

Abstract
Adenovirus (Ad) membrane penetration during cell entry is poorly understood. Here we show that antibodies which neutralize the membrane lytic activity of the Ad capsid protein VI interfere with Ad endosomal membrane penetration. In vitro studies using a peptide corresponding to an N-terminal amphipathic α-helix of protein VI (VI-Φ), as well as other truncated forms of protein VI suggest that VI-Φ is largely responsible for protein VI binding to and lysing of membranes. Additional studies suggest that VI-Φ lies nearly parallel to the membrane surface. Protein VI fragments membranes and induces highly curved structures. Further studies suggest that Protein VI induces positive membrane curvature. These data support a model in which protein VI binds membranes, inducing positive curvature strain which ultimately leads to membrane fragmentation. These results agree with previous observations of Ad membrane permeabilization during cell entry and provide an initial mechanistic description of a nonenveloped virus membrane lytic protein.
Keywords: nonenveloped virus, capsid, membrane, protein, cell entry
Introduction
Cell membranes are a major obstacle to viral infection of host cells. Unlike enveloped viruses, which fuse the viral envelope with cellular membranes to gain access to intracellular sites of replication, nonenveloped viruses must employ alternative mechanisms to cross this barrier (Tsai 2007). Our understanding of how nonenveloped viruses penetrate cellular membranes lags significantly behind that of analogous events occurring during enveloped virus cell entry. Adenoviruses are a family of nonenveloped dsDNA virus, which penetrate endosomal membranes during cell entry to gain access to the cytoplasm. However, the mechanisms involved in this event have yet to be fully defined.
To enter cells, Ads first bind to a primary attachment receptor via high affinity interactions between the virus fiber protein and the cell surface receptor (Chroboczek et al. 1995). For subgroup A, C, E and F, this receptor is the coxsackievirus and adenovirus receptor (Roelvink et al. 1998). For subgroup B and D viruses, this receptor has been found to be CD46, sialic acid or CD80/CD86 (Sakurai et al. 2007). After attachment, secondary engagement of αv integrins by the RGD motif of the viral penton base protein triggers clathrin mediated endocytosis of virions (Nemerow 2000).
For subgroup C viruses, disassembly of the viral capsid is initiated very soon after attachment, with the fiber reportedly released from the virion at the cell surface (Greber et al. 1993; Nakano et al. 2000). Endocytosis results in further disassembly of the Ad capsid proteins as additional proteins are shed from the virus (Greber et al. 1993). Uncoating correlates with acidification of the endosomal compartment (Svensson 1985; Greber et al. 1993) and is enhanced by interactions between the penton base and αv integrins (Wickham et al. 1993; Wickham et al. 1994). Studies employing a temperature sensitive mutant Ad2ts1, which possesses a hyperstable capsid when produced at the nonpermissive temperature due to lack of proteolytic capsid maturation, have demonstrated that, although this virus can attach and trigger endocytosis, it's failure to uncoat in endosomes prevents endosomal escape (Hannan et al. 1983; Greber et al. 1996). Additionally, while entry of wt Ad2 virions leads to eventual degradation of the interior capsid protein VI (pVI), by 2 hrs post infection, the precursor form of pVI present in the ts1 virions is not degraded during cell entry presumably because it is not released from the virion (Greber et al. 1993; Greber et al. 1996).
The exact mechanisms of Ad endosomal membrane penetration remain to be determined. While initial studies had implicated the penton base in the permeabilization of cellular membranes (Seth et al. 1984; Seth et al. 1985; Seth 1994), many of these studies were conducted prior to our understanding of the role for the penton base interactions with αv integrins. Perhaps the greatest evidence against a role for the penton base in direct permeabilization of endosomal membranes is the observation that incubation of cells with purified penton base at a concentration of 1 mg/ml did not directly permeabilize cell membranes but could actually prevent Ad2 virions from releasing [3]H-choline from cells (Wickham et al. 1993; Wickham et al. 1994). In contrast, one of the proteins released from the viral capsid during disassembly within endosomes, pVI, was recently found to possess >95% of the in vitro membrane lytic activity of the capsid (Wiethoff et al. 2005). This in vitro membrane lytic activity is pH independent, is possessed by both the precursor and the mature form of pVI, and is dependent upon an N-terminal predicted amphipathic α-helix.
Currently little structural information is available regarding pVI. Present at 342-369 copies per virion (van Oostrum et al. 1985; Lehmberg et al. 1999), pVI is expressed as a preprotein. This preprotein, facilitates nuclear accumulation of the major capsid protein, hexon, by mediating hexon import through nuclear pore complexes during virus assembly (Wodrich et al. 2003). Additionally, the C-terminus of prepVI serves as an allosteric activator of the virally encoded 23K cysteine protease which cleaves several capsid protein precursors during capsid maturation (Mangel et al. 1996). A tentative location of pVI underneath the 60 peripentonal hexons was originally suggested based on electron cryomicroscopy studies (Stewart et al. 1993). More recent studies using genetic and molecular modeling have suggested that pVI interactions with hexon occur in a large interior cavity of the hexon (Wodrich et al. 2003). This location was further supported by higher resolution structures determined by electron cryomicroscopy (Saban et al. 2005; Saban et al. 2006; Silvestry et al. 2009). These later studies have also suggested that density within the internal cavity of each of the 240 hexon proteins of the Ad capsid may correspond to pVI. Further studies should eventually provide a more definitive description of the location of pVI within Ad virions.
Our current studies demonstrate that pVI membrane lytic activity is required for efficient endosomal membrane penetration during Ad cell entry. Mechanistic studies indicate that the N-terminal amphipathic α helix is critical for pVI binding to and disrupting endosomal membrane. This helix alone is sufficient to mediate membrane disruption. The helix binds to lipid membranes, lying nearly parallel to the surface of the lipid bilayer. Disruption of liposomal membranes by pVI does not appear to involve the formation of pores with diameters less than 100 Å but rather a gross reorganization of the lipid bilayer resulting from the induction of positive membrane curvature. These studies support a role for pVI in Ad endosomal membrane penetration during cell entry, and represent a significant advance in our understanding of the mechanisms of nonenveloped virus penetration of cell membranes.
Materials and Methods
Materials
1-palmitoyl,2-oleoylphosphatidylcholine (POPC), 1-palmitoyl,2-oleoylphosphatidylserine (POPS), 1-palmitoyl,2-oleoylphosphatidylethanolamine (POPE), 1-Palmitoyl-2-Stearoyl(6′,7′-dibromo)-sn-Glycero-3-Phosphocholine 1-Palmitoyl-2-Stearoyl(9′,10′-dibromo)-sn-Glycero-3-Phosphocholine and 1-Palmitoyl-2-Stearoyl(11′,12′-dibromo)-sn-Glycero-3-Phosphocholine were purchased from Avanti Polar Lipids. Saporin and α-Lysophosphatidylcholine (lysoPC) were from Sigma, and N-fluoresceinyl-1, 2-sn-dihexadecylphosphatidylethanolamine (FITC-DHPE) sulforhodamine B, propidium iodide and alexafluor488-dextran were obtained from Invitrogen. All other reagents were from FisherBiotech. An E1, E3-deleted human adenovirus 5 containing a CMV-driven EGFP reporter gene was described previously (Wiethoff et al. 2005). HEK293 and Hela cells were obtained from ATCC.
