

Abstract
Objectives
The G-protein coupled receptor kinases GRK2 and GRK5 are important regulators of beta-adrenergic signaling. This study characterized single nucleotide polymorphisms in the GRK2 gene (ADRBK1)and determined if these and a GRK5 Gln41Leu polymorphism affect the blood pressure (BP)response to atenolol or hydrochlorothiazide or adverse cardiovascular outcomes in hypertensives.
Methods
ADRBK1 regions were sequenced for 48 individuals. Putative functional SNPs were tested for mRNA expression differences in 96 lymphoblastoid cell line samples and 12 leukocyte samples from hypertensives. BP response to atenolol and hydrochlorothiazide by ADRBK1 SNPs and GRK5 Gln41Leuwas tested in 418patients from the Pharmacogenomic Evaluation of Antihypertensive Responses (PEAR) using linear regression. The influence of ADRBK1 SNPs and GRK5 Gln41Leuon death, myocardial infarction or stroke in treated hypertensive patients was evaluated in a case-control cohort (1:3) of the International Verapamil SR/Trandolapril Study GENEtic Substudy (INVEST GENES) using logistic regression models.
Results
A novel ADRBK1 promoter SNP was not associated with differential GRK2 expression. GRK5 Leu41 decreased the risk for adverse cardiovascular outcomes independent of treatment strategy(adjusted odds ratio 0.535, 95% confidence interval 0.313 – 0.951, P = 0.0222)but was not associated with BP response to antihypertensive medication. An ADRBK1 SNP (rs1894111G>A) showed a signal for association with systolic and diastolic BP(SBP, DBP) response to hydrochlorothiazide in whites(DBP: −11.29±3.74 mmHg (G/A) vs. −4.26±4.79 mmHg (G/G), P = 0.0034 and SBP: −18.37±14.90 mmHg (G/A), −8.11±7.55 mmHg (G/G), P = 0.0191).
Conclusions
The GRK5 Leu41 allele protects from adverse cardiovascular outcomes in treated hypertensives.
Keywords: GRK5, GRK2, ADRBK1, polymorphism, hypertension, beta-blocker, atenolol, diuretic, hydrochlorothiazide
INTRODUCTION
β-blockers are commonly used agents for the treatment of hypertension and response varies widely among individuals.[1] The antihypertensive actions of β-blockers are mediated via antagonism of β-adenergic receptors. Adrenergic receptors mediate the physiological effects of the hormone epinephrine and the neurotransmitter norepinephrine via a complex network. Of particular relevance to this network are G-protein coupled receptor kinases (GRKs). Down regulation of β-adrenergic signaling is mediated via GRK2 and GRK5 that phosphorylate cardiac β-adrenergic receptors leading to β-arrest in recruitment and G-protein uncoupling.[2] Given the important role of GRK2 and GRK5 in β-adrenergic signaling, functional genetic polymorphisms in the genes coding for GRK2 (ADRBK1) and GRK5 (GRK5) might be important pharmacogenetic targets.
Recently, a non-synonymous single nucleotide polymorphism (SNP, A>T, rs17098707; Gln41Leu)in GRK5 was associated with differential survival in black heart failure patients, where β-blocker naïve patients who carried the variant minor allele had outcomes comparable to major allele homozygote patients treated with a β-blocker.[3]A second study had nearly identical findings in blackheart failure patients.[4] This variant is common in blacks(minor allele frequency, MAF = 0.308) but not in whites (MAF = 0.025) These findings in humans were supported by functional data where in Chinese hamster ovary cells co-transfected with β-adrenergic receptors, theGRK5 Leu41 cells showed greater agonist-promoted desensitization compared toGln41.[3] In addition, following isoproterenol infusion GRK5 Leu41 transgenic mice showed protective effects from left ventricular remodeling (left ventricular end diastolic dimension), effects that were not altered by β-blockade with propranolol.[3] These data suggest Leu41 creates a “physiologic β-blockade”. If the polymorphism creates a physiologic beta-blockade, the effect of genotyping may not be evident if all subjects evaluated are treated with a beta-blocker. Consistent with this theory, a healthy volunteer study did not find significant differences by GRK5 genotype in the negative chronotropic response to atenolol.[5] Thus, the data suggest this GRK5 variant may be clinically relevant, and the effects of the variant may be more evident in patients treated with drugs other than β-blockers. This polymorphism has not been studied in hypertensive patients and we sought to test genetic associations with response in two hypertensive populations where patients were randomized to either atenolol or an alternative therapy.
The data on GRK5 provide evidence for the potential role of GRK variants in cardiovascular disease or drug response. Given that GRK2 has historically been viewed as the more important GRK for regulation of cardiovascular β-receptors, we also sought to also explore variants in its gene (ADRBK1). Resequencing of exonic regions of ADRBK1 did not reveal non-synonymous SNPs or other coding region polymorphisms of functional importance.[3] However, the upstream region had not been sequenced. In addition, SNPs in the 3′-untranslated region (UTR) of ADRBK1 have been reported but not validated on the National Center for Biotechnology Information’s (NCBI) dbSNP database. Polymorphisms in both of these ADRBK1 regions could be are potentially functional by altering GRK2 expression. Thus, we also sought to identify and validate SNPs in these ADRBK1 regions to facilitate genetic association studies and to test potentially important SNPs in our clinical populations.
