

Abstract
Nitric oxide (NO) plays an important role in cell signalling and in the mammalian immune response to infection. On its own, NO is a relatively inert radical, and when it is used as a signalling molecule, its concentration remains within the picomolar range. However, at infection sites, the NO concentration can reach the micromolar range, and reactions with other radical species and transition metals lead to a broad toxicity. Under aerobic conditions, microorganisms cope with this nitrosative stress by oxidizing NO to nitrate (NO3−). Microbial hemoglobins play an essential role in this NO-detoxifying process. Under anaerobic conditions, detoxification occurs via a 2-electron reduction of two NO molecules to N2O. In many bacteria and archaea, this NO-reductase reaction is catalyzed by diiron proteins. Despite the importance of this reaction in providing microorganisms with a resistance to the mammalian immune response, its mechanism remains ill-defined. Because NO is an obligatory intermediate of the denitrification pathway, respiratory NO reductases also provide resistance to toxic concentrations of NO. This family of enzymes is the focus of this review. Respiratory NO reductases are integral membrane protein complexes that contain a norB subunit evolutionarily related to subunit I of cytochrome c oxidase (CcO). NorB anchors one high-spin heme b3 and one non-heme iron known as FeB, i.e., analogous to CuB in CcO. A second group of diiron proteins with NO-reductase activity is comprised of the large family of soluble flavoprotein A found in strict and facultative anaerobic bacteria and archaea. These soluble detoxifying NO reductases contain a non-heme diiron cluster with a Fe–Fe distance of 3.4 Å and are only briefly mentioned here as a promising field of research. This article describes possible mechanisms of NO reduction to N2O in denitrifying NO-reductase (NOR) proteins and critically reviews recent experimental results. Relevant theoretical model calculations and spectroscopic studies of the NO-reductase reaction in heme/copper terminal oxidases are also overviewed.
1 Nitric oxide reduction in biology
Hemoproteins play a dominant role throughout the chemistry of NO in biology. The production of NO from arginine by NO synthase depends on a heme cofactor, and regardless of whether NO is utilized as a secondary messenger, the regulatory domain of its target enzyme, guanylyl cyclase, also relies on a heme cofactor. Heme cofactors are equally crucial to the microbial response to toxic concentrations of NO by flavohemoglobins and other hemoglobin-like proteins under aerobic conditions. Here, we focus on a third branch of NO chemistry in biology: the reduction of NO to N2O by hemoproteins (eqn 1).
| (1) |
The reactions by which NO is produced by reduction of nitrite (NO2−) and consumed by reduction to nitrous oxide (N2O) are essential steps of the denitrification pathway. Respiratory NO reductases in true denitrifying organisms and in partial denitrifiers like the facultative anaerobes Neisseria gonorrhoeae and Neisseria meningitidis, provide these organisms with added resistance to toxic concentrations of NO.1-3
Denitrifying NO reductases (NOR) are integral membrane protein complexes for which there are no crystal structures yet available. The major subunit common to all denitrifying NORs, norB, is evolutionarily related to subunit I of cytochrome c oxidase (CcO). Specifically, the hydropathy pattern of CcO and the six histidine ligands of heme a, heme a3, and CuB appear to be conserved in NORs.4-6 Purification of NO reductases from a variety of denitrifying bacteria have confirmed that the NorB subunit (53 kDa) anchors one high-spin heme b3 and one non-heme iron now known as FeB, in analogy with CuB in CcO.5-9 Similar to the terminal oxidase superfamily, the NOR family exhibits variability in the electron-entry portion of the enzyme (Fig. 1).
Fig. 1.
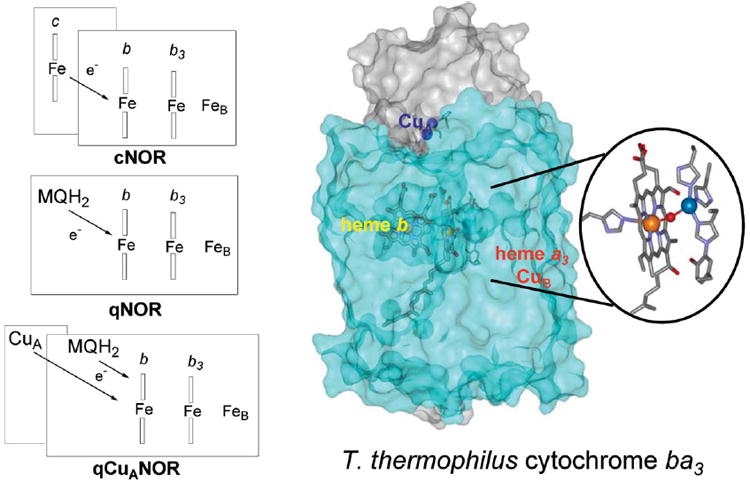
Schematic representations of 3 subfamilies of NO reductases, and the X-ray structure of T. thermophilus cytochrome ba3 (based on coordinates from ref. 83).
The most extensively characterized subfamily of denitrifying NO reductases contains an additional NorC subunit (17 kDa), which binds a heme c; its members are referred to as cNORs.8,10-12 NO reductases from Ralstonia eutropha and from the archaeon Pyrobaculum aerophilum are purified as single-component enzymes, which include quinol binding sites fused to their NorB subunits (84 kDa),13,14 and are thus referred to as qNORs. Most strikingly, in Bacillus azotoformans (B.a. qCuANOR) two branches of electron acceptors coexist, a menaquinol binding site on NorB and a dinuclear copper site, CuA, on subunit II.15-17
In contrast to terminal oxidases, which couple the reduction of O2 with translocation of protons across the membrane,18 the catalytic cycle of cNOR is non-electrogenic.19,20 Consequently, protons are supplied on the periplasmic side of the membrane, as are the electrons that originate from soluble and membrane-bound cytochrome c. Fully-reduced cNORs can complete two NO reductase catalytic cycles and have also been shown to reduce O2 to H2O.21-23 UV-vis monitoring of the reaction of O2 with fully-reduced cNOR reveals two major phases: one is assigned to O2-binding to heme b3 (k = 2 × 107 M−1 s−1), and the second corresponds to the oxidation of the low-spin heme b and c (k = 40 s−1), a process which is coupled with proton uptake.21,23
By analogy with the mechanism of O2 reduction in CcO and NOR, it is certainly reasonable to assume that NO binds at the dinuclear site composed by heme b3 and FeB, and evidence to support this view continues to accumulate. Furthermore, it is possible to envisage several possible catalytic mechanisms. After a brief description of spectroscopic signatures of iron–nitrosyl species and of putative models for the NO reductase catalytic cycle, the most pertinent spectroscopic information obtained to date and its interpretation in terms of catalytic relevance will be reviewed.
2 Spectroscopic signatures of heme iron–nitrosyl complexes
In contrast to CO and O2, NO coordinates both iron(ii) and iron(iii) species. In Enemark and Feltham formulism,24 which counts metal d-electrons plus the lone π* electron from the nitrosyl ligand, iron(ii)–NO and iron(iii)–NO complexes are represented as {FeNO}7 and {FeNO}6 species, respectively. Crystal structures of metal–nitrosyl porphyrin complexes have shown that {MNO}6 species adopt a linear M–N–O structure, while {MNO}7 species have bent M–N–O configurations.24-26 Ferrous hemoproteins typically exhibit a very high affinity for NO while the ferric forms bind NO with Kd values in the micromolar range.27-30 Because the concentration of NO during denitrification remains well below the micromolar range, it is assumed that the catalytic cycle of NO reductases is initiated by the binding of NO to an iron(ii) in the reduced form of the enzyme.
