

Abstract
Light sheet microscopy became a powerful tool in life sciences. Often, however, the sheet geometry is fixed, whereas it would be advantageous to adjust the sheet geometry to specimens of different dimensions. Therefore we developed an afocal cylindrical zoom lens system comprising only 5 lenses and a total system length of less than 160 mm. Two movable optical elements were directly coupled, so that the zoom factor could be adjusted from 1x to 6.3x by a single motor. Using two different illumination objectives we achieved a light sheet thickness ranging from 2.4 µm to 36 µm corresponding to lateral fields of 54 µm to 12.3 mm, respectively. Polytene chromosomes of salivary gland cell nuclei of C.tentans larvae were imaged in vivo to demonstrate the advantages in image contrast by imaging with different light sheet dimensions.
OCIS codes: (180.0180) Microscopy, (170.0170) Medical optics and biotechnology, (170.2520) Fluorescence microscopy, (170.2945) Illumination design
1. Introduction
Light sheet microscopy is a versatile tool for imaging of intact fluorescent biological objects in three dimensions. The fundamental principle of light sheet microscopy is the illumination of the sample by a thin, focused light sheet perpendicular to the detection axis. In this manner only the focal plane of the detection objective is illuminated and no out-of-focus fluorescence compromises the signal quality. The optical sectioning effect results in an excellent contrast and low photo-toxicity [1,2].
The light sheet technique is applicable to various specimens ranging from single cells to complete insect and mouse embryos [3–10]. These specimens cover several orders of magnitude in diameter and require widely differing light sheet geometries for appropriate illumination. Usually light sheet microscopes are therefore highly specialized to observe a certain specimen type. In most cases a fixed light sheet geometry limits the observable sample size. A complete mouse embryo requires a light sheet configuration that is several orders of magnitude larger than that required for a single cell nucleus [7,11]. However, a large lateral field-of-view automatically produces a large axial extension of the sheet due to the laws of Gaussian beam optics [12]. Similarly, very thin light sheets yield optimal optical sectioning but have small usable field-of-views. It is important to carefully adjust both parameters to achieve optimal imaging quality in each experiment. The focused beam waist is inversely proportional to the radius of the incident illumination beam. Hence, for a continuous and flexible adjustment of the focal waist an afocal cylindrical zoom beam expander can be used to modify the extension of the focus range. Neither power loss nor refractive distortions of the beam profile occur as it would happen by use of vertical and horizontal slit apertures for beam diameter definition [13].
Here we present a cylindrical zoom lens system for light sheet shaping, so that various specimens of different sizes can be observed using a single light sheet microscope. The system comprises only 5 commercially available cylindrical lenses and features a compact system length of 160 mm only. A zoom range from 1.0x to 6.3x magnification of the incident beam in one dimension is achieved. The system performance using different zoom factors was demonstrated by imaging fluorescently labeled nuclei and polytene chromosomes in salivary gland cells of Chironomus tentans larvae.
2. Results
2.1 Optical Setup
The light sheet microscopic setup is shown in Fig. 1 . Multiple laser lines (L1-L3 / λ0 = 488 nm, 532 nm and 638 nm / Sapphire-100, Coherent, Germany, LaNova50 Green, Lasos, Germany and Cube635-25C, Coherent, Germany) were combined by appropriate dichroic mirrors and were guided through an acousto-optical-tunable-filter (AOTF, AOTF.nC 1001, Opto-Electronics, France). The AOTF controlled the transmitted intensity and selected the desired wavelength. Behind the AOTF the light was coupled into a mono-mode fiber (kineFlex, Point Source, Hamble, UK) and guided to the microscope. Deviations from the Gaussian TEM00-mode were removed by a spatial filter, which consisted of two achromatic lenses (f = 30 mm) forming a 1:1 Keplerian telescope with a small pinhole (d = 25 µm) at the focal point.
Fig. 1.