Generating pVI single tryptophan mutants
To generate pVI containing single tryptophan residues, mutations were introduced in pET15bVI-N, a construct encoding residues 34-114. This region of pVI has only 3 tryptophans, therefore to obtain single tryptophan mutants, 2 out of the 3 tryptophan residues were mutated to phenylalanine using the QuickChange II site-directed mutagenesis kit (Strategene, La Jolla, CA). Three different mutants were generated using the following primers. Complementary primers are not shown. Altered nucleotides are indicated in bold.
W37 (W41/59F): VIW59F 5′ GGCAGCAAGGCCTTTAACAGCAGCACAGG 3′
VIW41F 5′ GCTGGGGCTCGCTGTTTAGCGGCATTAAAAATTTCG 3′
W41 (W37/59F): VIW59F 5′ GGCAGCAAGGCCTTTAACAGCAGCACAGG 3′
VIW37F 5′ ACAAGGCCTTCAGCTTTGGCTCGCTGTGGAGC 3′
W59 (W37/41F):VIW3741F 5′ GCTTTGGCTCGCTGTTTAGCGGCATTAAAAATTTCG 3′
The mutations were confirmed by sequencing, and the plasmids were used to overexpress proteins in E. coli.
Purification of recombinant proteins
Recombinant proteins were expressed in BL21(DE3) cells. Cultures inoculated with overnight culture were grown at 37°C, until they reached an optical density at 600 nm of 1.0. The NaCl concentration was then increased by adding an additional 0.9 g NaCl/L, and protein expression was induced by adding 1mM IPTG (isopropyl-α-D-thiogalactopyranoside) for 1 hr. Cells were pelleted, resuspended in cell lysis buffer (1% TritonX-100, 25 mM Phosphate, 150 mM NaCl pH 7.5, 0.5 mg/ml lysozyme, 0.1 mg/ml DNAse and 1mM PMSF (phenylmethylsulfonyl fluoride)), and soluble protein was isolated by centrifugation at 13,000 × g for 15 min at 4°C. Recombinant proteins were purified with Talon cobalt resin using the manufacturer's protocol (BD Biosciences). Proteins were extensively dialyzed into 25 mM Phosphate, 150 mM NaCl, and 10% (v/v) glycerol pH 7.5 before flash freezing in liquid nitrogen. Aliquots were stored at -80 °C until use.
Antibody inhibition of Ad membrane lytic activity
Rabbit polyclonal antiserum raised against purified preprotein VI was obtained with appropriate institutional IACUC approval. IgG from preimmune serum was purified by protein G chromatography. Affinity purified anti-pVI antibodies (αpVI) were obtained by chromatography using purified protein VI coupled to cyanogen bromide sepharose beads. The ability of αpVI to prevent protein VI membrane permeabilization was examined by incubating Hela cells with 25 μg/ml purified protein VI in the presence or absence of 1 mg/ml preimmune IgG or αpVI at 37 °C. The efficiency of protein VI membrane permeabilization was determined by using fluorescence microscopy to measure the percentage of cells which took up the fluorescent membrane impermeable nuclear stain, propidium iodide, 30 minutes after protein addition.
Antibody inhibition of Ad infectivity and endosomal membrane penetration
To determine the ability of αpVI to prevent Ad5 infection, Hela cells were preincubated with varying concentrations of preimmune IgG or αpVI for 6 hrs at 37 °C to saturate the endolysosomal system prior to infection with Ad5gfp (1 gfp-transducing unit per cell) in the continued presence of antibody. The percentage of gfp positive cells was determined by fluorescence microscopy 24 hrs post infection.
To determine whether αpVI prevents Ad5 endosomal escape, Hela cells were preincubated with 1 mg/ml of preimmune IgG or αpVI for 6 hrs at 37 °C to saturate the endolysosomal system prior to infection with increasing MOIs of Ad5gfp in the presence of 0.05mg/ml of the membrane impermeable ribosomal toxin, saporin (Sigma) and in the continued presence of antibody. Saporin dependent cell death in the presence of increasing concentrations of Ad5gfp was compared to that of saporin alone 24 hrs post infection using an MTT assay for cell viability. Data are presented as % cell viability compared to saporin alone.
Determining pVI in vitro membrane lytic activity
Liposomes containing POPC:POPS (75:25 mol%), and entrapped 100 mM sulforhodamine B (SulfoB) (Molecular Probes) were generated as previously described (Wiethoff et al. 2005). Liposomes containing entrapped SulfoB were separated from free dye using a Sephadex G-75 column, preequilibrated with 25mM HEPES, 150 mM NaCl buffer pH 7.5 (HBS). The liposome concentration was determined using a phosphate assay as previously described (Fiske et al. 1925).
Membrane lytic activity of recombinant pVI was determined by measuring SulfoB fluorescence dequenching upon release from liposomes. The liposomes were diluted in HBS to a final concentration of 10 μM. Different concentrations of pVI were then added to the liposomes and incubated for 20 min at 37° C. Fluorescence intensity was measured using the Cary Eclipse fluorescence spectrophotometer (Varian) with the excitation wavelength of 575 nm and emission wavelength of 590 nm. One hundred percent dye release was determined by adding Triton X-100 to the liposomes at a final concentration of 0.5% (w/v). The percentage of SulfoB released was calculated using the formula % SulfoB released = 100 × [Fmeas-F0)/(Ftx100-F0)], where Fmeas is the maximum fluorescence intensity measured, F0 is fluorescence intensity in absence of protein, and Ftx100 is the fluorescence intensity in the presence of 0.5% Triton X-100.
Analysis of pVI binding to membranes
Protein VI contains 4 tryptophans at residues 37, 41, 59 and 229. Binding to liposomes was assessed by monitoring changes in pVI intrinsic tryptophan fluorescence upon titration with increasing amounts of liposomes (POPC:POPS 75:25 mol%). This approach is routinely used for monitoring interactions between proteins and ligands, membranes or other proteins and relies on the assumption that the fractional spectral change in tryptophan fluorescence correlates directly with the amount of protein bound to its substrate (Eftink 1997). The fluorescence emission spectra from 300-480 nm of pVI in HBS and 37 °C was obtained by selective excitation of tryptophan at 295 nm. Increasing amounts of liposomes were added to pVI with mixing for 3 minutes and additional spectra were obtained. Spectra of buffer or an equivalent amount of liposomes alone were subtracted from the spectra of each protein/lipid mixture to obtain corrected spectra. The spectral center of mass, Iλ, for the emission spectra were determined using the Carey Eclipse software. Assuming that this spectral change in tryptophan fluorescence correlates with the amount of protein bound, the fractional saturation of binding sites, θ, was calculated using the following equation: θ = (Iλ(obs)- Iλ(0))/(Iλ(max)- Iλ(0)), where Iλ(obs) is the spectral center of mass for each protein/lipid ratio and Iλ(0) and Iλ(max) is the spectral center of mass for protein alone and the protein in the presence of saturating amounts of liposomes, respectively. Plotting θ versus protein/lipid molar ratios yielded the resulting binding isotherms.
Analysis of pVI membrane penetration using Giant Lipid Vesicles (GLV)
GLV were generated as described previously (Akashi et al. 1996), by mixing POPC, POPS and FITC-DHPE at a 70:25:5 mol ratio in chloroform. A thin lipid film was then generated on a glass tube by evaporating the chloroform with a stream of nitrogen gas. Residual chloroform was removed by placing the tube under vacuum for 6 hrs. The lipid film was then prehydrated with a stream of water saturated nitrogen gas for 25 min, followed by rehydration in 6 ml of HBS containing 0.1 M sucrose. The tube was then sealed with parafilm and incubated overnight at 37°C. GLVs were harvested as a flocculate near the top of the solution the next day and quantified by phosphate assay as described above. Typical preparations of GLVs are polydisperse with vesicle diameters ranging from 5 to 50 μm. To visualize pVI membrane lytic activity, recombinant pVI was incubated with GLV at a 1:100 (lipid:protein) ratio in HBS with 0.1 M glucose, on a glass slide. After 15 min the samples were analyzed by using an epifluorescence microscope.