METHODS
Resequencing of ADRBK1 regions and linkage disequilibrium (LD) structure
Human Variation Collections of the NIGMS Repository DNA samples (Coriell Institute for Medical Research, Camden, NJ, USA) from 24 whites and 24 blacks were sequenced. Sequencing was done in one direction (forward) and in the other direction (reverse) if the first run yielded a novel polymorphism. Sequencing reactions were carried out using a 3730 DNA Analyzer (Applied Biosystems). Oligo Primer Analysis Software (Version 6.71 Molecular Biology Insights, Cascade, CO, USA) was used to design polymerase chain reaction (PCR) oligonucleotides. Seven amplicons were designed to cover 2 kb in the ADRBK1 promoter region and 1.5kb downstream including the 3′-UTR. Sequencing chromatograms were analyzed using Sequencer 4.7(Gene Codes Corporation, Ann Arbor, MI, USA). The samples were also genotyped for known HAPMAP SNPs that are located in the ADRBK1 region but outside of the regions targeted for resquencing. Genotyping was done using pyrosequencing.[6] Following the manufacturer’s protocol, a pyrosequencing reaction was performed for sequence determination and allele designation in a Biotage PSQ HS 96 System(Biotage AB, Uppsala, Sweden)and data were captured with PSQ HS 96 SNP software. Hardy-Weinberg equilibrium (HWE)was tested separately by race at α=0.05 using a χ2 test with one degree of freedom. Pairwise tagging(r2 of 0.8) of identified and confirmed SNPs was used to identify the most efficient set of SNPs for genetic association testing. The analysis was done separately by race and visualization of LD was performed using Haploview 4.1.[7]
In silico SNP analysis and ADRBK1 expression
We performed in-silico analysis for all new and confirmed ADRBK1 SNPs to assess potential functionality using JASPAR[8]and FASTSNP[9]. SNPs predicted to occur in transcription factor binding sites were tested for differential expression in lymphoblastoid cell lines.
A total of 96 samples were obtained from the Coriell Institute for Medical Research (Human Variation Collections of the NIGMS Repository, Camden, NJ, USA). Total RNA was extracted from untreated lymphoblastoid cell lines corresponding to the DNA samples and analyzed using the Affymetrix U133 Plus 2.0 GeneChips (Affymetrix, Santa Clara, CA, USA)as previously discribed.[10] The Affymetrix U133 Pro2 probe set (201401_s_at) for ADRBK1 was used. Additionally, ADRBK1 expression in lymphocytes was measured in twelve samples with known ADRBK1 genotypes from the Pharmacogenomic Evaluation of Antihypertensive Responses (PEAR) study described below. The PAXgene Blood RNA System tube kit (Qiagen, Valencia, CA, USA) was used for RNA isolation following the manufacturer’s protocol. RNA concentrations were determined immediately (NanoDrop 1000 Spectrophotometer) and the samples were stored at −80 °C until assayed. RNA was converted into complementary DNA (cDNA) using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Real-time PCR was carried out in duplicate using the 7300 Real Time PCR system (Applied Biosystems) and specific probes for ADRBK1 (Hs00176395_m1) and ACTB (endogenous control). Threshold cycles (CT) were determined and expression was analyzed using the relative quantification method (RQ = 2(delta delta CT values)).
Clinical studies
Associations with clinical responses (i.e. blood pressure responses and cardiovascular outcomes) were studied in two prospective trials of hypertensive patients, called PEAR and International Verapamil SR/Trandolapril Study GENEtic Substudy (INVESTGENES).
The study design of PEAR has been previously described.[11] Briefly, recruitment of this study began in 2006 and includes enrollment of hypertensive patients at the University of Florida(Gainesville, FL, USA), the Mayo Clinic (Rochester, MN, USA) and Emory University (Atlanta, GA, USA). Study participants gave written informed consent, had an office DBP > 90 and < 110 mmHg, were of any race or ethnicity, and were between the ages of 17 and 65. They had no other cardiovascular disease, diabetes, renal or liver disease. Subjects are randomized to either hydrochlorothiazide(HCTZ)or atenolol with most subjects also studied for response to the combination. Blood pressure data were collected using home blood pressure monitors at baseline, end of monotherapy and the end of combination therapy. In PEAR, we tested the effect of GRK5 Gln41Leu and 16 ADRBK1 SNPs on blood pressure response to the atenolol and HCTZ.
The INVEST and INVEST GENES designs and patient characteristics have been published previously.[12–14] Briefly, patients with documented coronary artery disease and hypertension were randomized to one of two multi-drug hypertension treatment strategies, a calcium antagonist (verapamil SR) or a β-blocker (atenolol) strategy with addition of hydrochlorothiazide or trandolopril allowed in either arm of the study. Patients in INVEST were followed for an average of 2.8 ±0.7years, for the development of the primary study outcome, which was the first occurrence of death (all-cause), nonfatal MI or nonfatal stroke. To study the association of the ADRBK1 and GRK5 SNPs on cardiovascular outcomes, we selected a nested case-control cohort from the International Verapamil SR/Trandolapril Study GENEtic Substudy (INVESTGENES).