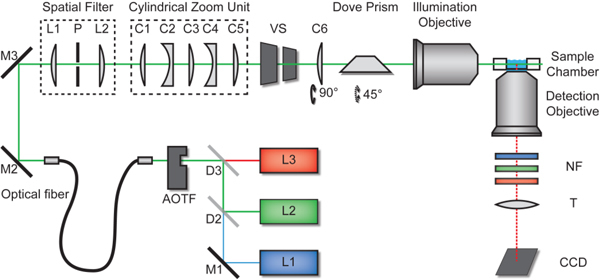
Light sheet microscope setup. Laser light (L1 - L3) was guided into the illumination beam path via dichroic beam splitters (D1 and D2), an optical fiber and mirrors. Laser wavelength and intensity was controlled by an acousto-optical-tunable-filter (AOTF). The two achromatic lenses L1 and L2 (f = 30 mm) and the pinhole P formed a spatial filter. The cylindrical zoom unit consisted of five lenses (C1 - C5). A further cylindrical lens (C6) oriented perpendicular to the zoom lens unit focused the beam into the back-focal-plane of the illumination objective, which focused the light sheet into the sample chamber. A vertical slit (VS) was used to control the width of the light sheet. The sample could be moved by a three-axis motorized sample scanner and the illumination objective was motorized along the optical axis. The detection unit comprised the detection objective, small bandwidth notch filters (NF), tube lens (T) and CCD-camera.
The key component of the illumination beam path was the cylindrical zoom unit, which comprised five cylindrical lenses (C1-C5, see Table 1 ; from Thorlabs, USA). The three convex lenses were fixed, and the two concave lenses were movable (Fig. 2a and Fig. 2b). The two concave lenses C2 and C4 were mounted on a micrometer stage and directly coupled. Thus the distance between the two lenses remained constant and the zoom factor Z could be modified by a single motor. A light sheet was generated inside a small water chamber by focusing the resulting elliptically shaped Gaussian beam with the illumination objective (10x Mitutoyo Plan Apochromat with NA 0.28) [7] or an achromatic lens (focal length 85 mm, Edmund Optics). Since the focused spot size was inversely proportional to the incoming beam diameter [12], the zoom unit regulated the light sheet thickness and length. The cylindrical lens C6 was arranged perpendicular to the zoom unit and affected only the width of the light sheet. The beam was focused into the back focal plane of the illumination objective, which therefore formed together with lens C6 a Keplerian telescope resulting in a collimated width (along the y-axis) of the light sheet inside the water chamber (Fig. 2c). A vertical slit (VS) was used to control the width (y-direction) of the light sheet from 20 µm to 120 µm full width at half maximum (FWHM).Fluorescence light emitted from the sample was collected by the detection objective lens and imaged onto a CCD camera (Clara, pixel size (6.45 µm)2 or iXonDU897, pixel size (16 µm)2, Andor Technologies, Belfast, Ireland). Excitation laser light was removed by Notch filters (NF, Semrock, Rochester, USA). Three motorized stages (M112-12s, PI, Karlsruhe, Germany) were used to adjust the sample chamber position in all three spatial dimensions, and a fourth stage was used for positioning the illumination objective along the optical axis. This was necessary because the air spacing between this objective and the water chamber changed when the sample chamber was moved in x-direction along the illumination axis. This caused a displacement of the focal position of the sheet of about 34 µm per 100 µm movement of the chamber that had to be corrected.
Table 1. Cylindrical lenses of the zoom unit.
| Lens [Thorlabs catalogue] | Focal length [mm] | |
|---|---|---|
| C1 | LJ1934L1 | 150 |
| C2 | LK1006L1 | −22.2 |
| C3 | LJ1075L1 | 25 |
| C4 | LK1085L1 | −20 |
| C5 | LJ1640L1 | 130 |
| C6 | LJ1934L1 | 150 |
Fig. 2.