Heme {FeNO}7 species are S = 1/2 species that can be reliably characterized by a combination of UV-vis, EPR and vibrational (resonance Raman (RR) and FTIR) spectroscopies. Specifically, 6-coordinate heme {FeNO}7 species display Soret absorbance at ca. 420 nm while 5-coordinate species exhibit blue-shifted Soret absorbance near 400 nm.31-36 EPR signals for both coordination numbers are centered around g = 2.0, but show distinct superhyperfine splitting: a 3-line hyperfine structure is typically associated with a 5-coordinate heme–nitrosyl, whereas the added N-ligand of a proximal histidine in the 6-coordinated species induces further splitting to produce a 9-line hyperfine structure.37,38 However, defining the coordination number of a {FeNO}7 species solely from EPR spectra may not be entirely foolproof. Indeed, in a recent study of NO binding to reduced heme/copper oxidase aa3 from Paracoccus denitrificans, the characteristic 3-line hyperfine structure was not assigned to a 5-coordinate heme a3 {FeNO}7 species, but rather to a 6-coordinate complex. In this case, the orientation of the Fe–N–O plane, with respect to the trans-imidazole plane, limits the involvement of the imidazole nitrogen p-orbital in the π-molecular orbital occupied by the unpaired electron, thus preventing further splitting of the 3-line hyperfine structure.39
In RR spectra, 6-coordinate {FeNO}7 species display ν3 and ν10 porphyrin core modes at 1500 and 1630 cm−1, respectively, while, in 5-coordinated species, these same modes are observed 5 to 10 cm−1 higher.36,40-42 The ν(Fe–NO) and ν(NO) modes can be identified with isotopic labeling and provide further confirmation of the coordination number of the heme iron. Specifically, the proximal ligand trans to the nitrosyl has been shown to intensify the back-donation of Fe dπ electrons into the nitrosyl π* orbital, thus resulting in higher ν(Fe–NO) and lower ν(NO) for 6-coordinate species.43 The distal pocket environment further modulates the extent of π-backbonding within the FeNO unit. While there are only a few fully characterized 6-coordinate {FeNO} models,25,26 extensive studies of carbonyl synthetic models have led to a convincing description of distal perturbations on π-backbonding in heme Fe–CO complexes.44-46 In hemoproteins, the control of the distal pocket environment on the extent of backbonding is equivalent in 6-coordinate {FeNO}7 species and Fe–CO complexes, as demonstrated by linear correlations between ν(NO)s and ν(CO)s, with slopes near unity for a series of distal pocket variants in myoglobin.43,47,48 As is known for carbonyl complexes, polar environments and hydrogen-bond donation to the nitrosyl group have been shown to promote greater backbonding by stabilizing electron density on NO, thus leading to lower ν(NO) frequencies. The analysis of low-frequency Fe–N–O vibrational modes are more elusive since the intrinsically bent geometry of the {FeNO}7 unit provides greater flexibility to the nitrosyl group and results in extensive mixing of the ν(Fe–NO) and δ(Fe–N–O) modes.49
Heme {FeNO}6 species can be viewed as S = 0 low-spin iron(ii)–NO+ complexes that are isoelectronic with iron(ii)–CO complexes. Accordingly, these species are EPR silent, and the Fe–N–O unit is usually linear with ν(NO) > 1900 cm−1. Both 6- and 5-coordinate {FeNO}6 porphyrinate models have been characterized, but in proteins, only 6-coordinate species are known.25,26 Electronic absorption spectra of {FeNO}6 in hemoproteins can be difficult to differentiate from those of their {FeNO}7 counterparts, and are sometimes strikingly similar.50 Porphyrin skeletal vibrations observed in the high-frequency range of RR spectra, obtained with Soret excitations support a low-spin description of {FeNO}6 complexes.30,50-54 The ν3 and ν10 porphyrin core modes are observed at higher frequencies in {FeNO}6 than in 6-coordinate {FeNO}7 species.30,50-54 Importantly, vibrational modes of the FeNO unit in {FeNO}6 species do not seem to correlate with those of {FeNO}7 and carbonyl complexes. In fact, ferric–nitrosyl complexes in P450cam, with various substrates bound in the distal pocket, reveal a direct correlation between ν(Fe–NO) and ν(NO) rather than the inverse correlation observed between ν(Fe–XO) and ν(X–O) (i.e., where X = N or C) in {FeNO}7 and carbonyl complexes.49,53 Recently, Rodgers and coworkers extended these studies of P450-like enzymes to include other porphyrin {FeNO}6 systems using density function theory calculations.55,56 Their results provide descriptions of higher-occupied molecular orbitals and their bonding characters, with respect to the Fe–NO and N–O bonds, in an effort to define relationships between observed vibrational frequencies, electronic structures, and reactivity.
3 Putative reaction mechanisms
The catalytic mechanism of NO reduction in dinuclear proteins can be divided into four steps: the initial coordination of NO at the diiron active site, the formation of an N–N bond, the cleavage of an N–O bond and the release of N2O. In terms of the first two steps, three possible mechanisms have been proposed (Fig. 2).
Fig. 2.
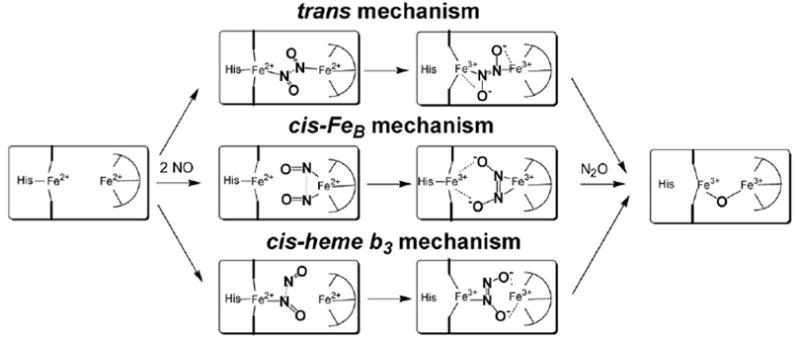
Putative mechanisms of NO reductase.
3.1 trans-Mechanism
The catalytic cycle of this mechanistic model is initiated by the binding of two NO molecules at the diiron(ii) cluster to form an iron–nitrosyl dimer [{FeNO}7]2 intermediate. The close proximity of the two metal–nitrosyl species promotes N–N bond formation either via an electrophilic attack by one nitrosyl at the nitrogen lone pair of the second nitrosyl, or via a radical coupling process, where the two nitrosyls combine to form a metal bound hyponitrite species (N2O2 2−). Several experimental observations support a trans-mechanism, including the characterization of an (FeCO)2 heme/non-heme dicarbonyl complex at the active site of B.a. qCuANOR.57 Also, rapid-freeze quench samples from single turnover reactions in cNOR have revealed EPR signals consistent with the trapping of S = 1/2 low-spin heme {FeNO}7 and S = 3/2 non-heme {FeNO}7 species.58
3.2 cis–FeB mechanism
This mechanism is characterized by the binding of two NO molecules at the non-heme iron center to form a dinitrosyl complex.59 Dinitrosyl iron complexes exhibit isotropic g = 2.0 EPR signals and can be described as {Fe(NO)2}9 species.60,61 In this mechanistic model, the role of the heme iron would be limited to electron transfer, although interactions with the putative hyponitrite complex may participate in the N–O bond cleavage. The cis mechanism has been strongly favored by Thomson and coworkers because it does not involve the formation of a heme b {FeNO}7 species, which they view as a potential dead-end product.62-65 Indeed, in many hemoproteins, ferrous–nitrosyl complexes are unreactive species.66-70 Redox titrations of cNOR by Thomson and coworkers have indicated that the redox potential of heme b3 is 200 mV more negative than that of the non-heme iron FeB, suggesting that a three-electron reduced state, where heme b3 remains oxidized, may be relevant to catalysis.64,65
3.3 cis-Heme b3 mechanism
In this mechanism, a heme b3 {FeNO}7 species forms before reacting with a second NO molecule. The lone electron pair of the metal-bound nitrosyl is then the target of an electrophilic attack by a free NO to generate a hyponitrite radical. The reduction of the N2O2− group to hyponitrite may occur subsequently to the formation of an asymmetrically-bridged diiron cluster. This reaction mechanism is exemplified in the Pd/Cu-catalyzed reduction of NO in aqueous solution where, in a rate limiting step, cuprous salt reduces the dinitrogen dioxide PdCl3(N2O2)2− to give PbCl4−, N2O, and water.71 The cis-heme b3 mechanism is also reminiscent of flavohemoglobin, if the initial {FeNO}7 species in NOR is seen as analogous to the initial superoxo complex {FeO2}8 in flavohemoglobin. This mechanism has been suggested for the NO reductase activity in terminal oxidases where a heme {FeNO}7 species accumulates in pseudo-steady state conditions.72,73
The timing of the N–O bond cleavage and protonation events that follow the N–N bond formation remain to be defined. Isomerization of putative transient hyponitrites to an O-coordinated species to promote the formation of a bridging oxo group and release of N2O may be energetically unfavorable.73 However, an O-coordination of the hyponitrite dianion to FeB could lead to N–O bond cleavage and formation of a FeB(VI)=O.74 Alternatively, protonation of the hyponitrite anion (or a hyponitrite radical precursor) by a catalytic acid could favor the heterolytic cleavage of the N–O bond.
4 Spectroscopic characterization of denitrifying NO reductases
4.1 The diiron cluster can accommodate an Fe–Fe distance ≤ 3.5 Å
As is often the case with heme-containing proteins, RR spectroscopy was instrumental in providing unequivocal support for the similarities between NO reductases and CcO and for the presence of a heme/non-heme dinuclear cluster at the active site of NO reductase. Similar to the heme a3 of CcO, RR spectra of fully reduced P.d. cNOR defined the heme b3 as a 5-coordinate high-spin heme. Moreover, the detection of an iron(ii)–NHis stretching mode at 218 cm−1 identified the heme b3 axial ligand as a neutral histidine side chain.11 In the oxidized enzyme, the presence of a μ-oxo-group bridging the two iron(iii) centers was revealed by isotopic exchange with 18O-labeled water.75 Comparison of the iron(iii)–O–iron(iii) stretching frequency observed in P.d. cNOR with other ν(Fe–O–Fe) vibrations in non-heme diiron models76 and a synthetic heme/non-heme synthetic model ([(5L)Feiii–O–Feiii–Cl]+)77 provided the first direct evidence of a diiron cluster with an Fe–Fe distance ≤ 3.5 Å (Fig. 3).75 Because the heme iron is 5-coordinate in both oxidized and reduced cNOR, and because a ν(Fe–NHis) was identified only in the reduced enzyme,11 we concluded that the μ-oxo bridge must be hydrolyzed upon reduction, allowing for the coordination of the proximal histidine to heme b3 and leaving an open coordination site facing FeB.75
Fig. 3.
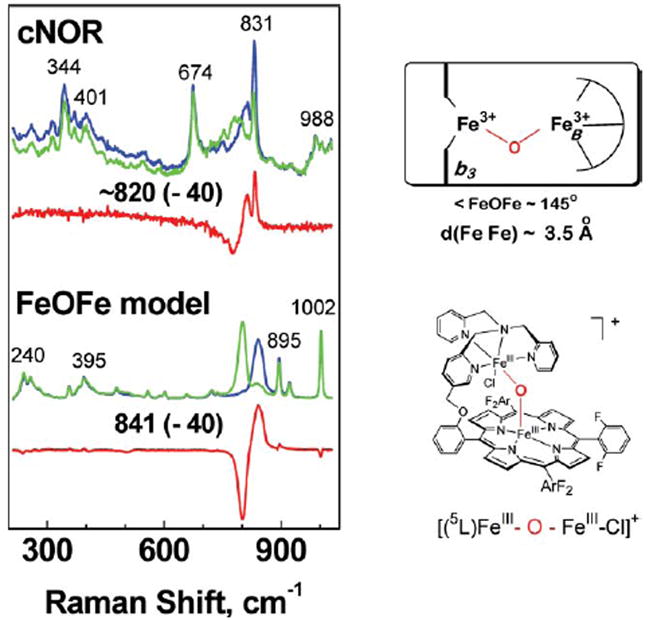
Resonance Raman characterization of heme/non-heme μ-oxo bridged diiron(iii) cluster in oxidized cNOR (in 16OH2 (blue), 18OH2 (green) and difference spectrum (red)), and in a synthetic model in CH3CN with 20% water (in 16OH2 (blue), 18OH2 (green) and difference spectrum (red)). The νas(Fe–O–Fe) modes are identified by their 40 cm−1 downshift upon exchange of the O-atom with 18O-labeled water. The schematic structures of the diiron clusters in cNOR and the model compound are also shown (adapted from ref. 75).