PEAR and INVEST had both similarities and differences. Both studies evaluated responses in hypertensive patients to β-blocker(atenolol) treatment vs. an alternative antihypertensive treatment. The primary response in PEAR was the BP response, whereas in INVEST it was adverse cardiovascular outcomes. Both populations are diverse, with large minority enrollment in both studies. PEAR patients were younger and at low risk for cardiovascular disease while INVEST-GENES patients were older and at substantial risk for adverse cardiovascular outcomes. Given the similarities and differences in these study populations, and differences in the primary response phenotype, we anticipate some SNPs might be associated with BP response but not caridiovascular outcomes (or vice versa), while other SNPs might be associated with both response phenotypes.
Genotyping
Taqman allelic discrimation (7300 Real Time PCR system Applied Biosystems, Foster City, CA, USA) was performed to genotype for the GRK5 Gln41Leu polymorphism. Genotyping for 16 ADRBK1 SNPs was performed using Illumina’s human cardiovascular disease (CVD) genotyping bead chip (IBC-chip, Illumina, San Diego, CA, USA), a custom SNP array (Illumina), and Taqman allelic discrimination. The lymphoblastoid cell lines were genotyped using pyrosequencing (-703 T/C) and Taqman allelic discrimination (rs4930416). Each SNP was tested for HWE as described above.
Statistical analysis
Four hundred eighteen PEAR patients were tested for association with home blood pressure response and the end of monotherapy was used as a study endpoint. Data were stratified a priori by race (blacks or whites)and by treatment arm, as blacks in PEAR responded differently to atenolol monotherapy than whites.[15] GRK5 Leu41 is only common in blacks (MAF = 0.308). Based on α=0.05, we calculated an 80% power to detect a 6 mmHg in systolic blood pressure (SBP) and a 3.5 mmHg difference in diastolic blood pressure (DBP), respectively, in black patients in each treatment arm. The difference (delta) between baseline and end of monotherapy was determined. GRK5 A/A homozygotes were compared with GRK5 Tallele carriers for the delta in home SBP and delta in home DBP, using unadjusted linear regression models. In addition, we performed models with age, sex, body mass index, and baseline SBP or DBP, respectively, as fixed effects. Because we wanted to confirm the protective effect of this SNP, P < 0.05 was considered statistically significant. ADRBK1 genotypes were compared using the same unadjusted linear regression models followed by adjusted models (including the cofactors described above). Adjusting for multiple comparisons, P < 0.003 (0.05/16) was considered significant. However, we also report on associations with P < 0.05 as this analysis is an exploratory effort to determine if studying the full cohort is justifiable once PEAR enrollment is completed. SNPs had to be associated with SBP and DBP reduction.
The INVEST case-control cohort was analyzed for allele risk estimates of primary study outcome (first occurrence of death from all-cause, nonfatal MI or nonfatal stroke) using logistic regression models(GRK5 A/A homozygotes vs. GRK5 Tallele carriers). The analysis was performed separately by race/ethnicity. Given that GRK5 Gln41Leu is a functional SNP further analysis was performed in all subjectsed if the point estimates for each racial group were in the same direction. The models controlled for age, sex, race/ethnicity(in the combined model only), previous MI, and prior heart failure as these five covariates were significant predictors of adverse outcomes in INVEST.[12] Further, we adjusted for body mass index, previous stroke or transient ischemic attack, history of peripheral vascular disease, smoking, diabetes, renal insufficiency, and coronary artery bypass graft surgery as these variables were significantly different between cases and controls. In addition, data were stratified a priori by treatment strategy (verapamil or atenolol) to test the effect in different treatment arms. Again, since considered a replication effort for the protective function of this SNP, P < 0.05 was considered statistically significant. Logistic regression models adjusting for the same factors described above were also used to determine the effects of ADRBK1 SNPs on the primary outcome of INVEST. For ADRBK1 SNPs, P < 0.003 was considered statistically significant. In this exploratory analysis P < 0.05 were also reported.
Mean ADRBK1 expression was compared between samples homozygous for the common variant and minor allele carriers using Student’s t-test. Data were analyzed using SAS JMP genomics 4.0 (SAS Institute, Cary, NC, USA).
RESULTS
Resequencing of ADRBK1 regions
We identified a novel T>C transition at position chr.11:66789808 (-703 relative to the transcription start site of ADRBK1). This SNP was confirmed by bidirectional sequencing and is relatively common in blacks(MAF = 0.326), but not whites (MAF = 0.021). We also confirmed several other SNPs (Table 1). ADRBK1 SNPs were more common in blacks with 14of 15 ADRBK1 SNPs having a MAF>0.05 in blacks. Figure 1 shows the LDplots in blacks and whites. The LD is low in both populations, thus, no efficient set of tagging SNPs to capture both populations could be found. In blacks, one bin with three SNPs was identified. The bin contains rs7128315, -703 T/C and rs948988 (correlation coefficient r2>0.8). Both software packages predicted a transcription factor binding site (NR2F1) for ADRBK1 -703 T/C. Thus, this SNP was studied in the expression studies.
Table 1.