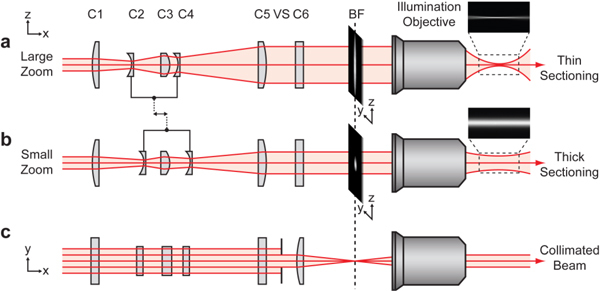
Cylindrical zoom beam expander. The configurations for a large zoom (a) and a small zoom (b) are shown. For a large zoom the beam diameter incident at the illumination objective is large in z-direction and hence the optical sectioning thickness is decreased compared to a small zoom. Lenses C2 and C4 are coupled and mounted together on a micrometer stage (not shown), so that a single motion readjusts the zoom factor. (c, top view) The incident beam diameter along y-direction is limited by a vertical slit aperture (VS), which defines the lateral width of the light sheet. The beam is focused into the back-focal-plane (BF) of the illumination objective by cylindrical lens C6. Exemplary images of the produced light sheets were shown on the right hand side (see section 2.3).
Furthermore, the position of the focus shifted for the different zoom configurations due to spherical aberration introduced by focusing with an air objective through glass walls into a water chamber. For the complete zoom range the focus shift was approximately 300 µm. This could also be corrected by an appropriate positioning of the illumination objective.
The setup was realized using a commercial inverted microscope (Nikon TI-U, Düsseldorf, Germany). Installation of the sheet illumination in a commercial microscope stage increased usability of the setup, and allowed to readily exchange detection objectives or to use different contrast modes.
2.2 Zoom lens system
The light sheet thickness was inversely proportional to the incoming beam diameter at the illumination objective [12], which was defined by the variable afocal cylindrical beam expander (Fig. 2). The design of the zoomable beam expander was based on work by Winterot et al. 2005 [14]. The optical layout of the zoom unit with important parameters like focal lengths and lens-to-lens distances were developed by ray tracing simulations with OSLO Light Version 6.4 (Lambda Research Corporation, Littleton, MA, USA) using only commercially available lenses. Variation of the zoom factor was achieved by a joint motion of two linked concave lenses, which could be moved over a total range of 25 mm. Table 2 shows exemplary distances between the various lenses for three different zoom positions. The installation length of the zoom lens unit was less than 160 mm, and the total zoom was designed to achieve a magnification range of 1x to 6.3x.To verify the theoretical ray tracing results we imaged the beam after passing the zoom lens system for different stage positions of the movable lenses. For this purpose a CCD camera was placed behind lens C5. From the recorded images (Fig. 3a ) we determined the diameter of the beam along the elongated axis by fitting the intensity profiles with a Gaussian function (Fig. 3b). The zoom factor Z was calculated by comparing the results to the incoming beam diameter, which was determined by the same method. We repeated the measurements five times and calculated a mean zoom factor for the different stage positions (Fig. 3c). The zoom factor ranged from Z = 1.00 ± 0.03 to Z = 6.3 ± 0.2. The measured curve was in excellent agreement with the ray tracing calculations.
Table 2. Lens to lens distance for three different zoom factors.
| Distance [mm] | 1x | 3x | 6x |
|---|---|---|---|
| C1 → C2 | 48.54 | 34.11 | 23.63 |
| C2 → C3 | 10.73 | 25.16 | 35.64 |
| C3 → C4 | 26.54 | 12.11 | 1.63 |
| C4 → C5 | 58.56 | 72.99 | 83.47 |
| C2 → C4 | 37.27 | 37.27 | 37.27 |
Fig. 3.