The μ-oxo bridged diiron(iii) cluster observed in P.d. cNOR is associated with a ligand-to-metal charge transfer absorption band at 595 nm. A lack of variation in both this absorption band and spectral features in the RR spectra suggest that the cluster is insensitive to pH changes between 6.0 and 9.0.75,78 Mediated redox potentiometry has shown that the heme b3 presents an unexpected low midpoint potential (Em = 60 mV), which allows for the formation of a three-electron reduced form of the enzyme where the heme b3 remains as the sole iron(iii) species.64 Room temperature absorption and MCD analyses of the three-electron reduced enzyme at different pH values suggest that, upon reduction of FeB, the μ-oxo bridge dissociates from FeB(ii) and the heme b3 iron(iii) rebinds the proximal histidine to form a 6-coordinate aquo/hydroxo complex with charge transfer bands at 635 and 605 nm, respectively.78 Not surprisingly, cyanide was shown to bind much more readily to heme b3 in the three-electron reduced form of the enzyme, where the displacement of the sixth aquo/hydroxo ligand is facile (e.g., as in metmyoglobin), than in the fully oxidized enzyme, where the μ-oxo group bridge between the two iron(iii) is quite stable.65 From these observations, Thomson and coworkers proposed that the μ-oxo bridged diiron(iii) cluster might represent a “closed” resting state of the enzyme, while the catalytic cycle may, in fact, be initiated by the binding of NO to the mixed valence heme b3(iii)–FeB(ii) cluster.64,65,78
Regardless of whether the diiron(iii) μ-oxo bridge cluster is part of the catalytic cycle of NO reductases, this structure reveals a metal–metal distance that is 1.5 Å shorter that that measured in the crystal structures of terminal oxidases between the iron of heme a3 and CuB.79-83 This significant distinction between the active sites of P.d. cNOR and CcO may explain their different reactivity toward NO. The iron–iron distance in NOR is more comparable with those measured for the carboxylate-bridged non-heme diiron clusters of the R2 subunit of E. coli ribonucleotide reductase, methane monooxygenase,84-86 and flavoprotein A.87 Interestingly, NO-reductase activity has been reported in all three of these diiron proteins.88-90 The characterization of fully-oxidized and fully-reduced P.d. cNOR makes clear that the proximal histidine is labile and suggests that the catalytic intermediate might include a pentacoordinated heme {FeNO}7 species. Structural characterizations of iron(ii)–nitrosyl porphyrin complexes by Scheidt and coworkers have shown that, as the proximal ligand is released, the iron is pulled further out of the porphyrin plane by 0.2 Å, while the Fe–N and N–O distances and the Fe–N–O angle decrease only slightly.25,26,91 The latter changes are consistent with a modest strengthening of both σ and π bonding in the FeNO unit. A recent crystal structure of nitrosyl–hemoglobin in the presence of the effector inositol hexaphosphate concurs with the iron displacement measured in model complexes.92 Specifically, in β-subunits where 6-coordinated {FeNO}7 species form, the iron remains 0.12 Å out of the porphyrin plane on the proximal side, however, in α-subunits where 5-coordinated {FeNO}7 species form, the iron is located 0.25 Å out of the porphyrin plane toward the distal side. At the dinuclear site of NO reductases, a 5-coordinate {FeNO}7 structure at the heme b3 would decrease the distance between the nitrosyl group and a second {FeNO}7 species at FeB, a change that may favor radical coupling and N–N bond formation.
4.2 The diiron site can accommodate two CO molecules
Carbon monoxide (CO) is an ideal probe to investigate the binding of exogenous diatomic ligands at the dinuclear site of NO reductases. Heme iron–carbonyl complexes are stable and exhibit a characteristic ν(C–O) between 1940 and 1980 cm−1 in RR and FTIR spectra.93 Moreover, heme iron–carbonyls are photolabile and can be used to probe the heme distal pocket. In myoglobin, for example, the photolyzed CO can be trapped in several docking sites within the distal pocket.94-96 These photolyzed CO molecules in proteinaceous docking sites display weak, but well-resolved, ν(C–O) near 2130 cm−1 in low-temperature FTIR spectra.97-99 In terminal oxidases, CO dissociates from heme a3 and coordinates to CuB. The CuB–CO complex can be trapped at cryogenic temperatures, and ‘dark’ minus ‘illuminated’ FTIR difference spectra exhibit a positive ν(CO) from the a3–CO complex and a negative ν(CO) between 2040–2070 cm−1 characteristic of CuB–CO.100-103
In 1998, Saraste and coworkers attempted to use this approach with P.d. cNOR and reported that FTIR difference spectra could only be observed between dark and continuous illumination at 234 K.104 The asymmetric absorption changes, centered around 1970 cm−1, were interpreted in terms of a ‘dark’ signal at 1977 cm−1, assigned to heme b3–CO, and a ‘light’ signal at 1963 cm−1, assigned to FeB–CO. However, these absorption changes may, in fact, originate from variations in populations of heme a3–CO conformers under continuous illumination rather than CO-coordination to FeB. FTIR difference spectra between P.d. cNOR–12CO and cNOR–13CO obtained at Oregon Health & Science University confirmed that only one CO molecule binds per diiron site.105 ‘Dark’ minus ‘illuminated’ FTIR difference spectra at 15 K exhibit a ‘dark’ ν(CO) at 1972 cm−1, assigned to heme b3–CO, and an ‘illuminated’ ν(CO) signal at 2120 cm−1 that is characteristic of non-coordinated CO docking at the hydrophobic site near the distal pocket.105 Thus, from these FTIR experiments, it remains unclear whether CO can bind FeB following photolysis from heme b3 in P.d. cNOR.
The CO photolysis in P.d. cNOR was also investigated at room temperature using time-resolved electronic absorption. These experiments revealed a very fast recombination rate (kon = 1.7 × 108 M−1 s−1), approximately three orders of magnitude faster than in terminal oxidases.106 In contrast, when Watmough and collaborators measured the rate constant for the initial binding of CO by stopped-flow spectroscopy, they observed a much slower rate (kon = 1.2 × 105 M−1 s−1).106 The authors suggested that binding of CO at the heme a3 requires the initial displacement of a distal heme ligand. The slow rebinding of this ligand, compared to CO, would explain the photolysis kinetics.106 However, RR studies identify the ferrous heme b3 as a 5-coordinate high-spin species,11,107 thus, the kinetic data may be consistent with the presence of a weakly interacting group that can adopt variable configurations rather than with the displacement of a true distal ligand of heme b3 (vide infra).
In sharp contrast with P.d. cNOR–CO, recent experiments with the CO complex of qCuANOR show that two CO molecules bind concomitantly at the diiron site of qCuANOR.57 Specifically, room temperature (12CO minus 13CO) FTIR difference spectra of qCuANOR–CO show two ν(CO) modes at 1972 and 2068 cm−1 (Fig. 4). The band at 1972 cm−1 (Δ13CO=−44 cm−1) corresponds to a heme–CO and is nearly identical to that observed in the heme–CO complex of P.d. cNOR.11,105,108 The ν(CO) at 2068 cm−1 (Δ13CO = −47 cm−1) is consistent with a non-heme iron–CO where the back-bonding donation from the iron dπ orbitals to the carbonyl π* orbitals is weakened compared to its heme–CO counterparts. The comparable integrated area of these two bands suggests that these two species are present in equivalent concentrations. These room temperature data may be interpreted in two ways: i) two CO molecules occupy the dinuclear active site, or ii) the active site contains only one CO molecule in a binding equilibrium between the heme and the non-heme irons. However, the comparison of the integrated areas of the room temperature signals with the total enzyme concentration favors the first alternative, and low-temperature photolysis experiments confirm this conclusion.57
Fig. 4.
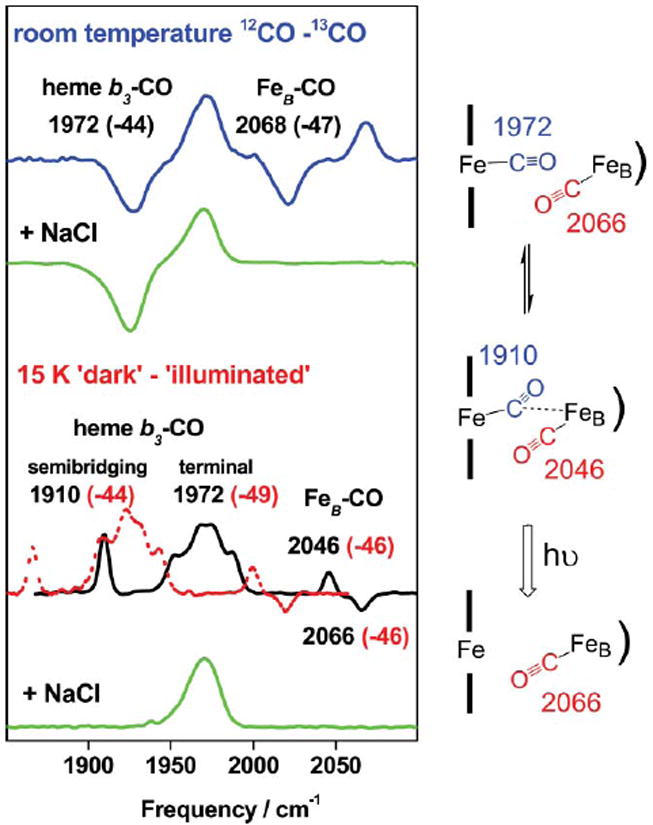
FTIR difference spectra of qCuANOR–CO. At room temperature (top traces), the ν(CO) are isolated using 12CO–13CO spectra (blue trace: no chloride added, green trace: 3 M chloride). At 15 K (bottom traces), ‘dark’ minus ‘illuminated’ spectra reveal photosensitive ν(CO) modes (black trace: 12CO; red trace: 13CO; and green trace: 12CO in the presence of 3 M NaCl). A schematic interpretation of the binding of CO in qCuA NOR is also shown (adapted from ref. 57).