New and confirmed ADRBK1 SNPs
| Chromosomal Position(*) | db SNP ID | Variant | Region | MAF Blacks | MAF Whites | MAF YRI** | MAF CEU** |
|---|---|---|---|---|---|---|---|
| chr11:66789652 | rs11605263 | C>T | 5′ upstream | 0 | 0.06 | 0 | 0.05 |
| chr11:66789725 | rs12286664 | G>A | 5′ upstream | 0.26 | 0 | 0.26 | 0 |
| chr11:66789808 | novel (-703) | T>C | 5′ upstream | 0.33 | 0.02 | ||
| chr11:66790842 | rs11227756 | C>A | exon 1(Ile/Ile) | 0.15 | 0.84 | 0.12 | 0.92 |
| chr11:66795188 | rs1894111 | C>T | intron | 0.19 | 0.05 | 0.2 | 0.03 |
| chr11:66797774 | rs7128315 | G>A | intron | 0.32 | 0 | 0.36 | 0 |
| chr11:66802048 | rs948988 | G>A | intron | 0.29 | 0 | 0.36 | 0 |
| chr11:66802154 | rs4930416 | A>C | intron | 0.06 | 0 | 0.05 | 0 |
| chr11:66803077 | rs12274774 | G>T | intron | 0.30 | 0 | 0.45 | 0.03 |
| chr11:66803824 | rs3730309 | T>C | intron | 0.38 | 0 | ||
| chr11:66803976 | rs3730310 | G>C | intron | 0.27 | 0 | ||
| chr11:66806699 | rs3730145 | T>G | intron | 0.43 | 0 | 0.44 | 0.03 |
| chr11:66806868 | rs2071007 | G>A | intron | 0.29 | 0.90 | 0.16 | 0.90 |
| chr11:66809042 | rs10896164 | G>A | intron | 0.30 | 0.88 | ||
| chr11:66809714 | rs4370946 | C>T | UTR | 0.20 | 0 |
Based on UCSD Golden Path
Minor allele frequencies from the International HapMap Project, if available
Abbreviations: GP golden path, CEU samples from Utah residents with ancestry from northern and western Europe, Ile Isoleucine, MAF minor allele frequency, SNP single nucleotide polymorphism, UTR untranslated region, YRI samples from Yoruba in Ibadan, Nigeria
Figure 1.
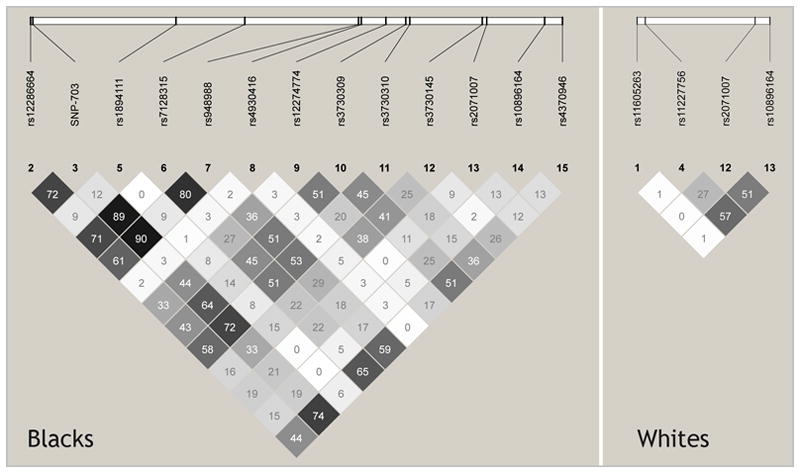
Haploview-generated linkage disequilibrium map of ADRBK1 SNPs in blacks and whites. The minimum minor allele frequency in each population was 5%. The numbers within boxes indicate the r2 values between the two corresponding SNPs. Haplotype blocks were defined by Haploview software with the default option of using the haplotype block definition used by Gabriel et al.[22] In blacks, three SNPs (rs7128315, -703 T/C and rs948988) have a correlation coefficient r2>0.8.
Blood pressure response
Baseline demographics of PEAR patients are shown in Table 2. Four hundred fifteen patients were successfully genotyped for the GRK5 Gln41Leu polymorphism. As previously reported, the GRK5 Leu allele was more common in blacks (MAF = 0.262)with 76(46%) variant carriers compared to whites (MAF = 0.011), where there were 5(2%) variant carriers. No white patient carried two copies of the Leu allele. The SNP did not deviate from HWE. Leu41 carriers did not respond differently to atenolol or hydrochlorothiazide in either race group when compared to Gln41Gln homozygote patients(Figure 2.). In addition, Leu41 carriers did not have different heart rate at baseline or show differential heart rate response to either drug (data not shown).
Table 2.
Baseline demographics of PEAR patients
| N=418 | |
|---|---|
| Age | 50.1±8.8 |
| Women | 236 (56.5) |
| White | 237 (56.7) |
| Black | 167 (40.0) |
| Asian | 5 (1.2) |
| Other/multiracial | 9 (2.1) |
| Duration of hypertension (years) | 8.1 (7.7) |
| Family history of hypertension | 329 (78.7) |
| Taking antihypertensive drug at entry | 317 (75.84) |
| BMI (kg/m2) | 31.0±5.7 |
| SBP (mmHg) | 146.0±10.8 |
| DBP (mmHg) | 93.6±6.4 |
| HR (beats/min) | 76.7±8.8 |
Abbreviations: BMI body mass index, DBP diastolic blood pressure (home), HR heart rate (home), SBP systolic blood pressure (home); Data are given as mean ± standard deviation, or N (%)
Figure 2.