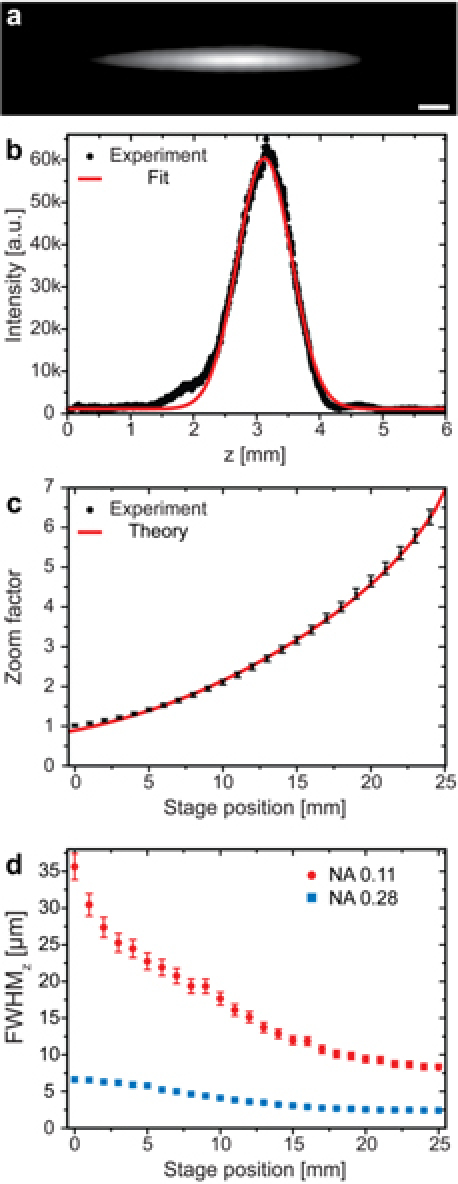
Measured zoom range of the cylindrical zoom lens and corresponding light sheet thickness. (a) Beam cross section behind the zoom unit Scale bar 100 µm. (b) Intensity profile along the central horizontal pixel line in the intensity profile shown in (a). The red line shows the Gaussian fit to the measured data. (c) The zoom factor was calculated for different stage positions of the movable zoom lenses by comparing output to input beam diameter. Standard deviation, 3% (error bars). The red line shows the expected result obtained by ray tracing calculations. (d) Measured optical sectioning thickness for an achromatic NA 0.28 (blue squares) and an NA 0.11 (full red dots) illumination objective lens. Standard deviation, 5% (error bars).
2.3 Optical sectioning thickness
The actual light sheet dimensions could directly be visualized by illumination and imaging of a homogeneously fluorescent sample such as ATTO647N dye in buffer solution as shown in Fig. 2. The lateral beam width could directly be obtained by imaging using a 10x objective lens. However, in order to determine the optical sectioning thickness the incident beam had to be rotated by 90°. This was conveniently achieved by placing a dove prism in front of the illumination objective. Rotating the prism by 45° resulted in a beam rotation of 90°. From images recorded in this configuration we deduced the axial extension of the light sheet by fitting a Gaussian function to the intensity values along each pixel line perpendicularto the optical axis. The minimal extension corresponded to the optical sectioning thick ness [7]. Five images were recorded for each stage position and the mean FWHM and its standard deviation were calculated. For the NA 0.28 illumination objective the FWHM of the optical sectioning range was found to be ΔzFWHM = 2.4 ± 0.1 µm (Z = 6.3) to ΔzFWHM = 6.6 ± 0.3 µm (Z = 1.0) for an excitation wavelength of λ0 = 638 nm (Fig. 3d). This corresponded to a lateral depth of focus of 54 µm to 410 µm. Generally, the optical sectioning thickness is dependent on the NA of the illumination optics. Therefore the optical sectioning thickness and the observable field of view can further be extended by altering the different illumination optics. With an illumination objective lens of 0.11 NA (focal length, 85 mm) the optical sectioning range was determined as ΔzFWHM = 8.3 ± 0.4 µm (Z = 6.3) to ΔzFWHM = 36 ± 2 µm (Z = 1.0) for an excitation wavelength of λ0 = 638 nm (Fig. 3d). The corresponding lateral depth of focus ranged from 650 µm to 12.3 mm.
2.4 Live imaging with different zoom configurations
To demonstrate the effect of the zoom unit in live biological specimen we imaged salivary gland cell nuclei of Chironomus tentans larvae. Salivary gland cells contain large nuclei with diameters of about 60 µm located up to 300 µm deep inside the salivary glands. Each gland cell nucleus contains four polytene chromosomes with a diameter of about 10 µm. Each chromosome comprises 8000 to 16000 perfectly aligned chromatids, which form a characteristic chromosome band structure [15]. To visualize this band structure we microinjected ATTO647N-labelled hrp36 into the nuclei of living gland cells. Hrp36 was bound to nascent mRNA [16] during mRNP formation, and therefore labelled active transcription sites. Use of a small zoom factor Z = 1.0 providing a large field-of-view and large optical sectioning thickness allowed a rapid localization of labelled polytene chromosomes. The light sheet extension along the y-axis was set to the maximum value of FWHM 120 µm (Fig. 4a and Fig. 4c). After locating the chromosomes we increased the zoom factor to Z = 6.3, what improved axial resolution and image contrast (Fig. 4c and Fig. 4d). The typical band structure of a polytene chromosome and a Balbiani Ring transcription site were well resolvable. The illumination shadows often encountered in light sheet microscopic images [17] were removed by a wavelet filter [18].