Determining tryptophan depth of membrane penetration
Quenching of tryptophan fluorescence by brominated phospholipids was used to determine the depth of tryptophan penetration into the lipid bilayer (Chattopadhyay et al. 1987; Ladokhin 1997). Liposomes containing 25 mol% POPS, 25 mol% POPC and 50 mol% brominated phosphatidylcholine (Br2-PC) were made as described above. Recombinant pVI single tryptophan mutants were incubated for 10 min at 37°C with brominated liposomes at a 1:100 (protein:lipid) ratio in HBS pH 7.5. The intensity of tryptophan fluorescence was measured at 325 nm upon excitation at 295 nm. The differences in quenching tryptophan fluorescence by the (6,7)-, (9,10)-, (11,12)-Br2-PC was used to calculate the location of the residue in the bilayer using two methods: the parallax method (Chattopadhyay et al. 1987) and distribution analysis (Ladokhin 1997). In the parallax method, the depth of the tryptophan residue was calculated using the formula:
where Zcf represents the distance of the fluorophore from the center of the bilayer, Lcl is the distance of the shallow quencher from the center of the bilayer, Lcl is the distance between the shallow and deep quencher, F1 is the fluorescence intensity in the presence of the shallow quencher, F2 is the fluorescence intensity in the presence of the deep quencher, and C is the concentration of quencher in molecules/Ǻ2. In the distribution analysis the depth of tryptophan residue was calculated by fitting the data to the equation:
where F0 represents the fluorescence intensity in the absence of the brominated phospholipids, Fh is the intensities measured as a function of the distance from the center of the lipid bilayer to the quencher h, c(h) is the concentration of the different quenchers, S is the area under the curve (measurement of quenching efficiency), σ is the dispersion (a measure of the distribution of the depth in the bilayer), hm is the most probable position of the fluorophore in the membrane, and h is the average bromine distances from the center of the bilayer, based on x-ray diffraction and taken to be 10.8, 8.3, and 6.3 for (6,7)-, (9,10)- and (11,12)-Br2-PC respectively. When equal concentrations of the Br-lipids are used, the c(h) value is unity.
Results
Protein VI is involved in Ad endosomal membrane penetration
Previous work has shown that Ad pVI possesses all of the in vitro membrane lytic activity observed for Ad5 virions (Wiethoff et al. 2005). To determine whether pVI plays a role in adenovirus endosomal membrane penetration, we generated rabbit polyclonal antibodies raised against recombinant pre-pVI. These antibodies were affinity purified against recombinant Ad5 pVI coupled to cyanogen bromide agarose beads. To demonstrate the ability of αpVI to prevent pVI disruption of cellular membranes, recombinant pVI was added to Hela cells in the presence of preimmune IgG or αpVI and the percentage of cells which stained positive with the membrane impermeable DNA intercalating dye, propidium iodide, were determined by fluorescence microscopy (Fig 1A). Incubation of cells with purified pVI in the presence of excess αpVI lead to >90% decrease in propidium iodide positive cells compared to cells treated with pVI in the presence of excess preimmune IgG.
Figure 1. αpVI antibodies inhibit protein VI membrane permeabilization and decrease adenovirus infectivity.

A) Hela cells were treated with purified pVI in the presence of either preimmune IgG or affinity-purified αpVI and the percentage of cell nuclei stained with the membrane impermeable dye, propidium iodide were quantified by fluorescence microscopy. B) Hela cells were preincubated with preimmune IgG (black bars) or affinity-purified anti-pVI (white bars) for 6 hr before infection with Ad5gfp in the continued presence of the antibody. Relative infectivity was calculated by determining the percentage of GFP positive cells 20 hr post-infection. C) Hela cells preincubated with antibodies as described above were infected with increasing MOIs of Ad5gfp in the presence of the ribosomal toxin saporin. Saporin mediated cytotoxicity was quantified by MTT assay 24 hrs post infection and % cell viability determined. Preimmune IgG (●), anti-pVI (○). Error bars represent the standard error of the mean for a minimum of 3 replicates.
Since polyclonal rabbit αpVI can inhibit pVI membrane lytic activity, we next assessed the ability of αpVI to block Ad5 infection. Since pVI is not exposed on the capsid exterior (Everitt et al. 1975; Saban et al. 2006; Silvestry et al. 2009) but becomes accessible upon virus uncoating in endosomes (Greber et al. 1993; Greber et al. 1996), endosomal compartments were saturated with antibody by incubating Hela cells with either preimmune IgG or αpVI for 6 hrs prior to infection with a replication defective E1, E3-deleted Ad, possessing a EGFP expression cassette (Ad5gfp). The percentage of gfp positive cells was then determined 24 hrs post-infection. A ∼2 and 5-fold decrease in gfp positive cells was observed when cells were incubated with the 0.4 and 1 mg/ml αpVI respectively compared to cells incubated with preimmune IgG, suggesting that pVI plays a role in virus entry (Fig 1B). To rule out an effect of αpVI on Ad5gfp cell binding, we also added antibodies to cells at the same time as Ad5gfp. Under these conditions, we did not observe a difference between Ad5gfp infectivity in cells incubated with preimmune IgG or αpVI (not shown). These data suggest that the decrease observed in Fig 1B is not due to αpVI interfering with Ad5 cell binding. To determine if this decreased transduction by Ad5gfp is due to αpVI antibodies inhibiting pVI disruption of endosomal membranes and, therefore, inhibiting virus release into cytosol, we looked at ability of αpVI to prevent Ad5gfp-mediated cytosolic translocation of a membrane impermeable ribosomal toxin, saporin. Endosomal compartments were again saturated with antibodies as described before, and cells were infected with Ad5gfp in the continued presence of antibody. At the time of infection, these cells were also treated with saporin, a membrane impermeable toxin, whose translocation into the cytosol is facilitated by cointernalization with the virus in endosomes, and subsequent virus mediated membrane disruption. Once in the cytosol, the toxin inhibits protein synthesis, ultimately resulting in cell death which is quantified via MTT assay. Preincubation with 1 mg/ml αpVI increased the amount of Ad5 necessary to induce a 50% decrease in cell viability by 5-fold compare to preimmune IgG, indicating that pVI antibodies decrease Ad5 mediated cytosolic translocation of saporin (Fig 1C). Thus, αpVI is able to prevent Ad membrane disruption and is also able to prevent endosomal membrane penetration by Ad5gfp.
