondrial permeability transition pore. Mol Cell Biol. 2000;20:5454–5468. doi: 10.1128/mcb.20.15.5454-5468.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saeki K, Yuo A, Okuma E, et al. Bcl-2 down-regulation causes autophagy in a caspase-independent manner in human leukemic HL60 cells. Cell Death Differ. 2000;7:1263–1269. doi: 10.1038/sj.cdd.4400759. [DOI] [PubMed] [Google Scholar]
- 26.Yanagisawa H, Miyashita T, Nakano Y, Yamamoto D. HSpin1, a transmembrane protein interacting with Bcl-2/Bcl-xL, induces a caspase-independent autophagic cell death. Cell Death Differ. 2003;10:798–807. doi: 10.1038/sj.cdd.4401246. [DOI] [PubMed] [Google Scholar]
- 27.Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci USA. 2003;100:15077–15082. doi: 10.1073/pnas.2436255100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qu X, Yu J, Bhagat G, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest. 2003;112:1809–1820. doi: 10.1172/JCI20039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Degenhardt K, Mathew R, Beaudoin B, et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell. 2006;10:51–64. doi: 10.1016/j.ccr.2006.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kufe D, Inghirami G, Abe M, Hayes D, Justi-Wheeler H, Schlom J. Differential reactivity of a novel monoclonal antibody (DF3) with human malignant versus benign breast tumors. Hybridoma. 1984;3:223–232. doi: 10.1089/hyb.1984.3.223. [DOI] [PubMed] [Google Scholar]
- 31.Gendler S, Taylor-Papadimitriou J, Duhig T, Rothbard J, Burchell JA. A highly immunogenic region of a human polymorphic epithelial mucin expressed by carcinomas is made up of tandem repeats. J Biol Chem. 1988;263:12820–12823. [PubMed] [Google Scholar]
- 32.Siddiqui J, Abe M, Hayes D, Shani E, Yunis E, Kufe D. Isolation and sequencing of a cDNA coding for the human DF3 breast carcinoma-associated antigen. Proc Natl Acad Sci USA. 1988;85:2320–2323. doi: 10.1073/pnas.85.7.2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Merlo G, Siddiqui J, Cropp C, et al. DF3 tumor-associated antigen gene is located in a region on chromosome 1q frequently altered in primary human breast cancer. Cancer Res. 1989;49:6966–6971. [PubMed] [Google Scholar]
- 34.Li Y, Liu D, Chen D, Kharbanda S, Kufe D. Human DF3/MUC1 carcinoma-associated protein functions as an oncogene. Oncogene. 2003;22:6107–6110. doi: 10.1038/sj.onc.1206732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ren J, Agata N, Chen D, et al. Human MUC1 carcinoma-associated protein confers resistance to genotoxic anti-cancer agents. Cancer Cell. 2004;5:163–175. doi: 10.1016/s1535-6108(04)00020-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wei X, Xu H, Kufe D. Human MUC1 oncoprotein regulates p53-responsive gene transcription in the genotoxic stress response. Cancer Cell. 2005;7:167–178. doi: 10.1016/j.ccr.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 37.Ren J, Bharti A, Raina D, Chen W, Ahmad R, Kufe D. MUC1 oncoprotein is targeted to mitochondria by heregulin-induced activation of c-Src and the molecular chaperone HSP90. Oncogene. 2006;25:20–31. doi: 10.1038/sj.onc.1209012. [DOI] [PubMed] [Google Scholar]
- 38.Leng Y, Cao C, Ren J, et al. Nuclear import of the MUC1-C oncoprotein is mediated by nucleoporin Nup62. J Biol Chem. 2007;282:19321–19330. doi: 10.1074/jbc.M703222200. [DOI] [PubMed] [Google Scholar]
- 39.Li Y, Bharti A, Chen D, Gong J, Kufe D. Interaction of glycogen synthase kinase 3β with the DF3/MUC1 carcinoma-associated antigen and β-catenin. Mol Cell Biol. 1998;18:7216–7224. doi: 10.1128/mcb.18.12.7216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li Y, Kuwahara H, Ren J, Wen G, Kufe D. The c-Src tyrosine kinase regulates signaling of the human DF3/MUC1 carcinoma-associated antigen with GSK3β and β-catenin. J Biol Chem. 2001;276:6061–6064. doi: 10.1074/jbc.C000754200. [DOI] [PubMed] [Google Scholar]
- 41.Ren J, Li Y, Kufe D. Protein kinase Cδ regulates function of the DF3/MUC1 carcinoma antigen in β-catenin signaling. J Biol Chem. 2002;277:17616–17622. doi: 10.1074/jbc.M200436200. [DOI] [PubMed] [Google Scholar]
- 42.Raina D, Ahmad R, Kumar S, et al. MUC1 oncoprotein blocks nuclear targeting of c-Abl in the apoptotic response to DNA damage. EMBO J. 2006;25:3774–3783. doi: 10.1038/sj.emboj.7601263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yamamoto M, Bharti A, Li Y, Kufe D. Interaction of the DF3/MUC1 breast carcinoma-associated antigen and β-catenin in cell adhesion. J Biol Chem. 1997;272:12492–12494. doi: 10.1074/jbc.272.19.12492. [DOI] [PubMed] [Google Scholar]