Fig. 4.
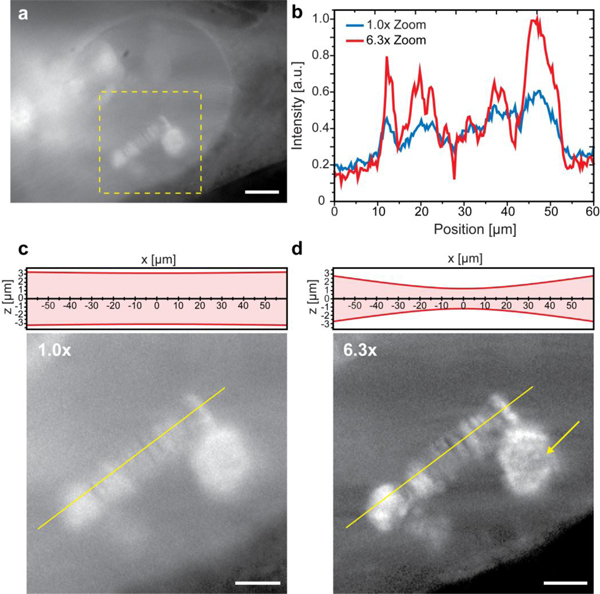
Live imaging with different zoom configurations. Polytene chromosomes of C. tentans larvae salivary gland cell nuclei imaged at different zoom factors. (a) Overview image at zoom 1.0x of the complete nucleus to locate the fluorescently labeled polytene chromosomes. Scale bar, 20 µm. (b) Intensity plots along the yellow lines in (c) and (d) demonstrated the improved contrast for a higher zoom factor yielding a smaller optical sectioning thickness. (c) and (d) Magnified views of the marked region in (a) at different zoom factors. The FWHM optical sectioning thickness along z was indicated above the images. The field-of-view in (d) was limited in comparison to (a) and (c), but the image contrast was increased revealing structures, which were hidden before. The typical band structures of the polytene chromosomes as well as the Balbiani Ring transcription site (yellow arrow) became well discernible. Scale bar 10 µm. Images were taken with 20x, 0.5 NA Nikon CFI Planfluor and imaged by a iXon EMCCD-camera with a 2.5x magnifier resulting in an effective pixel size of 320 nm.
3. Discussion
Light sheet microscopy has been shown to be advantageous for the imaging of various specimens ranging from single cell nuclei to complete fixed or living animal embryos. Such specimens differ significantly in size and therefore require greatly differing light sheet geometries for illumination. In this paper we introduced a variable light sheet concept comprising a cylindrical illumination zoom lens system and different illumination objective lenses. This concept provides the necessary flexibility to the illumination geometry of a light sheet microscope, because it allows the observation of biological samples with different sizes using a single instrument. It has several advantages over alternative beam shaping methods.
An alternative method for beam shaping of light sheets is to utilize slit apertures. By using rectangular slit apertures in the beam path [11,13,19] a significant amount of the illumination power is lost. Also, diffraction effects distort the excitation beam resulting in an uneven illumination profile and a reduced optical sectioning thickness. Diffractive effects are negligible for focal waists with FWHM values > 10 µm as it is the case in our setup for the light sheet width in y-direction.
A quite different approach to light sheet generation is digital scanned laser light-sheet fluorescence microscopy (DSLM) [20]. Here, a laser beam is focused to a single line and rapidly scanned up and down during image acquisition thus generating a virtual light sheet. The advantages of this technique are the uniform illumination intensity over the complete field-of-view and a simple adjustment of the light sheet width through computer controlled scanning mirrors. But in DSLM only the light sheet width is scanned and can be adjusted, but the optical sectioning thickness stays constant. This technique is especially suited to observe large specimen and can also be used for structured illumination for contrast enhancement [5],but the scan speed is to slow for applications like single molecule tracking, which require very high imaging rates [7,21].