The ‘dark’ minus ‘illuminated’ FTIR difference spectrum of qCuANOR–CO obtained at 15 K (Fig. 4) shows a positive cluster of bands centered around 1972 cm−1 that is assigned to comparable conformers of the photolabile heme–CO complex. A new band at 1910 cm−1 (Δ13CO = −44 cm−1), which is not observed at room temperature, is characteristic of a semi-bridging configuration between two metal ions.93 Accompanying this band is an S-signal with a positive band at 2046 cm−1 and a negative band at 2066 cm−1 (Fig. 4). The influence of temperature on the FTIR difference spectra shows the interdependence of these two signals. Indeed, as the temperature is raised to ~100 K, the 1972 cm−1 signal decreases as geminate rebinding begins to occur, but the 1910 cm−1 band and the 2046/2066 cm−1 S-signal are unchanged. We have assigned the S-signal to a downshift of the non-heme FeB–CO as it is perturbed by the photolysis of the semi-bridging heme–CO at 1910 cm−1 (Fig. 4).57 These results lend support to the trans-mechanistic model in which N–N bond formation is promoted by the binding of two NO molecules to form a [{FeNO}7]2 transient species.
Upon addition of chloride to qCuANOR–CO, the binding of CO at the FeB site is inhibited (Fig. 4), and the FTIR characterization of qCuANOR–CO resembles that of P.d. cNOR–CO. Similarly, in the heme/non-heme synthetic model [(6L)–Feii … Feii–(Cl)]+, CO binding occurs at the heme iron, but the non-heme iron(ii) does not bind CO, at least in part because of the chloride coordination.109 It remains unclear in the case of NOR, whether, chloride directly binds to FeB(ii) or whether its affect on FeB(ii) ligation is only allosteric, but control experiments suggest that different states of the diiron site cannot solely be explained by the effect of chloride.105,110 Thus, in both NOR proteins, there is clear experimental evidence for the presence of a yet unidentified ligand(s) that can adopt variable coordination geometries. At this time, it is tempting to speculate that a glutamate side chain at the diiron site might provide coordination flexibility to this site. Several glutamate residues are conserved in norB while absent from subunit I in terminal oxidases.4-6 In particular, conserved residues Glu198 and Glu202 (numbering from P. denitrificans norB) are located in transmembrane helix VI, which includes His194, a putative ligand to FeB. Moreover, the RR characterizations of the CO complexes of P.d. cNOR and qCuANOR suggest the presence of negatively charged residues in the distal pocket of heme b3.11,105
Richardson and coworkers have engineered expression systems from norB variants in P. denitrificans and E. coli and analyzed the NO reductase activity of membrane fragments and solubilized proteins.22 Three mutants of conserved glutamate residues were successfully expressed and purified: E198A and E202A, which are expected to be at or near the diiron active site, and E125A, which is predicted to be on the periplasmic surface. The location of Glu125, and the loss of NO reductase activity in E125A suggest that this residue may play a role in proton transfer from the periplasm to the active site. While E202A shows an NO-reductase activity comparable to the wild type protein, the E198A variant is inactive. These results suggest that E198 plays an important role at the diiron active site, either as a ligand to FeB or as a proton transfer group.
In non-heme diiron proteins, X-ray crystallography has revealed variations in the coordination of carboxylate groups that accompany changes in the oxidation state of the diiron cluster. For example, in the R2 subunit of E. coli ribonucleotide reductase and in methane monooxygenase, a coordinating glutamate side chain switches from bridging the two irons in the fully-reduced enzymes to terminally coordinating a single iron(iii) in the oxidized proteins.111-116 Variations in carboxylate coordination geometry also take place without changes in iron oxidation states. For example, interactions of regulatory subunits such as MMOB with the hydroxylase protein of methane monooxygenase and acyl carrier protein with Δ9-desaturase affect the spectroscopic characteristics of the diiron(ii) cluster at the active site.117-119 These observations led to the concept of carboxylate shifts controlling the number of open coordination sites and the range of Fe–Fe distances during catalytic turnover.120 Thus, in NOR, where glutamate side chains in the vicinity of coordinating histidine residues are known to be conserved, it is conceivable that carboxylate shifts might control the coordination of diatomic molecules at the heme/non-heme diiron site.
4.3 Spectroscopic studies of NO reactions with NOR
Using rapid-freeze quenching, Shiro and coworkers have shown that NO and fully-reduced cNOR react in a sub-millisecond time scale, as evidenced by the appearance of new EPR signals within 0.5 ms after mixing.58 One signal, at g = 4, is consistent with a non-heme S = 3/2 {FeNO}7 species, and another at g = 2.01, with a pronounced 3-line hyperfine structure, is characteristic of a heme S = 1/2 {FeNO}7 system. Shiro and coworkers estimate that these two nitrosyl signals represent 30% of the total diiron site concentration. In addition, they show that these species decay within 10 ms when their EPR signatures are replaced with those of ferric low-spin b and c hemes.58 The data have been interpreted in terms of a trans-mechanism where a [{FeNO}7]2 unit forms at the active site within the first ms after mixing.58
The observation of a g = 4 signal from a non-heme {FeNO}7 species and a g = 2.0 signal from a heme {FeNO}7 upon addition of NO to fully-reduced cNOR had been reported earlier in hand-mixing experiments,104 but their relevance to the native structure have been seriously questioned as these EPR signals appeared to increase under prolonged incubation.121 In contrast with hand-mixed experiments, the millisecond kinetics of the formation and decay of the EPR signals observed by Shiro and coworkers support the involvement of these species in the catalytic cycle of NOR.58 The interpretation of these EPR data may be revisited however, as one might expect a heme/non-heme [{FeNO}7]2 complex to be magnetically coupled to form an EPR-silent spin integer species (most likely antiferromagnetically coupled, i.e., an S = 1 system). For example, the major species formed by the reaction of NO with the reduced non-heme diiron site of protein R2 is an EPR-silent, antiferromagnetically-coupled [{FeNO}7]2 complex.88,122 Shiro and coworkers favor the formation of an [{FeNO}7]2 unit on the basis that the g = 4 signal of the reaction intermediate behaves differently, with respect to microwave power saturation, than in the CO-inhibited species, where the S = 0 low-spin iron(ii) carbonyl complex does not promote relaxation.58 However, an increase in relaxation would be expected whether or not NO binds to heme b3 since both heme {FeNO}7 and high-spin iron(ii) are S ≠ 0 species. An alternative to the authors’ proposal may be that within the first millisecond after exposure of fully-reduced cNOR to NO a mixture of [heme b3 Fe–NO/FeB] and [heme b3 Fe/FeB–NO] complexes can be produced. Indeed, if the metal centers are magnetically uncoupled upon complexation with a single NO molecule, both S = 3/2 and S = 1/2 {FeNO}7 species are expected to contribute to the EPR spectra. This alternative interpretation would imply that the two iron(ii) ions at the active site pocket have comparable binding affinity for NO, and that these {FeNO}7 complexes can be trapped by rapid-freeze quenched techniques. A [{FeNO}7]2 complex, on the other hand, might not be observable by EPR, or it might not accumulate in the course of a single turnover reaction. The concept of a mixture of {FeNO} complexes has support from the evolutionarily-related cytochrome ba3 oxidase from T. thermophilus, from which FTIR experiments have revealed an [heme b3 Fe–CO/CuB] ↔ [heme b3 Fe/CuB–CO] equilibrium.123 In addition, theoretical analyses of the in silico reduction of NO with simplified heme/copper and heme/non-heme dinuclear clusters calculate reaction-energy profiles where hyponitrite, rather than dinitrosyl complexes, are the observable intermediates.73,74 Thus, two CO molecules bound at the diiron site of NOR can be observed as the complex is stable in qCuANOR,57 but a [{FeNO}7]2 transient might not be a traceable intermediate if it does not accumulate in the course of the reaction.
NO can also react with fully-oxidized P.d. NOR,10,124 but this reaction only occurs at pH < 6, where acidic conditions presumably favor the hydrolysis and displacement of the μ-oxo bridge to allow NO coordination to heme b3.125 The heme b3 {FeNO}6 species displays a ν(Fe–NO) at 594 cm−1 and a ν(NO) at 1904 cm−1.124 These stretching frequencies are at the low range of those reported for 6-coordinate heme {FeNO}6 species with a proximal histidine ligand.30,50,51,54,126-128 Varotsis and coworkers124 noted that the low ν(NO) contrasts with the high ν(CO) associated with inhibition of π-backbonding in the negative polarity of the distal pocket.11 As discussed earlier (see section 2; spectroscopic signatures of heme iron–nitrosyl complexes), the influence of distal pocket environment on the characteristics of heme {FeNO}6 species remains poorly understood.55,56 It is also unclear whether or not the heme b3 {FeNO}6/FeB(iii) state is relevant to the catalytic cycle of NOR.