The N-terminal amphipathic α-helix is important for pVI membrane lytic activity
Currently there is no structural information regarding pVI. Secondary structure predictions suggest that the N-terminal 80 residues of mature pVI (residues 34-114) are predicted to form a stable α-helical domain (Fig 2A) (Wiethoff et al. 2005). The C-terminal domain is mostly disordered and contains a mixture of α-helical and β-structures. The membrane lytic activity of pVI was previously shown to be independent of pH and is strongly dependent on the presence of 20 residues at the N-terminus that are predicted to form an amphipathic α-helix (Wiethoff et al. 2005). To examine the importance of specific pVI domains for the observed membrane lytic activity, we initially used the heterologous overexpression of truncated versions of pVI in E. coli. Purified protein was used to assess the ability of various truncations in pVI to facilitate membrane disruption by monitoring the ability of purified forms of the proteins to mediate the release of an entrapped fluorophore (SulfoB) from the interior of model membranes in the form of liposomes (Fig 2B). Wild type pVI caused a dose dependent release of SulfoB from liposomes. The truncated 80 residue N-terminal domain (VI114Δ) possesses identical membrane lytic activity compared to the full length protein suggesting that the C-terminal 125 residues of pVI contribute significantly less to the in vitro membrane lytic activity. Equivalent membrane lytic activity was also observed for a 24 residue peptide corresponding to residues 34-54 and possessing an additional c-terminal tetralysine tag to enhance aqueous solubility suggesting that this helix was sufficient for in vitro membrane disruption. Truncation of pVI by removing residues 34-54 comprising the N-terminal amphipathic α-helix (VIΔ54) greatly reduces the membrane lytic activity of the protein, requiring ∼400 fold higher concentrations of protein to elicit similar SulfoB release as the mature form of the protein. Since equivalent amounts of the irrelevant protein, bovine serum albumin, do not induce significant SulfoB release, our data with the truncated VIΔ54 protein suggest that additional residues within pVI may also be involved in membrane interactions.
Figure 2. Adenovirus protein VI disruption of and binding to liposomes.

A) The predicted secondary structure of pVI with α-helices (cylinders) β-sheets (arrows) and unstructured (lines) secondary structures displayed. Recombinant forms of prepVI, pVI, VIΔ54, VI114Δ and VI34-54 used in studies of membrane binding and permeabilization. B) pVI membrane lytic activity was measured by quantifying the release of SulfoB from liposomes after treatment with increasing concentrations of the different pVI constructs. C) Membrane binding of the different pVI constructs. Increasing lipid concentrations were added to pVI constructs and changes in tryptophan fluorescence were used to determine the fractional saturation (θ) of pVI binding capacity as described in the materials and methods. Error bars represent the standard error of the mean for a minimum of 3 replicates. ■ protein VI ◆ VI34-54 ▲ VIΔ54 ○ VI114Δ. △ BSA
To further assess the role of the amphipathic α-helix in pVI membrane disruption, we examined the relative affinity of each protein construct and peptide for the same liposomal membranes. The binding of pVI to liposomes was assessed using changes in intrinsic tryptophan fluorescence upon association to membranes. By assuming that the relative change in tryptophan fluorescence upon binding liposomes was directly related to the amount of pVI bound to membranes, binding isotherms were generated and the fractional saturation of binding sites was plotted versus increasing lipid concentrations (Fig 2C). While pVI, VI114Δ and VI34-54 all possessed similar affinities as evidenced by dissociation constants between 2-4 μM, VIΔ54 membrane affinity was ∼600-fold lower. These results suggest that the N-terminal amphiphathic α-helix is important for membrane binding. Additionally, there is a strong correlation between the affinity of pVI for membranes and the in vitro membrane lytic activity.
Orientation of the pVI N-terminal amphipathic α-helix on model membranes
How this N-terminal amphipathic α-helix of pVI is oriented on the membrane upon binding is also of interest and could provide considerable insight into the mechanism of membrane disruption. Using the well documented distance-dependent quenching of tryptophan fluorescence by bromine atoms (Markello et al. 1985), we determined the depth of the conserved W37, W41, and W59 residues in lipid membranes containing brominated lipid which have bromine atoms covalently attached at specific positions on the lipid alkyl chains. Single tryptophan mutants of the 80 residue VI114Δ construct were made by mutating 2 out of the 3 tryptophans to phenylalanine since phenylalanine residues do not possess the fluorescent properties of tryptophan but have membrane binding properties most similar to tryptophan (Wimley et al. 1996). These mutants were shown to possess identical membrane lytic activity as wild type protein and they possessed similar secondary structure content as assessed by circular dichroism spectroscopy (Figs S1 and S2). Of note, VI114Δ possessed 87% α-helical content and 13% random coil in agreement with secondary structure predictions. Using this approach W37 and W41 were found to be 9.6 and 10.6 Å from the center of the bilayer by parallax analysis (Table 1). The distance from the center of the bilayer Zcf, was determined both via parallax and distribution analyses (Chattopadhyay et al. 1987; Ladokhin 1997). Interestingly, W59, which is outside the predicted N-terminal amphipathic helix was also found to interact with the membrane, being 9.9 Å from the center of the bilayer. As a control for the assay, we found that the single tryptophan of melittin was 10.9 Å from the center of the bilayer, which is in good agreement with previously published reports which position this tryptophan 10.8 Å from the bilayer center (Ghosh et al. 1997). Since W37 and W41 would be ∼6 Å apart in an α-helix, yet they are positioned at depths which differ by only ∼1 Å in the membrane, it is likely that this helix is positioned in an oblique orientation relative to the membrane surface and does not traverse the apolar region of the lipid bilayer. The angle between W37 and W41 would correspond to 10 degrees from the bilayer-water interface. A model for the orientation of this helix on membranes is shown in Fig 3.
Table 1. Depth of protein VI tryptophan residue penetration into lipid bilayers.
The distance of W37, W41 and W59 from the center of the lipid bilayer (Zcf) were calculated by distance dependent quenching of tryptophan fluorescence. The Zcf values were calculated for each tryptophan residue as described in the methods section using Parallax and Distribution analyses.
| Zcf (Ǻ) | ||
|---|---|---|
| Parallax | DA | |
| W37 | 9.6 ±0.2 | 9.7 ±0.2 |
| W41 | 10.6 ±0.3 | 10.6 ±0.3 |
| W59 | 10.3 ±0.5 | 10.5 ±0.7 |
Figure 3. Model for VI34-54 amphipathic α-helix orientation in lipid bilayer.
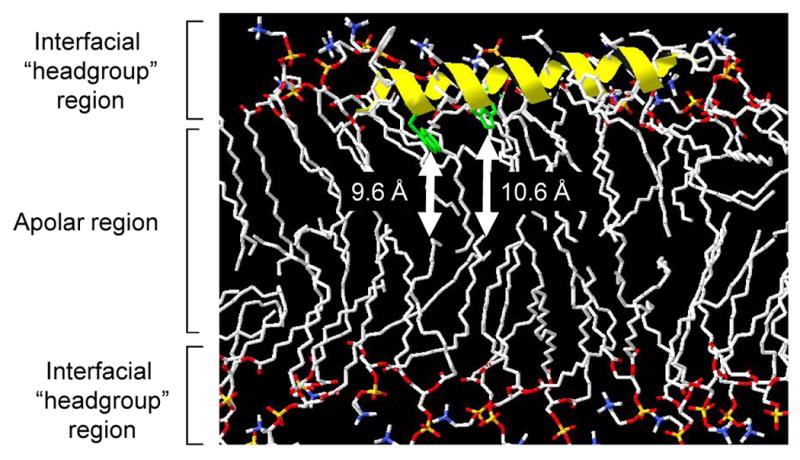
Residues 34-54 of Ad5 protein VI were modelled as an alpha-helix using swisspdb viewer and overlaid on the three dimensional structure of POPC lipids in the Lcα phase (POPC128a.pdb, downloaded from http://people.ucalgary.ca/∼tieleman/download.html) such that the distance of the center of W37 and W41 (green) were positioned at the distance from the center of the bilayer (Zcf) presented in Table 1 (indicated by white arrows).