We could already adapt our setup to a large variety of specimen due to a light sheet thickness ranging from 2.4 µm to 6.6 µm. The corresponding optimal field-of-view ranged from 54 µm to 410 µm. Axial and lateral illumination widths could be further extended by using illumination objective lenses with lower numerical apertures. We demonstrated this by replacing the 0.28 NA illumination objective by an 0.11 NA objective lens achieving light sheet thicknesses from 8.3 µm to 36 µm with a corresponding field-of-view of 650 µm to 12.3 mm. Since the used illumination objective lenses were designed to operate in air, the glass and water interface introduced a certain amount of spherical aberration. Thus it was not possible to translate the six-fold increase in beam diameter achievable in the zoom lens system into a corresponding reduction of the light sheet thickness. To take advantage of the full zoom range, either a very low NA objective, for which the spherical aberrations would be even less significant, or an especially corrected objective, which considers the unique illumination geometry, must be used [22]. In our current setup the width of the light sheet (y-direction) was adjustable in the range of 20 – 120 µm, which could easily be extended by lateral scanning or by choosing a focusing lens (C6) with a greater focal length, which would have to be traded against a greater system length for the illumination optics. Generally, reduction of the illumination field diminishes unwanted photon exposure and reduces light scattering.
Despite the encountered limitation we found the zoom lens concept to greatly expand the versatility of light sheet microscopes.
4. Author contributions
Conceived and designed the experiments: JGR, JHS, UK. Performed the experiments: JGR, JHS, TK. Analyzed the data: JGR. Wrote the paper: JGR and UK. Created the instrument concept: JGR. Developed the optical design: JGR.
Acknowledgments
This project was funded by grant of the German Research Foundation to U.K. (Ku 2474/7-1) and by the German Ministry for Economics and Technology (ZIM KF2297601AK9). Thanks to Eugen Baumgart for the shadow reduction in the images. Constructive discussions with Dr. Heinrich Spiecker and Volker Andresen from LaVision BioTec (Bielefeld, Germany) are gratefully acknowledged.
References and links
- 1.Vermot J., Fraser S. E., Liebling M., “Fast fluorescence microscopy for imaging the dynamics of embryonic development,” HFSP J 2(3), 143–155 (2008). 10.2976/1.2907579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reynaud E. G., Krzic U., Greger K., Stelzer E. H., “Light sheet-based fluorescence microscopy: more dimensions, more photons, and less photodamage,” HFSP J 2(5), 266–275 (2008). 10.2976/1.2974980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huisken J., Stainier D. Y., “Selective plane illumination microscopy techniques in developmental biology,” Development 136(12), 1963–1975 (2009). 10.1242/dev.022426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jährling N., Becker K., Schönbauer C., Schnorrer F., Dodt H. U., “Three-dimensional reconstruction and segmentation of intact Drosophila by ultramicroscopy,” Front Syst Neurosci 4, 1 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Keller P. J., Schmidt A. D., Santella A., Khairy K., Bao Z., Wittbrodt J., Stelzer E. H., “Fast, high-contrast imaging of animal development with scanned light sheet-based structured-illumination microscopy,” Nat. Methods 7(8), 637–642 (2010). 10.1038/nmeth.1476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Buytaert J. A., Dirckx J. J., “Tomographic imaging of macroscopic biomedical objects in high resolution and three dimensions using orthogonal-plane fluorescence optical sectioning,” Appl. Opt. 48(5), 941–948 (2009). 10.1364/AO.48.000941 [DOI] [PubMed] [Google Scholar]