Blood pressure (BP) response to atenolol (ATEN) and hydrochlorothiazide(HCTZ) by race and GRK5 Gln41Gln vs. GRK5 Leu-Carrier. Data are presented as mean change from baseline with standard error. Dark barsGRK5 Gln41Gln; light barsGRK5 Leu-Carrier; SBP systolic blood pressure; DBP diastolic blood pressure
Genotype data for 415 PEAR patients were available for all16 ADRBK1 SNPs. None of these SNPs had an association with BP response that met our predefined level for statistical significance. We report on the two strongest ADRBK1 SNPs for association with blood pressure response (0.003 < P < 0.05 Table 3). The SNPs were in HWE in both races and the trend was seen with response to hydrochlorothiazide. When compared to rs4930416 homozygote A/A black patients, heterozygote (rs4930416, A>C) blacks had similar blood pressure at baseline but greater blood pressure reduction with hydrochlorothiazide (Table 3). This SNP is not common in whites as only two white patients were heterozygotes (A/C) in PEAR. No patient was minor allele (C) homozygote (MAF blacks: 0.09). DBP response to atenolol also differed by rs4930416 genotype(Table 3). However, SBP reduction was not different.
Table 3.
ADRBK1 SNPs associated with difference in blood pressure response
| SNP | MAF black | MAF white | Race | Arm | Trait | ΔBP WT [mmHg] | ΔBP Het [mmHg] | P-value | adjusted P-value* |
|---|---|---|---|---|---|---|---|---|---|
| rs4930416 | 0.09 | 0 | black | ATEN | DBP | −3.11±6.51 | −7.48±5.84 | 0.0394 | 0.0413 |
| black | HCTZ | DBP | −6.35±6.58 | −11.32±6.34 | 0.0058 | 0.0089 | |||
| black | HCTZ | SBP | −11.29±9.76 | −16.84±8.83 | 0.0337 | 0.0492 | |||
| rs1894111 | 0.12 | 0.01 | white | HCTZ | DBP | −4.26±4.79 | −11.29±3.74 | 0.0016 | 0.0034 |
| white | HCTZ | SBP | −8.11±7.55 | −18.37±14.90 | 0.0055 | 0.0191 |
Abbreviations: ATEN atenolol, ΔBP delta blood pressure, DBP diastolic blood pressure, HCTZ hydrochlorothiazide, Het heterozygote, MAF minor allele frequency, NS not significant (P>0.05), SNP single nucleotide polymorphism, SBP systolic blood pressure, WT homozygote major allele; blood pressure data are given as mean ± standard deviation, no patient was homozygote variant for rs4930416 or rs1894111
adjusted for age, sex, body mass index and baseline blood pressure
There were also trends toward different DBP and SBP responses by ADRBK1 rs1894111 genotype in white patients receiving hydrochlorothiazide (Table 3). This SNP was not associated with altered blood pressure response to atenolol or response in blacks.
Cardiovascular outcomes
Baseline characteristics of the INVEST GENEScase-control cohort are shown in Table 4. DNA quantity and quality requirements are more stringent for the array genotyping. Thus, less samples (N = 1179) were successfully genotyped for ADRBK1 SNPs, although this slightly smaller cohort did not differ from the whole cohort on any demographics. Two ADRBK1 SNPs were excluded during the quality control procedure (rs12283002, rs1894111, SNP call rate<90%) and genotype data were available for 14 ADRBK1 SNPs. No ADRBK1 SNP was associated with cardiovascular outcomes, either overall, or in a treatment specific manner.
Table 4.
INVEST: Baseline characteristics case control cohort
| Variable | Cases (N=310) | Controls (N=948) |
|---|---|---|
| Age (years) | 71.4±9.8 | 70.3±9.2 |
| Women | 152 (49.0) | 476 (50.2) |
| White | 188 (60.6) | 574 (60.5) |
| Black | 44 (14.2) | 129 (13.6) |
| Hispanic | 77 (24.8) | 240 (25.3) |
| Other/multiracial | 1 (0) | 5 (0.5) |
| BMI (kg/m2) | 27.5±4.8 | 28.8±5.3 |
| Myocardial infarction | 112 (36.1) | 297 (31.3) |
| Stroke/TIA | 46 (14.8) | 67 (7.0) |
| Left ventricular hypertrophy | 57 (18.4) | 156 (16.5) |
| Heart failure (class I-III) | 33(10.6) | 34 (3.6) |
| Peripheral vascular disease | 54 (17.4) | 89 (9.4) |
| Smoker | 162 (52.3) | 422 (44.5) |
| Diabetes | 117 (37.7) | 257 (27.1) |
| Hypercholesterolemia | 192 (61.9) | 594 (62.6) |
| Renal impairment | 18 (5.8) | 18 (1.9) |
Abbreviations: BMI body mass index, DBP diastolic blood pressure, SBP systolic blood pressure, TIA transient ischemic attack; Data are given as mean ± standard deviation, or n (%)
GRK5 Gln41Leu genotypes were determined for1258 INVEST samples (310 cases, 948 controls, cases:controls=1:3). The GRK5 Leu allele was more common in blacks (MAF = 0.227) compared to Hispanics (MAF = 0.061) and whites (MAF = 0.021). Genotypes were in HWE in all three populations. When analyzed separately by race, the point estimates were similar in all groups, but did not reach statistical significance in any one group, likely the result of reduced power. The adjusted OR was 0.510 (95% CI 0.173–1.507) in whites, 0.481(95% CI 0.177–1.308) in Hispanics, and 0.423(95% CI 0.175–1.026, P = 0.073) in blacks. The consistency of association across race groups is not surprising for a known functional SNP. Based on the consistency between race groups, we conducted the prespecified combined analysis, and found GRK5 Gln41Leu was associated with altered risk for the primary outcome (Figure 3.) Leu-carriers had a 46.5% risk reduction compared to Gln41Gln homozygotes (adjusted odds ratio OR 0.535, 95% confidence interval CI 0.313 – 0.951, P = 0.0222). This effect was independent of treatment strategy(Figure 3).