Protein VI mediates membrane disruption by inducing positive membrane curvature
Since the above data suggest a superficial oblique orientation for the pVI N-terminal α-helix which is a key determinant of pVI membrane lytic activity, we considered that pVI may be inducing curvature stress in membranes leading to membrane disruption. To examine this possibility, we first determined the effects of pVI on membrane morphology using giant fluorescent lipid vesicles (GLV). The GLV membranes were labeled with 5 mol% fluoresceinylated lipid and observed by epifluorescence microscopy. Vesicles appear 5-50 μm in diameter and this morphology is unchanged upon addition of PBS (Fig 4, top). Vesicles incubated with VIΔ54 (1:100 protein:lipid) appeared similar to PBS treated vesicles (Fig 4, middle). Upon addition of protein VI to GLVs at a protein:lipid molar ratio of 1:100, the vesicles were fragmented into smaller structures with a greatly increased radius of curvature (Fig 4, bottom). Of note, reorganization of lipid membranes into tubular structures (arrows) appears to result from pVI addition. This data supports the hypothesis that pVI fragments membranes by inducing significant membrane curvature.
Figure 4. Protein VI fragmentation of giant lipids vesicles.

Fluoroscein-DHPE labelled giant lipid vesicles were incubated with PBS (TOP), VIΔ54 (MIDDLE) or protein VI (BOTTOM) for 15 minutes before visualization by epifluorescence microscopy. Protein-lipid ratios were 1:100 (mol/mol). Arrows indicate tubular lipid structures formed in the presence of protein VI.
The observation that pVI fragments membranes by inducing membrane curvature is in line with one proposed mechanism of membrane disruption by cationic antimicrobial peptides (Brogden 2005). These peptides have been shown to fragment membranes either by inducing positive membrane curvature or negative membrane curvature. To determine whether pVI induces positive or negative membrane curvature, we prepared liposomes entrapping SulfoB and containing increasing amounts of lipids which have a preference to adopt either positively (lysophosphatidylcholine, lysoPC) or negatively (phosphatidylethanolamine PE) curved membranes. If pVI membrane lytic activity involves the induction of positive membrane curvature, then we would expect the presence of lysoPC to enhance pVI membrane lytic activity while PE would inhibit pVI membrane lytic activity. Since we observed exactly these effects of lysoPC and PE on pVI membrane lytic activity (Fig 5), we conclude that pVI disrupts membranes by inducing positive membrane curvature. In further support of this observation, the primary sequence of the N-terminal amphipathic α-helix of pVI suggests that it fits recently identified criteria for helices which induce or associate with positively curved membranes (Drin et al. 2007). These helices typically possess a relatively small hydrophobic surface, few if any charged residues, and possess a hydrophilic surface which is composed mostly of short hydrophilic amino acids such as serine, threonine or glycine.
Figure 5. Positive curvature promoting lipids enhance protein VI membrane lytic activity.

POPC:POPS (75:25 mol%) (●) liposomes entrapping SulfoB and in which some POPC was replaced with 5 (△) or 10 (▲) mol% lysoPC or 15(□) or 25 (■) mol% of POPE were incubated with increasing amounts of protein VI. The %SulfoB released was determined as described in the materials and methods. Error bars represent the standard error of the mean for a minimum of 3 replicates.
Although results above demonstrate that pVI can fragment lipid membranes by inducing positive membrane curvature, they do not rule out the possibility that at lower ratios of pVI to lipid, pore stuctures are formed. In fact, the toroidal pore model for peptide induced membrane permeablization involves amphipathic α-helix-induced positive curvature in membrane lipids such that the pore channel is lined both by peptide and lipid headgroups (Epand et al. 1999; Brogden 2005). To determine whether pVI induced size selective pores in lipid membranes we compared the release of the 0.5 kDa SulfoB and 70 kDa FITC-dextran from liposomes incubated with increasing amounts of pVI at an overall lower ratio of protein to lipid (Fig 6). Previous studies have demonstrated that 70 kDa FITC-dextran has a hydrodynamic radius of 100 Å (Bohrer et al. 1979) and is unable to diffuse through membrane channels less than 50 Å in diameter (Ladokhin et al. 1997). If a size selective pore were formed at these lower ratios of pVI to lipid, then we would expect that a greater release of sulfoB compared to FITC-dextran would be observed. Since we observe comparable degrees of fluorophore release from vesicles by pVI, we conclude that pVI does not form pores capable of discriminating between 6 and 100 Å diameter molecules.
Figure 6. Protein VI does not induce size selective permeability of membranes.

POPC:POPS (75:25 mol%) liposomes (○) or the 70 kDa FITC-dextran (●) were incubated with increasing concentrations of protein VI. The liposome entrapped dye was then separated from the released dye by centrifugation and the released dye quantified by fluorescence spectroscopy as described in the methods. Determination of maximal dye release was achieved using triton X-100. Error bars represent the standard error of the mean for a minimum of 3 replicates.
Discussion
Like most viruses, Ads have conserved genomic size by encoding multiple functions in a single protein which may work during different phases of the viral life cycle. Adenovirus pVI functions during later stages of infection by facilitating virus assembly (Hannan et al. 1983; Webster et al. 1993; Ding et al. 1996; Wodrich et al. 2003). Amino acids between residues 48-74 and 233-239 of pVI mediate hexon binding and facilitate hexon nuclear import where capsid assembly occurs (Matthews et al. 1994; Matthews et al. 1995; Wodrich et al. 2003). The C-terminal 11 residues of pVI function late in the virus life cycle by activating the adenoviral 23K cysteine protease which cleaves several capsid proteins to trigger capsid maturation (Hannan et al. 1983; Webster et al. 1993; Ding et al. 1996). Most recently a role for pVI during cell entry has been proposed (Wiethoff et al. 2005). Our current data support a role for pVI in adenovirus escape from endosomes during cell entry. Furthermore, the major determinant of this activity of pVI appears to be a 20 residue putative N-terminal amphipathic helix.
It is well documented that adenovirus penetration of cell membranes occurs from within endosomal compartments (Brown et al. 1973; Svensson et al. 1984; Greber et al. 1993). To penetrate endosomal membranes, the adenovirus capsid must partially uncoat; a process that begins at the cell surface with the loss of the fiber protein and is enhanced through interactions with αv integrins (Wickham et al. 1994; Nakano et al. 2000). Within endosomes, acidification facilitates further uncoating (Svensson et al. 1984; Greber et al. 1993; Wiethoff et al. 2005). Previous studies of the membrane lytic activity of Ad capsids demonstrated that (Svensson et al. 1984; Greber et al. 1993; Wiethoff et al. 2005) membrane lytic activity is attributed to pVI (Wiethoff et al. 2005). These observations correlate with the inability of Ad2ts1, which possesses a hyperstable capsid and does not release pre-pVI upon endocytosis, to escape from endosomes (Greber et al. 1993; Greber et al. 1996). Additionally, capsid stabilization by defensins inhibits pVI release from the capsid and prevents Ad endosomal membrane penetration (Smith et al. 2008). Here we provide additional support for a role of pVI in endosomal membrane penetration by preventing this activity through the use of antibodies capable of neutralizing pVI membrane lytic activity. Our studies demonstrate that antibodies against protein VI must be present at relatively high concentrations to achieve significant inhibition of Ad cell entry and endosomal escape. However, since pVI is not exposed on the virus exterior but is released from the virus upon uncoating in endosomes, this observation is not unexpected. Similar neutralization of reovirus membrane penetration and poliovirus membrane penetration has been reported using antibodies against membrane disrupting proteins, μ1 and VP4, respectively. In these cases, however, pretreatment of reovirus with trypsin to generate ISVPs and expose μ1 (Hooper et al. 1996) or preincubation of poliovirus with VP4 antibodies at 37 °C for 1 hr prior to infection (Li et al. 1994) was required to expose μ1 and VP4 for neutralization. Such prior exposure of pVI would require the dissociation of the receptor binding fiber and penton base proteins from the capsid, an event which would prevent Ad5 infection by itself. Thus, while higher concentrations of αpVI can neutralize adenovirus infection, it seems implausible that these antibodies would impact infections, in vivo. This idea is supported by the lack of previous reports identifying neutralizing antibodies against pVI in clinical specimens. Nevertheless, neutralization of adenovirus infection by αpVI by blocking endosomal escape demonstrates a role for pVI in virus cell entry.