- 44.Huang L, Chen D, Liu D, Yin L, Kharbanda S, Kufe D. MUC1 oncoprotein blocks GSK3β-mediated phosphorylation and degradation of β-catenin. Cancer Res. 2005;65:10413–10422. doi: 10.1158/0008-5472.CAN-05-2474. [DOI] [PubMed] [Google Scholar]
- 45.Ahmad R, Raina D, Trivedi V, et al. MUC1 oncoprotein activates the IκB kinase β complex and constitutive NF-κB signaling. Nat Cell Biol. 2007;9:1419–1427. doi: 10.1038/ncb1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yin L, Kufe D. Human MUC1 carcinoma antigen regulates intracellular oxidant levels and the apoptotic response to oxidative stress. J Biol Chem. 2003;278:35458–35464. doi: 10.1074/jbc.M301987200. [DOI] [PubMed] [Google Scholar]
- 47.Yin L, Huang L, Kufe D. MUC1 oncoprotein activates the FOXO3a transcription factor in a survival response to oxidative stress. J Biol Chem. 2004;279:45721–45727. doi: 10.1074/jbc.M408027200. [DOI] [PubMed] [Google Scholar]
- 48.Huang L, Ren J, Chen D, Li Y, Kharbanda S, Kufe D. MUC1 cytoplasmic domain coactivates Wnt target gene transcription and confers transformation. Cancer Biol Ther. 2003;2:702–706. [PubMed] [Google Scholar]
- 49.Li Y, Ren J, Yu W-H, et al. The EGF receptor regulates interaction of the human DF3/MUC1 carcinoma antigen with c-Src and β-catenin. J Biol Chem. 2001;276:35239–35242. doi: 10.1074/jbc.C100359200. [DOI] [PubMed] [Google Scholar]
- 50.Ben-Ezra J, Johnson DA, Rossi J, Cook N, Wu A. Effect of fixation on the amplification of nucleic acids from paraffin-embedded material by the polymerase chain reaction. J Histochem Cytochem. 1991;39:351–354. doi: 10.1177/39.3.1704393. [DOI] [PubMed] [Google Scholar]
- 51.Mizushima N, Ohsumi Y, Yoshimori T. Autophagosome formation in mammalian cells. Cell Struct Funct. 2002;27:421–429. doi: 10.1247/csf.27.421. [DOI] [PubMed] [Google Scholar]
- 52.Yu L, Alva A, Su H, et al. Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science. 2004;304:1500–1502. doi: 10.1126/science.1096645. [DOI] [PubMed] [Google Scholar]
- 53.Kabeya Y, Mizushima N, Ueno T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Asanuma K, Tanida I, Shirato I, et al. MAP-LC3, a promising autophagosomal marker, is processed during the differentiation and recovery of podocytes from PAN nephrosis. FASEB J. 2003;17:1165–1167. doi: 10.1096/fj.02-0580fje. [DOI] [PubMed] [Google Scholar]
- 55.Schmid D, Pypaert M, Munz C. Antigen-loading compartments for major histocompatibility complex class II molecules continuously receive input from autophagosomes. Immunity. 2007;26:79–92. doi: 10.1016/j.immuni.2006.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tanida I, Minematsu-Ikeguchi N, Ueno T, Kominami E. Lysosomal turnover, but not a cellular level, of endogenous LC3 is a marker for autophagy. Autophagy. 2005;1:84–91. doi: 10.4161/auto.1.2.1697. [DOI] [PubMed] [Google Scholar]
- 57.Zhou G, Myers R, Li Y, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–1174. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Edinger AL, Thompson CB. Death by design: apoptosis, necrosis and autophagy. Curr Opin Cell Biol. 2004;16:663–669. doi: 10.1016/j.ceb.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 59.Nicotera P, Leist M, Ferrando-May E. Intracellular ATP, a switch in the decision between apoptosis and necrosis. Toxicol Lett. 1998:102–103. 139–142. doi: 10.1016/s0378-4274(98)00298-7. [DOI] [PubMed] [Google Scholar]
- 60.Blackburn RV, Spitz DR, Liu X, et al. Metabolic oxidative stress activates signal transduction and gene expression during glucose deprivation in human tumor cells. Free Radic Biol Med. 1999;26:419–430. doi: 10.1016/s0891-5849(98)00217-2. [DOI] [PubMed] [Google Scholar]
- 61.Lee YJ, Galoforo SS, Berns CM, et al. Glucose deprivation-induced cytotoxicity and alterations in mitogen-activated protein kinase activation are mediated by oxidative stress in multidrug-resistant human breast carcinoma cells. J Biol Chem. 1998;273:5294–5299. doi: 10.1074/jbc.273.9.5294. [DOI] [PubMed] [Google Scholar]
- 62.Kiffin R, Christian C, Knecht E, Cuervo AM. Activation of chaperone-mediated autophagy during oxidative stress. Mol Biol Cell. 2004;15:4829–4840. doi: 10.1091/mbc.E04-06-0477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yu L, Wan F, Dutta S, et al. Autophagic programmed cell death by selective catalase degradation. Proc Natl Acad Sci USA. 2006;103:4952–4957. doi: 10.1073/pnas.0511288103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vermeer PD, Einwalter LA, Moninger TO, et al. Segregation of receptor and ligand regulates activation of epithelial growth factor receptor. Nature. 2003;422:322–326. doi: 10.1038/nature01440. [DOI] [PubMed] [Google Scholar]