Figure 3.

Adjusted odds ratio for GRK5 Gln41Leu on probability of experiencing the INVEST primary outcome (first occurrence of death, nonfatal myocardial infarction, or stroke). Odds ratios smaller than 1 indicate lower likelihood of Leu allele carriers experiencing the INVEST primary outcome. Odds ratios greater than 1 indicate greater likelihood of Leu allele carriers experiencing the INVEST primary outcome. ALL entire cohort, BB β-blocker strategy, CCB calcium channel blocker strategy. Asterisk denotes P = 0.0222, Statistical comparisons are between patients who were Gln41Gln homozygote and patients who were Leu allele carriers. Analyses were adjusted for age, sex, race/ethnicity, previous myocardial infarction, prior heart failure; body mass index, previous stroke or transient ischemic attack, history of peripheral vascular disease, smoking, diabetes, renal insufficiency, and coronary artery bypass graft surgery
ADRBK1 expression
Forty-five samples were successfully genotyped for the novel ADRBK1 -703 T/C. Mean ADRBK1 expression was similar in ADRBK1 -703 T/T homozygotes (253.27±81.05, arbitrary units, AU) compared to C-carriers (238.96±43.01AU, P = 0.7715). These findings were consistent with the findings in PEAR. Here, mean ADRBK1 expression was not different between six ADRBK1 -703 T/T homozygotes (0.85 ± 0.21AU) and six C-carriers (0.81± 0.15, P = 0.7262 AU). These findings are consistent with the lack of clinical association in either PEAR or INVEST-GENES. To further explore the borderline blood pressure association with rs4930416, we tested for expression by rs4930416 genotype in 96Coriell samples. Among the 12 heterozygotes, average relative expression was 208.35±115.82 AUvs.215.08±96.96 AU in the 84 wildtype homozygotes (P = 0.7393). Due to the low allele frequency of rs189411, we did not test for expression differences since the population impact of this low frequency SNP is likely to be low, even if a real effect, and it would be difficult to identify a sufficient number of variant carriers to test expression differences.
DISCUSSION
In this study, GRK5 Leu41decreased the risk for the INVEST GENES primary outcome, but did not alter blood pressure and heart rate responses to either a beta-blocker or thiazide diuretic in PEAR. Two ADRBK1 SNPs trended towards significance with SBP and DBP response to hydrochlorothiazide in white PEAR patients. Anovel ADRBK1 promoter SNP did not change GRK2 expression, nor were there any clinical associations with it in either PEAR or INVEST-GENES.
This study was significant from several perspectives. First, the INVEST GENES data supportthe protective role of the GRK5 Leu41 allele that had been seen in heart failure patients, extending the finding to a different patient population, namely hypertensives with coronary artery disease.[3,4] GRK5 Leu-carriers had a lower risk for experiencing the INVEST GENES primary outcome (first occurrence of death, nonfatal MI, or nonfatal stroke). The effect was independent of treatment strategy. This lack of a pharmacogenetic interaction is consistent with the blood pressure response data where Leu-carriers had a similar response to either hydrochlorothiazide or atenolol when compared to Gln41Gln homozygotes. We observed a main effect on outcomes for the GRK5 Gln41Leu polymorphism. The lack of a treatment or pharmacogenetic effect in this hypertensive population could be viewed as different from the previous studies in heart failure patients. In those studies the protective effect of the Leu41 variant was evident only in blacks not treated with a beta-blocker, while outcomes by genotype were similar in beta-blocker treated heart failure patients.[3,4] Our data were consistent across race groups (whites, blacks and Hispanics), with nearly identical point estimates in all three populations, and was apparent in both beta-blocker and calcium channel blocker treated patients. Thus, the risk of the Gln41 allele was not offset by beta-blockers in hypertensives, as has been observed in two heart failure studies. Such differences between heart failure and hypertensive patients for a SNP involved in adrenergic signaling are perhaps not surprising. Specifically, heart failure leads to much higher levels of sympathetic activation than hypertension. Additionally treatment outcomes differ in the two disease states. In hypertension, calcium channel blockers have been shown to exert similar (or perhaps superior) reductions in cardiovascular outcomes compared to beta-blockers,[12,16]whereas in heart failure, beta-blockers have dramatic effects on reducing mortality while numerous studies of calcium channel blockers show they have no such benefit.[17–21] Thus treatment-related outcomes are quite different between hypertension and heart failure, so it is perhaps not surprising that the treatment-related nature of the findings in heart failure were not evident in a hypertensive population. The lack of differential BP response to both the beta-blocker and thiazide diuretic in the PEAR cohort also support that the treatment-related association observed in heart failure is related to the higher sympathetic state observed with that disease process. Nonetheless, these data and those from studies in heart failure, suggest the GRK5 Gln41Leu polymorphism has clinically important effects that influence outcomes in both disease states.