Currently, the molecular mechanisms of cell membrane disruption by capsid proteins of nonenveloped viruses is still poorly defined. Membrane destabilizing sequences from rotavirus (Dowling et al. 2000; Tihova et al. 2001; Golantsova et al. 2004), reovirus (Danthi et al. 2008; Danthi et al. 2008; Ivanovic et al. 2008), papillomaviruses (Kamper et al. 2006), picornaviruses (Prchla et al. 1995; Tosteson et al. 1997) and nodaviruses (Bong et al. 2000; Maia et al. 2006; Banerjee et al. 2009) have been reported, although the exact mechanisms of how they disrupt membranes are still under investigation. Our studies suggest that the N-terminal amphipathic helix of pVI binds to membrane surfaces in an oblique orientation and that this helix is sufficient to lyse membranes in vitro. Removal of this helix leads to a severe reduction in the membrane binding and membrane lytic capacity of pVI suggesting that this helix is the major determinant of pVI membrane lytic activity.
While reovirus μ1 and picornavirus VP1/VP4 are suggested to form pores to disrupt cell membranes, it has been proposed that Ad5 pVI disrupts membranes by fragmentation (Wiethoff et al. 2005). The oblique orientation of the N-terminal amphipathic helix observed in these studies is more in line with membrane fragmentation than it is with pore formation (Epand et al. 1999; Epand et al. 2000). This more severe reorganization of membranes via pVI could facilitate the translocation of a 90 nm diameter capsid across endosomal membranes. Additionally, Ad has been shown to facilitate the cytosolic translocation of other viruses such as a 25 nm diameter Minute Virus of Mice further supporting a gross reorganization of endosomal membranes by Ad (Farr et al. 2005). Our results suggest that pVI disruption of membranes does not likely involve the formation of pores with diameters less than 50 Å and it is unlikely that a 22 kDa or perhaps a 20 residue peptide would be able to induce stable pores with larger diameters. Our results using epifluorescence microscopy to examine the ability of pVI to reorganize fluorescently labeled giant lipid vesicles confirms previous observations that pVI fragments lipid membranes observed by negative stain TEM (Wiethoff et al. 2005). However, since negative stain TEM involves substantial dehydration of lipid membranes leading to potentially artifactual structures, our current results employing fully hydrated membranes in which only the lipid membranes and not potential protein aggregates are visualized provides greater confidence in the observation that pVI fragments membranes. Additionally, examination of pVI membrane lysis in solution has allowed us to visualize novel tubular structures possessing highly curved surfaces not previously observed by TEM. Although the biological significance of these tubules formed in the presence of pVI remain to be determined, overexpression of pVI in cells demonstrates that pVI associates with intracellular membranes, some of which appear to be part of a tubular endocytic network (Wodrich et al. 2010).
Numerous membrane lytic proteins and peptides exist in nature. These peptides often lyse membranes by induction of significant curvature stress. The amphipathic helix of pVI possesses a conserved primary sequence found in many membrane associated proteins which correlates with their association with positively curved membrane surfaces (Drin et al. 2007). Our data demonstrate that pVI membrane lytic activity is enhanced by membranes with an increased propensity to form positively curved membranes and is decreased for membranes with a greater propensity to adopt negatively curved structures. Thus, these observations along with observed association of pVI with highly curved membranes supports a model in which pVI lyses membranes by inducing positive curvature in target membranes. Parvoviral capsids deploy a phospholipase A2 enzyme which hydrolyzes phospholipids to release the positive curvature inducing lysolipids, although a role for curvature stress has yet to be reported for parvoviral escape from endosomes (Zadori et al. 2001; Farr et al. 2005; Lupescu et al. 2006). Furthermore, the flock house virus gamma peptide also induces membrane leakage but does not induce membrane fusion (Maia et al. 2006), an activity thought to rely on induction of negative curvature (Epand et al. 2000; Bertocco et al. 2003)
As a whole, our data shed new light into the mechanisms of membrane disruption by a nonenveloped virus. Adenoviruses appear to possess a novel method for disrupting endosomal membranes during cell entry compared to those previously described for reoviruses, picornaviruses or parvoviruses. Of the above mentioned nonenveloped viruses, adenovirus is best known for eliciting a proinflammatory response during cell entry. In fact, much of this inflammatory response is linked to the penetration of endosomal membranes (Muruve 2004; Zhu et al. 2007; Appledorn et al. 2008; Fejer et al. 2008). Recent observations link the proinflammatory response to certain intracellular bacteria with the membrane fragments generated during phagosomal escape (Dupont et al. 2009). While in vitro observations suggest that pVI fragments membranes, it is unclear whether membrane fragments are generated by adenovirus during cell entry. Further work is required to determine whether this proposed mechanism of adenovirus rupture of endosomal membranes contributes to the proinflammatory response.