5 NO reduction in terminal oxidases
The various reactions of NO with heme/copper terminal oxidases and their importance to the regulation process of O2-respiration have been recently reviewed.129-132 Briefly, the interaction of NO with the oxidized enzyme inhibits mitochondrial CcO via binding of NO to CuB(ii).133,134 The resulting copper–nitrosyl complex adopts a CuB(i)–NO+ configuration and produces nitrite upon hydration of the bound nitrosonium ion.129,135-137 In contrast, reduced terminal oxidases bind NO in a competitive fashion with O2 to form a heme a3 {FeNO}7 complex.138 In some terminal oxidases, experimental observations corroborate the concomitant binding of a second NO molecule at the CuB(i) site. For example, FTIR experiments by Caughey and coworkers on reduced bovine CcO exposed to NO revealed two ν(NO)s at 1610 and 1700 cm−1, which were assigned to heme a3-NO and CuB-NO, respectively.139 In addition, optical experiments monitoring rebinding dynamics in the picosecond time scale for the NO complex of reduced aa3 from P. denitrificans showed that geminate rebinding to the heme a3 site is predominant at high NO concentration. This behavior was assigned to an obstruction of the exit route for the photolyzed NO by a second NO bound to CuB.39 It is interesting to note that while these metal–nitrosyl Fe/Cu dimers in CcO and aa3 mimic the putative [{FeNO}7]2 intermediate in NOR, neither of these terminal oxidases show any NO reductase activity.
The fully-reduced caa3 and ba3 oxidases from Thermus thermophilus quickly react with NO to form a heme a3 {FeNO}7 complex, which decays to produce N2O with a turnover rate approximately 1% and 0.1% that of denitrifying NORs, respectively.138,140 Presumably, binding of a second NO to CuB(i) is slow and results in a facile turnover to form N2O.39,72 Thus, binding of a second NO molecule may occur in all oxidases, but the only catalytically competent dinitrosyl complexes form in T. thermophilus oxidases. All fully-reduced terminal oxidases react with NO to form a heme {FeNO}7 complex that can be spectroscopically characterized.130,131,141 While both 5-coordinate and 6-coordinate {FeNO}7 hemes have been reported in terminal oxidases,39,72,142 the coordination number of the heme iron does not appear to correlate with NO reductase activity.138,140
The heme iron–nitrosyl complex in T. thermophilus ba3 has been studied by time-resolved absorption spectroscopy and displays rebinding dynamics on the 15 ns time scale at room temperature.39 This relatively slow rebinding process suggests that the photolyzed-NO group binds transiently to another site, presumably CuB. Time-resolved step-scan FTIR spectra of the heme a3–NO complex in T. thermophilus ba3 after flash photolysis show no differential signals and indicate that rebinding occurs within the 5-μs time resolution of this technique.143 Using density function theory calculations, and within the context of a trans-mechanism, Varotsis and coworkers have proposed that the formation of a [heme a3-NO – CuB-NO] dinitrosyl complex leads to an N,N′-bridging hyponitrite intermediate.144 Using the same theoretical approach, Blomberg and coworkers favor a cis-heme a3 mechanism and a bridging coordination of the hyponitrite to form a 5-member ring with two O-atoms coordinated to CuB.73 Although only a few transition metal hyponitrite complexes have been structurally characterized to date,145-148 these complexes already demonstrate a wide diversity in coordination geometries. There is little doubt that the characterization of a hyponitrite intermediate in the reaction of fully-reduced ba3 with NO would constitute a major breakthrough toward understanding these catalytic reactions.
6 Conclusions and perspectives
Structural and functional similarities between NO reductases and the heme/copper terminal oxidases are now firmly established, and in view of the existing literature on terminal oxidases, the existence of different forms of NORs is not surprising. Indeed, resting and pulsed/fast forms of oxidized and reduced CcO are well documented.149 The effect of chloride on the family of terminal oxidases is notoriously complex and ranges from direct binding to cuprous CuB(i),150-152 to bridging the heme a3 iron(iii) and CuB(ii),149,153 to binding at heterotropic sites.149,154,155 With its poor NO reductase activity and access to its three-dimensional structure, T. thermophilus cytochrome ba3 provides an opportunity to gather spectroscopic information on NO reaction intermediates. Nevertheless, because of their different metal composition, what is learned from heme/copper terminal oxidase systems will not always be applicable to NORs. A clear definition of the catalytic cycle of NOR will require the combination of rapid-freeze quenching and multiple spectroscopic techniques.
Several carboxylate-bridged non-heme diiron proteins catalyze the reduction of NO to N2O, and the flavoprotein A family of enzymes has recently been exposed as detoxifying NO reductases.90,156,157 A recombinant form of flavoprotein A from Moorella thermoacetica, which exhibits comparable NO reductase activity to denitrifying NORs, was recently crystallized.87 In this protein, the active site contains a non-heme diiron cluster with two histidines per iron and two bridging ligands: a bidentate carboxylate group and a solvent molecule.87 Carboxylate groups have been shown to provide structural flexibility at the active sites of non-heme diiron proteins (for an extreme example, see recent crystal structures of nigerythrin, which reveal a 2 Å movement of one iron associated with a Glu ↔ His ligand “toggling” at one iron158). In denitrifying NOR, FeB may likewise recruit a glutamate side chain as a ligand, which, in turn, may allow the site to control the reactivity of {FeNO}7 species, regulate the catalytic cycle and prevent the formation of dead-end complexes through carboxylate shifts.
Acknowledgments
This work was supported by the National Institutes of Health (GM 074785). The contributions of students and colleagues at OHSU, Shen Lu, Hong-wei Huang, and I.-Jin Lin, are gratefully acknowledged. The author also expresses sincere thanks to his collaborators Simon de Vries, Kenneth D. Karlin, James A. Fee, and to members of their groups.
Biography
Pierre Moënne-Loccoz was born in France in 1964. He received his PhD in 1989 from Université Pierre et Marie Curie (Paris, France) for his work on Photosystem I and II of higher plants, under the supervision of Marc Lutz and Bruno Robert (CE Saclay, France). He continued to study photoactive proteins, first with Warner Peticolas at the University of Oregon and then with Mike Evans and Peter Heathcote at University College London. He then joined Thomas Loehr and Joann Sanders-Loehr at the Oregon Graduate Institute, where he started investigating O2-activation processes in heme and non-heme diiron proteins. He is now an Assistant Professor at the OGI School of Engineering at OHSU where he continues to work on O2 and NO chemistry in metalloproteins using a variety of spectroscopic techniques.

Footnotes
This paper was published as a part of a special issue on the chemistry and biochemistry of heme proteins.
References
- 1.Lissenden S, Mohan S, Overton T, Regan T, Crooke H, Cardinale JA, Householder TC, Adams P, O’Conner CD, Clark VL, Smith H, Cole JA. Mol Microbiol. 2000;37:839–855. doi: 10.1046/j.1365-2958.2000.02050.x. [DOI] [PubMed] [Google Scholar]