This study also comprehensively assessed whether genetic variations of ADRBK1 contribute to the variability in blood pressure response in hypertensive patients treated with a β-blocker. In addition, we investigated whether these genetic variations influence death, myocardial infarction or stroke in treated hypertensive patients. Our study identified a new polymorphism in the promoter region of ADRBK1 and provided comprehensive SNP data for the common SNPs in two major ancestral populations. Despite in silico predictions suggesting the novel SNP was in a critical transcription factor binding site, we were unable to show differences by genotype on β-ARK1 expression in lymphoblastoid cell lines or fresh lymphocytes. Gene expression is tissue specific and we cannot rule out that the -703 T/C SNP may lead to differential expression in different tissues(e.g. vasculature, myocardium). Consistent with a lack of association with expression, this SNP was not associated with antihypertensive response to atenolol nor with outcomes in atenolol treated hypertensive patients. Collectively, these data suggest this SNP is not important from a functional or clinical perspective. The ADRBK1 SNP rs1894111 had the strongest signal for associations with blood pressure response to hydrochlorothiazide in white hypertensive patients, although it did not meet our a priori p value for significance. The ADRBK1 SNP rs4930416 also had a borderline association; however, we did not detect expression differences by this SNP, although we cannot rule out that such differences might exist in other tissues. Thus, while ADRBK1 rs1894111and rs4930416may represent interesting signals, these SNPs need tested further in a larger population to understand their influence on response to atenolol and HCTZ.
In conclusion, GRK5 Leu-carrier status is protective in treated hypertensive patients with coronary artery disease, but does not influence blood pressure response to antihypertensives. These data also suggest that variation in ADRBK1 may have a minor influence on the antihypertensive response, although further studies in larger cohorts are needed to more completely assess the pharmacogenetic role of this gene.
Acknowledgments
SOURCES OF SUPPORT
PEAR: This work is supported by a grant from the National Institutes of Health (NIH, Bethesda, MD), grant GM074492, funded as part of the Pharmacogenetics Research Network.
INVEST: This study was supported by NIH grants HL 74730, HL69758, RR017568, and by a grant from Abbott Laboratories.
MTL was supported by the American Heart Association grant0615189B
Footnotes
Diclaimer: Although Dr. Zineh is an employee of FDA, the work in this manuscript does not necessarily reflect FDA views or policy. No official endorsement is intended or should be inferred.
References
- 1.Materson BJ, Reda DJ, Cushman WC, Massie BM, Freis ED, Kochar MS, et al. Single-drug therapy for hypertension in men. A comparison of six antihypertensive agents with placebo. The Department of Veterans Affairs Cooperative Study Group on Antihypertensive Agents. N Engl J Med. 1993;328:914–21. doi: 10.1056/NEJM199304013281303. [DOI] [PubMed] [Google Scholar]
- 2.Penela P, Murga C, Ribas C, Tutor AS, Peregrin S, Mayor F., Jr Mechanisms of regulation of G protein-coupled receptor kinases (GRKs) and cardiovascular disease. Cardiovasc Res. 2006;69:46–56. doi: 10.1016/j.cardiores.2005.09.011. [DOI] [PubMed] [Google Scholar]
- 3.Liggett SB, Cresci S, Kelly RJ, Syed FM, Matkovich SJ, Hahn HS, et al. A GRK5 polymorphism that inhibits beta-adrenergic receptor signaling is protective in heart failure. Nat Med. 2008;14:510–7. doi: 10.1038/nm1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cresci S, Kelly RJ, Cappola TP, Diwan A, Dries D, Kardia SL, et al. Clinical and genetic modifiers of long-term survival in heart failure. J Am Coll Cardiol. 2009;54:432–44. doi: 10.1016/j.jacc.2009.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kurnik D, Cunningham AJ, Sofowora GG, Kohli U, Li C, Friedman EA, et al. GRK5 Gln41Leu polymorphism is not associated with sensitivity to beta(1)-adrenergic blockade in humans. Pharmacogenomics. 2009;10:1581–7. doi: 10.2217/pgs.09.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ronaghi M, Uhlen M, Nyren P. A sequencing method based on real-time pyrophosphate. Science. 1998;281:363–365. doi: 10.1126/science.281.5375.363. [DOI] [PubMed] [Google Scholar]