- 7.Ritter J. G., Veith R., Veenendaal A., Siebrasse J. P., Kubitscheck U., “Light sheet microscopy for single molecule tracking in living tissue,” PLoS ONE 5(7), e11639 (2010). 10.1371/journal.pone.0011639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Preibisch S., Saalfeld S., Schindelin J., Tomancak P., “Software for bead-based registration of selective plane illumination microscopy data,” Nat. Methods 7(6), 418–419 (2010). 10.1038/nmeth0610-418 [DOI] [PubMed] [Google Scholar]
- 9.Reynaud E. G., Tomancak P., “Meeting report: first light sheet based fluorescence microscopy workshop,” Biotechnol. J. 5(8), 798–804 (2010). 10.1002/biot.201000177 [DOI] [PubMed] [Google Scholar]
- 10.Wohland T., Shi X., Sankaran J., Stelzer E. H., “Single plane illumination fluorescence correlation spectroscopy (SPIM-FCS) probes inhomogeneous three-dimensional environments,” Opt. Express 18(10), 10627–10641 (2010). 10.1364/OE.18.010627 [DOI] [PubMed] [Google Scholar]
- 11.Saghafi S., Becker K., Jährling N., Richter M., Kramer E. R., Dodt H. U., “Image enhancement in ultramicroscopy by improved laser light sheets,” J Biophotonics 3(10-11), 686–695 (2010). 10.1002/jbio.201000047 [DOI] [PubMed] [Google Scholar]
- 12.B. E. A. Saleh, and M. C. Teich, Fundamentals of Photonics (John Wiley & Sons, 2007). [Google Scholar]
- 13.Dodt H. U., Leischner U., Schierloh A., Jährling N., Mauch C. P., Deininger K., Deussing J. M., Eder M., Zieglgänsberger W., Becker K., “Ultramicroscopy: three-dimensional visualization of neuronal networks in the whole mouse brain,” Nat. Methods 4(4), 331–336 (2007). 10.1038/nmeth1036 [DOI] [PubMed] [Google Scholar]
- 14.J Winterot, P. Huettel, “Microscope with an afocal zoom system,” EP1544653A1 (2005).
- 15.Wieslander L., “The Balbiani ring multigene family: coding repetitive sequences and evolution of a tissue-specific cell function,” Prog. Nucleic Acid Res. Mol. Biol. 48, 275–313 (1994). 10.1016/S0079-6603(08)60858-2 [DOI] [PubMed] [Google Scholar]
- 16.Wurtz-T E., Kiseleva G., Nacheva A., Alzhanova-Ericcson A., Rosén, Daneholt B., “Identification of two RNA-binding proteins in Balbiani ring premessenger ribonucleoprotein granules and presence of these proteins in specific subsets of heterogeneous nuclear ribonucleoprotein particles,” Mol. Cell. Biol. 16(4), 1425–1435 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huisken J., Stainier D. Y., “Even fluorescence excitation by multidirectional selective plane illumination microscopy (mSPIM),” Opt. Lett. 32(17), 2608–2610 (2007). 10.1364/OL.32.002608 [DOI] [PubMed] [Google Scholar]
- 18.Münch B., Trtik P., Marone F., Stampanoni M., “Stripe and ring artifact removal with combined wavelet--Fourier filtering,” Opt. Express 17(10), 8567–8591 (2009). 10.1364/OE.17.008567 [DOI] [PubMed] [Google Scholar]
- 19.Kalchmair S., Jährling N., Becker K., Dodt H. U., “Image contrast enhancement in confocal ultramicroscopy,” Opt. Lett. 35(1), 79–81 (2010). 10.1364/OL.35.000079 [DOI] [PubMed] [Google Scholar]
- 20.Keller P. J., Schmidt A. D., Wittbrodt J., Stelzer E. H., “Reconstruction of zebrafish early embryonic development by scanned light sheet microscopy,” Science 322(5904), 1065–1069 (2008). 10.1126/science.1162493 [DOI] [PubMed] [Google Scholar]
- 21.Wang X., Wohland T., Korzh V., “Developing in vivo biophysics by fishing for single molecules,” Dev. Biol. 347(1), 1–8 (2010). 10.1016/j.ydbio.2010.08.004 [DOI] [PubMed] [Google Scholar]
- 22.Ritter J. G., Veith R., Siebrasse J. P., Kubitscheck U., “High-contrast single-particle tracking by selective focal plane illumination microscopy,” Opt. Express 16(10), 7142–7152 (2008). 10.1364/OE.16.007142 [DOI] [PubMed] [Google Scholar]