- 2.Householder TC, Fozo EM, Cardinale JA, Clark VL. Infect Immun. 2000;68:5241–5246. doi: 10.1128/iai.68.9.5241-5246.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anjum MF, Stevanin TM, Read RC, Moir JW. J Bacteriol. 2002;184:2987–2993. doi: 10.1128/JB.184.11.2987-2993.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Castresana J, Lübben M, Saraste M, Higgins DG. EMBO J. 1994;13:2516–2525. doi: 10.1002/j.1460-2075.1994.tb06541.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van der Oost J, de Boer AP, de Gier JW, Zumft WG, Stouthamer AH, van Spanning RJ. FEMS Microbiol Lett. 1994;121:1–9. doi: 10.1111/j.1574-6968.1994.tb07067.x. [DOI] [PubMed] [Google Scholar]
- 6.Zumft WG, Braun C, Cuypers H. Eur J Biochem. 1994;219:481–490. doi: 10.1111/j.1432-1033.1994.tb19962.x. [DOI] [PubMed] [Google Scholar]
- 7.Heiss B, Frunzke K, Zumf WG. J Bacteriol. 1989;171:3288–3297. doi: 10.1128/jb.171.6.3288-3297.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wasser IM, de Vries S, Moënne-Loccoz P, Schröder I, Karlin KD. Chem Rev. 2002;102:1201–1234. doi: 10.1021/cr0006627. [DOI] [PubMed] [Google Scholar]
- 9.Averill BA. Chem Rev. 1996;96:2951–2965. doi: 10.1021/cr950056p. [DOI] [PubMed] [Google Scholar]
- 10.Girsch P, de Vries S. Biochim Biophys Acta. 1997;1318:202–216. doi: 10.1016/s0005-2728(96)00138-7. [DOI] [PubMed] [Google Scholar]
- 11.Moënne-Loccoz P, de Vries S. J Am Chem Soc. 1998;120:5147–5152. [Google Scholar]
- 12.Zumft WG. J Inorg Biochem. 2005;99:194–215. doi: 10.1016/j.jinorgbio.2004.09.024. [DOI] [PubMed] [Google Scholar]
- 13.Cramm R, Pohlmann A, Friedrich B. FEBS Lett. 1999;460:6–10. doi: 10.1016/s0014-5793(99)01315-0. [DOI] [PubMed] [Google Scholar]
- 14.de Vries S, Strampraad MJ, Lu S, Moënne-Loccoz P, Schröder I. J Biol Chem. 2003;278:35861–35868. doi: 10.1074/jbc.M300857200. [DOI] [PubMed] [Google Scholar]
- 15.Suharti, Strampraad MJ, Schröder I, de Vries S. Biochemistry. 2001;40:2632–2639. doi: 10.1021/bi0020067. [DOI] [PubMed] [Google Scholar]
- 16.Suharti, Heering HA, de Vries S. Biochemistry. 2004;43:13487–13495. doi: 10.1021/bi0488101. [DOI] [PubMed] [Google Scholar]
- 17.Suharti, de Vries S. Biochem Soc Trans. 2005;33:130–133. doi: 10.1042/BST0330130. [DOI] [PubMed] [Google Scholar]
- 18.Ferguson-Miller S, Babcock GT. Chem Rev. 1996;96:2889–2907. doi: 10.1021/cr950051s. [DOI] [PubMed] [Google Scholar]
- 19.Shapleigh JP, Payne WJ. J Bacteriol. 1985;163:837–840. doi: 10.1128/jb.163.3.837-840.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hendriks JH, Jasaitis A, Saraste M, Verkhovsky MI. Biochemistry. 2002;41:2331–2340. doi: 10.1021/bi0121050. [DOI] [PubMed] [Google Scholar]
- 21.Fujiwara T, Fukumori Y. J Bacteriol. 1996;178:1866–1871. doi: 10.1128/jb.178.7.1866-1871.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Butland G, Spiro S, Watmough NJ, Richardson DJ. J Bacteriol. 2001;183:189–199. doi: 10.1128/JB.183.1.189-199.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Flock U, Watmough NJ, Ädelroth P. Biochemistry. 2005;44:10711–10719. doi: 10.1021/bi050524h. [DOI] [PubMed] [Google Scholar]
- 24.Enemark JH, Feltham RD. Coord Chem Rev. 1974;13:339–406. [Google Scholar]
- 25.Scheidt WR, Ellison MK. Acc Chem Res. 1999;32:350–359. [Google Scholar]
- 26.Wyllie GR, Scheidt WR. Chem Rev. 2002;102:1067–1090. doi: 10.1021/cr000080p. [DOI] [PubMed] [Google Scholar]
- 27.Hoshino M, Ozawa K, Seki H, Ford PC. J Am Chem Soc. 1993;115:9568–9575. [Google Scholar]
- 28.Abu-Soud HM, Ichimori K, Presta A, Stuehr DJ. J Biol Chem. 2000;275:17349–17357. doi: 10.1074/jbc.M000050200. [DOI] [PubMed] [Google Scholar]
- 29.Laverman LE, Wanat A, Oszajca J, Stochel G, Ford PC, van Eldik R. J Am Chem Soc. 2001;123:285–293. doi: 10.1021/ja001696z. [DOI] [PubMed] [Google Scholar]
- 30.Wang J, Lu S, Moënne-Loccoz P, Ortiz de Montellano PR. J Biol Chem. 2003;278:2341–2347. doi: 10.1074/jbc.M211131200. [DOI] [PubMed] [Google Scholar]
- 31.Yoshimura T, Ozaki T. Arch Biochem Biophys. 1984;229:126–135. doi: 10.1016/0003-9861(84)90137-1. [DOI] [PubMed] [Google Scholar]
- 32.Yoshimura T, Suzuki S, Nakahara A, Iwasaki H, Masuko M, Matsubara T. Biochemistry. 1986;25:2436–2442. [Google Scholar]
- 33.Coletta M, Boffi A, Ascenzi P, Brunori M, Chiancone E. J Biol Chem. 1990;265:4828–4830. [PubMed] [Google Scholar]
- 34.Stone JR, Marletta MA. Biochemistry. 1994;33:5636–5640. doi: 10.1021/bi00184a036. [DOI] [PubMed] [Google Scholar]
- 35.Decatur SM, Franzen S, DePillis GD, Dyer RB, Woodruff WH, Boxer SG. Biochemistry. 1996;35:4939–4944. doi: 10.1021/bi951661p. [DOI] [PubMed] [Google Scholar]
- 36.Reynolds MF, Parks RB, Burstyn JN, Shelver D, Thorsteinsson MV, Kerby RL, Roberts GP, Vogel KM, Spiro TG. Biochemistry. 2000;39:388–396. doi: 10.1021/bi991378g. [DOI] [PubMed] [Google Scholar]
- 37.Palmer G. In: The porphyrins. ch. 6 Dolphin D, editor. IV. Academic Press; New York: 1979. [Google Scholar]
- 38.Hille R, Olson JS, Palmer G. J Biol Chem. 1979;254:12110–12120. [PubMed] [Google Scholar]
- 39.Pilet E, Nitschke W, Rappaport F, Soulimane T, Lambry JC, Liebl U, Vos MH. Biochemistry. 2004;43:14118–14127. doi: 10.1021/bi0488808. [DOI] [PubMed] [Google Scholar]
- 40.Tsubaki M, Yu NT. Biochemistry. 1982;21:1140–1144. doi: 10.1021/bi00535a005. [DOI] [PubMed] [Google Scholar]
- 41.Deinum G, Stone JR, Babcock GT, Marletta MA. Biochemistry. 1996;35:1540–1547. doi: 10.1021/bi952440m. [DOI] [PubMed] [Google Scholar]
- 42.Andrew CR, George SJ, Lawson DM, Eady RR. Biochemistry. 2002;41:2353–2360. doi: 10.1021/bi011419k. [DOI] [PubMed] [Google Scholar]
- 43.Vogel KM, Kozlowski PM, Zgierski MZ, Spiro TG. J Am Chem Soc. 1999;121:9915–9921. [Google Scholar]
- 44.Kerr EA, Mackin HC, Yu NT. Biochemistry. 1983;22:4373–4379. doi: 10.1021/bi00288a005. [DOI] [PubMed] [Google Scholar]
- 45.Li XY, Spiro TG. J Am Chem Soc. 1988;110:6024–6033. doi: 10.1021/ja00226a017. [DOI] [PubMed] [Google Scholar]
- 46.Ray GB, Li X-Y, Ibers JA, Sessler JL, Spiro TG. J Am Chem Soc. 1994;116:162–176. [Google Scholar]
- 47.Park ES, Thomas MR, Boxer SG. J Am Chem Soc. 2000;122:12297–12303. [Google Scholar]
- 48.Coyle CM, Vogel KM, Rush TS, 3rd, Kozlowski PM, Williams R, Spiro TG, Dou Y, Ikeda-Saito M, Olson JS, Zgierski MZ. Biochemistry. 2003;42:4896–4903. doi: 10.1021/bi026395b. [DOI] [PubMed] [Google Scholar]
- 49.Hu S, Kincaid JR. J Am Chem Soc. 1991;113:9760–9766. [Google Scholar]
- 50.Ding XD, Weichsel A, Andersen JF, Shokhireva TK, Balfour C, Pierik AJ, Averill BA, Montfort WR, Walker FA. J Am Chem Soc. 1999;121:128–138. [Google Scholar]
- 51.Benko B, Yu NT. Proc Natl Acad Sci U S A. 1983;80:7042–7046. doi: 10.1073/pnas.80.22.7042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hu S, Kincaid JR. J Biol Chem. 1993;268:6189–6193. [PubMed] [Google Scholar]
- 53.Obayashi E, Tsukamoto K, Adachi S, Takahashi S, Nomura M, Iizuka T, Shoun H, Shiro Y. J Am Chem Soc. 1997;119:7807–7816. [Google Scholar]
- 54.Tomita T, Haruta N, Aki M, Kitagawa T, Ikeda-Saito M. J Am Chem Soc. 2001;123:2666–2667. doi: 10.1021/ja001431k. [DOI] [PubMed] [Google Scholar]
- 55.Linder DP, Rodgers KR, Banister J, Wyllie GR, Ellison MK, Scheidt WR. J Am Chem Soc. 2004;126:14136–14148. doi: 10.1021/ja046942b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Linder DP, Rodgers KR. Inorg Chem. 2005;44:1367–1380. doi: 10.1021/ic049045h. [DOI] [PubMed] [Google Scholar]
- 57.Lu Suharti S, de Vries S, Moënne-Loccoz P. J Am Chem Soc. 2004;126:15332–15333. doi: 10.1021/ja045233v. [DOI] [PubMed] [Google Scholar]