- 7.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–5. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 8.Sandelin A, Alkema W, Engstrom P, Wasserman WW, Lenhard B. JASPAR: an open-access database for eukaryotic transcription factor binding profiles. Nucleic Acids Res. 2004;32:D91–4. doi: 10.1093/nar/gkh012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yuan HY, Chiou JJ, Tseng WH, Liu CH, Liu CK, Lin YJ, et al. FASTSNP: an always up-to-date and extendable service for SNP function analysis and prioritization. Nucleic Acids Res. 2006;34:W635–41. doi: 10.1093/nar/gkl236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moyer AM, Salavaggione OE, Wu TY, Moon I, Eckloff BW, Hildebrandt MA, et al. Glutathione s-transferase p1: gene sequence variation and functional genomic studies. Cancer Res. 2008;68:4791–801. doi: 10.1158/0008-5472.CAN-07-6724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Johnson JA, Boerwinkle E, Zineh I, Chapman AB, Bailey K, Cooper-DeHoff RM, et al. Pharmacogenomics of antihypertensive drugs: Rationale and design of the Pharmacogenomic Evaluation of Antihypertensive Responses (PEAR) study. Am Heart J. 2009;157:442–9. doi: 10.1016/j.ahj.2008.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pepine CJ, Handberg EM, Cooper-DeHoff RM, Marks RG, Kowey P, Messerli FH, et al. A calcium antagonist vs a non-calcium antagonist hypertension treatment strategy for patients with coronary artery disease. The International Verapamil-Trandolapril Study (INVEST): a randomized controlled trial. Jama. 2003;290:2805–16. doi: 10.1001/jama.290.21.2805. [DOI] [PubMed] [Google Scholar]
- 13.Beitelshees AL, Gong Y, Wang D, Schork NJ, Cooper-Dehoff RM, Langaee TY, et al. KCNMB1 genotype influences response to verapamil SR and adverse outcomes in the INternational VErapamil SR/Trandolapril STudy (INVEST) Pharmacogenet Genomics. 2007;17:719–29. doi: 10.1097/FPC.0b013e32810f2e3c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Johnson AD, Gong Y, Wang D, Langaee TY, Shin J, Cooper-Dehoff RM, et al. Promoter polymorphisms in ACE (angiotensin I-converting enzyme) associated with clinical outcomes in hypertension. Clin Pharmacol Ther. 2009;85:36–44. doi: 10.1038/clpt.2008.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Johnson JA, Gong Y, Bailey KR, Cooper-Dehoff RM, Chapman AB, Turner ST, et al. Hydrochlorothiazide and Atenolol Combination Antihypertensive Therapy: Effects of Drug Initiation Order. Clin Pharmacol Ther. 2009 doi: 10.1038/clpt.2009.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dahlof B, Sever PS, Poulter NR, Wedel H, Beevers DG, Caulfield M, et al. Prevention of cardiovascular events with an antihypertensive regimen of amlodipine adding perindopril as required versus atenolol adding bendroflumethiazide as required, in the Anglo-Scandinavian Cardiac Outcomes Trial-Blood Pressure Lowering Arm (ASCOT-BPLA): a multicentre randomised controlled trial. Lancet. 2005;366:895–906. doi: 10.1016/S0140-6736(05)67185-1. [DOI] [PubMed] [Google Scholar]
- 17.The effect of diltiazem on mortality and reinfarction after myocardial infarction. The Multicenter Diltiazem Postinfarction Trial Research Group. N Engl J Med. 1988;319:385–92. doi: 10.1056/NEJM198808183190701. [DOI] [PubMed] [Google Scholar]
- 18.Goldstein RE, Boccuzzi SJ, Cruess D, Nattel S. Diltiazem increases late-onset congestive heart failure in postinfarction patients with early reduction in ejection fraction. The Adverse Experience Committee; and the Multicenter Diltiazem Postinfarction Research Group. Circulation. 1991;83:52–60. doi: 10.1161/01.cir.83.1.52. [DOI] [PubMed] [Google Scholar]
- 19.Barjon JN, Rouleau JL, Bichet D, Juneau C, De Champlain J. Chronic renal and neurohumoral effects of the calcium entry blocker nisoldipine in patients with congestive heart failure. J Am Coll Cardiol. 1987;9:622–30. doi: 10.1016/s0735-1097(87)80057-8. [DOI] [PubMed] [Google Scholar]
- 20.Packer M, O’Connor CM, Ghali JK, Pressler ML, Carson PE, Belkin RN, et al. Effect of amlodipineon morbidity and mortality in severe chronic heart failure. Prospective Randomized Amlodipine Survival Evaluation Study Group. N Engl J Med. 1996;335:1107–14. doi: 10.1056/NEJM199610103351504. [DOI] [PubMed] [Google Scholar]
- 21.Elkayam U, Amin J, Mehra A, Vasquez J, Weber L, Rahimtoola SH. A prospective, randomized, double-blind, crossover study to compare the efficacy and safety of chronic nifedipine therapy with that of isosorbide dinitrate and their combination in the treatment of chronic congestive heart failure. Circulation. 1990;82:1954–61. doi: 10.1161/01.cir.82.6.1954. [DOI] [PubMed] [Google Scholar]
- 22.Gabriel SB, Schaffner SF, Nguyen H, Moore JM, Roy J, Blumenstiel B, et al. The structure of haplotype blocks in the human genome. Science. 2002;296:2225–9. doi: 10.1126/science.1069424. [DOI] [PubMed] [Google Scholar]