Supplementary Material
Acknowledgments
We would like to acknowledge funding from the NIH to C.M.W (AI082430, and a subcontract from HL054352 (awarded to Glen R. Nemerow), and O.M. (AI007508). H.W. acknowledges financial support from a CNRS idea grant (PEPS 2009) and support from the Aquitaine region.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Citations
- Akashi K, Miyata H, et al. Preparation of giant liposomes in physiological conditions and their characterization under an optical microscope. Biophys J. 1996;71(6):3242–50. doi: 10.1016/S0006-3495(96)79517-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appledorn DM, Patial S, et al. Adenovirus vector-induced innate inflammatory mediators, MAPK signaling, as well as adaptive immune responses are dependent upon both TLR2 and TLR9 in vivo. J Immunol. 2008;181(3):2134–44. doi: 10.4049/jimmunol.181.3.2134. [DOI] [PubMed] [Google Scholar]
- Banerjee M, Khayat R, et al. Dissecting the functional domains of a nonenveloped virus membrane penetration peptide. J Virol. 2009;83(13):6929–33. doi: 10.1128/JVI.02299-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertocco A, Formaggio F, et al. Design and function of a conformationally restricted analog of the influenza virus fusion peptide. J Pept Res. 2003;62(1):19–26. doi: 10.1034/j.1399-3011.2003.00063.x. [DOI] [PubMed] [Google Scholar]
- Bohrer MP, Deen WM, et al. Influence of molecular configuration on the passage of macromolecules across the glomerular capillary wall. J Gen Physiol. 1979;74(5):583–93. doi: 10.1085/jgp.74.5.583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bong DT, Janshoff A, et al. Membrane partitioning of the cleavage peptide in flock house virus. Biophys J. 2000;78(2):839–45. doi: 10.1016/S0006-3495(00)76641-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brogden KA. Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat Rev Microbiol. 2005;3(3):238–50. doi: 10.1038/nrmicro1098. [DOI] [PubMed] [Google Scholar]
- Brown DT, Burlingham BT. Penetration of host cell membranes by adenovirus 2. J Virol. 1973;12(2):386–96. doi: 10.1128/jvi.12.2.386-396.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattopadhyay A, London E. Parallax method for direct measurement of membrane penetration depth utilizing fluorescence quenching by spin-labeled phospholipids. Biochemistry. 1987;26(1):39–45. doi: 10.1021/bi00375a006. [DOI] [PubMed] [Google Scholar]
- Chroboczek J, Ruigrok RW, et al. Adenovirus fiber. Curr Top Microbiol Immunol. 1995;199(Pt 1):163–200. doi: 10.1007/978-3-642-79496-4_10. [DOI] [PubMed] [Google Scholar]
- Danthi P, Coffey CM, et al. Independent regulation of reovirus membrane penetration and apoptosis by the mu1 phi domain. PLoS Pathog. 2008;4(12):e1000248. doi: 10.1371/journal.ppat.1000248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danthi P, Kobayashi T, et al. Reovirus apoptosis and virulence are regulated by host cell membrane penetration efficiency. J Virol. 2008;82(1):161–72. doi: 10.1128/JVI.01739-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding J, McGrath WJ, et al. Crystal structure of the human adenovirus proteinase with its 11 amino acid cofactor. Embo J. 1996;15(8):1778–83. [PMC free article] [PubMed] [Google Scholar]
- Dowling W, Denisova E, et al. Selective membrane permeabilization by the rotavirus VP5* protein is abrogated by mutations in an internal hydrophobic domain. J Virol. 2000;74(14):6368–76. doi: 10.1128/jvi.74.14.6368-6376.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drin G, Casella JF, et al. A general amphipathic alpha-helical motif for sensing membrane curvature. Nat Struct Mol Biol. 2007;14(2):138–46. doi: 10.1038/nsmb1194. [DOI] [PubMed] [Google Scholar]
- Dupont N, Lacas-Gervais S, et al. Shigella phagocytic vacuolar membrane remnants participate in the cellular response to pathogen invasion and are regulated by autophagy. Cell Host Microbe. 2009;6(2):137–49. doi: 10.1016/j.chom.2009.07.005. [DOI] [PubMed] [Google Scholar]
- Eftink MR. Fluorescence methods for studying equilibrium macromolecule-ligand interactions. Methods Enzymol. 1997;278:221–57. doi: 10.1016/s0076-6879(97)78013-3. [DOI] [PubMed] [Google Scholar]
- Epand RM, Epand RF. Modulation of membrane curvature by peptides. Biopolymers. 2000;55(5):358–63. doi: 10.1002/1097-0282(2000)55:5<358::AID-BIP1009>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Epand RM, Vogel HJ. Diversity of antimicrobial peptides and their mechanisms of action. Biochim Biophys Acta. 1999;1462(1-2):11–28. doi: 10.1016/s0005-2736(99)00198-4. [DOI] [PubMed] [Google Scholar]
- Everitt E, Lutter L, et al. Structural proteins of adenoviruses. XII. Location and neighbor relationship among proteins of adenovirion type 2 as revealed by enzymatic iodination, immunoprecipitation and chemical cross-linking. Virology. 1975;67(1):197–208. doi: 10.1016/0042-6822(75)90417-1. [DOI] [PubMed] [Google Scholar]
- Farr GA, Zhang LG, et al. Parvoviral virions deploy a capsid-tethered lipolytic enzyme to breach the endosomal membrane during cell entry. Proc Natl Acad Sci U S A. 2005;102(47):17148–53. doi: 10.1073/pnas.0508477102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fejer G, Drechsel L, et al. Key role of splenic myeloid DCs in the IFN-alphabeta response to adenoviruses in vivo. PLoS Pathog. 2008;4(11):e1000208. doi: 10.1371/journal.ppat.1000208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiske CH, Subbarrow Y. The colorimetric determination of phosphorous. J Biol Chem. 1925;66:374–389. [Google Scholar]
- Ghosh AK, Rukmini R, et al. Modulation of tryptophan environment in membrane-bound melittin by negatively charged phospholipids: implications in membrane organization and function. Biochemistry. 1997;36(47):14291–305. doi: 10.1021/bi971933j. [DOI] [PubMed] [Google Scholar]
- Golantsova NE, Gorbunova EE, et al. Discrete domains within the rotavirus VP5* direct peripheral membrane association and membrane permeability. J Virol. 2004;78(4):2037–44. doi: 10.1128/JVI.78.4.2037-2044.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greber UF, Webster P, et al. The role of the adenovirus protease on virus entry into cells. Embo J. 1996;15(8):1766–77. [PMC free article] [PubMed] [Google Scholar]
- Greber UF, Willetts M, et al. Stepwise dismantling of adenovirus 2 during entry into cells. Cell. 1993;75(3):477–86. doi: 10.1016/0092-8674(93)90382-z. [DOI] [PubMed] [Google Scholar]
- Hannan C, Raptis LH, et al. Biological and structural studies with an adenovirus type 2 temperature-sensitive mutant defective for uncoating. Intervirology. 1983;19(4):213–23. doi: 10.1159/000149363. [DOI] [PubMed] [Google Scholar]
- Hooper JW, Fields BN. Monoclonal antibodies to reovirus sigma 1 and mu 1 proteins inhibit chromium release from mouse L cells. J Virol. 1996;70(1):672–7. doi: 10.1128/jvi.70.1.672-677.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanovic T, Agosto MA, et al. Peptides released from reovirus outer capsid form membrane pores that recruit virus particles. Embo J. 2008;27(8):1289–98. doi: 10.1038/emboj.2008.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamper N, Day PM, et al. A membrane-destabilizing peptide in capsid protein L2 is required for egress of papillomavirus genomes from endosomes. J Virol. 2006;80(2):759–68. doi: 10.1128/JVI.80.2.759-768.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladokhin AS. Distribution analysis of depth-dependent fluorescence quenching in membranes a practical guide. Methods Enzymol. 1997;278:462–73. doi: 10.1016/s0076-6879(97)78024-8. [DOI] [PubMed] [Google Scholar]
- Ladokhin AS, Selsted ME, et al. Sizing membrane pores in lipid vesicles by leakage of co-encapsulated markers: pore formation by melittin. Biophys J. 1997;72(4):1762–6. doi: 10.1016/S0006-3495(97)78822-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmberg E, Traina JA, et al. Reversed-phase high-performance liquid chromatographic assay for the adenovirus type 5 proteome. J Chromatogr B Biomed Sci Appl. 1999;732(2):411–23. doi: 10.1016/s0378-4347(99)00316-3. [DOI] [PubMed] [Google Scholar]