- 58.Kumita H, Matsuura K, Hino T, Takahashi S, Hori H, Fukumori Y, Morishima I, Shiro Y. J Biol Chem. 2004;279:55247–55254. doi: 10.1074/jbc.M409996200. [DOI] [PubMed] [Google Scholar]
- 59.Ye RW, Averill BA, Tiedje JM. Appl Environ Microbiol. 1994;60:1053–1058. doi: 10.1128/aem.60.4.1053-1058.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bryar TR, Eaton DR. Can J Chem. 1992;70:1917–1926. [Google Scholar]
- 61.Wang X, Sundberg EB, Li L, Kantardjieff KA, Herron SR, Lim M, Ford PC. Chem Commun. 2005:477–479. doi: 10.1039/b412086h. [DOI] [PubMed] [Google Scholar]
- 62.Butler CS, Seward HE, Greenwood C, Thomson AJ. Biochemistry. 1997;36:16259–16266. doi: 10.1021/bi971481a. [DOI] [PubMed] [Google Scholar]
- 63.Watmough NJ, Cheesman MR, Butler CS, Little RH, Greenwood C, Thomson AJ. J Bioenerg Biomembr. 1998;30:55–62. doi: 10.1023/a:1020507511285. [DOI] [PubMed] [Google Scholar]
- 64.Grönberg KL, Roldán MD, Prior L, Butland G, Cheesman MR, Richardson DJ, Spiro S, Thomson AJ, Watmough NJ. Biochemistry. 1999;38:13780–13786. doi: 10.1021/bi9916426. [DOI] [PubMed] [Google Scholar]
- 65.Grönberg KL, Watmough NJ, Thomson AJ, Richardson DJ, Field SJ. J Biol Chem. 2004;279:17120–17125. doi: 10.1074/jbc.M400824200. [DOI] [PubMed] [Google Scholar]
- 66.Moore EG, Gibson QH. J Biol Chem. 1976;251:2788–2794. [PubMed] [Google Scholar]
- 67.Traylor TG, Sharma VS. Biochemistry. 1992;31:2847–2849. doi: 10.1021/bi00126a001. [DOI] [PubMed] [Google Scholar]
- 68.Brucker EA, Olson JS, Ikeda-Saito M, Phillips GN., Jr Proteins: Struct, Funct, Genet. 1998;30:352–356. [PubMed] [Google Scholar]
- 69.Zhao Y, Brandish PE, Ballou DP, Marletta MA. Proc Natl Acad Sci U S A. 1999;96:14753–14758. doi: 10.1073/pnas.96.26.14753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sharma VS, Magde D. Methods. 1999;19:494–505. doi: 10.1006/meth.1999.0892. [DOI] [PubMed] [Google Scholar]
- 71.MacNeil JH, Berseth PA, Bruner EL, Perkins TL, Wadia Y, Westwood G, Trogler WC. J Am Chem Soc. 1997;119:1668–1675. [Google Scholar]
- 72.Pinakoulaki E, Stavrakis S, Urbani A, Varotsis C. J Am Chem Soc. 2002;124:9378–9379. doi: 10.1021/ja0271633. [DOI] [PubMed] [Google Scholar]
- 73.Blomberg ML, Blomberg MRA, Siegbahn PEM. Biochem Biophys Acta. 2006;1757:31–46. [Google Scholar]
- 74.Blomberg LM, Blomberg MR, Siegbahn PE. Biochim Biophys Acta. 2006;1757:240–252. doi: 10.1016/j.bbabio.2006.04.008. [DOI] [PubMed] [Google Scholar]
- 75.Moënne-Loccoz P, Richter O-MH, Huang HW, Wasser IM, Ghiladi RA, Karlin KD, de Vries S. J Am Chem Soc. 2000;122:9344–9345. [Google Scholar]
- 76.Sanders-Loehr J, Wheeler WD, Shiemke AK, Averill BA, Loehr TM. J Am Chem Soc. 1989;111:8084–8093. [Google Scholar]
- 77.Martens CF, Murthy NN, Obias HV, Karlin KD. Chem Commun. 1996:629–630. [Google Scholar]
- 78.Field SJ, Prior L, Roldán MD, Cheesman MR, Thomson AJ, Spiro S, Butt JN, Watmough NJ, Richardson DJ. J Biol Chem. 2002;277:20146–20150. doi: 10.1074/jbc.M112202200. [DOI] [PubMed] [Google Scholar]
- 79.Iwata S, Ostermeier C, Ludwig B, Michel H. Nature. 1995;376:660–669. doi: 10.1038/376660a0. [DOI] [PubMed] [Google Scholar]
- 80.Tsukihara T, Aoyama H, Yamashita E, Tomizaki T, Yamaguchi H, Shinzawa-Itoh K, Nakashima R, Yaono R, Yoshikawa S. Science. 1995;269:1069–1074. doi: 10.1126/science.7652554. [DOI] [PubMed] [Google Scholar]
- 81.Tsukihara T, Aoyama H, Yamashita E, Tomizaki T, Yamaguchi H, Shinzawa I-K, Nakashima R, Yaono R, Yoshikawa S. Science. 1996;272:1136–1144. doi: 10.1126/science.272.5265.1136. [DOI] [PubMed] [Google Scholar]
- 82.Ostermeier C, Harrenga A, Ermler U, Michel H. Proc Natl Acad Sci U S A. 1997;94:10547–10553. doi: 10.1073/pnas.94.20.10547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Soulimane T, Buse G, Bourenkov GP, Bartunik HD, Huber R, Than ME. EMBO J. 2000;19:1766–1776. doi: 10.1093/emboj/19.8.1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Feig AL, Lippard SJ. Chem Rev. 1994;94:759–805. [Google Scholar]
- 85.Solomon EI, Tuczek F, Root DE, Brown CA. Chem Rev. 1994;94:827–856. [Google Scholar]
- 86.Wallar BJ, Lipscomb JD. Chem Rev. 1996;96:2625–2658. doi: 10.1021/cr9500489. [DOI] [PubMed] [Google Scholar]
- 87.Silaghi-Dumitrescu R, Kurtz DM, Jr, Ljungdahl LG, Lanzilotta WN. Biochemistry. 2005;44:6492–6501. doi: 10.1021/bi0473049. [DOI] [PubMed] [Google Scholar]
- 88.Haskin CJ, Ravi N, Lynch JB, Münck E, Que L., Jr Biochemistry. 1995;34:11090–11098. doi: 10.1021/bi00035a014. [DOI] [PubMed] [Google Scholar]
- 89.Coufal DE, Tavares P, Pereira AS, Hyunh BH, Lippard SJ. Biochemistry. 1999;38:4504–4513. doi: 10.1021/bi9823378. [DOI] [PubMed] [Google Scholar]
- 90.Silaghi-Dumitrescu R, Coulter ED, Das A, Ljungdahl LG, Jameson GN, Huynh BH, Kurtz DM., Jr Biochemistry. 2003;42:2806–2815. doi: 10.1021/bi027253k. [DOI] [PubMed] [Google Scholar]
- 91.Wyllie GR, Schulz CE, Scheidt WR. Inorg Chem. 2003;42:5722–5734. doi: 10.1021/ic034473t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chan NL, Kavanaugh JS, Rogers PH, Arnone A. Biochemistry. 2004;43:118–132. doi: 10.1021/bi030172j. [DOI] [PubMed] [Google Scholar]
- 93.Nakamoto K. Infrared and Raman spectroscopy of inorganic and coordination compounds. 5. John Wiley and Sons Inc.; New York: 1997. [Google Scholar]
- 94.Schlichting I, Berendzen J, Phillips GN, Jr, Sweet RM. Nature. 1994;371:808–812. doi: 10.1038/371808a0. [DOI] [PubMed] [Google Scholar]
- 95.Teng TY, Srajer V, Moffat K. Nat Struct Biol. 1994;1:701–705. doi: 10.1038/nsb1094-701. [DOI] [PubMed] [Google Scholar]
- 96.Hartmann H, Zinser S, Komninos P, Schneider RT, Nienhaus GU, Parak F. Proc Natl Acad Sci U S A. 1996;93:7013–7016. doi: 10.1073/pnas.93.14.7013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Alben JO, Beece D, Bowne SF, Doster W, Eisenstein L, Frauenfelder H, Good D, McDonald JD, Marden MC, Moh PP, Reinisch L, Reynolds AH, Shyamsunder E, Yue KT. Proc Natl Acad Sci U S A. 1982;79:3744–3748. doi: 10.1073/pnas.79.12.3744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Fiamingo FG, Alben JO. Biochemistry. 1985;24:7964–7970. doi: 10.1021/bi00348a019. [DOI] [PubMed] [Google Scholar]
- 99.Ansari A, Berendzen J, Braunstein D, Cowen BR, Frauenfelder H, Hong MK, Iben IE, Johnson JB, Ormos P, Sauke TB, Scholl R, Schulte A, Steinbach PJ, Vittitow J, Young RD. Biophys Chem. 1987;26:337–355. doi: 10.1016/0301-4622(87)80034-0. [DOI] [PubMed] [Google Scholar]
- 100.Alben JO, Moh PP, Fiamingo FG, Altschuld RA. Proc Natl Acad Sci U S A. 1981;78:234–237. doi: 10.1073/pnas.78.1.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Woodruff WH, Dallinger RF, Antalis TM, Palmer G. Biochemistry. 1981;20:1332–1338. doi: 10.1021/bi00508a045. [DOI] [PubMed] [Google Scholar]
- 102.Einarsdóttir Ó, Killough PM, Fee JA, Woodruff WH. J Biol Chem. 1989;264:2405–2408. [PubMed] [Google Scholar]
- 103.Dyer RB, Einarsdóttir Ó, Killough PK, López-Garriga JJ, Woodruff WH. J Am Chem Soc. 1989;111:7657–7659. [Google Scholar]
- 104.Hendriks J, Warne A, Gohlke U, Haltia T, Ludovici C, Lübben M, Saraste M. Biochemistry. 1998;37:13102–13109. doi: 10.1021/bi980943x. [DOI] [PubMed] [Google Scholar]
- 105.Lu S. Ph. D. thesis, Department of Environmental & Biomolecular Systems, OGI School of Science&Engineering, Oregon Health&Science University. 2005. Characterization of nitrosyl, azido, and carbonyl complexes in diiron enzymes and their implications for O2 and NO activation. [Google Scholar]
- 106.Hendriks JH, Prior L, Baker AR, Thomson AJ, Saraste M, Watmough NJ. Biochemistry. 2001;40:13361–13369. doi: 10.1021/bi011428t. [DOI] [PubMed] [Google Scholar]
- 107.Koutsoupakis K, Stavrakis S, Soulimane T, Varotsis C. J Biol Chem. 2003;278:14893–14896. doi: 10.1074/jbc.M210293200. [DOI] [PubMed] [Google Scholar]
- 108.Pinakoulaki E, Varotsis C. Biochemistry. 2003;42:14856–14861. doi: 10.1021/bi035289m. [DOI] [PubMed] [Google Scholar]
- 109.Wasser IM, Huang HW, Moënne-Loccoz P, Karlin KD. J Am Chem Soc. 2005;127:3310–3320. doi: 10.1021/ja0458773. [DOI] [PubMed] [Google Scholar]
- 110.Suharti . Ph. D. thesis, Department of Biotechnology, Delft University of Technology. 2005. qCuANOR a novel bifunctional nitric oxide reductase from Bacillus azotoformans. [Google Scholar]
- 111.Rosenzweig AC, Frederick CA, Lippard SJ, Nordlund P. Nature. 1993;366:537–543. doi: 10.1038/366537a0. [DOI] [PubMed] [Google Scholar]
- 112.Rosenzweig AC, Nordlund P, Takahara PM, Frederick CA, Lippard SJ. Chem Biol. 1995;2:409–418. [PubMed] [Google Scholar]
- 113.Whittington DA, Sazinsky MH, Lippard SJ. J Am Chem Soc. 2001;123:1794–1795. doi: 10.1021/ja0031725. [DOI] [PubMed] [Google Scholar]
- 114.Nordlund P, Sjöberg B-M, Eklund H. Nature. 1990;345:593–598. doi: 10.1038/345593a0. [DOI] [PubMed] [Google Scholar]
- 115.Nordlund P, Eklund H. J Mol Biol. 1993;232:123–164. doi: 10.1006/jmbi.1993.1374. [DOI] [PubMed] [Google Scholar]
- 116.Logan DT, Su XD, Aberg A, Regnstrom K, Hajdu J, Eklund H, Nordlund P. Structure. 1996;4:1053–1064. doi: 10.1016/s0969-2126(96)00112-8. [DOI] [PubMed] [Google Scholar]
- 117.Pulver S, Froland WA, Fox BG, Lipscomb JD, Solomon EI. J Am Chem Soc. 1993;115:12409–12422. [Google Scholar]
- 118.Pulver SC, Froland WA, Lipscomb JD, Solomon EI. J Am Chem Soc. 1997;119:387–395. [Google Scholar]
- 119.Fox BG, Lyle KS, Rogge CE. Acc Chem Res. 2004;37:421–429. doi: 10.1021/ar030186h. [DOI] [PubMed] [Google Scholar]
- 120.Rardin RL, Tolman WB, Lippard SJ. New J Chem. 1991;15:417–430. [Google Scholar]
- 121.Sakurai T, Sakurai N, Matsumoto H, Hirota S, Yamauchi O. Biochem Biophys Res Commun. 1998;251:248–251. doi: 10.1006/bbrc.1998.9451. [DOI] [PubMed] [Google Scholar]
- 122.Lu S, Libby E, Saleh L, Xing G, Bollinger JM, Jr, Moënne-Loccoz P. J Biol Inorg Chem. 2004;9:818–827. doi: 10.1007/s00775-004-0582-8. [DOI] [PubMed] [Google Scholar]
- 123.Koutsoupakis K, Stavrakis S, Pinakoulaki E, Soulimane T, Varotsis C. J Biol Chem. 2002;277:32860–32866. doi: 10.1074/jbc.M204943200. [DOI] [PubMed] [Google Scholar]
- 124.Pinakoulaki E, Gemeinhardt S, Saraste M, Varotsis C. J Biol Chem. 2002;277:23407–23413. doi: 10.1074/jbc.M201913200. [DOI] [PubMed] [Google Scholar]
- 125.Sakurai T, Nakashima S, Kataoka K, Seo D, Sakurai N. Biochem Biophys Res Commun. 2005;333:483–487. doi: 10.1016/j.bbrc.2005.05.149. [DOI] [PubMed] [Google Scholar]
- 126.Wang YN, Averill BA. J Am Chem Soc. 1996;118:3972–3973. [Google Scholar]
- 127.Miller LM, Pedraza AJ, Chance MR. Biochemistry. 1997;36:12199–12207. doi: 10.1021/bi962744o. [DOI] [PubMed] [Google Scholar]
- 128.Mukai M, Ouellet Y, Ouellet H, Guertin M, Yeh SR. Biochemistry. 2004;43:2764–2770. doi: 10.1021/bi035798o. [DOI] [PubMed] [Google Scholar]
- 129.Torres J, Cooper CE, Wilson MT. J Biol Chem. 1998;273:8756–8766. doi: 10.1074/jbc.273.15.8756. [DOI] [PubMed] [Google Scholar]
- 130.Cooper CE. Trends Biochem Sci. 2002;27:33–39. doi: 10.1016/s0968-0004(01)02035-7. [DOI] [PubMed] [Google Scholar]
- 131.Brunori M, Giuffrè A, Forte E, Mastronicola D, Barone MC, Sarti P. Biochim Biophys Acta. 2004;1655:365–371. doi: 10.1016/j.bbabio.2003.06.008. [DOI] [PubMed] [Google Scholar]
- 132.Brunori M, Forte E, Arese M, Mastronicola D, Giuffrè A, Sarti P. Biochim Biophys Acta. 2006;1757:1144–1154. doi: 10.1016/j.bbabio.2006.05.011. [DOI] [PubMed] [Google Scholar]
- 133.Stevens TH, Brudvig GW, Bocian DF, Chan SI. Proc Natl Acad Sci U S A. 1979;76:3320–3324. doi: 10.1073/pnas.76.7.3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Brudvig GW, Stevens TH, Chan SI. Biochemistry. 1980;19:5275–5285. doi: 10.1021/bi00564a020. [DOI] [PubMed] [Google Scholar]
- 135.Torres J, Cooper CE, Sharpe M, Wilson MT. J Bioenerg Biomembr. 1998;30:63–69. doi: 10.1023/a:1020559528124. [DOI] [PubMed] [Google Scholar]
- 136.Torres J, Sharpe MA, Rosquist A, Cooper CE, Wilson MT. FEBS Lett. 2000;475:263–266. doi: 10.1016/s0014-5793(00)01682-3. [DOI] [PubMed] [Google Scholar]
- 137.Butler C, Forte E, Maria Scandurra F, Arese M, Giuffrè A, Greenwood C, Sarti P. Biochem Biophys Res Commun. 2002;296:1272–1278. doi: 10.1016/s0006-291x(02)02074-0. [DOI] [PubMed] [Google Scholar]
- 138.Brunori M, Giuffrè A, Sarti P, Stubauer G, Wilson MT. Cell Mol Life Sci. 1999;56:549–557. doi: 10.1007/s000180050452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Zhao XJ, Sampath V, Caughey WS. Biochem Biophys Res Commun. 1994;204:537–543. doi: 10.1006/bbrc.1994.2492. [DOI] [PubMed] [Google Scholar]
- 140.Forte E, Urbani A, Saraste M, Sarti P, Brunori M, Giuffrè A. Eur J Biochem. 2001;268:6486–6491. doi: 10.1046/j.0014-2956.2001.02597.x. [DOI] [PubMed] [Google Scholar]
- 141.Brown GC. Biochim Biophys Acta. 2001;1504:46–57. doi: 10.1016/s0005-2728(00)00238-3. [DOI] [PubMed] [Google Scholar]
- 142.Pearce LL, Bominaar EL, Hill BC, Peterson J. J Biol Chem. 2003;278:52139–52145. doi: 10.1074/jbc.M310359200. [DOI] [PubMed] [Google Scholar]
- 143.Pinakoulaki E, Ohta T, Soulimane T, Kitagawa T, Varotsis C. J Am Chem Soc. 2005;127:15161–15167. doi: 10.1021/ja0539490. [DOI] [PubMed] [Google Scholar]
- 144.Ohta T, Kitagawa T, Varotsis C. Inorg Chem. 2006;45:3187–3190. doi: 10.1021/ic050991n. [DOI] [PubMed] [Google Scholar]
- 145.Bau R, Sabherwal IH, Burg AB. J Am Chem Soc. 1971;93:4926–4928. [Google Scholar]
- 146.Arulsamy N, Bohle DS, Imonigie JA, Levine S. Angew Chem, Int Ed. 2002;41:2371–2373. doi: 10.1002/1521-3773(20020703)41:13<2371::AID-ANIE2371>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 147.Bottcher HC, Graf M, Mereiter K, Kirchner K. Organometallics. 2004;23:1269–1273. [Google Scholar]
- 148.Bottcher HC, Wagner C, Kirchner K. Inorg Chem. 2004;43:6294–6299. doi: 10.1021/ic0496424. [DOI] [PubMed] [Google Scholar]
- 149.Moody AJ. Biochim Biophys Acta. 1996;1276:6–20. doi: 10.1016/0005-2728(96)00035-7. [DOI] [PubMed] [Google Scholar]
- 150.Scott RA, Li PM, Chan SI. Ann N Y Acad Sci. 1988;550:53–58. doi: 10.1111/j.1749-6632.1988.tb35322.x. [DOI] [PubMed] [Google Scholar]
- 151.Li PM, Gelles J, Chan SI, Sullivan RJ, Scott RA. Biochemistry. 1987;26:2091–2095. doi: 10.1021/bi00382a005. [DOI] [PubMed] [Google Scholar]
- 152.Ralle M, Verkhovskaya ML, Morgan JE, Verkhovsky MI, Wikström M, Blackburn NJ. Biochemistry. 1999;38:7185–7194. doi: 10.1021/bi982885l. [DOI] [PubMed] [Google Scholar]
- 153.Fabian M, Skultety L, Brunel C, Palmer G. Biochemistry. 2001;40:6061–6069. doi: 10.1021/bi010059y. [DOI] [PubMed] [Google Scholar]
- 154.Li W, Palmer G. Biochemistry. 1993;32:1833–1843. doi: 10.1021/bi00058a018. [DOI] [PubMed] [Google Scholar]
- 155.Orii Y, Mogi T, Sato-Watanabe M, Hirano T, Anraku Y. Biochemistry. 1995;34:1127–1132. doi: 10.1021/bi00004a004. [DOI] [PubMed] [Google Scholar]
- 156.Gardner AM, Helmick RA, Gardner PR. J Biol Chem. 2002;277:8172–8177. doi: 10.1074/jbc.M110471200. [DOI] [PubMed] [Google Scholar]
- 157.Gomes CM, Giuffrè A, Forte E, Vicente JB, Saraiva LM, Brunori M, Teixeira M. J Biol Chem. 2002;277:25273–25276. doi: 10.1074/jbc.M203886200. [DOI] [PubMed] [Google Scholar]
- 158.Iyer RB, Silaghi-Dumitrescu R, Kurtz DM, Jr, Lanzilotta WN. J Biol Inorg Chem. 2005;10:407–416. doi: 10.1007/s00775-005-0650-8. [DOI] [PubMed] [Google Scholar]