- Li Q, Yafal AG, et al. Poliovirus neutralization by antibodies to internal epitopes of VP4 and VP1 results from reversible exposure of these sequences at physiological temperature. J Virol. 1994;68(6):3965–70. doi: 10.1128/jvi.68.6.3965-3970.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupescu A, Bock CT, et al. Phospholipase A2 activity-dependent stimulation of Ca2+ entry by human parvovirus B19 capsid protein VP1. J Virol. 2006;80(22):11370–80. doi: 10.1128/JVI.01041-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maia LF, Soares MR, et al. Structure of a membrane-binding domain from a non-enveloped animal virus: insights into the mechanism of membrane permeability and cellular entry. J Biol Chem. 2006;281(39):29278–86. doi: 10.1074/jbc.M604689200. [DOI] [PubMed] [Google Scholar]
- Mangel WF, Toledo DL, et al. Characterization of three components of human adenovirus proteinase activity in vitro. J Biol Chem. 1996;271(1):536–43. doi: 10.1074/jbc.271.1.536. [DOI] [PubMed] [Google Scholar]
- Markello T, Zlotnick A, et al. Determination of the topography of cytochrome b5 in lipid vesicles by fluorescence quenching. Biochemistry. 1985;24(12):2895–901. doi: 10.1021/bi00333a012. [DOI] [PubMed] [Google Scholar]
- Matthews DA, Russell WC. Adenovirus protein-protein interactions: hexon and protein VI. J Gen Virol. 1994;75(Pt 12):3365–74. doi: 10.1099/0022-1317-75-12-3365. [DOI] [PubMed] [Google Scholar]
- Matthews DA, Russell WC. Adenovirus protein-protein interactions: molecular parameters governing the binding of protein VI to hexon and the activation of the adenovirus 23K protease. J Gen Virol. 1995;76(Pt 8):1959–69. doi: 10.1099/0022-1317-76-8-1959. [DOI] [PubMed] [Google Scholar]
- Muruve DA. The innate immune response to adenovirus vectors. Hum Gene Ther. 2004;15(12):1157–66. doi: 10.1089/hum.2004.15.1157. [DOI] [PubMed] [Google Scholar]
- Nakano MY, Boucke K, et al. The first step of adenovirus type 2 disassembly occurs at the cell surface, independently of endocytosis and escape to the cytosol. J Virol. 2000;74(15):7085–95. doi: 10.1128/jvi.74.15.7085-7095.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemerow GR. Cell receptors involved in adenovirus entry. Virology. 2000;274(1):1–4. doi: 10.1006/viro.2000.0468. [DOI] [PubMed] [Google Scholar]
- Prchla E, Plank C, et al. Virus-mediated release of endosomal content in vitro: different behavior of adenovirus and rhinovirus serotype 2. J Cell Biol. 1995;131(1):111–23. doi: 10.1083/jcb.131.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roelvink PW, Lizonova A, et al. The coxsackievirus-adenovirus receptor protein can function as a cellular attachment protein for adenovirus serotypes from subgroups A, C, D, E, and F. J Virol. 1998;72(10):7909–15. doi: 10.1128/jvi.72.10.7909-7915.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saban SD, Nepomuceno RR, et al. CryoEM structure at 9A resolution of an adenovirus vector targeted to hematopoietic cells. J Mol Biol. 2005;349(3):526–37. doi: 10.1016/j.jmb.2005.04.034. [DOI] [PubMed] [Google Scholar]
- Saban SD, Silvestry M, et al. Visualization of alpha-helices in a 6-angstrom resolution cryoelectron microscopy structure of adenovirus allows refinement of capsid protein assignments. J Virol. 2006;80(24):12049–59. doi: 10.1128/JVI.01652-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai F, Kawabata K, et al. Adenovirus vectors composed of subgroup B adenoviruses. Curr Gene Ther. 2007;7(4):229–38. doi: 10.2174/156652307781369137. [DOI] [PubMed] [Google Scholar]
- Seth P. Adenovirus-dependent release of choline from plasma membrane vesicles at an acidic pH is mediated by the penton base protein. J Virol. 1994;68(2):1204–6. doi: 10.1128/jvi.68.2.1204-1206.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seth P, Fitzgerald D, et al. Evidence that the penton base of adenovirus is involved in potentiation of toxicity of Pseudomonas exotoxin conjugated to epidermal growth factor. Mol Cell Biol. 1984;4(8):1528–33. doi: 10.1128/mcb.4.8.1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seth P, Willingham MC, et al. Binding of adenovirus and its external proteins to Triton X-114. Dependence on pH. J Biol Chem. 1985;260(27):14431–4. [PubMed] [Google Scholar]
- Silvestry M, Lindert S, et al. Cryoelectron microscopy structure of the adenovirus type 2 temperature sensitive mutant 1 reveals insight into the cell entry defect. J Virol. 2009 doi: 10.1128/JVI.00331-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JG, Nemerow GR. Mechanism of adenovirus neutralization by Human alpha-defensins. Cell Host Microbe. 2008;3(1):11–9. doi: 10.1016/j.chom.2007.12.001. [DOI] [PubMed] [Google Scholar]
- Stewart PL, Fuller SD, et al. Difference imaging of adenovirus: bridging the resolution gap between X-ray crystallography and electron microscopy. Embo J. 1993;12(7):2589–99. doi: 10.1002/j.1460-2075.1993.tb05919.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svensson U. Role of vesicles during adenovirus 2 internalization into HeLa cells. J Virol. 1985;55(2):442–9. doi: 10.1128/jvi.55.2.442-449.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svensson U, Persson R. Entry of adenovirus 2 into HeLa cells. J Virol. 1984;51(3):687–94. doi: 10.1128/jvi.51.3.687-694.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tihova M, Dryden KA, et al. Localization of membrane permeabilization and receptor binding sites on the VP4 hemagglutinin of rotavirus implications for cell entry. J Mol Biol. 2001;314(5):985–92. doi: 10.1006/jmbi.2000.5238. [DOI] [PubMed] [Google Scholar]
- Tosteson MT, Chow M. Characterization of the ion channels formed by poliovirus in planar lipid membranes. J Virol. 1997;71(1):507–11. doi: 10.1128/jvi.71.1.507-511.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai B. Penetration of nonenveloped viruses into the cytoplasm. Annu Rev Cell Dev Biol. 2007;23:23–43. doi: 10.1146/annurev.cellbio.23.090506.123454. [DOI] [PubMed] [Google Scholar]
- van Oostrum J, Burnett RM. Molecular composition of the adenovirus type 2 virion. J Virol. 1985;56(2):439–48. doi: 10.1128/jvi.56.2.439-448.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster A, Kemp G. The active adenovirus protease is the intact L3 23K protein. J Gen Virol. 1993;74(Pt 7):1415–20. doi: 10.1099/0022-1317-74-7-1415. [DOI] [PubMed] [Google Scholar]
- Wickham TJ, Filardo EJ, et al. Integrin alpha v beta 5 selectively promotes adenovirus mediated cell membrane permeabilization. J Cell Biol. 1994;127(1):257–64. doi: 10.1083/jcb.127.1.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham TJ, Mathias P, et al. Integrins alpha v beta 3 and alpha v beta 5 promote adenovirus internalization but not virus attachment. Cell. 1993;73(2):309–19. doi: 10.1016/0092-8674(93)90231-e. [DOI] [PubMed] [Google Scholar]
- Wiethoff CM, Wodrich H, et al. Adenovirus protein VI mediates membrane disruption following capsid disassembly. J Virol. 2005;79(4):1992–2000. doi: 10.1128/JVI.79.4.1992-2000.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wimley WC, White SH. Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nat Struct Biol. 1996;3(10):842–8. doi: 10.1038/nsb1096-842. [DOI] [PubMed] [Google Scholar]
- Wodrich H, Guan T, et al. Switch from capsid protein import to adenovirus assembly by cleavage of nuclear transport signals. Embo J. 2003;22(23):6245–55. doi: 10.1093/emboj/cdg614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wodrich H, Henaff D, et al. A Capsid-Encoded PPxY-motif Facilitates Adenovirus Entry. PLoS Pathog. 2010;6(3) doi: 10.1371/journal.ppat.1000808. online_3/19/2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zadori Z, Szelei J, et al. A viral phospholipase A2 is required for parvovirus infectivity. Dev Cell. 2001;1(2):291–302. doi: 10.1016/s1534-5807(01)00031-4. [DOI] [PubMed] [Google Scholar]
- Zhu J, Huang X, et al. Innate immune response to adenoviral vectors is mediated by both Toll-like receptor-dependent and -independent pathways. J Virol. 2007;81(7):3170–80. doi: 10.1128/JVI.02192-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.